 图 1
(a) Ag3PO4, (b) BiPO4/Ag3PO4的SEM照片
Figure 1.
SEM images of (a) Ag3PO4 and (b) BiPO4/Ag3PO4
图 1
(a) Ag3PO4, (b) BiPO4/Ag3PO4的SEM照片
Figure 1.
SEM images of (a) Ag3PO4 and (b) BiPO4/Ag3PO4
引用本文:
王昭华, 蔡琼瑶, 叶挺铭, 郭英娜, 耿直, 杨霞, 于洪斌. p-n异质结BiPO4/Ag3PO4的制备及其增强的模拟太阳光活性[J]. 无机化学学报,
2017, 33(7): 1196-1204.
doi:
10.11862/CJIC.2017.140

Citation: WANG Zhao-Hua, CAI Qiong-Yao, YE Ting-Ming, GUO Ying-Na, GENG Zhi, YANG Xia, YU Hong-Bin. BiPO4/Ag3PO4 p-n Heterojunction with Enhanced Photocatalytic Activity under Simulated Sunlight Irradiation[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(7): 1196-1204. doi: 10.11862/CJIC.2017.140
Citation: WANG Zhao-Hua, CAI Qiong-Yao, YE Ting-Ming, GUO Ying-Na, GENG Zhi, YANG Xia, YU Hong-Bin. BiPO4/Ag3PO4 p-n Heterojunction with Enhanced Photocatalytic Activity under Simulated Sunlight Irradiation[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(7): 1196-1204. doi: 10.11862/CJIC.2017.140
p-n异质结BiPO4/Ag3PO4的制备及其增强的模拟太阳光活性
摘要:
采用一步水热合成法制备了BiPO4、Ag3PO4和BiPO4/Ag3PO4复合光催化剂,通过X射线粉末衍射(XRD)、X射线光电子能谱(XPS)、扫描电子显微镜(SEM)、紫外-可见漫反射(UV-Vis DRS)等表征手段对其组成结构、形貌及光吸收性质进行了表征,结果表明Ag3PO4呈块状结构,BiPO4则分布在其表面,形成的BiPO4/Ag3PO4复合光催化剂具有单斜相和立方晶相结构,带边吸收拓宽至571 nm。以甲基橙和加替沙星为目标污染物,考察了BiPO4/Ag3PO4复合光催化剂在模拟太阳光照射下的降解矿化能力,结果表明复合催化剂比单一催化剂的降解矿化能力更强,稳定性更好。此外,自由基捕获实验表明空穴是该光催化过程中的主要活性物种,·O2-次之。p-n异质结的形成使BiPO4/Ag3PO4复合光催化剂具有较强的电子空穴分离能力是光催化活性提高的主要原因,这与光电流和电化学阻抗谱测试结果相一致。基于以上结果,文中对BiPO4/Ag3PO4光催化降解有机污染物的机理进行了推测。
-
关键词:
- BiPO4/Ag3PO4
- / 光催化
- / 模拟太阳光
- / 甲基橙
- / 加替沙星
English
BiPO4/Ag3PO4 p-n Heterojunction with Enhanced Photocatalytic Activity under Simulated Sunlight Irradiation
Abstract:
BiPO4, Ag3PO4 and BiPO4/Ag3PO4 heterostructure photocatalysts were synthesized by one-step hydro-thermal method.X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), scanning electron microscope (SEM) and UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS) techniques were used to characterize morphology, crystal structure, and absorption spectrum.The SEM results showed that the BiPO4/Ag3PO4 sample was in blocky structure and BiPO4 distributed on the surface of Ag3PO4.The XRD of BiPO4/Ag3PO4 sample displayed monoclinic and cubic crystal structure.The UV-Vis DRS spectra results indicated that the BiPO4/Ag3PO4 sample displayed obviously red shifts compared to BiPO4 and the light absorption range broaden to 571 nm.Moreover, the photocatalytic activity was evaluated by photodegradation of methyl orange (MO) and gatifloxacin solution under simulated sunlight irradiation.BiPO4/Ag3PO4 exhibits higher degradation and mineralized activities and the better stability on degradation of methyl orange and gatifloxacin than the single BiPO4 and Ag3PO4.Moreover, the test of active radical showed that the h+ was the main active radical in the degradation process, ·O2- was weaker.The enhanced photocatalytic activity and stability of BiPO4/Ag3PO4 was mainly attributes to the formation of p-n heterojunction interface in BiPO4/Ag3PO4 which facilitated the transfer and separation of photogenerated electron-hole pairs confirmed by the results of transient photocurrent response and electrochemical impedance spectroscopy measurement.Based on the above results, the photocatalytic mechanism for organics degradation over BiPO4/Ag3PO4 was proposed.
-
Key words:
- BiPO4/Ag3PO4
- / photocatalytic
- / simulated sunlight
- / methyl orange
- / gatifloxacin
-
0 引言
光催化技术作为一种新兴、节能、高效的污染治理技术,被认为是目前最有应用前景的环境保护和能源转换技术。传统的TiO2带隙较宽(3.2 eV),只能利用太阳光中4%的紫外光部分。随着环境污染和能源危机越来越严重,清洁能源越来越为大家所关注。因此,开发高效的太阳光响应型催化剂成为光催化领域研究热点之一。目前研究较多的具有太阳光响应的光催化剂有改性TiO2[1]、BiVO4[2]、WO3[3]、Ag3PO4[4]等。根据Ye等的研究发现,Ag3PO4半导体在可见光下能高效地催化降解有机污染物,其活性远高于BiVO4及商品化的WO3[5]。Ag3PO4禁带宽度较窄,带隙能为2.36~2.45 eV,能吸收波长小于530 nm的太阳光,在波长小于480 nm时的量子产率极高,光生电子和空穴分离效率较高,有着优异的催化活性[6-7]。但是Ag3PO4在水中的溶解度相对较大,在光催化的过程中会发生光腐蚀现象,限制了Ag3PO4的进一步推广应用。构建异质结构,进行半导体复合被认为是有效抑制Ag3PO4光腐蚀,提高光催化活性的方法之一,研究者们进行了系列研究,如设计具有核壳结构的Ag3PO4@g-C3N4复合材料,其可见光下照射亚甲基蓝溶液30 min降解效率高达97%,而Ag3PO4仅降解了69%,复合材料在五次循环后仍能保持81%的降解活性[8],还有Ag3PO4/MoS2复合光催化剂构建Z-scheme体系[9]和将Ag3PO4量子点负载到InVO4/BiVO4晶体表面[10]等。BiPO4在紫外线照射下,对染料的降解效果比商业化P25(TiO2)要更加优越[11]。然而,BiPO4带隙较宽(3.85~3.93 eV)[12-13],对可见光的利用率很低。目前,拓宽BiPO4的可见光响应范围的途径主要是掺杂窄带隙的半导体形成复合材料,已报道的有AgBr/BiPO4[12]、BiOI/BiPO4[14]等,对其光催化降解活性都有显著提高。
本研究采用水热法实现窄带隙半导体Ag3PO4对BiPO4负载,从而降低了BiPO4的禁带宽度,Ag3PO4与BiPO4形成的异质结构可有效分离光生电子与空穴,进一步提高模拟太阳光光催化降解效率及Ag3PO4的稳定性。采用典型染料甲基橙溶液和抗生素加替沙星为目标污染物进行光催化降解及矿化能力研究,文中对催化剂的激发原理,光催化降解活性及稳定性提高的原因进行了深入探讨。
1 实验方法
1.1 光催化剂的制备
称取0.239 4 g五水合硝酸铋(Bi(NO3)3·5H2O)和0.547 9 g硝酸银(AgNO3)溶解于2 mL 2 mol·L-1的HNO3,依次加入0.561 8 g十二水合磷酸氢二钠(Na2HPO4·12H2O)和20 mL去离子水,避光搅拌10 min后用2 mol·L-1 NaOH调节pH=7.0,继续搅拌30 min。然后将混合物转移到含聚四氟乙烯内衬的不锈钢水热釜中,以2 ℃·min-1的升温速率,升温至180 ℃,并于该温度下保持12 h。待反应釜自然冷却至室温后取出,离心,水洗固体物质3次,去除杂质,离心后的产物在80 ℃下恒温干燥8 h。黄色粉末即为BiPO4和Ag3PO4质量比为1:3的BiPO4/Ag3PO4复合光催化剂。BiPO4和Ag3PO4也按照同样的方法制备。实验中所用的试剂和药品均为分析纯,未经进一步纯化。
1.2 光催化剂的表征
采用日本理学公司生产的D/max 2000 X-射线粉末衍射仪用Cu Kα射线(λ=0.154 16 nm,U=40 kV,I=30 mA)从10°扫描至80°分析样品的晶相结构;利用德国里奥公司生产的1530VP扫描电子显微镜来获得光催化剂的SEM微观图像;使用美国瓦里安生产的Cary 500 UV-VIS-NIR紫外-可见-近红外光谱仪对光催化剂进行吸收光谱测试;采用英国Kratos公司生产的VG-ADES 400型X射线光电子能谱仪对光催化剂组成进行分析;采用上海辰华仪器公司生产的电化学工作站CHI660E进行了光电流测试,CHI650D测试了电化学阻抗谱,分析催化剂的电子传输效率和光生载流子的分离情况。
1.3 光催化降解实验
采用北京泊菲莱科技有限公司生产的PLS-SXE 300 W氙灯作为外置光源,平均光强度为5 mW·cm-2,光斑直径为63 mm,发射波长为模拟太阳光光谱(320~680 nm)进行光催化降解实验。自制石英反应器直径为80 mm,用于盛放反应液,其顶端与光源距离约15 cm。将所制备的催化剂样品(100 mg)和甲基橙溶液(20 μg·mL-1,100 mL)放置于反应器中,避光搅拌30 min以达到吸附-脱附平衡。开启模拟太阳光光源,进行辐照,整个催化过程中持续搅拌。反应液温度通过外部的冷却循环水保持在20 ℃。每隔一定时间,取出约3 mL反应液,经离心(8 000 r·min-1,10 min)后对上层清液进行紫外分光光度计检测,选取甲基橙最大吸收波长464 nm处测其吸光度。可见光光催化活性实验在模拟太阳光光源中加入420 nm滤光片后进行,其它步骤测试方法与前面一致。采用日本岛津公司的TOC-VCPN总有机碳分析仪测试不同时间反应液中的总有机碳含量。通过美国VARIAN公司生产的SpectrAA-220FS型火焰原子吸收分光光度计对催化剂循环使用4次后的降解溶液中的银离子含量进行了检测。
光催化降解加替沙星实验与降解甲基橙实验类似,只需将甲基橙替换为加替沙星(20 μg·mL-1,100 mL)。采用日本岛津高效液相色谱测定加替沙星溶液的浓度变化。具体检测条件为:LC-18色谱柱,流动相为乙腈与0.025 mol·L-1 Na2HPO4(用磷酸调节pH值至3.2) 以体积比25:75混合的溶液,柱温35 ℃,流速为0.8 mL·min-1,流动相使用前进行超声脱气。采用紫外检测器,检测波长为293 nm。
光催化降解效率η用公式(1) 计算,其中C0为目标污染物的初始浓度,Ct为目标降解物在t时刻的浓度。
2 结果与讨论
2.1 BiPO4/Ag3PO4复合型光催化剂的表征
图 1为Ag3PO4、BiPO4/Ag3PO4的SEM照片,可以观察到图 1(a)中Ag3PO4呈多面体块状结构,尺寸为1~3 μm。由图 1(b)可知,在块状Ag3PO4上,片状BiPO4分布在其表面。
图 1
(a) Ag3PO4, (b) BiPO4/Ag3PO4的SEM照片
Figure 1.
SEM images of (a) Ag3PO4 and (b) BiPO4/Ag3PO4
样品的晶相结构可通过X射线粉末衍射测试获得,在图 2中可以观察到BiPO4样品在14.56°,20.04°,29.40°,31.28°处的衍射峰,与标准卡片(JCPDS No.80-209) 对比可知,分别对应于单斜相独居石结构BiPO4的(101),(011),(120),和(012) 晶面[15]。相同条件下制备的Ag3PO4中位于20.88°,33.28°,36.56°处的衍射峰分别对应于立方相Ag3PO4的(110),(210) 和(211) 晶面(JCPDS No.06-0505)[15]。BiPO4/Ag3PO4复合催化剂的衍射峰更加尖锐,半峰宽更窄,表明结晶度较高,可分别在图中找到归属于BiPO4和Ag3PO4的特征衍射峰,其中29.05°处的衍射峰强度明显增强,并发生了少许位移,这表明该复合材料BiPO4与Ag3PO4之间不是简单的物理混合。
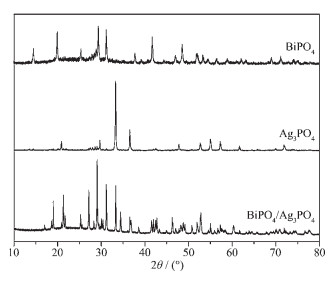 图 2
所制备样品的XRD图
Figure 2.
XRD patterns of as prepared samples
图 2
所制备样品的XRD图
Figure 2.
XRD patterns of as prepared samples
图 3对比研究了BiPO4、Ag3PO4、BiPO4/Ag3PO4复合光催化剂的光吸收特性。由图 3可知BiPO4的带边吸收在321 nm,只在紫外光区有吸收,Ag3PO4的带边吸收拓宽至555 nm,在整个可见光区都有吸收,BiPO4/Ag3PO4复合光催化剂在紫外光和可见光区域的吸收强度与Ag3PO4、BiPO4的相比都有较大程度的增强,带边吸收拓宽至571 nm。进一步根据公式(2) 计算所制备样品的带隙能,其中λg为波长,Eg为带隙能。根据各物质吸收波长,可以得出Ag3PO4、BiPO4、BiPO4/Ag3PO4的带隙能分别为2.23、3.86、2.17 eV。因此,BiPO4/Ag3PO4复合光催化剂更易被模拟太阳光所激发。
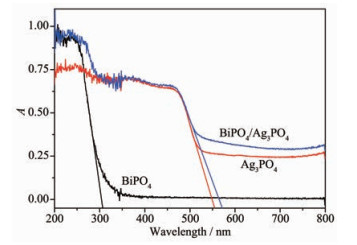 图 3
所制备样品的UV-Vis DRS谱图
Figure 3.
UV-Vis DRS spectra of as prepared samples
图 3
所制备样品的UV-Vis DRS谱图
Figure 3.
UV-Vis DRS spectra of as prepared samples
2.2 光催化降解及矿化研究
工业生产染料产生的废水中含有大量的染料,染料有着色度高,毒性强,有机物含量高等特点,对环境和人类健康有着潜在危害[16]。加替沙星是典型的第四代新型氟喹诺酮类抗生素,主要用于呼吸道、泌尿道等部位的急、慢性感染,也广泛应用于养殖业和食品加工业[17]。但是动物对抗生素的吸收利用能力较差,除了少部分会残留在体内之外,大部分会以原药和代谢产物排出体外,并进入到城市污水处理系统[18]。本文以染料甲基橙和抗生素加替沙星为目标分子,考察了BiPO4、Ag3PO4、BiPO4/Ag3PO4复合光催化剂在模拟太阳光照射下的光催化降解活性。
由图 4(a)可知,甲基橙溶液在不添加催化剂经模拟太阳光30 min照射时,甲基橙基本不发生光解。BiPO4为催化剂时降解活性较差,相同照射时间下,甲基橙仅降解了6.4%,因为BiPO4带隙能较宽为3.86 eV,难以被模拟太阳光所激发。纯Ag3PO4在模拟太阳光下表现出一定的活性,降解10 min时效率达到82.0%。BiPO4/Ag3PO4复合光催化剂在此时降解效率高达95.3%,模拟太阳光下光催化降解甲基橙活性的催化剂顺序依次为:BiPO4/Ag3PO4 > Ag3PO4 > BiPO4 > 直接光解。为评价BiPO4/Ag3PO4在可见光下的光催化降解能力,对模拟太阳光光源加入420 nm的滤光片(Visible light),照射10 min后,甲基橙的光催化降解效率为84.6%。图 4(b)对比了模拟太阳光和可见光下甲基橙溶液降解的动力学,符合准一级反应动力学方程ln(Ct /C0)=-kt,反应速率常数k分别为0.242 1和0.192 9 min-1,模拟太阳光照射下的反应速率常数为可见光下的1.26倍,表明模拟太阳光中的紫外部分对光催化活性有一定贡献,但是可见区的光谱响应是其活性的主要来源。该结果与紫外可见漫反射吸收光谱结果相一致。TOC是评价催化剂对有机污染物矿化能力的重要指标[19],因而进行了甲基橙的矿化研究。采用BiPO4/Ag3PO4作为光催化剂时,延长模拟太阳光照射时间至4 h,甲基橙的矿化率达到45.71%,而相同照射时间下Ag3PO4作为光催化剂时,甲基橙矿化率为37.77%,如图 4(c)所示。经过N-H键断裂,苯环裂解,脱N等一系列过程,甲基橙分子逐渐分解,TOC含量降低,最终将转化为CO2和H2O等小分子[20]。
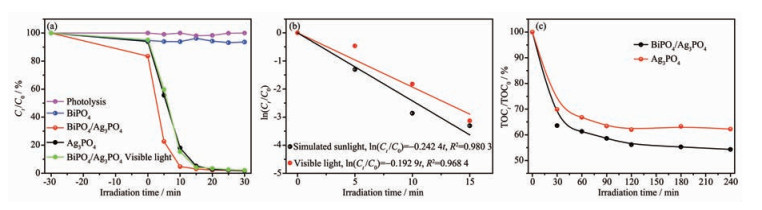 图 4
(a)甲基橙在模拟太阳光照射下的光催化降解曲线, (b)甲基橙在模拟太阳光和可见光下的降解动力学曲线及(c)甲基橙在模拟太阳光照射下TOC变化曲线
Figure 4.
(a) Degradation of methyl orange, (b) Kinetics of degradation of methyl orange under simulated sunlight and visible light irradiation, (c) TOC changes on the degradation of methyl orange under simulated sunlight
图 4
(a)甲基橙在模拟太阳光照射下的光催化降解曲线, (b)甲基橙在模拟太阳光和可见光下的降解动力学曲线及(c)甲基橙在模拟太阳光照射下TOC变化曲线
Figure 4.
(a) Degradation of methyl orange, (b) Kinetics of degradation of methyl orange under simulated sunlight and visible light irradiation, (c) TOC changes on the degradation of methyl orange under simulated sunlight
选用无色的典型抗生素加替沙星为目标污染物,不加光催化剂,对其进行直接光解实验,在模拟太阳光照射5 min时其浓度未发生变化,表明加替沙星在模拟太阳光下此时间内能够稳定的存在。由图 5(a)可知,Ag3PO4、BiPO4/Ag3PO4复合光催化剂催化降解加替沙星3 min时,两者的降解效率分别达到88.9%、95.7%,而此条件下BiPO4降解体系基本未表现出降解活性,该降解规律与甲基橙的降解规律一致。进一步对加替沙星的降解过程进行动力学拟合,由图 5(b)可知加替沙星模拟太阳光下的降解过程符合准一级反应动力学方程。BiPO4/Ag3PO4作为催化剂时反应速率常数k为0.915 0 min-1,约为Ag3PO4(k=0.665 2 min-1)的1.4倍、BiPO4(k=0.012 2 min-1)的75倍。
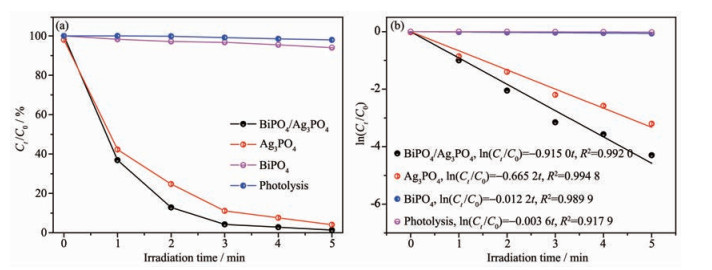 图 5
加替沙星在模拟太阳光下的(a)光催化降解曲线(b)降解动力学曲线
Figure 5.
(a) Degradation curve and (b) the kinetics of degradation of gatifloxacin under the simulated sunlight irradiation
图 5
加替沙星在模拟太阳光下的(a)光催化降解曲线(b)降解动力学曲线
Figure 5.
(a) Degradation curve and (b) the kinetics of degradation of gatifloxacin under the simulated sunlight irradiation
2.3 BiPO4/Ag3PO4稳定性分析
甲基橙降解的循环实验结果如图 6所示,经过4次循环后Ag3PO4作为催化剂时,降解甲基橙能力明显下降,30 min光照后的甲基橙降解效率由第1次的98.2%,降低为第4次的4.6%,此时溶液中银离子浓度为96.88 μg·mL-1。而经4次循环后BiPO4/Ag3PO4复合催化剂的光催化降解能力也有所下降,但降解效率仍能达到22.1%,银离子含量也明显降低,为59.92 μg·mL-1。溶液中的银离子含量越高表明催化剂光腐蚀越严重越不稳定,因而可知BiPO4对Ag3PO4负载有助于提高Ag3PO4的稳定性。这是由于BiPO4/Ag3PO4形成p-n异质结后,较有效地促进了电子的转移,减少了Ag3PO4中银离子与电子的相互作用,从而在一定程度上抑制了Ag3PO4的光腐蚀。
 图 6
Ag3PO4和BiPO4/Ag3PO4模拟太阳光照射下降解甲基橙的循环实验
Figure 6.
Cycling runs of Ag3PO4 and BiPO4/Ag3PO4 for the degradation of methyl orange under the simulated sunlight irradiation
图 6
Ag3PO4和BiPO4/Ag3PO4模拟太阳光照射下降解甲基橙的循环实验
Figure 6.
Cycling runs of Ag3PO4 and BiPO4/Ag3PO4 for the degradation of methyl orange under the simulated sunlight irradiation
进一步对循环前后催化剂中Ag3d的XPS谱图进行对比,如图 7(a)所示,BiPO4/Ag3PO4中Ag3d两个峰的结合能分别由循环前的367.88、373.83 eV变为循环后的367.84、373.79 eV,表明样品中有部分Ag3PO4被光腐蚀生成了单质银,这也是活性下降的主要原因。而图 7(b)中Ag3PO4的Ag3d结合能分别由367.76、373.77 eV变为367.67、373.68 eV。对比可知循环前后Ag3PO4中Ag3d的结合能位移比循环前后BiPO4/Ag3PO4中Ag3d的结合能的位移更加明显,表明Ag3PO4光腐蚀生成单质银更严重。通过计算,循环后BiPO4/Ag3PO4中Ag0含量占总银量的28.0%,而Ag3PO4作为催化剂的体系中有43.8%的Ag0存在,说明BiPO4/Ag3PO4在降解过程中催化剂表面形成的Ag0更少[21],与循环实验中BiPO4/Ag3PO4的光催化活性和稳定性比Ag3PO4的更好这一结果相一致。
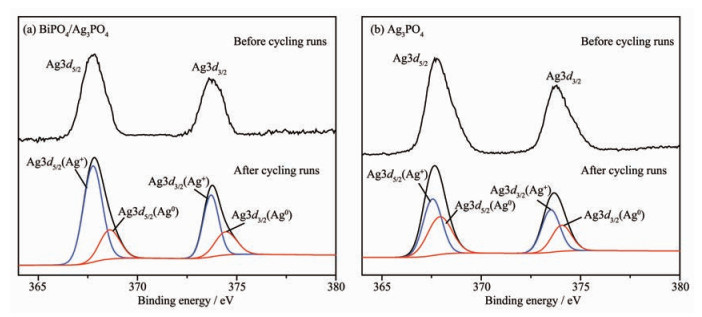 图 7
循环前后Ag3d XPS谱图(a)BiPO4/Ag3PO4和(b)Ag3PO4
Figure 7.
XPS spectra of Ag3d from (a) BiPO4/Ag3PO4 and (b) Ag3PO4 before and after cycling runs
图 7
循环前后Ag3d XPS谱图(a)BiPO4/Ag3PO4和(b)Ag3PO4
Figure 7.
XPS spectra of Ag3d from (a) BiPO4/Ag3PO4 and (b) Ag3PO4 before and after cycling runs
2.4 BiPO4/Ag3PO4光催化活性物种的检测
以BiPO4/Ag3PO4为催化剂,分别在降解甲基橙的体系中加入叔丁醇,EDTA-2Na和对苯醌作为羟基自由基、空穴和超氧自由基的捕获剂。如图 8所示,向光催化反应体系加入1 mmol·L-1叔丁醇(t-BuOH)来捕获羟基自由基(·OH),与未加入t-BuOH的反应体系相比,甲基橙的降解速率变化不大。说明BiPO4/Ag3PO4复合光催化剂在光催化降解过程中只产生了少量的·OH,·OH不是光催化反应的主要活性物种。当向光催化体系中加入空穴(h+)捕获剂1 mmol·L-1 EDTA-2Na时,光催化剂的催化活性明显受到抑制,甲基橙在模拟太阳光照射下降解效率仅为3.8%,基本上不降解;引入超氧自由基(·O2-)捕获剂1 mmol·L-1对苯醌(BQ)时,光催化剂的催化活性也有所下降,甲基橙的降解速率减小,降解效率为30.1%。综上所述,BiPO4/Ag3PO4光催化剂在光催化过程中产生的最主要的活性物种是空穴(h+),其次是超氧自由基(·O2-),即污染物在降解过程中直接通过空穴(h+)和超氧自由基(·O2-)进行氧化。
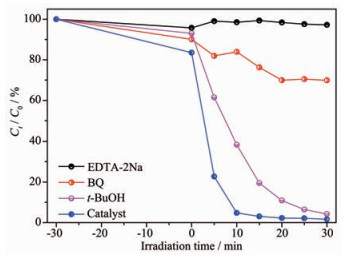 图 8
不同自由基捕获下BiPO4/Ag3PO4降解甲基橙曲线
Figure 8.
Methyl orange degradation curves of BiPO4/Ag3PO4 under different radical scavenger
图 8
不同自由基捕获下BiPO4/Ag3PO4降解甲基橙曲线
Figure 8.
Methyl orange degradation curves of BiPO4/Ag3PO4 under different radical scavenger
2.5 光电化学性质测试
电子与空穴的分离效率是光催化反应的关键,半导体光催化剂的导带产生的光生电子能够形成光电流,而留下空穴在价带上。因此,光电流信号越强代表光生电子与空穴的分离效率越高[22]。对所制备样品进行了光电化学性质研究,结果如图 9(a)所示,当光源关闭时光电流强度极低,而当光源打开后则显著增加到0.2 mA·cm-2左右。其中BiPO4/Ag3PO4的光电流强度最高为0.235 mA·cm-2,表明BiPO4/Ag3PO4电极中光生电子与空穴的分离能力最强。为了进一步揭示光催化机制,进行了电化学阻抗谱测试研究界面电荷分离效率。如图 9(b)所示,BiPO4/Ag3PO4的Nynquist曲线的圆弧半径比Ag3PO4和BiPO4的要小,圆弧半径越小意味着光生电子-空穴对越有效的分离和电极-电解质界面上的电荷迁移效率越高[23-25]。因而BiPO4/Ag3PO4复合体系较之单一体系其光催化活性有所提高。
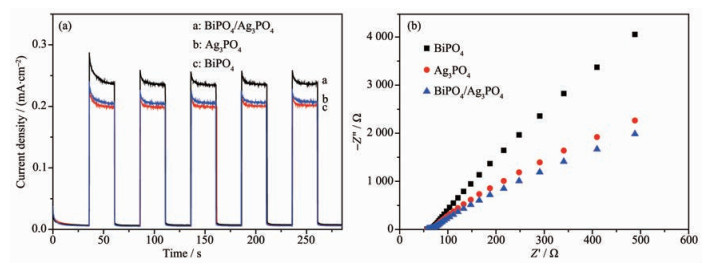 图 9
BiPO4, Ag3PO4和BiPO4/Ag3PO4的(a)光电流和(b)电化学阻抗谱测试
Figure 9.
(a) Transient photocurrent density responses and (b) electrochemical impedance spectroscopy of BiPO4, Ag3PO4 and BiPO4/Ag3PO4
图 9
BiPO4, Ag3PO4和BiPO4/Ag3PO4的(a)光电流和(b)电化学阻抗谱测试
Figure 9.
(a) Transient photocurrent density responses and (b) electrochemical impedance spectroscopy of BiPO4, Ag3PO4 and BiPO4/Ag3PO4
2.6 BiPO4/Ag3PO4光催化机理探讨
根据以上实验结果,分析BiPO4/Ag3PO4复合催化剂在模拟太阳光下具有较高的光催化活性,首先归因于p型Ag3PO4和n型BiPO4间形成了p-n异质结,其次与PO43-的特性也有关。根据公式(3)(4) 计算所制备的Ag3PO4和BiPO4的导带和价带位置。
其中,EVB是半导体催化剂的价带能,ECB是其导带能,X是其电负性,Ee是自由电子在氢电极电势下的能量值(4.5 eV),Eg是半导体禁带宽度。Ag3PO4和BiPO4的X值分别为5.96 eV和6.49 eV[26]。由图 3已知Ag3PO4、BiPO4的带隙能(Eg)分别为2.23、3.86 eV。因此,计算得Ag3PO4的价带边为2.58 eV,导带边为0.34 eV;BiPO4的价带边为3.92 eV,导带边为0.06 eV。
据此,磷酸银和磷酸铋的能带结构如图 10所示。半导体的导带和价带的位置决定了光生空穴与光生电子对的分离程度,最终决定了光催化活性。p型Ag3PO4的费米能级靠近其价带边,n型BiPO4的费米能级靠近其导带边,Ag3PO4的费米能级低于BiPO4的费米能级,两者未结合前结构为嵌套式,如图 10(a)所示。当p型Ag3PO4和n型BiPO4结合后,p区和n区电子空穴未达到平衡,BiPO4电子向Ag3PO4扩散,而Ag3PO4空穴向BiPO4扩散,而在BiPO4留下固定的正电荷,在Ag3PO4留下固定的负电荷,这些电荷产生了从n型BiPO4指向p型Ag3PO4的内建电场。当电子和空穴的扩散达到平衡后,Ag3PO4与BiPO4达到了统一费米能级。此时,如图 10(b)所示,Ag3PO4的能带上移,BiPO4的能带下移,Ag3PO4的导带边高于BiPO4的导带边,形成了交互式的p-n异质结构,更有利于光生电子-空穴对的分离[26]。
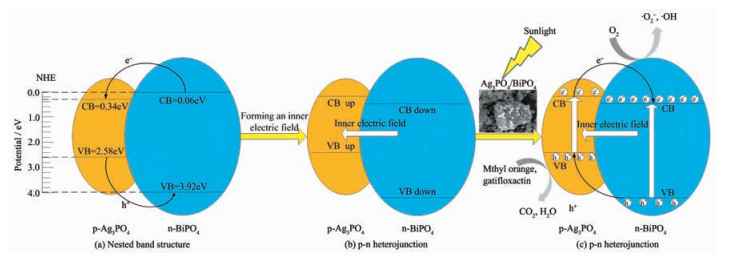 图 10
(a) BiPO4嵌套式结构,(b)BiPO4/Ag3PO4 p-n异质结形成,(c)BiPO4/Ag3PO4受光激发后电子传递路径
Figure 10.
(a) Nested band strctyre of BiPO4 and Ag3PO4 (b) contacted formation of p-n heterojunction and (c) electron teansport paths of BiPO4/Ag3PO4 after photoexcition
图 10
(a) BiPO4嵌套式结构,(b)BiPO4/Ag3PO4 p-n异质结形成,(c)BiPO4/Ag3PO4受光激发后电子传递路径
Figure 10.
(a) Nested band strctyre of BiPO4 and Ag3PO4 (b) contacted formation of p-n heterojunction and (c) electron teansport paths of BiPO4/Ag3PO4 after photoexcition
当复合催化剂受到模拟太阳光照射时,如图 10(c)所示,p型Ag3PO4被光激发,价带上的电子跃迁到导带,从而形成电子-空穴对。由于内建电场的作用,p型Ag3PO4的电子会穿过p-n异质结进入到n型BiPO4,之后,在内部电场的作用下,p型Ag3PO4的光生电子会有效地发生界面转移到n型BiPO4区域,并迅速被氧分子捕获,生成·O2-或·OH,因而实现了光生电子与空穴的有效分离。与此同时,n型BiPO4的空穴也穿过p-n异质结进入到p型Ag3PO4区域。迁移的空穴与Ag3PO4自身受光激发形成的空穴在p型Ag3PO4的价带上累积,迅速将吸附在催化剂表面的甲基橙、加替沙星分子氧化,经过4 h光催化降解实验,总有机碳降解率达到45.71%,进一步延长光照时间,有机物分子最终将矿化为CO2、H2O等无机小分子,这与自由基捕获实验证明空穴是氧化有机污染物的关键的结果一致。
此外,Pan等[11, 27]研究发现PO43-有利于光催化过程的进行。首先,PO43-存在大量的电子云重叠,光生电子受到其排斥,而光生空穴被吸引集聚到PO43-附近,这为光生电子-空穴对的分离提供了有利条件。其次,PO43-有较强的P-O共价键,PO43-很难形成氧化价键,减少了光生电子空穴对复合的概率。最后,PO43-有较强的亲水性,因此,H2O易被吸附在光催化剂表面,被空穴氧化生成·OH,·OH进一步与甲基橙、加替沙星等污染物发生作用。
3 结论
采用水热合成法一步成功制备了p-n异质结BiPO4/Ag3PO4光催化剂,其在模拟太阳光光照10 min后,甲基橙降解效率高达95.3%,加替沙星在光照3 min后降解率为95.7%,而经过4次循环后甲基橙的降解效率仍能达到22.1%,表现出比单一催化剂更高的光催化降解活性和稳定性。复合光催化剂光谱吸收拓宽至571 nm,带隙能降低为2.17 eV,使其更易被模拟太阳光所激发,在p-n异质结的作用下,实现了电子-空穴对的有效分离,进而空穴对有机物有效降解,此外PO43-的特性也是复合光催化剂活性提高的原因之一。
-
-
[1]
匡元江, 古瑶, 郭丽梅, .高等学校化学学报, 2015, 36(6):1174-1179 http://www.cjcu.jlu.edu.cn/CN/abstract/abstract25706.shtmlKUANG Yuan-Jiang, GU Yao, GUO Li-Mei, et al. Chem. J. Chinese Universities, 2015, 36(6):1174-1179 http://www.cjcu.jlu.edu.cn/CN/abstract/abstract25706.shtml
-
[2]
刘坤杰, 常志东, 李文军, .无机化学学报, 2011, 27(8):1465-1470 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20110802&journal_id=wjhxxbcnLIU Kun-Jie, CHANG Zhi-Dong, LI Wen-Jun, et al. Chinese J. Inorg. Chem., 2011, 27(8):1465-1470 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20110802&journal_id=wjhxxbcn
-
[3]
Yao S Y, Zhang X, Qu F Y, et al. J. Alloys Compd., 2016, 689:570-574 doi: 10.1016/j.jallcom.2016.08.025
-
[4]
Xie Y, Huang Z H, Zhang Z J, et al. Appl. Surf. Sci., 2016, 389:56-66 doi: 10.1016/j.apsusc.2016.07.088
-
[5]
Yi Z G, Ye J H, Kikugawa N, et al. Nat. Mater., 2010, 9(7):559-564 doi: 10.1038/nmat2780
-
[6]
Bi Y P, Ouyang S X, Umezawa N, et al. J. Am. Chem. Soc., 2011, 133(17):6490-6492 doi: 10.1021/ja2002132
-
[7]
Shekofteh-Gohari M, Habibi-Yangjeh A. Ceram. Int., 2016, 42(14):15224-15234 doi: 10.1016/j.ceramint.2016.06.158
-
[8]
Liu L, Qi Y H, Lin S L, et al. Appl. Catal., B, 2016, 183:133-141 doi: 10.1016/j.apcatb.2015.10.035
-
[9]
Zhu G S, Zhang L, Jiang B, et al. Appl. Surf. Sci., 2016, 377:99-108 doi: 10.1016/j.apsusc.2016.03.143
-
[10]
Lin X, Guo X Y, Shi W L, et al. Catal. Commun., 2015, 71:21-27 doi: 10.1016/j.catcom.2015.08.010
-
[11]
Pan C S, Zhu Y F. Environ. Sci. Technol., 2010, 44(14):5570-5574 doi: 10.1021/es101223n
-
[12]
Xu H, Xu Y G, Li H M, et al. Dalton Trans., 2012, 41(12):3387-3394 doi: 10.1039/c2dt11969b
-
[13]
尹延峰, 周锋, 詹溯, .无机化学学报, 2016, 32(3):483-490 doi: 10.11862/CJIC.2016.065YIN Yan-Feng, ZHOU Feng, ZHAN Su, et al. Chinese J. Inorg. Chem., 2016, 32(3):483-490 doi: 10.11862/CJIC.2016.065
-
[14]
Cao J, Xu B Y, Lin H L, et al. Chem. Eng. J., 2013, 228:482-488 doi: 10.1016/j.cej.2013.05.008
-
[15]
Wu S Y, Zhang H, Wu Y Y, et al. Ceram. Int., 2014, 40(9):14613-14620 doi: 10.1016/j.ceramint.2014.06.047
-
[16]
王会丽, 赵越, 马乐宽, .高等学校化学学报, 2016, 37(2):335-341 doi: 10.7503/cjcu20150544WANG Hui-Li, ZHAO Yue, MA Le-Kuan, et al. Chem. J. Chinese Universities, 2016, 37(2):335-341 doi: 10.7503/cjcu20150544
-
[17]
Jiang Z M, Li G Y, Zhang M X. Sens. Actuators B, 2016, 228:59-65 doi: 10.1016/j.snb.2016.01.013
-
[18]
Seifrtová M, Nováková L, Lino C, et al. Anal. Chim. Acta, 2009, 649(2):158-179 doi: 10.1016/j.aca.2009.07.031
-
[19]
Lü Y H, Pan C S, Ma X G, et al. Appl. Catal., B, 2013, s138-139(28):26-32
-
[20]
Yang Y X, Guo W, Guo Y N, et al. J. Hazard. Mater., 2014, 271:150-159 doi: 10.1016/j.jhazmat.2014.02.023
-
[21]
Zhang J, Yu K, Yu Y, et al. J. Mol. Catal. A:Chem, 2014, 391(1):12-18
-
[22]
Xiang Q, Yu J, Jaroniec M. J. Phys. Chem. C, 2011, 115(15):7355-7363 doi: 10.1021/jp200953k
-
[23]
Liu L, Qi Y H, Hu J S, et al. Mater. Lett., 2015, 158:278-281 doi: 10.1016/j.matlet.2015.06.034
-
[24]
Chen D M, Wang K W, Ren T Z, et al. Dalton Trans., 2014, 43:13105-13114 doi: 10.1039/C4DT01347F
-
[25]
Liang L H, Lin S L, Liu L, et al. Appl. Catal., B, 2015, 164:192-203 doi: 10.1016/j.apcatb.2014.08.048
-
[26]
Mohaghegh N, Rahimi E, Gholami M R. Mater. Sci. Semicond. Process., 2015, 39:506-514 doi: 10.1016/j.mssp.2015.05.066
-
[27]
Huang K, Lü Y H, Zhang W, et al. Mater. Res., 2015, 18(5):939-945 doi: 10.1590/1516-1439.346614
-
[1]
-

图 1 (a) Ag3PO4, (b) BiPO4/Ag3PO4的SEM照片
Figure 1 SEM images of (a) Ag3PO4 and (b) BiPO4/Ag3PO4

图 4 (a)甲基橙在模拟太阳光照射下的光催化降解曲线, (b)甲基橙在模拟太阳光和可见光下的降解动力学曲线及(c)甲基橙在模拟太阳光照射下TOC变化曲线
Figure 4 (a) Degradation of methyl orange, (b) Kinetics of degradation of methyl orange under simulated sunlight and visible light irradiation, (c) TOC changes on the degradation of methyl orange under simulated sunlight

图 5 加替沙星在模拟太阳光下的(a)光催化降解曲线(b)降解动力学曲线
Figure 5 (a) Degradation curve and (b) the kinetics of degradation of gatifloxacin under the simulated sunlight irradiation

图 6 Ag3PO4和BiPO4/Ag3PO4模拟太阳光照射下降解甲基橙的循环实验
Figure 6 Cycling runs of Ag3PO4 and BiPO4/Ag3PO4 for the degradation of methyl orange under the simulated sunlight irradiation

图 7 循环前后Ag3d XPS谱图(a)BiPO4/Ag3PO4和(b)Ag3PO4
Figure 7 XPS spectra of Ag3d from (a) BiPO4/Ag3PO4 and (b) Ag3PO4 before and after cycling runs

图 8 不同自由基捕获下BiPO4/Ag3PO4降解甲基橙曲线
Figure 8 Methyl orange degradation curves of BiPO4/Ag3PO4 under different radical scavenger

图 9 BiPO4, Ag3PO4和BiPO4/Ag3PO4的(a)光电流和(b)电化学阻抗谱测试
Figure 9 (a) Transient photocurrent density responses and (b) electrochemical impedance spectroscopy of BiPO4, Ag3PO4 and BiPO4/Ag3PO4
-


 下载:
下载:









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 2
- 文章访问数: 2249
- HTML全文浏览量: 289
 下载:
下载:
