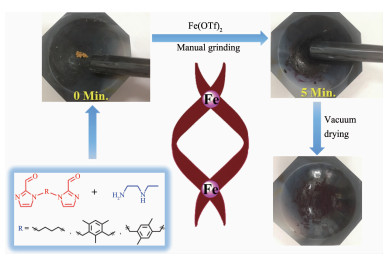
 图 1
铁 (Ⅱ) 配合物的无溶剂研磨合成示意图
Figure 1.
Schematic representation of the synthesis of iron (Ⅱ) complexes under solvent-free grinding conditions
图 1
铁 (Ⅱ) 配合物的无溶剂研磨合成示意图
Figure 1.
Schematic representation of the synthesis of iron (Ⅱ) complexes under solvent-free grinding conditions
引用本文:
韩王康, 田蕾, 徐宗利, 朱威, 李志华, 李涛, 顾志国, 李在均. 无溶剂研磨合成双核席夫碱配合物及多组分自分类行为[J]. 无机化学学报,
2017, 33(4): 550-559.
doi:
10.11862/CJIC.2017.069

Citation: HAN Wang-Kang, TIAN Lei, XU Zong-Li, ZHU Wei, LI Zhi-Hua, LI Tao, GU Zhi-Guo, LI Zai-Jun. Self-Sorting of Binuclear Schiff-Base Complexes under Solvent-Free Grinding Conditions[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(4): 550-559. doi: 10.11862/CJIC.2017.069
Citation: HAN Wang-Kang, TIAN Lei, XU Zong-Li, ZHU Wei, LI Zhi-Hua, LI Tao, GU Zhi-Guo, LI Zai-Jun. Self-Sorting of Binuclear Schiff-Base Complexes under Solvent-Free Grinding Conditions[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(4): 550-559. doi: 10.11862/CJIC.2017.069
无溶剂研磨合成双核席夫碱配合物及多组分自分类行为
摘要:
利用手动研磨法在无溶剂条件下通过1,4-二(咪唑-2-甲醛)丁烷(A)、2,4-二亚甲基-二(咪唑-2-甲醛)-三甲基苯(B)、2,5-二亚甲基-二(咪唑-2-甲醛)-对-二甲苯(C)中的任一组分与N-乙基乙二胺(D)和三氟甲磺酸亚铁组装合成3个Fe(Ⅱ)席夫碱配合物1、2和3。X射线单晶衍射表明,配合物1结晶于空间群Cmca,2和3结晶于空间群P21/c。1~3中每个Fe(Ⅱ)与配体中6个氮原子配位形成变形八面体FeN6配位环境;每个双臂六齿配体螯合2个亚铁离子,而每个亚铁离子被2条配体包裹形成Fe2L2型双核配合物。基于以上配合物的合成,我们详细研究了混合多组分间的固态自分类行为。研究表明,多组分[D+A+B+Fe2+]或[D+A+C+Fe2+]发生自恋型自分类;而多组分[D+B+C+Fe2+]发生群体型自分类,形成了一个由2个不同类型配体构筑的Fe2LL'杂配型配合物4,同时X射线单晶衍射确证了4的结构。此外,通过与溶液中自分类的对比,我们发现该体系固态自分类表现出更为高效的特点。
-
关键词:
- 自分类
- / 无溶剂研磨
- / 双核Fe (Ⅱ) 配合物
- / 席夫碱
English
Self-Sorting of Binuclear Schiff-Base Complexes under Solvent-Free Grinding Conditions
Abstract:
Three binuclear iron (Ⅱ) imidazole Schiff-base complexes were constructed by one-pot multicomponent assembly of di (imidazole aldehyde)(A~C), 2-aminoethyl (ethyl) amine (D) and Fe (OTf)2 under solvent-free grinding conditions. X-ray crystallography revealed that the three complexes crystallized in different space groups of Cmca for 1, P21/c for 2~3. And the molecular structures of 1~3 all display Fe2L2 arrangement. In complexes 1~3, two ligand strands wrap around two Fe2+ ions and the Fe2+ ions are coordinated to chelating moieties from two imidazole nitrogen atoms and four amine nitrogen atoms, resulting in the pseudo octahedral FeN6 coordination geometry. Taking advantage of the facile approach above, we then investigated the discrimination abilities of the multicomponent assembly process under the solvent-free condition when different components were mixed together (a mixture of D, two kinds of di (imidazole aldehyde) of A~C and Fe (OTf)2). In this assemble system, combinations[D+A+B+Fe2+] or[D+A+C+Fe2+] adopt narcissistic self-sorting with only two species, while social self-sorting took place in the combination[D+B+C+Fe2+] forming the single heteromer 4, which was composed of two different ligands and exhibited Fe2LL' arrangement. The structure of 4 was further verified by the X-ray analysis by preparing the single crystal of the heteromer 4. To compare the efficiency of solvent-free grinding solid self-sorting against the solution self-sorting, the conventional solution-based self-sorting reactions were also performed using the same starting materials with equal amount. In consequence of the complexity of 1H NMR peaks of mixture, the selectivity is poor in solution. Overall, compared with self-sorting in solution, the solvent-free grinding self-sorting is more efficient due to the certain restrictions of molecular movement.
-
Key words:
- self-sorting
- / solvent-free grinding
- / binuclear iron (Ⅱ) complexes
- / Schiff-base ligands
-
0 引言
近年来,通过小分子模块的组装来构筑复杂的超分子金属配合物体系已经获得了很大的进展[1]。对于多组分组装行为,竞争性构筑模块之间的相互作用最终可能导致自分类 (Self-sorting) 现象的发生。分子自分类描述的是多组分组装过程中分子在复杂混合体系中有效识别“异己”与“自己”的能力[2]。分子的自分类行为作为重要的分子行为准则之一,代表着混合系统从混乱无序到简单有序的过程。自分类行为的产生主要是因为相互作用的行为分子本身固有的空间尺寸和位阻、立体几何形貌、取代基电荷性质以及反应活性等因素的差异。对于金属配合物而言,诸多因素会影响最终的自分类,比如配体的尺寸、齿合度、配位角度以及分子间作用力 (氢键、π-π堆积、C-H…π作用) 等[3]。通常来讲,这种选择性的组装行为包括2类:自恋型自分类 (narcissistic self-sorting) 和群体型自分类 (social self-sorting)。自恋型自分类是指由相同的构筑单元通过识别出与其相同的部分从而形成单一配体组成的同型配合物;而群体型自分类是指具有相似大小尺寸的不同结构的配体选择性组装得到杂配型结构[4-5]。1993年Lehn课题组发现长度不同的同系物配体与金属离子配位时,其混合组装体系具有很高的自分类性,报道了第一例以结构多样性导向的配合物自分类组装[6]。此后,陆续有各种关于超分子配合物多组分组装过程中自分类现象的文献报道,而这些研究多为自恋型自分类,关于群体型自分类的文献报道还是相对较少[7]。一般来说,群体型自分类获得的杂配型配合物结构对称性低于自恋型自分类得到的同型配合物,这使得群体型自分类现象相对较为少见。另一方面,设计形成杂配型结构的配合物有利于提高分子多样性。然而,任意设计构筑金属-有机杂配型化合物仍然极具挑战性[8]。杂配型配合物研究相对较少的主要原因可能在于其具有不同供体性质的配体动态配位过程很难控制[9]。
无论是自恋型还是群体型自分类,目前的研究基本局限在溶液介质中,而无溶剂条件下的多组分自分类行为报道还相对较少[10]。在固态化学合成中,机械化学合成法 (mechanochemical synthesis,MC) 作为一种较为新型的材料制备与合成方法,已经越来越受到研究者的广泛关注,并且逐步应用到医药合成、材料开发和废物处理等多个领域。一方面,相对于液相反应,机械合成由于没有溶剂的参与,反应条件大为不同,因此对于相同的原料通过这种方法进行合成有可能得到新的分子或者新型材料;另一方面合成过程也相对较为简单,只需机械研磨即可。在过去几年间,研究者已经通过实验成功证明利用手动研磨合成法 (MC) 可以形成金属-配体配位键和共价键[11]。因此,机械化学合成法为无溶剂条件下的多组分自分类行为研究提供了一个平台。
本文在“一锅法”无溶剂手动研磨条件下通过咪唑双醛与N-乙基乙二胺 (D) 和三氟甲磺酸亚铁组装合成3个Fe2L2型席夫碱配合物1、2和3,如图 1所示。实验证明在机械研磨下,本文所涉及的多组分体系可以很容易地进行组装反应,即咪唑双醛、多胺和亚铁离子可以快速地反应形成C=N亚胺键和Fe-N配位键。基于配合物1~3的成功合成,我们利用无溶剂研磨法详细研究了固态条件下混合体系的自分类行为,以核磁共振手段来观察自分类情况,并与溶液中的自分类行为进行对比。有趣的是,在无溶剂研磨固态自分类组装过程中分离得到一个新的Fe2LL′杂配型化合物4,并通过X射线单晶衍射确证了4的结构。本文的研究结果为后续关于超分子配合物固态条件下分子组装和自分类行为研究提供了一个新的思路。
图 1
铁 (Ⅱ) 配合物的无溶剂研磨合成示意图
Figure 1.
Schematic representation of the synthesis of iron (Ⅱ) complexes under solvent-free grinding conditions
1 实验部分
1.1 仪器与试剂
FTLA2000-104红外光谱仪,扫描范围4 000~500 cm-1;AVANCE Ⅲ 400 MHz全数字化核磁共振谱仪;Elementar Corporation Vario EL Ⅲ元素分析仪;TU1900型双光束紫外可见分光光度计;SMART APEX Ⅱ DUO CCD衍射仪。1,4-二 (咪唑-2-甲醛) 丁烷 (A) 的合成按照文献中的方法进行[12]。咪唑-2-甲醛;1,4-二溴丁烷;2,4-双 (氯甲基) 三甲基苯;2,5-双 (氯甲基)-对-二甲苯;N-乙基乙二胺 (D);三氟甲磺酸亚铁;N,N-二甲基甲酰胺;无水碳酸钾;乙酸乙酯;氯化钾;无水硫酸镁;乙腈;无水乙醚;所用试剂均为分析纯。
1.2 合成
1.2.2 2,5-二亚甲基-二 (咪唑-2-甲醛)-对-二甲苯 (C) 的合成
与B的合成方法相似,用2,5-双 (氯甲基)-对-二甲苯替换2,4-双 (氯甲基) 三甲基苯 (产率:72 %)。1H NMR (CD3CN,400 MHz):δ 9.75(s,2H,H-C=O),7.31(s,2H,imidazole),7.25(s,2H,py),6.57(s,1H,imi-dazole),5.58(s,4H,-CH2-N),2.20(s,6H,CH3-phenyl)(Fig.S2b);IR (KBr,cm-1):3 113,3 091,2 923,2 840,1 686,1 673,1 509,1 476,1 458,1 406,1 339,1 302,1 262,1 236,1 160,1 037,918,871,785,769,751,705,692,684,631(Fig.S1);元素分析按C18H18N4O2计算值 (%):C 67.07,H 5.63,N 17.38;实测值 (%):C 66.80,H 5.82,N 17.5。
1.2.1 2,4-二亚甲基-二 (咪唑-2-甲醛)-三甲基苯 (B) 的合成
称取2-咪唑甲醛 (2.402 3 g,25 mmol),2,4-双 (氯甲基) 三甲基苯 (2.171 3 g,10 mmol),无水碳酸钾 (2.764 2 g,20 mmol),加入到100 mL圆底烧瓶中,向反应体系中添加40 mL N,N-二甲基甲酰胺,在氮气保护下于323 K持续搅拌3 d。反应结束后冷却至室温并过滤,滤液加15 mL水,然后用60 mL乙酸乙酯萃取3次 (3×20 mL)。收集有机层并用饱和氯化钾水溶液洗涤,接着用无水硫酸镁干燥,最后旋转蒸发去除溶剂。所得粗产品在40 ℃真空干燥箱中干燥得到浅黄色固体,乙酸乙酯重结晶得到浅黄色针状晶体。(产率:81%)。1H NMR (CD3CN,400 MHz):δ 9.82(s,2H,H-C=O),7.18(s,2H,imidazole),6.74(s,2H,imidazole),5.67(s,4H,-CH2-N),7.14(s,1H, phenyl), 2.09(s, 3H, CH3-phenyl), 2.28(d, 6H,CH3-phenyl)(Fig.S2a);IR (KBr,cm-1):3 114,2 972,2 922,2 835,2 799,1 690,1 466,1 483,1 407,1 340,1 330,804,793,780,764,754(Fig.S1);元素分析按C19H20N4O2计算值 (%):C 67.84,H 5.99,N 16.66;实测值 (%):C 67.80,H 5.86,N 16.49。
1.2.3 配合物[Fe2(L1)2](CF3SO3)4 (1) 的合成
分别称取A(123.13 mg,0.5 mmol)、D(88.20 mg,1 mmol) 和三氟甲磺酸亚铁Fe (OTf)2(176.99 mg,0.5 mmol) 至玛瑙研钵中,持续研磨5 min,混合固体颜色逐渐由黄色完全转变为深紫红色。将上述所得粗产品用无水乙醚 (3×10 mL) 洗涤,除去少量可能未反应完全的物质,然后将样品放置于40 ℃真空干燥箱中干燥,最终得到紫红色粉末。将最终产物溶解于乙腈中,过滤除去少量杂质,再将滤液转移至试管中,用无水乙醚扩散法培养晶体。一周后,在试管壁上析出紫黑色条状晶体 (产率:78%)。UV-Vis (CH3CN) λmax/nm:203, 297, 555(Fig.S4);1H NMR (CD3CN, 400 MHz):δ 10.54(s,2H5),7.01(s,2H4),6.29(s,2H3),5.63(s,4H2),5.04(s,4H6),4.50(s,4H7),4.18(d,J=12.8 Hz,4H8),3.62(s,4H1),2.96(s,6H9)(Fig.S3a);IR (KBr,cm-1):3 480,3 235,2 961,1 560,1 441,1 273,1 161,1 023,647;元素分析按C46H71F12Fe2N17O12S4计算值 (%):C 36.30,H 4.70,N 15.64;实测值 (%):C 36.63,H 4.88,N 15.67。
1.2.7 X射线单晶衍射
单晶衍射测试在173(2) K温度下,使用APEX Ⅱ DUO CCD衍射仪测定。采用石墨单色化的Mo Kα射线 (λ=0.071 073 nm),以ω扫描方式收集数据。晶胞参数用SMART软件还原并采用SAINT软件[13]进行数据精修。实验过程中设置ω扫描步进为0.30°,衍射强度数据经SADABS软件校正[14]。晶体结构运用SHELX-97程序采用直接法解出并基于F 2对所有非氢原子坐标及其各向异性参数进行了全矩阵最小二乘法精修,氢原子由理论加氢法得到[15]。化合物1结构测定尝试过多次,但结果都是高角度没有衍射或衍射弱,数据参数比较低,wR2值偏高。各晶体结构中都存在不同程度的无序,对于晶体结构中无序处理的方法参照文献[16]进行。配合物的晶体学数据见表 1,键长键角列于表S1。
 表 1
配合物1~4的晶体学数据
Table 1.
Summary of crystallographic data for complexes 1~4
表 1
配合物1~4的晶体学数据
Table 1.
Summary of crystallographic data for complexes 1~4
1 2 3 4 Formula C44H68F12Fe2N16O11S4 C58H80F 12Fe2N16O12S4 C56H76F 12Fe2N16O12S4 C 57H78F12Fe2N 16O12S4 Formula weight 1 465.08 1 661.32 1 633.27 1 647.29 Crystal system Orthorhombic Monoclinic Monoclinic Monoclinic Space group Cmca P21/c P21/c P21/c a / nm 3.535 4(3) 1.090 04(8) 0.970 95(6) 1.017 2(11) b/ nm 2.533 43(19) 1.879 99(15) 1.942 81(13) 1.912 1(2) c/ nm 1.454 73(11) 2.159 47(15) 2.112 80(11) 2.122 56(19) β/(°) 90 117.290(3) 109.004(3) 111.197(5) V/ nm3 13.029 7(17) 3.932 8(5) 3.768 3(4) 3.850 3(7) Dc/ (g·cm3) 1.494 1.403 1.439 1.421 Z 8 2 2 2 F(000) 6 048 1 720 1 688 1 704 θ range / (°) 2.85~24.50 3.02~25.36 2.98~25.40 1.48~25.00 μ / mm-1 0.672 0.567 0.590 0.578 Index range -40 ≤ h ≤ 41,
-28 ≤ k ≤ 29,
-15 ≤ l ≤ 16-12 ≤ h ≤ 16,
-22 ≤ k ≤ 22,
-24 ≤ l ≤ 26-11 ≤ h ≤ 11,
-23 ≤ k ≤ 23,
-25 ≤ l ≤ 25-6 ≤ h ≤ 12,
-22 ≤ k ≤ 22,
-25 ≤ l ≤ 21Reflections collected 23 886 22 160 25 021 19 951 Independent reflections 5 437(Rint=0.078 2) 7 067(Rint=0.051 1) 6 749(Rint=0.073 4) 6 629(Rint=0.082 1) G0F (F2) 1.357 1.038 1.158 1.047 R1a (I > 2σ(I)) 0.126 9 0.058 6 0.069 6 0.099 7 wR2b (I > 2σ(I)) 0.3123 0.161 4 0.185 6 0.251 8 R1a (all data) 0.178 5 0.095 6 0.1344 0.253 2 wR2b (all data) 0.339 1 0.179 4 0.223 2 0.293 3 a R1=∑||Fo|-|Fc||/∑|Fo|; b wR2=[∑w(Fo2-Fc2)2/∑w(Fo2)]1/2 表 1 配合物1~4的晶体学数据
Table 1. Summary of crystallographic data for complexes 1~4CCDC:1447337, 1; 1447338, 2; 1447339, 3; 1447340, 4。
1.2.6 配合物[Fe2(L2L3)](CF3SO3)4 (4) 的合成
配合物4是在混合组分[D+B+C+Fe2+]无溶剂研磨自分类组装过程中形成的,具体步骤如下:分别称取B(84.09 mg, 0.25 mmol), C(80.59 mg, 0.25 mmol),D(88.20 mg,1 mmol) 和三氟甲磺酸亚铁Fe (OTf)2(176.99 mg,0.5 mmol) 至玛瑙研钵中,持续研磨5 min,混合固体颜色逐渐由黄色完全转变为深紫红色。将上述所得粗产品用无水乙醚 (3×10 mL) 洗涤,除去少量可能未反应完全的物质,然后将样品放置于40 ℃真空干燥箱中干燥,最终得到紫红色粉末。将最终产物溶解于乙腈中,过滤除去少量杂质,再将滤液转移至试管中,用无水乙醚扩散法培养晶体。一周后,在试管壁上析出紫黑色条状晶体 (产率:59%)。UV-Vis (CH3CN) λmax/nm:206,297,556(Fig.S4);1H NMR (CD3CN,400 MHz):δ 10.91(s,2H6′),10.72(d,J=40.4 Hz,2H6),7.12(m,2H2, 11),6.75(s,4H5),6.33(m,J=19.1 Hz,2H5′), 6.28 (m,4H4, 4′),5.74(m,4H8),5.64(m,4H8′),5.23(s,4H8),5.10(s,6H9, 9′),3.77(s,6H13),3.66(s,6H1),2.93(s,4H10, 12),2.38(d,J=29.3 Hz,4H10′)(Fig.S3d);IR (KBr,cm-1):3 463,3 236, 2 974, 1 452, 1 268, 1 155,1 029,635;元素分析按C57H78F12Fe2N16O12S4计算值 (%):C 41.56,H 4.77,N 13.60;实测值 (%):C 41.72,H 4.85,N 13.73。
1.2.5 配合物[Fe2(L3)2](CF3SO3)4 (3) 的合成
与配合物1的合成方法相似,用C替换A,可获得配合物3。同时利用乙醚扩散法可获得单晶衍射用的紫黑色条状晶体 (产率:78%)。UV-Vis (CH3CN)λmax/nm:206,298,555(Fig.S4);1H NMR (CD3CN,400 MHz):δ 10.62(s,2H6),6.66(s,2H2),6.56(s,2H5),6.32(s, 2H4), 5.70(m,J=14.9 Hz,4H3),5.61(m,4H7),5.23(m,4H8), 5.09(s,4H9),3.62(s,6H1),3.02(d,J=40.4 Hz,6H10)(Fig.S3c);IR (KBr,cm-1):3 475,3 236,2 974,1 554,1 453,1 268,1 155,1 029,642 cm-1;元素分析按C56H76F12Fe2N16O12S4计算值 (%):C 41.18,H 4.69,N 13.72;实测值 (%):C 42.23,H 4.76,N 13.83。
1.2.4 配合物[Fe2(L2)2](CF3SO3)4 (2) 的合成
与配合物1的合成方法相似,用B替换A,可获得配合物2。同时利用乙醚扩散法可获得单晶衍射用的紫黑色条状晶体 (产率:65%)。UV-Vis (CH3CN) λmax/nm:206, 297, 556(Fig.S4);1H NMR (CD3CN, 400 MHz):δ 10.90(s,2H7),7.14(m,2H2),6.36(s,2H6),6.25(s,2H5),5.78(d,J=12.65 Hz,4H4),5.57(m,4H8),5.32(m,4H9),5.11(s,4H10),3.78(s,3H1),2.92(s,6H3),2.33(s,6H11)(Fig.S3b);IR (KBr,cm-1):3 493,3 236,2 968,2 354,1 560,1 447,1 268,1 154,1 035,642;元素分析按C58H80F12Fe2N16O12S4计算值 (%):C 41.93,H 4.85,N 13.49;实测值 (%):C 42.06,H 4.92,N 13.53。
2 结果与讨论
2.1 配合物1~3的无溶剂研磨合成
采取无溶剂研磨合成法,按照物质的量之比 (1:2:1) 称取咪唑双醛 (B或C或D)、多胺A和三氟甲磺酸亚铁至玛瑙研钵中,持续研磨可以观察到固体颜色开始由黄色转变为深紫红色。这代表在研磨过程中C=N亚胺键和Fe-N配位键的形成,且金属中心处于低自旋态[17]。保持研磨5 min后可以发现固体颜色完全转变为深紫红色,这意味着此时多组分自组装已经基本反应完全。然后将上述粗产品用无水乙醚洗涤除去少量可能未反应完全的物质即为目标产物1~3。相比溶液合成法,无溶剂机械研磨合成反应更加迅速,反应时间更短。之所以机械研磨法如此高效, 我们推测可能存在以下2个原因:(1) 产物中的C=N亚胺键和Fe-N配位键都较为容易形成。经过对起始组分的研磨,机械作用使得分子松动,提高了分子的移动性,同时摩擦能够瞬间产生热量给予反应体系一定能量,促使C=N键和Fe-N键的快速形成。(2) 本体系中所选用的反应物组分之一多胺在常温时呈现液态,在“一锅法”固态研磨多组分自组装过程中,液态多胺有利于活化固态混合物的反应,进一步加速了自组装反应的进行。
2.2 红外光谱分析
为了证明固态研磨下配合物的成功合成,对各原料组分、配合物1~3的研磨产品和对应的单晶化合物红外光谱进行对比分析 (图 2)。从图中可以看出原料组分咪唑双醛在1 680 cm-1附近的C=O双键伸缩振动吸收峰消失,而在1 560 cm-1附近出现了新的吸收峰,代表有新的共价键生成,为C=N双键伸缩振动吸收峰,这也暗示着研磨过程中席夫碱配体的生成;另外,C=N双键伸缩振动吸收峰的位置相比文献已报道的结果更偏向于低波数,说明此时亚铁离子与化合物已经成功配位,且金属中心处于低自旋态[18]。在1 260和638 cm-1左右出现的吸收峰则为阴离子OTf-的特征吸收峰。最后,对比配合物1~3的研磨产品和对应的单晶化合物红外光谱图,我们发现各红外特征吸收峰基本吻合。红外分析的结果也进一步证明了利用机械研磨法成功得到目标产物。
 图 2
配合物1~3各自对应的咪唑双醛、多胺、无溶剂研磨产品和单晶化合物的红外对比图
Figure 2.
FT-IR spectra of corresponding di (imidazole aldehyde), diamine, crystalline products and crude products obtained under solvent-free grinding condition
图 2
配合物1~3各自对应的咪唑双醛、多胺、无溶剂研磨产品和单晶化合物的红外对比图
Figure 2.
FT-IR spectra of corresponding di (imidazole aldehyde), diamine, crystalline products and crude products obtained under solvent-free grinding condition
2.3 晶体结构分析
为了直观说明席夫碱配合物的结构,我们测定了相应化合物的X射线单晶结构,晶体结构信息和部分键长键角见表 1和表S1。此外,研磨产物与单晶产物通过粉末X射线衍射证实两者结构上的一致性 (图S5)。X射线单晶衍射测试结果表明,如图 3所示,3个配合物在空间单元结构上非常相似,均为Fe2L2型配合物。每个结构基元中包含1个[Fe2L2]4+阳离子和4个OTf-阴离子。从图 3可知,配合物1~3的差别仅在于配体中2个咪唑环之间的桥连基团,配合物1的桥连基团为柔性烷基链-C4H8-,而配合物2、3的桥连基团则是半刚性结构的苄环取代基。配合物1结晶于正交晶系的Cmca空间群,配合物2、3结晶于单斜晶系中的P21/c空间群。3个配合物分子中亚铁离子与2条配体带中的2个咪唑N原子以及多胺组分中的4个N原子发生螯合作用最终形成了变形八面体FeN6配位环境。每个双臂六齿配体螯合2个亚铁离子,而每个亚铁离子被2条配体包裹形成Fe2L2型双核配合物。它们的Fe-N平均键长分布在0.196~0.199 nm范围内,说明此时配合物1~3的金属中心均处于低自旋态[17-18]。对配合物1,单个分子中Fe…Fe距离为0.964 nm,且配体末端咪唑基团向相反方向延伸形成“Z”字形 (图S6a),双 (咪唑-亚胺) 丁烷连结基团采取邻位交叉-邻位交叉的构型方式与铁 (Ⅱ) 进行配位,且2个咪唑环所在平面夹角为41.30°(图S7a)。配合物2、3中,单个分子中的Fe…Fe距离分布在1.197~1.211 nm范围内。与配合物1有所不同,配合物2、3配体带末端的2个咪唑环延伸方向相同,形成了2个相同的“C”字形配体带 (图S6b),配合物2、3采取对位交叉-对位交叉的构型方式,末端的2个咪唑环所在平面夹角分布在93.91°~120.42°范围内 (图S7b、c)。导致配合物1与配合物2、3结构上差异的原因,可能在于配合物1配体中的柔性烷基链取代基被替换成半刚性的苄环基团后,极大地影响了配体带的扭曲程度。另外在空间堆积上,配合物1所有分子的Fe原子连线形成一维折线链 (图S8a)。配合物2和3则有所不同 (图S8b、c),从它们沿a轴形成的晶体堆积图可以明显看出,配合物2、3分子排列方式完全一致,呈现“风车”状,均由两组相同的分子列组成。此外,配合物1~3中OTf-阴离子按一定规律填充在不同分子之间。这些抗衡阴离子的存在可以有效地使整个超分子化合物更加稳定,因为OTf-阴离子可以与[Fe2L2]4+通过H…O、H…F两类氢键相互作用力使双核亚铁配合物分子排列有序化。
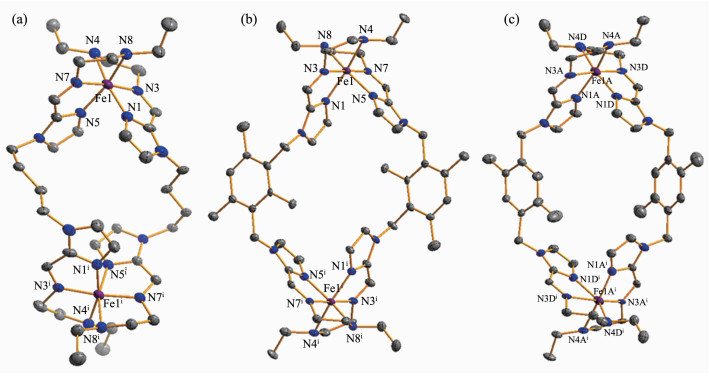 图 3
配合物1 (a)、2 (b) 和3 (c) 的分子椭球图
Figure 3.
Perspective drawings of complexes 1 (a), 2 (b) and 3 (c) showing the atom numbering
图 3
配合物1 (a)、2 (b) 和3 (c) 的分子椭球图
Figure 3.
Perspective drawings of complexes 1 (a), 2 (b) and 3 (c) showing the atom numbering
2.4 固态自分类研究
基于无溶剂条件下成功研磨合成配合物1~3以及对相应配合物结构的确定,我们对该体系在无溶剂固态条件下组装过程中的自我识别能力展开了进一步的研究。此外,本文中咪唑双醛与多胺构成的席夫碱体系,在“一锅法”组装合成过程中C=N亚胺键和Fe-N配位键同时形成,这对于研究具有多个相似同等组分的混合体系组装过程,探讨生成同配体型配合物还是生成不同配体构筑的杂配型结构是十分合适且具有一定意义的。
为此,本文将多个同等组分混合在一起,构成3个混合体系,即[D+A+B+Fe2+]、[D+A+C+Fe2+]和[D+B+C+Fe2+]。在研钵中手动研磨3个组分的混合物,Fe (OTf)2(2eq)、N-乙基乙二胺D(4eq) 和咪唑双醛A~C中的任意2个 (各1eq)。研磨获得的粗产品用无水乙醚洗涤,经真空干燥,最终可得到紫红色粉末。红外分析证实了混合组分在无溶剂研磨条件下亚铁咪唑席夫碱配合物的形成 (图S9)。称取一定量处理后的样品转移至核磁管中,添加0.6 mL氘代乙腈,待样品完全溶解后立即对其进行1H NMR测试,结果如图 4所示。从图 4的1H NMR对比图可以看出,多组分混合物[D+A+B+Fe2+]的氢谱中,在10.54和10.90处出现的2组单峰正好与化合物1和2中席夫碱配体H-C=N质子的2个单峰信号相对应。同样,混合物[D+A+C+Fe2+]在化学位移为10.54和10.62处也均出现了与化合物1和3中对应的H-C=N质子信号峰。这说明多组分混合物[D+A+B+Fe2+]和[D+A+C+Fe2+]经过研磨均通过自我识别、单元模块组装生成了2种配合物,即发生了自恋型自分类。然而有意思的是,对混合组分[D+B+C+Fe2+]却发生了不同的自分类行为。其核磁谱图与单一化合物的核磁谱图并没有出现很好的对应性,其中较为明显的是在10.91和10.72处分别出现了1个单峰信号和1个裂分后的两重峰,在7.12附近出现2个较为明显的信号峰,同时在其它位置也出现了一些信号峰的加宽和偏移,这与单一配合物2和3中的信号峰无法对应。这表明混合组分[D+B+C+Fe2+]经过机械化学反应之后各单元组分之间选择性自组装生成了一种新的化合物。然而,组装过程中单元模块间的自我识别能力,即自分类现象并不受合成过程中不同的原料混合顺序的影响。为了进一步论证上述的自分类行为,我们将混合组分[D+B+C+Fe2+]经过机械研磨和化学反应之后获得的产物溶解于乙腈中,利用单晶培养技术获得该产物的衍射用单晶,同时利用X射线单晶衍射测试表征了该化合物的单晶结构。X射线单晶衍射结果表明,新生成的配合物4依旧是双核结构,与配合物2和3结晶于相同的空间群P21/c,在配位模式以及空间构型上与配合物2和3没有明显差异,但是配合物4是由2个不同类型的配体构筑形成的Fe2LL′杂配型配合物 (图 5),这也进一步证明了混合组分[D+B+C+Fe2+]在固态研磨组装过程中发生了群体型自分类。之所以会发生这一现象,究其原因可能在于咪唑双醛B和C具有相同的构型以及它们极其相似的连结基团,仅在连接基团苯环上的取代位置存在微弱的差异。因此,混合组分[D+B+C+Fe2+]的自分类行为表现为一个群体行为,导致杂配型配合物4的出现。这一基于半刚性配体的群体型自分类,也为目前少有的多组分组装形成超分子配合物过程中群体型自分类行为的研究增添了一个重要的实例。
 图 4
(a) 自分类体系示意图:无溶剂条件下研磨混合物[D+A+B+Fe2+],[D+A+C+Fe2+]和[D+B+C+Fe2+]; (b) 化合物1(三角形)、2(圆点)、3(菱形) 及自分类化合物[D+B+C+Fe2+]、[D+A+B+Fe2+]和[D+A+C+Fe2+]的氢谱 (4 mmol·L-1, 400 MHz, CD3CN, 298 K)
Figure 4.
(a) Schematic diagram of self-sorting: [D+A+B+Fe2+], [D+A+C+Fe2+] and [D+B+C+Fe2+] under the solvent-free grinding conditions; (b) 1H NMR spectra of self-sorted complex formation (4 mmol·L-1, 400 MHz, CD3CN, 298 K) of 1 (triangle), 2 (dot) and 3 (rhombus), a mixture of [D+B+C+Fe2+], [D+A+B+Fe2+] and [D+A+C+Fe2+]
图 4
(a) 自分类体系示意图:无溶剂条件下研磨混合物[D+A+B+Fe2+],[D+A+C+Fe2+]和[D+B+C+Fe2+]; (b) 化合物1(三角形)、2(圆点)、3(菱形) 及自分类化合物[D+B+C+Fe2+]、[D+A+B+Fe2+]和[D+A+C+Fe2+]的氢谱 (4 mmol·L-1, 400 MHz, CD3CN, 298 K)
Figure 4.
(a) Schematic diagram of self-sorting: [D+A+B+Fe2+], [D+A+C+Fe2+] and [D+B+C+Fe2+] under the solvent-free grinding conditions; (b) 1H NMR spectra of self-sorted complex formation (4 mmol·L-1, 400 MHz, CD3CN, 298 K) of 1 (triangle), 2 (dot) and 3 (rhombus), a mixture of [D+B+C+Fe2+], [D+A+B+Fe2+] and [D+A+C+Fe2+]
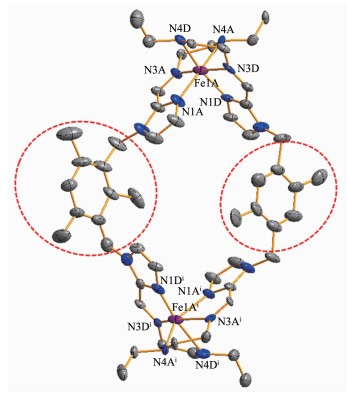 图 5
配合物4的分子椭球图
Figure 5.
Perspective drawings of complexes 4 showing the atom numbering
图 5
配合物4的分子椭球图
Figure 5.
Perspective drawings of complexes 4 showing the atom numbering
为了将固态自分类与溶液中的自分类进行比较,本文选用与上述相同的起始组分,各组分按照对应的量进行称取,用氘代乙腈溶解,在323 K环境下加热反应7 h后,移取适量反应后的溶液至核磁管中,进行1H NMR测试,结果如图S9所示。溶液环境下,[D+A+B+Fe2+]和[D+A+C+Fe2+]的核磁谱图分别显示在δ=8附近均出现了一组新的很强的信号峰,同时部分峰也出现加宽的现象,这都表明可能有未知的新化合物生成。与此相比,在本文所选用的体系中无溶剂研磨自分类组装所得核磁谱图中各信号峰与相应配合物的信号峰对应性较好,这表明无溶剂研磨条件下生成单一化合物,比溶液态自分类表现出更为高效的特点。对于组分[D+B+C+Fe2+],溶液条件下混合多组分组装过程的1H NMR谱图中各峰值与单一配合物2和3的谱图峰值相对应,这说明群体型自分类在溶液态和无溶剂研磨条件下均能有效的发生。之所以固态自分类与溶液中的自分类存在差异,我们认为原因如下:(1) 相比溶液态自分类,固态自分类由于缺乏大量溶剂,使得反应物小分子运动受到很大限制,在研磨过程中也可以迅速给予反应物较大的能量,在达到反应所需要的活化能之后便会形成产物。若参与反应物为液体状,可以加速反应物分子的活化,同时使得C=N亚胺键和Fe-N配位键在几分钟内迅速形成。(2) 此外,固态时晶体分子排列更加紧密且更有规律性。而溶液状态下分子与分子间相互作用相对较弱,在加热过程中反应物小分子在溶剂中加速运动往往会导致一些副产物的形成。因此,固态反应与液态反应相比具有更高的效率和更好的选择性。
3 结论
本文利用无溶剂手动研磨合成方法成功设计合成了一系列双核Fe (Ⅱ) 咪唑席夫碱配合物。基于固态研磨组装合成方法,我们对该体系的固态自分类行为进行了详细研究。结果表明,在该体系下既可以发生自恋型自分类,也可以发生群体型自分类,并且通过群体型自分类还获得了一个新的杂配型配合物。这为现有已经报道的多组分组装过程中自分类现象的研究提供了一种全新的思路,对后续的进一步研究具有一定借鉴意义。
-
-
[1]
(a) Cook T R, Stang P J. Chem. Rev., 2015, 115(15):7001-7045
(b) McConnell A J, Wood C S, Neelakandan P P, et al. Chem. Rev., 2015, 115(15):7729-7793
(c) Castilla A M, Ramsay W J, Nitschke J R. Acc. Chem. Res., 2014, 47(7):2063-2073
(d) Ward M D, Raithby P R. Chem. Soc. Rev., 2013, 42(4):1619-1636
(e) Giuseppone N. Acc. Chem. Res., 2012, 45(12):2178-2188
(f) Nitschke J R. Acc. Chem. Res., 2007, 40(2):103-112
(g) Smulders M M J, Riddell I A, Browne C, et al. Chem. Soc. Rev., 2013, 42(4):1728-1754 -
[2]
(a) He Z, Jiang W, Schalley C A. Chem. Soc. Rev., 2015, 44(3):779-789
(b) Yang L, Tan X, Wang Z, et al. Chem. Rev., 2015, 115(15):7196-7239
(c) Herrmann A. Chem. Soc. Rev., 2014, 43(6):1899-1933
(d) Zheng B, Wang F, Dong S, et al. Chem. Soc. Rev., 2012, 41(5):1621-1636
(e) Chakrabarty R, Mukherjee P S, Stang P J. Chem. Rev., 2011, 111(11):6810-6918
(f) Maeda C, Kamada T, Aratani N, et al. Coord. Chem. Rev., 2007, 251(21):2743-2752 -
[3]
(a) Saha M L, De S, Pramanik S, et al. Chem. Soc. Rev., 2013, 42(16):6860-6909
(b) Sun Q F, Iwasa J, Ogawa D, et al. Science, 2010, 328(5982):1144-1147
(c) Holloway L R, Young M C, Beran G J O, et al. Chem. Sci., 2015, 6(8):4801-4806
(d) Gütz C, Hovorka R, Stobe C, et al. Eur. J. Org. Chem., 2014, (1):206-216 -
[4]
(a) Gidron O, Jirásek M, Trapp N, et al. J. Am. Chem. Soc., 2015, 137(39):12502-12505
(b) Gütz C, Hovorka R, Struch N, et al. J. Am. Chem. Soc., 2014, 136(33):11830-11838
(c) Acharyya K, Mukherjee S, Mukherjee P S. J. Am. Chem. Soc., 2012, 135(2):554-557
(d) Wang W, Zhang Y, Sun B, et al. Chem. Sci., 2014, 5(12):4554-4560
(e) Singh A S, Sun S S. Chem. Commun., 2012, 48(59):7392-7394
(f) Yan L L, Tan C H, Zhang G L, et al. J. Am. Chem. Soc., 2015, 137(26):8550-8555 -
[5]
(a) Tomimasu N, Kanaya A, Takashima Y, et al. J. Am. Chem. Soc., 2009, 131(34):12339-12343
(b) Klotzbach S, Beuerle F. Angew. Chem. Int. Ed., 2015, 54(35):10356-10360
(c) Benkhäuser C, Lützen A. Beilstein J. Org. Chem., 2015, 11(1):693-700
(d) Mukhopadhyay P, Wu A, Isaacs L. J. Org. Chem., 2004, 69(19):6157-6164 -
[6]
Baxter P, Lehn J M, DeCian A, et al. Angew. Chem. Int. Ed., 1993, 32(1):69-72 doi: 10.1002/(ISSN)1521-3773
-
[7]
(a) Safont-Sempere M M, Fernández G, Wurthner F. Chem. Rev., 2011, 111(9):5784-5814
(b) Jiménez A, Bilbeisi R A, Ronson T K, et al. Angew. Chem., Int. Ed., 2014, 53(18):4556-4560
(c) Bloch W M, Abe Y, Holstein J J, et al. J. Am. Chem. Soc., 2016, 138(41):13750-13755 -
[8]
(a) Mayoral M J, Rest C, Schellheimer J, et al. Chem. Eur. J., 2012, 18(49):15607-15611
(b) Smulders M M J, Jiménez A, Nitschke J R. Angew. Chem. Int. Ed., 2012, 51(27):6681-6685 -
[9]
(a) De S, Mahata K, Schmittel M. Chem. Soc. Rev., 2010, 39(5):1555-1575
(b) Ronson T K, Roberts D A, Black S P, et al. J. Am. Chem. Soc., 2015, 137(45):14502-14512
(c) Saha M L, Neogi S, Schmittel M. Dalton Trans., 2014, 43(10):3815-3834 -
[10]
(a) Giri C, Sahoo P K, Puttreddy R, et al. Chem. Eur. J., 2015, 21(17):6390-6393
(b) Chen P N, Lai C C, Chiu S H. Org. Lett., 2011, 13(17):4660-4663 -
[11]
(a) Biswal B P, Chandra S, Kandambeth S, et al. J. Am. Chem. Soc., 2013, 135(14):5328-5331
(b) Friščić T, Reid D G, Halasz I, et al. Angew. Chem. Int. Ed., 2010, 49(4):712-715
(c) Tireli M, Kulcsár M J, Cindro N, et al. Chem. Commun., 2015, 51(38):8058-8061
(d) Das G, Shinde D B, Kandambeth S, et al. Chem. Commun., 2014, 50(84):12615-12618
(e) Bowmaker G A. Chem. Commun., 2013, 49(4):334-348
(f) Loots L, Wahl H, Van Der Westhuizen L, et al. Chem. Commun., 2012, 48(94):11507-11509 -
[12]
Ren D H, Qiu D, Pang C Y, et al. Chem. Commun., 2015, 51(4):788-791 doi: 10.1039/C4CC08041F
-
[13]
SAINT-Plus, Version 6.02, Bruker Analytical X-ray System:Madison, WI, 1999.
-
[14]
Sheldrick G M. SADABS, Bruker Analytical X-ray Systems:Madison, WI, 1996.
-
[15]
(a) Sheldrick G M. SHELXTL-97, Program for X-ray Crystal Structure Solution and Refinement, University of Göttingen, Germany, 1997.
(b) Sheldrick G M. Acta Crystallogr. Sect. A, 2008, 64(1):112-122 -
[16]
(a) Sluis P V D, Spek A L. Acta Crystallogr. Sect. A, 1990, A46:194-201
(b) Müller P, Herbst-Irmer R, Spek A L, et al. Crystal Structure Refinement-a Crystallographer's Guide to SHELXL. New York:Oxford University Press, 2006:63-96 -
[17]
(a) Brooker S. Chem. Soc. Rev., 2015, 44(10):2880-2892
(b) Hagiwara H, Okada S. Chem. Commun., 2016, 52(4):815-818
(c) Qin L F, Pang C Y, Han W K, et al. Dalton Trans., 2016, 45(17):7340-7348 -
[18]
(a) Herber R, Casson L M. Inorg. Chem., 1986, 25(6):847-852
(b) Zilverentant C L, Van Albada G A, Bousseksou A, et al. Inorg. Chim. Acta, 2000, 303(2):287-290
(c) Sunatsuki Y, Kawamoto R, Fujita K, et al. Inorg. Chem., 2009, 48(18):8784-8795
-
[1]
-

图 1 铁 (Ⅱ) 配合物的无溶剂研磨合成示意图
Figure 1 Schematic representation of the synthesis of iron (Ⅱ) complexes under solvent-free grinding conditions

图 2 配合物1~3各自对应的咪唑双醛、多胺、无溶剂研磨产品和单晶化合物的红外对比图
Figure 2 FT-IR spectra of corresponding di (imidazole aldehyde), diamine, crystalline products and crude products obtained under solvent-free grinding condition

图 3 配合物1 (a)、2 (b) 和3 (c) 的分子椭球图
Figure 3 Perspective drawings of complexes 1 (a), 2 (b) and 3 (c) showing the atom numbering
30% ellipsoids;All hydrogen atoms and anions are omitted for clarity; Symmetry transformations used to generate equivalent atoms: i x, -y+2, -z+1 for 1; i-x+1, -y+1, -z+1 for 2; i-x+1, -y+1, -z for 3

图 4 (a) 自分类体系示意图:无溶剂条件下研磨混合物[D+A+B+Fe2+],[D+A+C+Fe2+]和[D+B+C+Fe2+]; (b) 化合物1(三角形)、2(圆点)、3(菱形) 及自分类化合物[D+B+C+Fe2+]、[D+A+B+Fe2+]和[D+A+C+Fe2+]的氢谱 (4 mmol·L-1, 400 MHz, CD3CN, 298 K)
Figure 4 (a) Schematic diagram of self-sorting: [D+A+B+Fe2+], [D+A+C+Fe2+] and [D+B+C+Fe2+] under the solvent-free grinding conditions; (b) 1H NMR spectra of self-sorted complex formation (4 mmol·L-1, 400 MHz, CD3CN, 298 K) of 1 (triangle), 2 (dot) and 3 (rhombus), a mixture of [D+B+C+Fe2+], [D+A+B+Fe2+] and [D+A+C+Fe2+]

图 5 配合物4的分子椭球图
Figure 5 Perspective drawings of complexes 4 showing the atom numbering
30% ellipsoids; All hydrogen atoms and anions are omitted for clarity; Symmetry transformations used to generate equivalent atoms for 4: i-x+1, -y, -z+1
表 1 配合物1~4的晶体学数据
Table 1. Summary of crystallographic data for complexes 1~4
1 2 3 4 Formula C44H68F12Fe2N16O11S4 C58H80F 12Fe2N16O12S4 C56H76F 12Fe2N16O12S4 C 57H78F12Fe2N 16O12S4 Formula weight 1 465.08 1 661.32 1 633.27 1 647.29 Crystal system Orthorhombic Monoclinic Monoclinic Monoclinic Space group Cmca P21/c P21/c P21/c a / nm 3.535 4(3) 1.090 04(8) 0.970 95(6) 1.017 2(11) b/ nm 2.533 43(19) 1.879 99(15) 1.942 81(13) 1.912 1(2) c/ nm 1.454 73(11) 2.159 47(15) 2.112 80(11) 2.122 56(19) β/(°) 90 117.290(3) 109.004(3) 111.197(5) V/ nm3 13.029 7(17) 3.932 8(5) 3.768 3(4) 3.850 3(7) Dc/ (g·cm3) 1.494 1.403 1.439 1.421 Z 8 2 2 2 F(000) 6 048 1 720 1 688 1 704 θ range / (°) 2.85~24.50 3.02~25.36 2.98~25.40 1.48~25.00 μ / mm-1 0.672 0.567 0.590 0.578 Index range -40 ≤ h ≤ 41,
-28 ≤ k ≤ 29,
-15 ≤ l ≤ 16-12 ≤ h ≤ 16,
-22 ≤ k ≤ 22,
-24 ≤ l ≤ 26-11 ≤ h ≤ 11,
-23 ≤ k ≤ 23,
-25 ≤ l ≤ 25-6 ≤ h ≤ 12,
-22 ≤ k ≤ 22,
-25 ≤ l ≤ 21Reflections collected 23 886 22 160 25 021 19 951 Independent reflections 5 437(Rint=0.078 2) 7 067(Rint=0.051 1) 6 749(Rint=0.073 4) 6 629(Rint=0.082 1) G0F (F2) 1.357 1.038 1.158 1.047 R1a (I > 2σ(I)) 0.126 9 0.058 6 0.069 6 0.099 7 wR2b (I > 2σ(I)) 0.3123 0.161 4 0.185 6 0.251 8 R1a (all data) 0.178 5 0.095 6 0.1344 0.253 2 wR2b (all data) 0.339 1 0.179 4 0.223 2 0.293 3 a R1=∑||Fo|-|Fc||/∑|Fo|; b wR2=[∑w(Fo2-Fc2)2/∑w(Fo2)]1/2  下载: 导出CSV
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 7
- 文章访问数: 1874
- HTML全文浏览量: 230
 下载:
下载:
