 图1
半导体光解水制氢基本过程示意图
Figure1.
Scheme of the basic processes in photocatalytic water splitting using semiconductors
图1
半导体光解水制氢基本过程示意图
Figure1.
Scheme of the basic processes in photocatalytic water splitting using semiconductors
引用本文:
谢英鹏, 王国胜, 张恩磊, 张翔. 半导体光解水制氢研究:现状、挑战及展望[J]. 无机化学学报,
2017, 33(2): 177-209.
doi:
10.11862/CJIC.2017.030

Citation: XIE Ying-Peng, WANG Guo-Sheng, ZHANG En-Lei, ZHANG Xiang. Photocatalytic Hydrogen Evolution from Water Splitting Using Semiconductors:Advance, Challenge and Prospects[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(2): 177-209. doi: 10.11862/CJIC.2017.030
Citation: XIE Ying-Peng, WANG Guo-Sheng, ZHANG En-Lei, ZHANG Xiang. Photocatalytic Hydrogen Evolution from Water Splitting Using Semiconductors:Advance, Challenge and Prospects[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(2): 177-209. doi: 10.11862/CJIC.2017.030
半导体光解水制氢研究:现状、挑战及展望
English
Photocatalytic Hydrogen Evolution from Water Splitting Using Semiconductors:Advance, Challenge and Prospects
Abstract:
Achieving energy conversion from solar to clean hydrogen through water splitting reaction photo-catalyzed by a semiconductor is one of the ultimate ways to solve mankind's energy and environmental crisis. The key to this goal is the development of a wide spectral responsive, and efficient photocatalyst. To date, engineering band gap and crystal facet, constructing semiconductor heterostructures and loading cocatalysts were adopted to expand absorbance range and improve photocatalytic avtivity of semiconductors. In this paper, we have introduced the basic principles and reviewed the research advances of photocatalytic water splitting. The paper is focused on the challenges and bottlenecks in improving the photocatalytic activity of semiconductors, and some coping strategies are also proposed based on relative group's research.
-
Key words:
- solar energy
- / semiconductors
- / photocatalytic
- / water splitting
- / hydrogen energy
-
0 引言
1972年日本科学家Fujishima和Honda等首次利用金红石二氧化钛单晶光电极实现了光电催化分解水制氢[1](称为Honda-Fujishima效应)。随后,科研人员又利用粉体半导体材料实现了光催化分解水制氢。无论光电催化还是光催化分解水制氢都是将太阳能转化成氢能,上述过程既实现了对可再生能源太阳能的利用,获得的氢能在使用过程中又避免了化石能源利用过程所带来的环境问题,因而被认为是最理想、最清洁的能源利用方式。
因外加电场的有无,光电催化和光催化分解水在反应过程上略有差异,但在这两个体系中核心的部分都是半导体材料的利用。本文以粉体光催化分解水制氢体系为例,首先介绍了半导体光解水过程的基本原理,接着简要概述了已开发的半导体光催化材料,并概述了半导体光解水体系的研究现状和面临的挑战;随后,介绍了提高半导体材料光催化活性的方法,同时分析了各种策略存在的挑战和瓶颈问题,并结合本课题组与其他研究小组的相关研究工作提出可能的应对策略;最后对半导体光解水制氢未来的发展进行了展望。
针对半导体光解水制氢这一研究领域,国内外很多研究学者都已做了很好的总结[2-13]。本文的目的是希望依据课题组多年来从事半导体光解水制氢研究所获得的对该领域的理解,提出该领域面临的瓶颈问题和解决问题的可行策略。因此,在介绍该领域研究进展时只做总体的概述、原理的分析和经典案例的介绍,不做具体研究案例的罗列(文中会指出相应的综述性论文,以供读者参考)。
1 半导体光解水制氢
1.1 水的分解
热力学上,氢气的燃烧反应是一个自发过程,其标准吉布斯自由能的变化(ΔG⊖)是-237 kJ·mol-1,如反应式(1)所示。而作为氢气燃烧逆反应的水的分解反应则是非自发过程,反应需要克服一个很大的能垒(反应式(2))。若将分解水的反应看作一个电池反应,则在标准状态下,系统的吉布斯自由能的变化等于对外所作的最大非膨胀功,即ΔG⊖=Wf, max,如果非膨胀功只有电功,则ΔG⊖=-nEF,式中n为电池输出电荷的物质的量,单位为mol,E为可逆电池的电动势,单位为伏特(V),F是法拉第常数(96 500 C·mol-1)。据此公式,可得依据水的分解反应组成的电池的电动势为-1.229 V,E为负值说明水的分解反应是非自发的。因此标准状态下若想实现水的分解反应,需要在电解池中进行,且至少加电压1.229 V。在实际利用电解池电解水的过程中,由于存在电极的极化作用所引起的过电势,无论在酸或碱的溶液中,水的分解电压都是在1.7 V左右,即所用的电压大于自发电池的电动势。
1.2 半导体的能带与光吸收
依据固体能带理论,在半导体材料中,电子填满了一些能量较低的能带,称为满带,最上面的满带称为价带;价带上面有一系列的空带,最下面的空带称为导带;价带和导带之间能态密度为零的区间称为禁带,并将导带的最低点和价带的最高点的能量之差称为带隙(禁带宽度),其单位是电子伏特(eV,1 eV=1.6×10-19 J),用符号Eg表示。光照可以激发半导体价带的电子到导带,形成电子-空穴对,这个过程称为半导体的本征光吸收。光子的能量是普朗克常量(h=6.63×10-34 J·s)和电磁辐射频率(ν/Hz)的乘积(E=hν),所以要实现半导体的本征光吸收,光子的能量要等于或大于半导体的禁带宽度,即hν≥Eg。因为有频率和波长的换算公式ν=c/λ(其中c是光在真空中的速度,c=3.0×108 m·s-1),所以就可变换出吸收光的波长与半导体禁带宽度的关系:hc/λ≥Eg,其中当hc/λ=Eg时所对应的光的波长称为半导体的本征吸收边。如果本征吸收边的波长以纳米(nm)为单位,则Eg的绝对数值ε=[(6.63×10-34 J·s)×(3.0×1017 nm·s-1)]/[λ×(1.6×10-19 J)]≈1 240/λ。
此外,半导体光解水的目的是要实现对太阳能的利用(太阳光谱能量的分布:紫外光,200~400 nm,能量约占太阳光谱总能量的5%;可见光,400~760 nm,能量约占太阳光谱总能量的45%;红外光,760~2 500 nm,能量约占太阳光谱总能量的50%)。遗憾的是,太阳光谱中红外光激发的半导体材料不能用于光解水过程(见1.4节论述)。因此,为在光解水反应中利用太阳能,开发具有可见光响应的半导体材料是必须的。
1.3 半导体光解水制氢过程
概括来说,半导体光解水制氢过程可分为3步(图 1):(1)半导体受光激发。半导体吸收能量等于或大于自身带隙的光子,价带中的电子被激发到导带中,在导带中就多了带负电荷的电子(e-),在价带中则留下带正电荷的空位(称之为空穴,h+),因两者成对出现,称之为光生电子-空穴对(即光生载流子);(2)光生载流子的复合与迁移。由于热振动或其他因素大部分光生电子和空穴会快速的复合掉(释放出光或热),只有少部分的光生载流子会由体相迁移到表面(迁移过程伴随着复合过程);(3)表面反应。到达表面的光生载流子仍有一部分会在表面发生复合,另一部分则被半导体表面吸附的水分子捕获,从而引发水的分解反应。
图1
半导体光解水制氢基本过程示意图
Figure1.
Scheme of the basic processes in photocatalytic water splitting using semiconductors
所有半导体材料都可以完成上述光解水过程中的步骤(1)和(2)历程,但仅有部分半导体可以完成步骤(3)水的分解,这是因为水的分解取决于电子-空穴的还原-氧化能力。电子-空穴的还原-氧化能力则取决于半导体材料的导带底和价带顶的位置:导带底的位置高于(或负于)氢的电极电势,则说明光致产生的电子有足够的还原能力来还原水成氢气;价带顶的位置低于(或正于)氧的电极电势,则说明光致产生的空穴有足够的氧化能力来氧化水成氧气。图 2中列举了一些半导体材料的能带结构示意图,并与氢和氧的电极电势相对比。需要说明的是,氢和氧电极电势的数值与溶液的pH值直接相关(根据能斯特公式,当pH2=p⊖,T=298 K时,φ(H+/H2)=-0.059 16pH;当pO2=p⊖,T=298 K时,φ(O2/H2O)=1.229-0.059 16pH),图 2中标明的电极电势为pH=0时的数值。
1.4 全分解水与半分解水
利用半导体材料将水同时分解为氢气和氧气的反应可以称为全分解水反应(反应式(2))[2]。要想实现光催化全分解水的过程,半导体的能带结构要包含水的分解电势,即导带底要高于水的还原电势,价带顶要低于水的氧化电势。且由于过电势的影响,半导体的带隙要大于水的理论分解电势(1.23 V),一般应在2 eV以上,对应的光吸收范围则在600 nm以下。由图 2可以看到,并不是所有半导体材料的能带结构都能满足全分解水的要求。
为了更好的研究半导体材料的光解水特性,在实际的实验过程中,研究人员更多的是对分解水的半反应进行研究,也就是在牺牲剂条件下进行光催化分解水反应[2, 14]。分解水的2个半反应如式(3)和(4)所示,当研究产氢半反应时,可以向溶液中加入甲醇、乳酸、三乙醇胺、硫化钠和亚硫酸钠等比水更容易被氧化的物质,以快速的消耗掉半导体上的光生空穴,加速光生电子的还原过程;当研究产氧半反应时,可以加入比水更容易被还原的物质(如硝酸银或碘酸钠),以快速的消耗掉半导体上的光生电子,加速光生空穴的氧化过程。
需要说明的是,既使某个半导体材料在牺牲剂条件下具备产氢和产氧的能力,也不能说明该材料就可以全分解水,这是因为牺牲剂条件下半导体材料光生载流子的有效分离在纯水条件下不一定能实现。也就是说,全分解水需要半导体材料同时满足反应的热力学(光生载流子能氧化和还原水)和动力学(光生载流子能有效分离)要求,且动力学要求是反应过程中的关键因素。
1.5 能量转换效率
在光解水反应中最常用的效率是表观量子效率,即参与反应电子数和入射光子数的比值:
φ=参与反应的电子数/入射光子数×100%
在光解水反应过程中,需要两个电子才能生成一个氢分子,所以表观量子效率也可表示为:
φ=(生成氢气的分子数×2)/入射光子数×100%
其中,入射光子数可通过辐照仪测得。
除了表观量子效率,太阳能-氢能转换效率可以更好的评估太阳能利用的程度,其表示式为:
η=生成氢气的化学能/入射太阳能×100%
其中,生成氢气的化学能等于生成氢气的速率(mol·h-1)与其在标准状态下的生成吉布斯自由能(237 kJ·mol-1)之积;入射太阳能等于太阳光辐射强度和辐射面积之积,如当大气质量(air mass,AM)为1.5时,即典型晴天时太阳光照射到一般地面的情况,其辐射强度为1 kW·m-2。
光解水反应中的表观量子效率和太阳能-氢能转化效率可以通过吸收波长对应起来,如图 3所示。根据推算,太阳能-氢能的转换效率至少要达到10%以上才具有工业化应用价值。要实现10%的太阳能-氢能转换效率,由图 3可以看到[12],半导体材料的光吸收范围要在600 nm以上、且表观量子效率要达到60%以上。遗憾的是,到目前为止仍没有材料能够达到对太阳能-氢能转换效率的最低要求。
2 半导体光催化材料的开发
2.1 无机化合物半导体
自Fujishima和Honda开启了半导体光催化这一研究领域以来,研究人员的工作重心就一直集中在开发和研究光催化材料上。40多年以来,人们基于元素周期表,已经找出了数百种能够用于光催化过程的光催化材料,且绝大多数光催化材料为无机化合物半导体,如金属氧化物、硫化物、氮化物、磷化物及其复合物等。如图 4所示[2],在已知的能够用于光催化过程的半导体材料的元素组成有以下的特点:利用具有d0或d10电子结构的金属元素和非金属元素构成半导体的基本晶体结构,并决定其能带结构;碱金属、碱土金属或镧系元素可以参与上述半导体晶体结构的形成,但对其能带结构几乎无影响;一些金属离子或非金属离子可以作为掺杂元素对半导体的能带结构进行调控;贵金属元素一般作为助催化剂使用[7]。
根据组成半导体化合物的金属离子(阳离子)的电子特性,单一光催化材料可以分为两大类,一类是金属离子的d电子轨道处于无电子填充状态(d0),如Ti4+、Zr4+、Nb5+、Ta5+和W6+;另一类是金属离子的d电子轨道处于满电子填充状态(d10),如In3+、Ga3+、Ge4+、Sn4+、和Sb5+。与d0金属离子相配的非金属元素主要是氧元素,它们之间组合成的氧化物如TiO2[15-16]、ZrO2[17]、Nb2O5[18]、Ta2O5[19]和WO3[20]都是被广泛应用的光催化剂。如前所述,一些碱金属、碱土金属或其它金属离子可以引入到上述化合物中组成一些盐类,并且这些盐类也被证明具有良好的光催化能力,如钛酸盐:SrTiO3[21], A2Ti6O13(A=Na,K,Rb)[22-23],BaTi4O9[24], A2La2Ti3O10(A=K, Rb, Cs)[25], K2Ti4O9[26];铌酸盐:A4Nb6O17(A=K,Rb)[27],Sr2Nb2O7[28],Cs2Nb4O11[29],Ba5Nb4O15[30];钽酸盐:ATaO3(A=Na,K)[31-32],MTa2O6(M=Ca,Sr,Ba)[33],Sr2Ta2O7[28],ACa2Ta3O10(A=H,Na,Ca)[34],A2SrTa2O7·nH2O(A=H,K,Rb)[35],K3Ta3B2O12[36],Ba5Ta4O15[37];钨酸盐:Ag2W2O7[38], Bi2W2O6[39], CuWO4[40], SnWO4[41], ZnWO4[42], ZrW2O8[43], Na2W4O13[44];以及钒酸盐:Ag3VO4[45],BiVO4[46]。与d10金属离子相配的非金属元素主要也是氧元素,它们之间组合成的氧化物如In2O3[47],Ga2O3[48],GeO2[49]和SnO2[50]也都被应用于光催化反应中。d10金属离子也可组成具有光催化活性的盐类, 如铟酸盐:MIn2O4(M=Ca,Sr)[51-52],AInO2(A=Li,Na)[53],LaInO3[54];镓酸盐:ZnGa2O4[55],锗酸盐:Zn2GeO4[56];锡酸盐:M2SnO4(M=Ca,Sr)[51];以及锑酸盐NaSbO3[51]。
依据组成半导体材料的非金属元素(阴离子)的类型,单一光催化剂又可分为氧化物,硫化物,氮化物,氧氮化物和氧硫化物等。除了上段提到的氧化物之外,CuO[57],Cu2O[58],Fe2O3[59]等也是常见的光催化材料。硫化物除了简单的CdS[60]和ZnS[61]可作为光催化材料外,一些多元硫化物也可用于光催化作用中,如ZnIn2S4[62],AgInZn7S9[63],AgGa0.9In0.5ZnS2[64]和Cu0.25Ag0.25In0.5ZnS2[65]。硫化物大多需要在有牺牲剂(通常为Na2S和Na2SO3)参与的情况下才能分解水制氢,并且都有很高的催化效率,但因为硫化物存在的光腐蚀效应,即使有牺牲剂存在,硫化物在长时间的光催化反应中也不稳定,因此解决硫化物光催化材料的稳定性是一个重要的研究方向。氮化物应用于光分解水反应的有Ge3N4[66]、Ta3N5[67]等,其中Ge3N4是第一种被报道的具有全分解水能力的非金属氧化物。一般稳定的金属氧化物带隙相对较大,只能吸收紫外光,而氧氮金属化合物大多具有较小的带隙,具有可见光吸收的特性,并且展现出较高的光催化活性,如TaON就具有较高的光解水产氧的催化活性(在牺牲剂存在下)[68]。虽然金属硫化物由于光腐蚀效应不能进行光解水产氧,但金属氧硫化物Sm2Ti2O5S2却可以光解水产氧[69]。
2.2 聚合物半导体
2009年王心晨等报道了一种完全由非金属元素组成的聚合物半导体材料g-C3N4[70],该材料具有类似石墨的层状结构,C、N原子通过sp2杂化形成一个高度离域的π共轭电子能带结构,禁带宽度为2.7 eV,并且导带底在氢的氧化还原电位之上,价带顶在氧的氧化还原电位之下。因此,g-C3N4可以在牺牲剂(如三乙醇胺或硝酸银)存在下光催化分解水产氢或产氧。令人惊喜的是,最近的研究表明g-C3N4经过修饰后可以实现全分解水,譬如:(1)王心晨等通过原位光沉积方法获得了可以全分解水的Pt-PtOx/g-C3N4[71],其中Pt作为产氢助催化剂,PtOx作为产氧助催化剂;(2)康振辉等发现由碳量子点和g-C3N4组成的复合材料可以通过两步双电子步骤实现全分解水[72]:(ⅰ) 2H2O → H2O2+H2(g-C3N4光催化);(ⅱ) 2H2O2 → 2H2O+O2(碳量子点催化),且其在AM 1.5G太阳光模拟器照射下全分解水的太阳能-氢能转换效率达到了2%;(3)陈等则通过Na离子掺杂对g-C3N4进行了去质子化[73],从而抑制了光解水过程中H2O2的产生,实现了一步全分解水;(4) g-C3N4还可与其他半导体材料配合实现Z机制全分解水[74-75](有关Z机制全分解水的讨论详见3.2节)。
由于聚合物的材料特性,g-C3N4展现出比表面积小、产生的光生载流子的激子结合能高且复合严重等特点,这都不利于其在光解水制氢中的应用,因而各种提高其光催化活性的方法被报道[76-79]。这些方法主要有:(1)制备方法的改进。王心晨等利用手性介孔氧化硅为模板制备了螺旋g-C3N4纳米棒,该螺旋纳米棒展现出特殊的光学特性和很好的光催化活性[80]。王心晨等以HNO3对g-C3N4进行质子化和解聚合作用,可以获得稳定的g-C3N4胶体悬浮液,从而可以制备出结合性能很好的g-C3N4薄膜电极[81]。刘岗等利用g-C3N4具有层状结构的特点,以空气为氧化剂,通过对体相g-C3N4进行简单的二次热处理可将其剥离成厚度只有2 nm左右的纳米片,并大幅提高其光催化活性[82]。(2)掺杂。刘岗等利用硫掺杂和随之引起的量子尺寸效应对g-C3N4的能带结构进行调控,使其光解水制氢效率提高8倍左右[83]。王心晨等发现通过非金属元素B、I对g-C3N4进行掺杂也可提高其光催化活性[84-85]。(3)与半导体复合。g-C3N4可与多种半导体材料组成Ⅱ型半导体异质结构,从而提高其光催化活性[76-79];(4)王心晨等通过共聚合方式把特定的有机官能团(如苯环、吡啶、噻吩等)嫁接到g-C3N4的骨架中,制备出一系列π共轭体系连续可调的g-C3N4基聚合物半导体光催化剂[86-89]。
共轭聚合物半导体具有可调的组分和电子结构[90],某些能带结构合适的共轭聚合物半导体材料也可被应用于光解水制氢反应中,譬如,苯并噻二唑聚合物[91]、二苯并噻吩聚合物[92]、甲亚胺聚合物[93]等都展现出良好的光解水产氢性能。
2.3 单质半导体
除了2.1和2.2节中的无机化合物半导体和聚合物半导体以外,最近的研究表明一些具有可见光吸收的单质元素(Si[94],P[95],B[96],Se[97]和S[98])也具有一定的光催化活性,这丰富了光催化材料家族,但这些单质光催化剂的催化活性都很低,还需要进一步研究以提高它们的光催化效率。
3 半导体光解水体系的研究现状与挑战
3.1 一步全分解水的研究进展与挑战
一步全分解水指的是在单一光催化材料上同时进行产氢和产氧反应。目前报道较多的能进行一步全分解水的材料主要是负载了助催化剂的宽带隙半导体,且大都是在紫外光照射下进行反应。这是因为:(1)紫外光激发宽带隙半导体所产生的电子-空穴具有高的还原-氧化能力,易于突破全分解水的热力学限制;(2)助催化剂可以促进半导体材料光生电子-空穴的有效分离,且有时可以抑制全分解水的逆反应(氢气和氧气化合成水)。研究表明,溶液的pH值对材料的全分解水性能影响较大,某些材料体系在碱性环境中更有利于全分解水的进行:Takata等发现[99],加入碳酸钠后大部分体系的全分解水性能都有所提高;Ikeda等报道了在KOH水溶液(0.1 mol·L-1)中NiO/K2La2Ti3O10全分解水效率可以达到30%[100]。负载NiO助催化剂的La-NaTaO3光催化剂在NaOH溶液中、270 nm单色光下的一步全分解水量子效率可以达到56%[101],是紫外光照射下、一步全分解水的标志性研究成果。
其他能进行一步全分解水的宽带隙半导体材料主要是包含d0或d10金属离子的氧化物及其盐类(如SrTiO3、K2La2Ti3O10、K4Nb6O17、Ba5Nb4O15、Cs2Nb4O11、K3Ta3B2O12、Ba5Ta4O15等),相关综述文章中已进行了很好的总结[2-3],在此就不再一一罗列。钽酸钡盐混合物Ba5Ta4O15/Ba3Ta5O15/BaTa2O6[102]、宽带隙固溶体Bi0.5Y0.5VO4[103]等在紫外光下也展现出很好的全分解水性能。研究发现,利用晶体晶面的各向异性可以实现光生载流子的有效分离(见4.1.3.2节),譬如在18个晶面暴露的SrTiO3中,在各向异性的晶面上分别负载产氢和产氧助催化剂后,可以大幅提高其全分解水效率[104]。
少数窄带隙半导体材料在可见光激发下也可进行一步全分解水反应,如钽基半导体TaON[105]、LaMgxTa1-xO1+3xN2-3x[106]、In0.9Ni0.1TaO4[107]、CaTaO2N[108]等[109],以及铜基半导体Cu2O[110]、CuFeO2[99]等。负载Rh2-x CrxO3助催化剂的GaN-ZnO固溶体在420~440 nm可见光照射下的全分解水表观量子效率达到2.5%[111],这在光催化分解水研究领域是一个里程碑工作;对GaN-ZnO固溶体组成进行优化后,这一表观量子效率可达5.9%[112]。GaN-ZnO可以被制备出有极性晶面暴露的纳米棒,并应用于可见光全分解水[113]。除GaN-ZnO之外,ZnGeN2-ZnO也可在可见光下实现全分解水[114]。Ge3N4是首个被发现具有全分解水活性的氮化物单体材料[115],最近的研究表明g-C3N4在恰当的修饰条件下也可以实现一步全分解水(如在2.2节中所述)。除了自身能带结构适合可见光全分解水外,半导体材料还可以通过以下两种方式获得可见光全分解水能力:(1)对宽带隙半导体掺杂。Nb掺杂AgTaO3后降低了其导带低位置,使得AgTa0.7Nb0.3O3在可见光下能够全分解水[116];(2)适当增大窄带隙半导体的带隙。利用量子尺寸效应,抬升了Co3O4量子点的导带低位置,从而使其满足了全分水的热力学需求[117]。
下面对一步全分解水研究中的几个关键问题进行说明。单一半导体材料的全分解水反应始终存在一个挑战性难题没有得到解答,即很多半导体的能带结构满足热力学上全分解水的要求,但是实际上并不能实现全分解水。譬如,具有相似带隙的锐钛矿相、金红石相和板钛矿相TiO2,只有金红石相展现出全分解水的性能[118]。最近,李灿等通过对不同晶相TiO2全分解水反应的深入研究发现[119]:(1)热力学方面,锐钛矿相和板钛矿相TiO2的价带附近存在很多浅束缚态能级直接影响了其氧化水的能力,当强光照处理去掉浅束缚态能级后即可实现全分解水,而金红石相TiO2价带附近无任何浅束缚态能级,具有足够的能量实现全分解水;(2)动力学方面,不同物相TiO2的氧化水过程经历了不同的反应中间体路径,锐钛矿相和板钛矿相TiO2的光生空穴生成羟基自由基,通过羟基自由基的进一步耦合才能放出O2,而金红石相TiO2的光生空穴则经过超氧自由基的中间体物种,更容易直接生成O2。该研究工作再次说明,热力学和动力学因素相互协同共同影响光催化全分解水过程。
更多的实验发现,要想实现一步全分解水反应,负载助催化剂对于绝大多数半导体材料是必需的,这说明在全分解水过程所面临的热力学和动力学限制中,突破动力学的限制是关键。助催化剂的结构对半导体全分解水的性能有显著影响,如核壳结构的Rh/Cr2O3就要比混合氧化物Rh2-xCrxO3对GaN-ZnO固溶体全分解水性能的提升作用要大[120];MoS2和N掺杂石墨烯组成的p-n结助催化剂将CdS的全分解水性能大幅提高[121]。有关助催化剂的研究将在4.2.2节中做详细介绍。针对全分解水过程,需要说明的是产氢助催化剂的负载是关键;且在全分解水过程中应用最广谱的产氢助催化剂是NiO[2-3],而非牺牲剂条件下最广谱的产氢助催化剂Pt。这是因为助催化剂除了作为反应的活性中心之外,也可作为逆反应的活性中心,因此,具有催化氢气和氧气化合作用的Pt等贵金属催化剂就不适合做全分解水的助催化剂。
全分解水所产生的H2和O2量并不是随着时间呈线性增加,而是表现出抛物线下降的趋势。这是因为:(1)全分解水反应是在密闭真空系统下进行的,产生的H2和O2量随着反应时间延长而增加,从而增加了逆反应;(2)光催化剂随着反应时间延长而失活。研究发现,氮化物和氧氮化物光催化剂的失活主要是由于O2氧化带来的氮元素流失所造成的。在负载产氢助催化剂的基础上,再负载产氧助催化剂(Mn3O4、RuO2、IrO2等)既可以提高全分解水的效率,也可以有效避免氮化物和氧氮化物光催化剂的失活。
3.2 Z机制体系全分解水的研究进展与挑战
为了解决一步全分解水难度大、效率低的问题,可以采用两步全分解水的策略,该过程是将产氢光催化剂与跟它能级匹配的产氧光催化剂相耦合(两者之间能级错排),并配以氧化-还原电对(如IO3-/I-和Fe3+/Fe2+),三者组成Z机制体系进行全分解水[122](图 5)。在Z机制体系中,产氢光催化剂上的光生电子还原H2O生成H2,其上的光生空穴则将氧化-还原电对中的还原态离子氧化(如将I-氧化成IO3-);产氧光催化剂上的光生空穴氧化H2O生成O2,其上的光生电子将氧化-还原电对中的氧化态离子还原(如将IO3-还原成I-)。由此,在Z机制体系中,氧化-还原电对可以实现氧化态和还原态的循环,在该电对的循环过程中,实现了水的全分解。Z机制全分解水的优点是:(1)将两种不能全分解水的光催化剂耦合在一起实现了全分解水,是一条解决大多数半导体材料不能全分解水问题的可行途径;(2)在Z机制体系中,两种半导体材料的能带结构只要能满足分解水半反应的要求就行,这为开发可见光全分解水体系提供了可能。
目前已报道的Z机制全分解水体系中,产氢光催化剂主要是Ta基(TaON、CaTaO2N、BaTaO2N等)和Ti基(SrTiO3等)半导体以及它们的改性材料,产氧光催化剂主要是WO3和BiVO4,氧化-还原电对主要是IO3-/I-和Fe3+/Fe2+ [122, 124]。其中代表性的Z机制全分解水体系是:Pt-ZrO2/TaON作为产氢光催化剂、Pt-WO3作为产氧光催化剂、IO3-/I-作为氧化还原电对,该体系在420 nm单色光下全分解水的表观量子效率可达6.3%[125];Pt-MgTa2O6-xNy /TaON作为产氢光催化剂、PtOx-WO3作为产氧光催化剂、IO3-/I-作为氧化还原电对,该体系在420 nm单色光下全分解水的表观量子效率达到目前文献报道的最高值6.8%[126]。
虽然理论上可以任意将能级匹配的产氢和产氧光催化剂搭配氧化-还原电对组成Z机制全分解水体系,但是能组成Z机制全分解水体系的半导体材料并不是很多。这主要是因为在Z机制体系中光解水反应与氧化-还原电对反应的竞争引起的,以配有IO3-/I-氧化-还原电对的Z机制体系为例,具有强氧化能力的产氧光催化剂上的空穴既能氧化H2O,也能氧化I-,而且氧化I-在热力学上更容易;而具有强还原能力的产氢光催化剂上的电子既能还原H2O,也能还原IO3-,而且还原IO3-在热力学上更容易。所以上述反应的竞争会大大降低全分解水的效率,甚至根本不能全分解水,只是实现了对IO3-/I-电对的氧化还原循环。研究发现,WO3上的光生电子可以实现对IO3-的优先还原[125],使得产氢光催化剂上的光生电子主要参与H2O的还原反应,从而触发整个Z机制反应过程。虽然WO3对IO3-的优先还原的机理还不太明确,但WO3已经被证明是最适合用于Z机制体系的产氧光催化剂,其可以与多种具有可见光响应的产氢光催化剂相耦合,从而实现可见光下的全分解水。
除了氧化-还原电对的竞争反应以外,Z机制全分解水过程还受到pH值和助催化剂的影响。譬如,在以Fe3+/Fe2+作为氧化-还原电对的Z机制体系中,较低的pH值(~2.4)是必需的,以抑制Fe3+的水解[127-128]。助催化剂对于Z机制的顺利运行也起到关键作用,且也要为产氢光催化剂和产氧光催化剂分别负载产氢助催化剂(Pt、Ni、Rh)和产氧助催化剂(RuO2、IrO2)。
为避免氧化-还原电对在Z机制全分解水体系中的副作用,Kudo等建立了无氧化-还原电对参与的Z机制全分解水体系。在该体系中,以Ru-SrTiO3:Rh和BiVO4分别作为产氢和产氧光催化剂[129]。实验发现,该体系全分解水性能受pH值(最佳值在pH=3.5)影响很大。这是因为:Ru-SrTiO3:Rh和BiVO4的等电点分别为pH=4和pH=2,则在pH=3.5的溶液中,Ru-SrTiO3:Rh表面带正电荷,BiVO4表面带负电荷,二者受静电吸引作用而有了较紧密的接触(图 6a),从而触发了两者之间的Z型电子转移(图 6b)。需要说明的是,在上述Z机制体系中,Rh元素的存在是必需的,Rh作为电子转移介质可以认为在其中起到了氧化-还原电对的作用。
为了区分有氧化-还原电对参与的Z机制体系,上述没有氧化-还原电对参与的Z机制体系可称之为矢量Z机制体系。矢量Z机制体系将在4.2.1.1中做进一步介绍。
3.3 牺牲剂条件下分解水制氢的研究进展与挑战
鉴于光催化全分解水面临的巨大挑战,到目前为止研究最多的分解水反应是在牺牲剂条件下进行的,特别是牺牲剂条件下的分解水制氢。有研究学者认为牺牲剂条件下的分解水制氢也有可能实现工业化应用,这需要两个前提:(1)太阳能-氢能转换效率足够高;(2)牺牲剂廉价易得(如Na2S和Na2SO3),或者以污染物为牺牲剂(如偶氮染料、H2S等)。
40多年以来,研究人员提出并发展了多种策略以提高分解水制氢的效率,本文将在第4节中介绍这些策略的研究现状并讨论各个策略所面临的挑战。为简略表示,在接下来的论述中“牺牲剂条件下的光催化分解水制氢/制氧”均表述为“光解水制氢/制氧”,而同时产生氢气和氧气的分解水过程表述为全分解水。
3.4 牺牲剂条件下光解水制氧的研究进展与挑战
因为氧化水的反应是分解水的速决步骤,所以牺牲剂条件下的分解水制氧研究有助于更好的理解半导体材料的氧化特性,以促进全分解水(一步过程和Z机制)反应的研究。光解水制氧与光解水制氢同样受到热力学和动力学上的双重限制,且光解水制氧比光解水制氢困难的多,这主要是由于以下原因造成的[130]:(1)氧化水的反应需要2个水分子和4个电子参与,这需要光催化剂具有足够深的价带以提供强氧化能力的空穴,且光催化剂还应具有容纳多电子的能力;(2)光解水制氧的反应时间是秒级(0.27 s)远远长于毫秒级的光解水制氢反应;(3)产生的O2分子容易吸附到光催化剂表面的氧空位上,从而抑制了制氧反应的持续进行;(4)氧化水过程中产物复杂,容易生成中间产物,譬如生成H2O2、O2-等,这也限制了O2的生成。
由于上述原因的限制,使得能够稳定进行光解水制氧且效率较高的半导体材料并不多,而这些半导体材料往往表现出以下特点:(1)具有由金属元素s或p轨道与O2p轨道杂化而成的价带的半导体材料。在这一类半导体中,由于轨道间的互斥作用,使得杂化所形成的价带比较离散,有利于光生空穴的扩散与转移。譬如,BiVO4(由Bi6s和O2p轨道杂化组成价带)[131]、Zn3V2O8(由Zn3d和O2p轨道杂化组成价带)[132]等,而具有由Ag4d和O2p轨道杂化组成价带的Ag3PO4则展现出很高的光解水制氧效率[133];(2)具有共角MO6八面体结构的半导体材料。在这一类半导体中,相邻的MO6八面体通过共用顶角氧原子而联结形成晶体,这也为载流子转移提供了通道,譬如拥有WO6八面体结构的WO3和BiWO6[134]就是很好的光解水制氧材料。上述光解水制氧材料所具有的特殊电子结构和原子结构还可以体现在半导体材料的特定晶面上,譬如,研究表明WO3的(200)和(020)晶面[135]、BiVO4的(040)晶面[136]在氧化水反应中具有重要作用。
除了半导体材料自身具有光解水制氧能力外,负载助催化剂也是提高光解水制氧效率的关键(有关助催化剂的论述详见4.2.2节),常用的光催化分解水制氧助催化剂有贵金属氧化物RuO2和IrO2。钴、锰系列氧化物在电催化分解水制氧过程中展现出很高的效率,近年来它们也作为助催化剂被广泛应用于光催化分解水制氧。g-C3N4由于存在自身氧化的问题,使得其光催化制氧效率很低,王心晨等通过负载钴系列助催化剂(分子钴[137]、Co3O4[138]、CoSe[139]、Co(OH)2[140])可使g-C3N4的光解水制氧效率大幅提高。
最近的研究表明石墨烯可以显著改善半导体材料的光解水制氧特性[141],这主要表现在以下几个方面:(1)扩展半导体材料的光吸收。当石墨烯与TiO2相结合时,石墨烯中的p电子可以与TiO2中的表面Ti原子结合形成Ti-O-C键,从而扩展了TiO2的光吸收范围[142-144];(2)石墨烯作为优良的导电材料可以极大的促进半导体材料(如WO3[145]、BiWO6[146]、Fe2O3[147]等)中的光生载流子转移。(3)石墨烯作为载体,在制备石墨烯/半导体异质结构时可以控制半导体的形貌和粒径[148],以及显著增大异质结构的比表面积[149];(4)提高半导体材料的光催化稳定性。Ag3PO4虽然具有优异的光解水制氧性能,但也存在着光腐蚀现象,而当Ag3PO4与石墨烯复合后,其光催化稳定性得到显著增强[150-151]。
过去几十年以来,相比于光解水制氢取得的丰硕成果,光解水制氧的研究成果略显单薄,这在某种程度上也限制了光催化全分解水的研究。因此,加强光解水制氧领域的研究是很有必要的。
4 提高半导体光解水性能的研究现状与挑战
依据光解水过程中对半导体材料的要求,大多数单一光催化材料在光解水应用中都面临着热力学和动力学上的双重限制,所以几十年以来科研人员为突破此双重限制进行了大量的机理和实验上的研究。到目前为止,突破口主要体现在两个方面:一是通过对半导体的能带结构进行调控,以主要突破热力学上的限制;二是通过构建复合材料(如半导体异质结构和助催化剂的负载)来加速半导体上光生载流子的转移和分离,以主要突破动力学上的限制。下面将分别介绍这些调控策略、改性方法的作用机理及其所取得的重要研究进展,并对他们所面临的挑战进行讨论。
4.1 半导体能带调控策略与挑战
4.1.1 掺杂与空位调控
追求具有可见光吸收的光催化剂是光催化应用的必然选择。除了利用本身具有可见光吸收的半导体材料以外,对带隙较大的半导体进行掺杂也是开发可见光响应的光催化材料的重要策略。掺杂的主要特征是破坏了晶体原子排列的周期性,引起晶体周期势场的畸变,其结果是在禁带中引进新的电子态,称为缺陷态或杂质态,这些缺陷态或杂质态就可以引起宽带隙半导体材料对可见光的吸收。
4.1.1.1 金属元素掺杂
最初是通过金属元素掺杂来实现宽带隙半导体对可见光的吸收。依据半导体能带理论,因掺杂金属元素价态和半导体中金属元素价态的差异,金属元素对半导体材料进行掺杂后可在半导体的带隙中产生施主能级或受主能级。因能级对电子束缚作用的强弱,施主(或受主)能级存在深能级或浅能级两种状态。如图 7所示,浅施主能级存在于半导体导带下方(图 7a),浅受主能级存在于半导体价带上方(图 7b),而深施主能级在半导体能带中靠近价带一侧(图 7c),深受主能级在半导体能带中靠近导带一侧(图 7d)。电子会在施主能级(或价带)与导带(或受主能级)间发生跃迁,其中由浅施主能级向导带的跃迁(或由价带向浅受主能级的跃迁)为浅跃迁,而电子由深施主能级向导带的跃迁(或由价带向深受主能级的跃迁)为深跃迁。因浅跃迁或深跃迁所要跨过的能垒都要小于半导体的本征带隙,所以可见光都可以激发浅跃迁、且大多数情况下也都可以激发深跃迁。
 图7
金属掺杂对半导体能带结构的影响: (a)掺杂形成浅施主能级; (b)掺杂形成浅受主能级;
(c)掺杂形成深施主能级; (d)掺杂形成深受主能级
Figure7.
Effects of metal doping on band structure of semiconductors: (a) shallow donor level,
(b) shallow acceptor level, (c) deep donor level and (d) deep acceptor level formed by metal doping
图7
金属掺杂对半导体能带结构的影响: (a)掺杂形成浅施主能级; (b)掺杂形成浅受主能级;
(c)掺杂形成深施主能级; (d)掺杂形成深受主能级
Figure7.
Effects of metal doping on band structure of semiconductors: (a) shallow donor level,
(b) shallow acceptor level, (c) deep donor level and (d) deep acceptor level formed by metal doping
早在1982年,Borgarello等就发现Cr掺杂的TiO2就能够实现在可见光照射下(400~550 nm)的全分解水反应[152]。此后,所有的过渡金属元素都被用来掺杂到宽带隙半导体材料中,以期望扩展其可见光吸收并提高其可见光活性。实验结果表明,金属掺杂确实可以在宽带隙半导体能带中引入杂质能级,这些杂质能级也能够带来可见光的吸收,但却不一定带来可见光下光催化效率的提高。针对这一问题,目前被广泛接受的解释是金属掺杂剂有可能会成为半导体材料内光生电子和空穴的复合中心(特别是形成的深杂质能级),从而不能提高(甚至降低)半导体材料的可见光活性;且半导体材料的可见光活性受掺杂元素种类、掺杂方法(化学合成、气氛热处理、磁控溅射等)、掺杂量、掺杂位置(取代位或间隙位)以及掺杂分布(体相或表面)的影响。
4.1.1.2 非金属元素掺杂
受制于金属掺杂的瓶颈问题,自2001年Asahi等报道了氮掺杂TiO2具有可见光活性以来,非金属掺杂研究逐渐成为主流[153],在过去十几年关于非金属掺杂的研究包括合成、表征、机理研究与性能等,取得了重要进展。其中以非金属掺杂的TiO2研究最为广泛,下面我们就以TiO2为例介绍非金属掺杂。
根据半导体能带理论,半导体的能带是组成原子的原子轨道通过杂化形成的新的分子轨道。而绝大多数过渡金属氧化物(包括TiO2)的导带与价带主要分别由金属的3d轨道与氧的2p轨道构成。再依据分子轨道理论,当用一种电负性比氧低的元素取代了TiO2晶格中的部分氧原子后,这用作掺杂剂的元素的电子轨道与氧的2p轨道杂化形成的新的分子轨道的能量要比氧的2p轨道的能量低,也就是说,这新的分子轨道所形成的价带顶要比氧的2p轨道形成的价带顶高。而构成导带的金属钛的3d轨道没有变化,由此可见,通过电负性比氧低的元素的掺杂,使得二氧化钛的导带底不变,价带顶被提高了,从而减小了TiO2的带隙。实际上,绝大多数的非金属元素的电负性都要比氧小,都可以用来实现通过掺杂减小TiO2带隙的目的(非金属元素的Paul电负性,B:2.04;C:2.55;N:3.04;O:3.44;F:3.98;Si:1.90;P:2.19;S:2.58;Cl:3.16;Br:2.96;I:2.66)。此外,一个合适的掺杂剂元素除了有电负性的要求之外,它的离子半径应该与氧离子的半径相近,以实现原子的替代掺杂。在众多的非金属元素掺杂中,氮掺杂的研究最为深入,相应提出的机理也最多。而最有争议的机理在于氮掺杂引起的可见光吸收,除了上述Asahi等依据杂化理论提出氮掺杂使TiO2价带顶提高、带隙减小引起可见光吸收以外,Irie等提出可见光吸收是位于价带顶之上N2p轨道形成的孤立局域化能级的贡献[154],而另一些研究人员认为氮掺杂所伴生的位于导带底的氧空位局域化能级带来了可见光吸收,氮掺杂只是起到稳定氧空位的作用[155]。
非金属掺杂对半导体能带结构的改变同样受到掺杂元素种类、掺杂方法、掺杂量、掺杂位置以及掺杂分布的影响。譬如,我们在最近的研究中发现B掺杂TiO2微米球的能带结构受到B元素掺杂分布(B元素位于TiO2微米球核心或表面)的显著影响,且这种影响直接带来了TiO2微米球在光解水产氢和产氧性能上的翻转[156]。
依据半导体的禁带宽度与吸收光的波长的关系,带隙减小就对应着本征吸收边的红移。而在大多数的氮掺杂TiO2中,TiO2的本征吸收边并没有整体的红移,而是在长波长处出现了肩膀状的吸收,如图 8b中ⅴ,ⅵ曲线所展示的P25型TiO2和氮掺杂P25的吸收光谱。这种肩膀状吸收的原因可能是由于氮原子只是在P25的表层取代了部分氧原子,氮原子轨道与氧原子轨道之间的杂化作用非常弱,使得氮原子轨道无法参与价带的形成,不能有效的抬高价带顶,只能在带隙中形成一些孤立的能级。而如果氮原子能够在TiO2体相和表面均匀的掺杂,那么氮原子轨道与氧原子轨道之间的杂化作用就会很强,从而能够有效的提高TiO2的价带顶。为了实现这种理想的均相氮掺杂,刘岗等选用了钛酸碱金属盐(Cs0.68Ti1.83O4)和其质子化钛酸(H0.68Ti1.83O4)作为掺杂基体。如图 8a所示,Cs0.68Ti1.83O4和H0.68Ti1.83O4都是纤铁矿结构,基本结构单元是由共边的两层TiO6八面体网格嵌套而成的纳米片。在钛酸盐结构中,这些纳米片被碱金属离子或质子隔离,片与片之间形成层内空间。而正是这种特有的层状结构为氮原子在整个体相的扩散和掺杂提供了便利,如图 8b中ⅰ,ⅱ,ⅲ,ⅳ曲线所示,氮掺杂的Cs0.68Ti1.83O4和H0.68Ti1.83O4的光吸收边都出现了整体的红移[157]。这种均相掺杂的策略可以应用到其它具有层状结构的半导体材料,但对大多数不具有层状结构的半导体材料却无能为力。
 图8
(a) 纤铁矿型钛酸盐的晶体结构示意图; (b)不同样品的吸收光谱: (ⅰ) Cs0.68Ti1.83O4; (ⅱ) Cs0.68Ti1.83O4-xNx; (ⅲ) H0.68Ti1.83O4; (ⅳ) H0.68Ti1.83O4-xNx; (ⅴ)商业P25型二氧化钛; (ⅵ)氮掺杂P25; (b)中的插图是(ⅰ), (ⅱ), (ⅲ)和(ⅳ) 4个样品的光学照片[157]
Figure8.
(a) Crystal structure model of typical lepidocrocite-type titanate; (b) UV-visible light absorption spectra of (ⅰ) Cs0.68Ti1.83O4, (ⅱ) Cs0.68Ti1.83O4-xNx, (ⅲ) H0.68Ti1.83O4, (ⅳ) H0.68Ti1.83O4-xNx, (ⅴ) commercial P25 titania, and (ⅵ) nitrogen-doped P25; Inset is the photos of samples (ⅰ), (ⅱ), (ⅲ) and (ⅳ)[157]
图8
(a) 纤铁矿型钛酸盐的晶体结构示意图; (b)不同样品的吸收光谱: (ⅰ) Cs0.68Ti1.83O4; (ⅱ) Cs0.68Ti1.83O4-xNx; (ⅲ) H0.68Ti1.83O4; (ⅳ) H0.68Ti1.83O4-xNx; (ⅴ)商业P25型二氧化钛; (ⅵ)氮掺杂P25; (b)中的插图是(ⅰ), (ⅱ), (ⅲ)和(ⅳ) 4个样品的光学照片[157]
Figure8.
(a) Crystal structure model of typical lepidocrocite-type titanate; (b) UV-visible light absorption spectra of (ⅰ) Cs0.68Ti1.83O4, (ⅱ) Cs0.68Ti1.83O4-xNx, (ⅲ) H0.68Ti1.83O4, (ⅳ) H0.68Ti1.83O4-xNx, (ⅴ) commercial P25 titania, and (ⅵ) nitrogen-doped P25; Inset is the photos of samples (ⅰ), (ⅱ), (ⅲ) and (ⅳ)[157]
为了实现在非层状半导体材料中可见光吸收的整体红移,刘岗等在最近的工作中又提出了梯度掺杂的策略[158]。如图 9a所示,通过水热方法和随后的热处理过程原位获得了具有间隙B梯度掺杂的锐钛矿TiO2微米球(B的掺杂量由TiO2微米球表面到体相逐渐减小)。由于B元素位于TiO2晶格中的间隙位,弱化了晶格中的Ti-O键,使得随后的氮掺杂过程中氮元素更容易取代晶格氧。如图 9b,c所示,具有梯度B/N元素掺杂的锐钛矿TiO2显现出特有的红色,其光吸收边也扩展到大约700 nm,实现了对可见光谱的全吸收。红色TiO2中B/N的梯度掺杂和相应的能带结构示意图见图 9d。这种通过间隙原子弱化金属原子与氧(M-O)的键合实现替代晶格氧的掺杂原子进入体相的新方法,成功突破了在非层状结构材料中实现掺杂原子体相掺杂的难题。
 图9
(a) 红色TiO2微米球的扫描电镜照片; (b)红色TiO2样品的光学照片; (c)白色和红色TiO2的吸收光谱(d)红色TiO2的梯度能带结构示意图[158]
Figure9.
(a) SEM image of a red TiO2 microsphere; (b) optical photograph of the prepared red TiO2 sample; (c) UV-visible absorption of the white TiO2 and red TiO2; (d) schematic of band structures of the red TiO2 with a bandgap gradient[158]
图9
(a) 红色TiO2微米球的扫描电镜照片; (b)红色TiO2样品的光学照片; (c)白色和红色TiO2的吸收光谱(d)红色TiO2的梯度能带结构示意图[158]
Figure9.
(a) SEM image of a red TiO2 microsphere; (b) optical photograph of the prepared red TiO2 sample; (c) UV-visible absorption of the white TiO2 and red TiO2; (d) schematic of band structures of the red TiO2 with a bandgap gradient[158]
4.1.1.3 空位
对半导体进行异质原子的掺杂通常也会在材料晶格中引入空位。空位是半导体材料中的本征点缺陷之一,譬如氧化物中的氧空位、氮化物中的氮空位等。空位可以在半导体的带隙中引入缺陷能级(譬如,氧空位的缺陷能级位于导带下方),从而引起半导体材料的可见光吸收。有关空位(特别是氧空位[159-160])的研究非常多,且大多数情况下空位的存在能够提高半导体材料的催化活性,例如,氮空位的引入可以有效提高C3N4的光解水产氢性能[161-162]。此外,缺陷能级的存在甚至可以使绝缘体SiO2(带隙,>8 eV)具有了光解水制氢的能力[163]。
长久以来,研究人员普遍认为结晶度高的半导体材料有助于光生载流子的传输和转移,从而适于光催化应用;结晶度差的(无定形态或无序化状态)的半导体材料因具有大量的缺陷,容易形成光生载流子的复合中心,而不适合光催化应用。但也有研究表明,无定形态或无序化状态的半导体材料也可应用于光催化领域。例如,2011年陈小波等报道了表面无序化的TiO2具有很好的光解水产氢性能[164]。在该工作中,TiO2纳米晶体(图 10a)经高压、长时间的氢气热处理后,会在晶体表面造成大量的氧空位缺陷(图 10b),形成表面无序化状态,这使得TiO2的光吸收范围可以拓展到红外区域,也使得TiO2纳米晶体由白色变成了黑色。实验测试(图 10c)和理论计算(图 10d)表明,在无序状态下,O2p轨道和Ti3d轨道在TiO2价带上方杂化成连续的中间能带,而不是传统的由氧空位带来的位于导带下方的缺陷能级,这使得TiO2的带隙由3.3 eV大幅降低到1.54 eV。
 图10
(a) 氢气处理前(白色TiO2)和(b)氢气处理后(黑色TiO2)TiO2纳米晶体的高分辨透射电镜照片; (c)黑色和白色TiO2的XPS价带谱; (d)表面无序化处理后的黑色TiO2与未处理的白色TiO2的态密度示意图[164]
Figure10.
(a) and (b) HRTEM images of TiO2 nanocrystals before and after hydrogenation, respectively; (c) Valence-band XPS spectra of the white and black TiO2 nanocrystals; (d) Schematic illustration of the DOS of disorder-engineered black TiO2 nanocrystals, as compared to that of unmodified TiO2 nanocrystals[164]
图10
(a) 氢气处理前(白色TiO2)和(b)氢气处理后(黑色TiO2)TiO2纳米晶体的高分辨透射电镜照片; (c)黑色和白色TiO2的XPS价带谱; (d)表面无序化处理后的黑色TiO2与未处理的白色TiO2的态密度示意图[164]
Figure10.
(a) and (b) HRTEM images of TiO2 nanocrystals before and after hydrogenation, respectively; (c) Valence-band XPS spectra of the white and black TiO2 nanocrystals; (d) Schematic illustration of the DOS of disorder-engineered black TiO2 nanocrystals, as compared to that of unmodified TiO2 nanocrystals[164]
刘岗等在最近的研究中也发现,无定形态C3N4的光吸收范围由石墨态(晶态)C3N4的460 nm提高到680 nm(图 11a),其带隙也由石墨态C3N4的2.82 eV减小为1.90 eV(图 12b)。且相比于石墨态C3N4,无定形态C3N4的可见光分解水产氢性能得到大幅提高[165]。
4.1.1.4 掺杂与空位调控的挑战
通过掺杂或制造空位在半导体的带隙中引入杂质能级或缺陷能级,这是宽带隙半导体扩展可见光吸收最基本和重要的手段。但是该方法存在两个瓶颈问题:(1)降低半导体的导带或抬高其价带可以缩小其带隙,但同时也降低了半导体光生电子或空穴的还原或氧化能力;(2)半导体带隙中的局域化能级带来可见光吸收的同时,也很容易成为光生载流子的复合中心(譬如,现在一般粗略的认为表面氧空位可以促进光生载流子转移,提高半导体材料的光催化活性;而体相氧空位则作为光生载流子的复合中心,不利于光催化过程)。因此在利用掺杂或空位对半导体的能带结构进行调控时既要扩大材料的吸光范围,又要兼顾材料的氧化还原能力,更要避免复合中心的出现。
对半导体进行掺杂或制造空位的目的是提高半导体的可见光催化活性,如前所述,这一目标的实现受到掺杂元素种类、掺杂量(空位浓度)、掺杂(空位)位置、掺杂(空位)分布等多种因素的影响。虽然基于第一性原理的理论计算可以很好的展示各影响因素对半导体能带结构的影响,但遗憾的是,到目前为止仍没有明确、统一的理论体系去回答这一基本问题:什么样的掺杂(空位)可以带来半导体可见光催化活性的提高?这说明有关掺杂与空位的研究仍然没有深入到问题的本质。
在TiO2氧空位的研究中,普遍认为:(1) TiO2中同时存在氧空位和Ti3+两种点缺陷(也可认为是Ti3+自掺杂),且Ti3+的存在平衡了TiO2的电性并增加了氧空位的稳定性;(2)还原TiO2的可见光吸收主要是由于氧空位或Ti3+在导带下方形成了缺陷能级或Ti3+带隙态。然而,最近杨学明等通过双光子光电子能谱实验测量和理论计算发现在还原TiO2中,Ti3+离子3d轨道由于姜-泰勒效应分裂成为带隙态和激发态,且他们认为还原TiO2的可见光吸收来源于Ti3+离子带隙态到激发态的跃迁(局域的d→d跃迁)[166]。
上述案例说明,虽然有关掺杂与空位的理论计算已经非常成熟,但没有实验结果的支持,使得理论计算结果仍有一定的局限性。因此,依靠实验技术的进步,实现对半导体的基态-激发态电子结构的准确测量,有望成为解决有关掺杂与空位本质问题的关键突破口。
4.1.2 固溶体调控
4.1.2.1 固溶体光解水材料
除了掺杂与空位以外,两种或多种半导体形成固溶体也是一种有效的改变半导体能带结构的方法。相比于掺杂(或空位)对半导体能带结构的改变,固溶体半导体最大的优势是(图 12):(1)随着组分比例的变化,固溶体的带隙和带边位置同时可调;(2)固溶体半导体的可见光吸收为带边吸收,而非大多数掺杂(或空位)带来的拖尾吸收。
 图12
(a) (AgIn)xZn2(1-x)S2固溶体(ZnS-AgInS2)的紫外-可见吸收光谱, 其中x的值分别为: (ⅰ) 0, (ⅱ) 0.17, (ⅲ) 0.22,
(ⅳ) 0.29, (ⅴ) 0.33, (ⅵ) 0.40, (ⅶ) 0.5和(ⅷ) 1; (b) (AgIn)xZn2(1-x)S2固溶体、ZnS和AgInS2的能带结构示意图[63]
Figure12.
(a) UV-vis light absorption spectra of (AgIn)xZn2(1-x)S2 solid solutions; the values of x are (ⅰ) 0, (ⅱ) 0.17, (ⅲ) 0.22,
(ⅳ) 0.29, (ⅴ) 0.33, (ⅵ) 0.40, (ⅶ) 0.5, and (ⅷ) 1; (b) Band structures of (AgIn)xZn2(1-x)S2 solid solutions, ZnS, and AgInS2[63]
图12
(a) (AgIn)xZn2(1-x)S2固溶体(ZnS-AgInS2)的紫外-可见吸收光谱, 其中x的值分别为: (ⅰ) 0, (ⅱ) 0.17, (ⅲ) 0.22,
(ⅳ) 0.29, (ⅴ) 0.33, (ⅵ) 0.40, (ⅶ) 0.5和(ⅷ) 1; (b) (AgIn)xZn2(1-x)S2固溶体、ZnS和AgInS2的能带结构示意图[63]
Figure12.
(a) UV-vis light absorption spectra of (AgIn)xZn2(1-x)S2 solid solutions; the values of x are (ⅰ) 0, (ⅱ) 0.17, (ⅲ) 0.22,
(ⅳ) 0.29, (ⅴ) 0.33, (ⅵ) 0.40, (ⅶ) 0.5, and (ⅷ) 1; (b) Band structures of (AgIn)xZn2(1-x)S2 solid solutions, ZnS, and AgInS2[63]
目前应用于光解水制氢的固溶体半导体材料依据组成阴离子的种类可分为三大类:(1)硫化物固溶体,如CdS-ZnS[167-168]、ZnS-AgInS2[63, 169]、ZnS-CuInS2[170]、ZnS-AgInS2-CuInS2[65, 171]等;(2)氧化物固溶体,如Y2WO6-Bi2WO6[172]、BiVO4-DyVO4[173]等;(3)氧氮化物固溶体,如GaN-ZnO[174]、ZnGeN2-ZnO[175]等。其中,由日本Domen课题组发展的GaN-ZnO固溶体是光解水制氢领域的明星材料,其有两大特点:(1)两种只能吸收紫外光的宽带隙半导体(GaN,3.4 eV;ZnO,3.2 eV)形成了能吸收可见光的窄带隙固溶体(GaN-ZnO,2.6~2.8 eV);(2) GaN-ZnO固溶体自身没有光催化活性,在负载助催化剂后不但可以实现可见光下全分解水,且其表观量子效率达到了较高的数值5.9%[112]。
有关GaN-ZnO固溶体可见光吸收的起源,Domen等给出了两种可能的解释:(1) GaN-ZnO固溶体的价带由N2p、Zn3d和O2p杂化而成,由于N2p-Zn3d轨道间的互斥作用,从而抬高了GaN-ZnO固溶体的价带(相比于由N2p(或O2p)构成的GaN(或ZnO)的价带),缩小了带隙,带来了可见光的吸收(图 13a)[176];(2)通过光致发光激发谱和吸收谱的测试认为在GaN为主的GaN-ZnO固溶体中,Zn在价带上方形成了受主能级(类似于Zn掺杂GaN的情况),Zn杂质能级的存在引起了GaN-ZnO可见光的吸收[177](图 13b)。
 图13
(a) GaN、ZnO和GaN-ZnO固溶体的能带结构示意图[176]; (b) GaN、Zn掺杂GaN、和GaN-ZnO固溶体(GaN为主)的杂质能级结构示意图[177]
Figure13.
(a) Schematic illustration of band structures of GaN, ZnO, and their solid solution[176]; (b) Expected energy level diagram for impurity levels in undoped GaN, Zn-doped GaN, and GaN-rich (Ga1-xZnx)(N1-xOx) solid solutions[177]
图13
(a) GaN、ZnO和GaN-ZnO固溶体的能带结构示意图[176]; (b) GaN、Zn掺杂GaN、和GaN-ZnO固溶体(GaN为主)的杂质能级结构示意图[177]
Figure13.
(a) Schematic illustration of band structures of GaN, ZnO, and their solid solution[176]; (b) Expected energy level diagram for impurity levels in undoped GaN, Zn-doped GaN, and GaN-rich (Ga1-xZnx)(N1-xOx) solid solutions[177]
此外,Domen课题组也对影响GaN-ZnO固溶体光催化性能的各要素进行了深入研究,他们发现氮化条件(氮化温度、氮化时间和氨气纯度)、前驱体(α-Ga2O3+ZnO;β-Ga2O3+ZnO;γ-Ga2O3+ZnO;ZnGa2O4;ZnGa2O4+ZnO等)、前驱体粒径和Zn/Ga原子比都会对GaN-ZnO固溶体的全分解水性能产生重要影响[176]。
4.1.2.2 固溶体光催化材料的挑战
通过GaN-ZnO固溶体的案例可以看出,固溶体半导体材料作为光解水制氢光催化剂的最大优势是其物理特性的可调变性,而正是这种可调变性带来了其光催化应用的巨大差异,也成为其作为光催化材料最大的挑战。因此,优化合成方法以精确调控固溶体的物理特性、实验结合理论计算阐明相关特性的物理本质是推动固溶体光催化材料发展的关键。
4.1.3 晶面调控
因为光催化反应都在材料的表面进行,所以光催化材料的表面结构就直接影响着反应物分子的吸附、光生载流子的转移以及产物分子的脱附,最终影响着光催化材料的催化活性。因此针对光催化材料的表面调控,特别是合成具有高能晶面暴露的光催化材料成为近年来研究的热点,中国科学院金属研究所刘岗团队在该领域进行了相关的研究工作[178-182],并在综述性论文中阐述了晶面控制的基本原理和应用实例[10-11],在此就不再一一累述,只重点介绍半导体材料晶面中存在的两种特有效应:晶面电子结构效应和晶面光生载流子分离效应(应属4.2节内容范畴,因由晶面引起,故在此一并介绍)。
4.1.3.1 晶面电子结构效应
半导体材料不同暴露晶面上的原子结构有很大的差异,而这种原子排列的差异必然引起电子结构的差异。或者说,对于不同晶面暴露的半导体材料,它们的体相电子结构是一样的,而表面电子结构会因暴露晶面的不同而有差异。半导体材料的吸收光谱是由材料的能带结构决定的,所以半导体材料的表面电子结构差异也必然会反映在它的吸收光谱上,刘岗等在TiO2材料中观察到了这种现象[179]。如图 14a所示,{101}晶面暴露比例为82%的锐钛矿TiO2的吸收光谱相对于{001}晶面暴露比例为72%的锐钛矿TiO2的吸收光谱有了明显的蓝移,进一步的分析发现,两种不同晶面暴露的TiO2的价带顶没有变化,只是{101}晶面占优的TiO2的导带底相比于{001}晶面占优的TiO2的导带底略有上移。需要说明的是,虽然上述两种锐钛矿TiO2的颗粒尺寸存在着很大的差异,但纳米尺寸的TiO2(~100 nm)还远没有达到TiO2发生量子尺寸效应的尺寸(2 nm)。而且,刘岗等进一步合成出了尺寸可比的微米尺度的三种不同晶面占优的(分别是{001}、{101}和{010})锐钛矿TiO2晶体[180],通过对3种晶体的吸收光谱表征也发现了它们之间存在的能带结构差异(图 14b),且这种差异直接带来光催化活性上的高低。
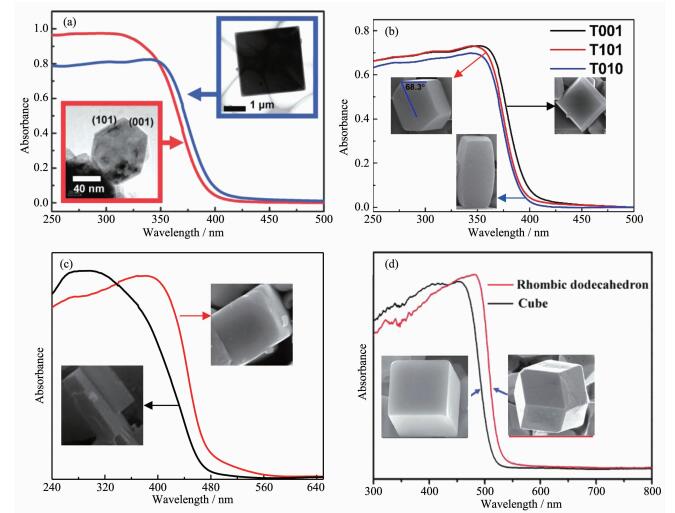 图14
(a) 纳米尺寸{101}晶面暴露比例为82%和微米尺寸{001}晶面暴露比例为72%的锐钛矿TiO2晶体的吸收光谱[179]; (b) {001}、{101}和{010}晶面分别占优、微米尺寸的锐钛矿TiO2晶体的吸收光谱[180]; (c)亚微米尺寸的类立方体和长方体WO3的紫外-可见吸收光谱[135]; (d)亚微米尺寸的菱形十二面体和立方体形貌的Ag3PO4晶体的吸收光谱[176]
Figure14.
(a) UV-visible absorption spectra of nanosized anatase crystals with 82% {101} facets and micron-sized anatase crystals with 72% {001} facets[179]; (b) UV-visible absorption spectra of T001, T101 and T010[180]; (c) UV-visible absorption spectra of quasi-cubic-like WO3 crystals and rectangular sheet-like WO3 crystals[135]; (d) UV-vis diffusive reflectance spectra of Ag3PO4 sub-microcrystals with rhombic dodecahedrons and cubes morphologies[176]
图14
(a) 纳米尺寸{101}晶面暴露比例为82%和微米尺寸{001}晶面暴露比例为72%的锐钛矿TiO2晶体的吸收光谱[179]; (b) {001}、{101}和{010}晶面分别占优、微米尺寸的锐钛矿TiO2晶体的吸收光谱[180]; (c)亚微米尺寸的类立方体和长方体WO3的紫外-可见吸收光谱[135]; (d)亚微米尺寸的菱形十二面体和立方体形貌的Ag3PO4晶体的吸收光谱[176]
Figure14.
(a) UV-visible absorption spectra of nanosized anatase crystals with 82% {101} facets and micron-sized anatase crystals with 72% {001} facets[179]; (b) UV-visible absorption spectra of T001, T101 and T010[180]; (c) UV-visible absorption spectra of quasi-cubic-like WO3 crystals and rectangular sheet-like WO3 crystals[135]; (d) UV-vis diffusive reflectance spectra of Ag3PO4 sub-microcrystals with rhombic dodecahedrons and cubes morphologies[176]
随后,谢英鹏等又合成出暴露晶面为{002},{020}和{200}的、2种不同形貌WO3单斜晶体,其中在类立方体WO3中3种晶面的暴露比例相等,而在长方体WO3中则是{002}晶面占优。吸收光谱(图 14c)和XPS价带谱分析发现,WO3晶体由于暴露晶面比例的不同而引起能带结构的差异:长方体WO3的价带边和导带边相比于立方体WO3分别上移了0.22和0.3 eV,且这种差异带来了WO3光催化氧化还原特性的逆转[135]。在其他研究组的工作中,也可以看到由暴露晶面不同带来的材料表面电子结构上的差异。如图 14d所示,叶金花等合成出了亚微米尺度的分别具有菱形十二面体和立方体形貌的Ag3PO4晶体[183],其中菱形十二面体Ag3PO4的暴露晶面全都是{110},而立方体Ag3PO4的暴露晶面全都是{100},这也导致了它们在吸收光谱上的差异。
由以上的案例可知,通过暴露晶面的不同可以对半导体材料的表面电子结构进行有效的调控,可以将这种现象称之为晶面电子结构效应。该效应相比于其他半导体能带调控方法的优势在于:(1)不会带来杂质(缺陷)能级,避免了形成载流子复合中心;(2)可以在不改变半导体体相能带结构的前提下,去调节表面的氧化还原特性,可以避免可见光吸收扩展和氧化还原能力降低之间的矛盾。
4.1.3.2 晶面光生载流子分离效应
Ohno等在2002年就发现光沉积的方法可以将Pt和PbO2(Pt4++e- → Pt;Pb2++H2O+h+ → PbO2)负载到TiO2的不同晶面上[184]。如图 15所示,在锐钛矿TiO2单晶颗粒上,Pt大多被光还原沉积在TiO2的{110}晶面,而PbO2大多被光氧化沉积在TiO2的{011}晶面。这说明光生电子和空穴分别富集在TiO2的{110}和{011}晶面上,或者说晶面对于光生载流子具有分离效应。
在最近的研究中,李灿等利用晶面光生载流子分离效应分别在BiVO4的{010}和{110}晶面负载了还原助催化剂Pt和氧化助催化剂MnOx,从而使BiVO4的光解水产氧性能有了数量级的提升[185](图 16)。
 图16
(a) 光沉积Pt-MnOx-BiVO4颗粒的扫描电镜图片;
(b) BiVO4系列样品的光解水产氧性能, 其中助催化剂的负载量为0.1%, imp为浸渍法负载, P.D.为光沉积法负载[185]
Figure16.
(a) SEM images of Pt and MnOx deposited rutile BiVO4 particle; (b) photocatalytic water oxidation performance of BiVO4 (imp, impregnation method; P.D., photo-deposition method; the contents of the deposited cocatalysts are all 0.1%)[185]
图16
(a) 光沉积Pt-MnOx-BiVO4颗粒的扫描电镜图片;
(b) BiVO4系列样品的光解水产氧性能, 其中助催化剂的负载量为0.1%, imp为浸渍法负载, P.D.为光沉积法负载[185]
Figure16.
(a) SEM images of Pt and MnOx deposited rutile BiVO4 particle; (b) photocatalytic water oxidation performance of BiVO4 (imp, impregnation method; P.D., photo-deposition method; the contents of the deposited cocatalysts are all 0.1%)[185]
晶面光生载流子分离效应的起源可以归为材料晶面电子结构的差异,即不同晶面间能带结构的差异驱使光生载流子在晶面间迁移,从而造成了电子和空穴的空间分离。最近,李灿等通过表面光电压谱观察到BiVO4粒子上不同晶面的表面电势差异:在光激发下,BiVO4不同晶面存在不同的空间电荷层内建电场,这种电场的存在使BiVO4{011}和{010}晶面间具有70倍差别的空穴迁移各向异性[186]。而且他们通过表面光电压成像系统,首次实现了单个光催化剂粒子不同晶面的光生电荷的光电成像,进一步验证了BiVO4晶面上的光生载流子分离效应。
4.1.3.3 晶面效应在光解水应用中的挑战
半导体材料暴露的特定晶面上具有不饱和配位的原子、表面台阶和表面重构等物理特性,这些特性带来了晶面吸附、晶面电子结构和晶面光生载流子分离的各向异性。因此,半导体的晶面效应在光解水应用中被给予很高的期望。但是,实验结果却发现具有规则形貌和特定晶面暴露的半导体材料的光解水性能往往还不如没有规则形貌的。这可能是由以下原因造成的。半导体的表面原子结构是晶面特性各向异性的起源,而实验获得的特定晶面暴露的半导体材料往往不会具备这些晶面在理论上所具有的表面原子结构。这是因为要想获得特定晶面暴露的半导体材料,就需要在合成过程中抑制晶体自然的择优生长方向,譬如,为了制备TiO2{001}高能晶面就需要抑制晶体沿(001)晶向的生长。为了抑制晶体自然的择优生长,这需要在合成过程中添加合适的化学物质(如酸根离子和表面活性剂等)以降低高能晶面的表面能。在合成TiO2晶体过程中,F-离子的存在可以有效降低{001}晶面的表面能[187],从而可以制备出{001}晶面大比例暴露的TiO2晶体。然而,化学物质吸附在半导体材料的高能晶面上以降低其表面能是通过饱和高能晶面上的悬键(不饱和配位原子)而实现的。因而,表面悬键的缺失大大抑制了这些高能晶面暴露的半导体材料的光解水性能。
4.2 促进载流子分离的方法与挑战
半导体光生载流子的有效转移和分离是获得高光催化活性的必要条件。下面就被广泛研究的几种促进载流子分离的方法做简要概述,并对他们存在的问题进行讨论。
4.2.1 构建半导体异质(相)结
4.2.1.1 Ⅱ型半导体异质结
半导体材料上的光生电子和空穴大部分在体相扩散过程中或转移到表面后复合掉,只有极少数的电子和空穴去发生还原和氧化反应,这是限制半导体材料光催化活性的一个最根本因素。对单一材料而言,其表面的缺陷(如空位等)会成为电子或空穴的捕获位,从而使光生电子和空穴发生分离,但分离出的电子和空穴也是有限的。为了获得更多的分离开的电子和空穴,研究人员在过去几十年中利用了复合半导体之间的载流子转移。图 17展示了这些年来所发展的不同机制的半导体之间载流子转移的过程,下面我们分别介绍。
 图17
(a) 敏化过程, (b) Ⅱ型半导体异质结构, (c) p-n结和(d)矢量Z机制中的载流子转移过程
Figure17.
Charge-carrier transfer process in sensitization (a), traditional (b), p-n junction (c) and vectorial Z-scheme (d) mechanisms
图17
(a) 敏化过程, (b) Ⅱ型半导体异质结构, (c) p-n结和(d)矢量Z机制中的载流子转移过程
Figure17.
Charge-carrier transfer process in sensitization (a), traditional (b), p-n junction (c) and vectorial Z-scheme (d) mechanisms
图 17a展示了半导体敏化过程中的载流子转移,敏化过程主要是为了扩大复合半导体材料的光吸收范围。一般是将窄带隙且导带底位置较高的半导体(如Si[188]、CdS[189]、CdSe[190]、ZnTe[191]、CuO[192]、Cu2O[193]、Fe2O3[194]等)与宽带隙且导带底位置较低的半导体(如TiO2、ZnO等)相复合。在这种复合半导体材料中,能被可见光激发的窄带隙半导体导带上的电子在能级差的驱动下转移到宽带隙半导体的导带上,从而实现了宽带隙半导体材料对可见光的利用。
如果将两种能级错排的半导体材料结合在一起组成Ⅱ型半导体异质结构,它们之间的载流子转移如图 17b所示。电子在能差的作用下由具有高导带底位置的半导体向具有低导带底位置的半导体转移,而空穴则由具有低价带顶位置的半导体向具有高价带顶位置的半导体转移,从而促进了光生电子和空穴的空间分离。到目前为止,绝大多数的半导体材料都可以找到与之能级相匹配的另一种半导体材料,并组成这种Ⅱ型半导体异质结构,而且Ⅱ型能级排列也是最常用的半导体组合方式,其组成的半导体异质结构也是最多的。例如TiO2/ZnO[195]、ZnS/ZnO[196]、SnO2/ZnO[197]、TiO2/WO3[198]、C3N4/WO3[199]、CdS/TiO2/WO3[200]等。
如果组成Ⅱ型半导体异质结构的半导体材料分别为p型和n型且二者的带边排列如图c所示,那么在p-n结中存在的内建电场会进一步加大由能带差带来的载流子转移,从而更大程度的促进半导体上光生载流子的分离,此类的半导体异质结构有p-In2O3/n-ZnO[201],p-CaFe2O4/n-ZnO[202],p-NiO/n-ZnO[203]和p-ZnO/n-TiO2[204]等。
上述的Ⅱ型半导体异质结构可以促进半导体上的光生载流子分离,但也存在着不利的因素,即载流子都是向低能级的位置转移,从而降低了光生电子和空穴的还原氧化能力。如果能够实现光生载流子分离的同时还保持它们的还原氧化能力,将有利于半导体异质结构在光催化中的应用。为此,研究人员在一些特定Ⅱ型半导体异质结构中(譬如,CdS/ZnO[205]、WO3/CaFe2O4[206]、CdS/WO3[207]、BiOI/g-C3N4[208]等)提出了矢量Z机制载流子转移的概念(图 17d)。以CdS/ZnO为例,在矢量Z机制载流子转移过程中,ZnO上的光生电子与CdS上的光生空穴在界面处相复合,从而保留了还原能力更强的CdS上的光生电子和氧化能力更强的ZnO上光生空穴(图 18a)。
 图18
(a) CdS/ZnO中的矢量Z机制和(b) CdS/Au/ZnO中的矢量Z机制载流子转移过程[210];
(c) Ru修饰的SrTiO3:La, Rh/Au/BiVO4:Mo全分解水示意图[218]
Figure18.
Schematic of (a) the vectorial Z-scheme charge-carrier transfer process in the CdS/ZnO heterostructure and (b) the vectorial Z-scheme charge-carrier transfer process in the CdS/Au/ZnO heterostructure[210]; (c) Schematic of overall water splitting on the Ru-modified SrTiO3:La, Rh/Au/BiVO4:Mo sheet[218]
图18
(a) CdS/ZnO中的矢量Z机制和(b) CdS/Au/ZnO中的矢量Z机制载流子转移过程[210];
(c) Ru修饰的SrTiO3:La, Rh/Au/BiVO4:Mo全分解水示意图[218]
Figure18.
Schematic of (a) the vectorial Z-scheme charge-carrier transfer process in the CdS/ZnO heterostructure and (b) the vectorial Z-scheme charge-carrier transfer process in the CdS/Au/ZnO heterostructure[210]; (c) Schematic of overall water splitting on the Ru-modified SrTiO3:La, Rh/Au/BiVO4:Mo sheet[218]
需要说明的是,在具有矢量Z机制载流子转移的Ⅱ型半导体异质结构中,传统的Ⅱ型载流子转移过程(即载流子由高能级向低能级转移)仍然可以发生,且两种载流子转移过程是相互竞争的。研究人员发现,如果在Ⅱ型半导体异质结构的界面处引入适当的电子转移介质(图 18b),譬如贵金属(Au[209-211]、Ag[212]、Rh[129, 213])的纳米颗粒和石墨烯[214-217],则可以有效的加速Ⅱ型半导体异质结构中的矢量Z机制载流子转移过程,从而大幅提高其光催化活性。令人振奋的是,矢量Z机制体系可应用于光催化全分解水。最近Domen等设计并组装了以La、Rh共掺杂SrTiO3、Mo掺杂BiVO4和Au为结构单元的片层状SrTiO3:La, Rh/Au/BiVO4:Mo光催化剂(图 18c),该催化剂经Ru修饰后,可实现可见光下(λ>420 nm)1.1%的太阳能-氢能转换效率,其在419 nm单色光下的表观量子效率可达30%[218],这是光解水领域的又一里程碑工作。
4.2.1.2 Ⅰ型半导体异质结
相比于Ⅱ型半导体异质结构在光催化过程中的广泛应用,Ⅰ型半导体异质结构因其嵌套式对应的能带结构(窄带隙半导体的导带底和价带顶都位于宽带隙半导体的禁带中,图 19a)不利于光生载流子的空间分离,故很少被应用于光催化过程。特别是在Ⅰ型半导体核-壳结构中,如果成核的是窄带隙半导体,成壳的是宽禁带半导体,那么在光照条件下,核半导体上的光生电子和空穴都会很难转移出去,造成了电子和空穴的聚集,这种情况可以称之为电子限域效应(图 19b)。
 图19
(a) Ⅰ型半导体异质结构和(b) Ⅰ型半导体核-壳结构的能带结构示意图
Figure19.
Band alignments of (a) type-Ⅰ and (b) core-shell type-Ⅰ semiconductor heterostructures
图19
(a) Ⅰ型半导体异质结构和(b) Ⅰ型半导体核-壳结构的能带结构示意图
Figure19.
Band alignments of (a) type-Ⅰ and (b) core-shell type-Ⅰ semiconductor heterostructures
电子限域效应的应用一般在光学研究中,这是因为限域效应造成了电子和空穴的聚集,从而大大增加了它们复合的机率,使得它们的发光效率大幅提高。如果能够将Ⅰ型半导体核-壳结构中被限域的电子或空穴由核中转移到壳层,实现光生载流子在核-壳间的空间分离,那么就可将该结构材料应用于光催化反应中。到目前为止,有两种方式可以实现核中限域载流子的转移。
第一种方式是通过电子隧穿效应来实现的。电子隧穿效应是指低能电子穿过高能势垒的现象,因为它需要利用量子力学的波函数来加以解释,所以也称之为量子隧穿效应。用量子力学的观点来看,电子具有波动性,其运动用波函数描述,而波函数遵循薛定谔方程,从薛定谔方程的解就可以知道电子在各个区域出现的概率密度,从而能进一步得出电子穿过势垒的概率,该隧穿概率随着势垒宽度的增加而指数衰减。因此,要想实现Ⅰ型半导体核-壳结构中电荷由核到壳的有效隧穿,壳层的厚度要在几个纳米以内。譬如,在由CdSe/ZnS核-壳量子点和钴肟络合物组成的光解水制氢催化剂中(图 20)[219],CdSe中的光生电子就要通过隧穿ZnS壳层才能转移到钴肟配合物上。针对这种Ⅰ型半导体核-壳量子点内的类Ⅱ型载流子转移,也有研究人员直接从波函数的角度加以理解(如在CdSe/CdS核-壳量子点内,电子在核-壳中是离域化的,而空穴被局域在核中[220])。
对于达不到电子隧穿效应要求的Ⅰ型半导体核-壳结构,可以通过缺陷能级的帮助来实现类Ⅱ型载流子转移。在最近的研究中,谢英鹏等利用前驱体反应在热力学上的先后顺序来控制产物的生成顺序,以ZnO、醋酸镉(Cd(CH3COO)2)和硫脲(CH4N2S)分别为锌源、镉源和硫源,用一步水热方法制备出了CdS-ZnS核-壳样品,且ZnS壳层具有孔结构(图 21)[221]。
通过时间分辨荧光光谱等实验手段可以确认:由于在ZnS中存在着由Zn空位和间隙S等带来的缺陷能级(价带上方的受主能级),使得CdS上的光生空穴可以转移到ZnS的缺陷能级上,而CdS上的光生电子被限域在核内(图 22a),从而在CdS-ZnS核-壳间形成了类Ⅱ型载流子转移。这种载流子在CdS-ZnS核-壳间的空间分离以及ZnS壳层特有的介孔结构使得CdS-ZnS核-壳材料具有很高的光解水产氢性能(图 22b);且由于ZnS壳层对CdS的保护作用,抑制了CdS的光腐蚀,CdS-ZnS核-壳材料也表现出很好的光催化稳定性。
通过上述的案例可以看出,Ⅰ型半导体核-壳结构应用于光解水制氢具有明显的优势:(1)窄带隙半导体的核可有效利用可见光;(2)电子限域效应为核上提供了大量可参与反应的载流子;(3)在光生载流子实现空间分离的过程中,光生电子的还原能力不会降低。通过隧穿效应转移出的电子甚至有可能到达壳层半导体更高的导带上,从而提高其还原能力;(4)壳层的存在可以有效保护内核,免受光腐蚀的影响,特别是对于以硫化物半导体为核的核-壳结构。
4.2.1.3 半导体异相结
除了构建不同半导体材料组成的异质结构,在同种半导体、不同晶相间构建异相结也可以有效提高光生载流子的空间分离,从而提高其光解水活性。李灿等就发现由锐钛矿和金红石相构成的TiO2异相结(图 23a)可以有效提高光解水制氢性能[222]。最近,他们发现α-Ga2O3和β-Ga2O3之间形成的异相结可以实现全分解水反应[223]。这些异相结都是通过热处理诱使TiO2或Ga2O3发生相变而形成的,因此获得的异相结非常致密,这也是他们具有高光催化活性的重要原因。因为半导体不同晶相间能带结构的差异,TiO2或Ga2O3异相结之间会实现类似于Ⅱ型半导体异质结构中载流子的转移和分离(图 23b)。
4.2.1.4 半导体异质(相)结的挑战
依据半导体间能带结构的差异构建半导体异质(相)结以实现载流子的空间分离,从而提高半导体材料的光解水性能。这是固体能带理论在半导体光解水领域的最成功应用,异质(相)结理论在指导光催化材料的设计上具有非常高的广谱性。即使如此,异质(相)结理论在光解水应用中也还有很大的发展空间。
如前所述,Ⅱ型半导体异质结如能实现直接(矢量)Z机制载流子转移,高能量的光生电子和空穴将会被保留,这对光解水制氢具有非常重要的意义。然而,直接(矢量)Z机制载流子转移还没有得到理论上很好的解释。我们认为,在CdS-ZnO异质结中直接(矢量)Z机制载流子转移过程和传统的Ⅱ型载流子转移过程应该是同时存在的,且处于相互竞争的关系。那么在什么情况下,直接(矢量)Z机制载流子转移过程将会占优?此外,如何匹配(包括能带匹配、晶型匹配等)半导体才能实现矢量Z机制载流子转移?这两个问题如果能得到理论上的正确的解释和指导,相信半导体光解水领域的发展将会跃上新的台阶。
杂质或缺陷能级在单体光催化材料中虽可以扩展半导体的吸光范围,但也存在成为光生载流子复合中心的弊端。然而,这个单体光催化材料中的瓶颈问题却可能成为解决Ⅰ型半导体异质结在光解水应用中的钥匙(如前述的CdS-ZnS核-壳结构)。Ⅰ型半导体异质结为宽带隙半导体在可见光下的光解水应用提供了可能,但其中的很多关键问题仍需深入的研究。
4.2.2 负载助催化剂
4.2.2.1 助催化剂的作用
对于一个有效的光催化反应,除了用于吸收光能产生电子-空穴对的半导体作为主体光催化剂外,负载在主体光催化剂表面的助催化剂常常是必不可少的。虽然助催化剂的负载量(相对于主体催化剂的重量比)一般很小(< 3%),而且本身对光催化反应只有很小的活性或者没有活性,但是它能大大提高主体光催化剂的活性。因而随着光催化研究的不断深入,助催化剂的研究日渐引起人们的重视。
助催化剂一般作为电子或空穴的捕获点来加速主体光催化剂中电子-空穴对的分离,同时该电子或空穴的捕获点就可作为光催化还原或氧化反应中的活性中心。助催化剂除了能促进半导体表面的光生载流子分离,合适的助催化剂还能降低氧化或还原反应中的过电位,使得光催化反应所需的能量降低。例如,通过对Co-Pi负载的BiVO4进行光电化学测试发现(图 24),Co-Pi负载后使BiVO4的开路电压由0.3 V降低到了-0.05 V,大大降低了氧化反应的过电势[224]。再者,在光解水反应中,合适的助催化剂能够降低气体从催化剂表面脱离的逸出能,同时还可抑制气体在催化剂表面的逆反应,从而有助于光催化反应的进行。
4.2.2.2 助催化剂的分类
常见的助催化剂有贵金属(如Pt、Pd、Ru、Rh、Au、Ag、Ir等)[2, 4, 225-226]、氧化物(如RuO2[227]、RhO2[2]、NiO[228]、IrO2[2]、Cu2O[229]、CuO[230-231]、Mn3O4[232]等)、硫化物(如MoS2[233]、WS2[234]、NiS[235]、FeS[236]、Ag2S[237]、CoS[238]、CuS[239]、PdS[240]等)和复合物(如Ni/NiO[241]、RhxCr1-xO3[111]、Rh/Cr2O3[242]等)。对于以上的助催化剂现在没有一个统一的标准对其归类,一般习惯于把对光解水产氢效率的提高大于对光解水产氧效率的提高的归为产氢助催化剂(如Pt、Pd、NiO等),相反的归为产氧助催化剂(如RuO2、IrO2、Mn3O4等)。其它对光解水产氢或产氧效率的提高差不多的助催化剂一般不予区分。
助催化剂的主要作用是促进半导体表面的光生电子和空穴的分离,根据助催化剂与半导体光催化剂之间载流子转移机制的不同,助催化剂则可分为金属类、半导体类以及由这两类组成的复合型助催化剂。下面分别对这3类助催化剂的作用机制和研究进展进行介绍。其中,有关金属-半导体之间作用机制的讨论参考了刘恩科等编著的《半导体物理学》(第七版)中第七章的部分内容[243]。
4.2.2.3 金属类助催化剂
金属类助催化剂指具有金属导电性质的助催化剂,如上节中提到的贵金属类助催化剂,而且由于贵金属的氧化物(如RuO2、RhO2、IrO2)也具有非常好的导电性,一般也将它们归为金属类助催化剂。当金属类助催化剂负载在半导体催化剂上时,它们之间就会发生一些相互作用。从能级的观点来看,要使电子从金属或半导体中逸出,就必须做功。因为金属或半导体内的绝大多数电子都比体外电子处于较低的能级,则体外真空中静止电子的能量与该物体费米能级之差称之为逸出功或功函数(金属的逸出功,Wm;半导体的逸出功,Ws)。显然,逸出功越大,电子越不容易离开物体。
当金属与n型半导体接触时,如果n型半导体的逸出功小于金属的逸出功(Ws < Wm),这种费米能级的差别意味着在金属内部和半导体导带相对应的那部分能级上,电子的密度要小于半导体导带的电子密度,因此当它们接触时,电子便从半导体向金属扩散,结果使金属带负电,半导体带正电。这使得在半导体表面形成一个正的空间电荷区,其中电场方向由体内指向表面,它使半导体表面电子的能量高于体内,能带向上弯曲。这个向上弯曲的能带对电子形成一个阻止其由半导体向金属扩散的势垒,既是肖特基表面势垒,如图 25a所示。图中E0表示真空中静止电子的能量,EF表示金属和半导体拉平的费米能级,EC、EV分别表示半导体的导带底和价带顶的能级。金属与n型半导体的肖特基势垒形成后,当被光辐照时,半导体价带上的电子就被激发到导带上,这时导带上的电子能量虽然低于肖特基势垒的顶端,但仍有一定的概率穿过这个势垒到达金属(隧穿效应),穿透的概率与电子的能量和势垒高度有关。可见电子的能量越高、势垒的高度越低,电子越容易穿透。当势垒高度小于一个临界值时,势垒对于电子是完全透明的,电子可以直接穿过它(电子在半导体与金属之间离域化)。
若n型半导体的逸出功大于金属的逸出功(Ws>Wm),这种费米能级的差别意味着在金属内部和半导体导带相对应的那部分能级上,电子的密度大于半导体导带的电子密度,于是当两者接触时,电子便从金属向半导体扩散,结果使金属表面带正电,n型半导体表面带负电,在半导体表面形成负的空间电荷区。其中电场方向由表面指向体内,能带向下弯曲。这里电子浓度比体内大得多,因而是一个高电导的区域。这样的半导体和金属的接触没有形成势垒,称之为欧姆接触,如图 25b所示。其中χ为电子亲和能,它表示要使半导体导带底的电子逸出体外所需要的最小能量(χ=E0-EC)。金属与n型半导体的欧姆接触形成后,当半导体被光激发后,导带上的电子就很容易在空间电场的作用下转移到金属上。
当金属和p型半导体接触时,形成肖特基表面势垒的条件正好与n型的相反。若Ws < Wm,能带向上弯曲,形成p型欧姆接触;若Ws>Wm,能带向下弯曲,造成空穴的势垒,形成p型肖特基表面势垒。
在光解水制氢实际应用中,大多数半导体材料都属于n型,且其逸出功一般都小于金属类助催化剂的逸出功,即半导体与金属形成肖特基接触。根据上面的分析,用不同的金属与半导体形成的肖特基接触,其势垒高度应当直接随金属逸出功而变化(对于同一种半导体,χ将保持一定的值)。但因半导体表面态的存在(对应的能级称为表面能级),这种变化有时候并不是很明显(费米能级钉扎效应)。无论势垒高度是否随金属逸出功而变化,在大多数金属类助催化剂负载的半导体材料体系中,肖特基势垒是载流子转移过程中必须要克服的。
由以上对金属和半导体接触情况的分析可知,金属和半导体间的相互作用是复杂的,而且对于同一种金属或半导体材料而言,材料的特性(如颗粒尺寸、暴露晶面、元素价态等)具有可变性,这也使得金属和半导体间的相互作用是可变的。鉴于这些复杂性和可变性,使得金属类助催化剂在光催化应用中受到多种因素的影响。下面我们以具有广谱性应用的贵金属Pt为例,介绍金属类助催化剂在光催化反应中的影响因素。
(1) 颗粒尺寸的影响。按照现有的固体物理学知识,要在形成肖特基接触的半导体与金属间实现载流子的转移,只能通过电子隧穿效应或者电子离域化方式,这就要求金属类助催化剂的尺寸要控制在超细纳米尺度上(< 10 nm)。按传统催化理论,金属纳米颗粒的尺寸越小,其比表面积和表面能越高,催化活性也越高。特别是近年来发展起来的单原子催化概念已经在传统催化领域展现出极佳的应用效果。上述理论在光解水制氢应用中也得到了验证,譬如,立方体Pt(4 nm)-CdS的光解水制氢性能是立方体Pt(8 nm)-CdS的2倍[244]。但需要注意的是,传统的金属催化过程并不涉及光催化过程中的半导体与金属之间载流子转移。随着金属纳米颗粒的尺寸减小,因量子尺寸效应,金属的功函数会增大,则半导体与金属之间的肖特基势垒高度增加,这将加大载流子由半导体向金属转移的难度(不利于光催化过程),但同时转移到金属上的载流子也很难再回到半导体上,从而延长了载流子的寿命(有利于光催化过程)。因此,金属类助催化剂在光催化应用中是不是越小越好?是否存在颗粒尺寸的最优临界值?还是像在传统催化领域一样,单原子金属具有更优的催化性能?这些问题都有待于实验结果的验证。
(2) 暴露晶面的影响。如同半导体材料一样(见4.1.3节),金属纳米颗粒暴露的不同晶面间也存在原子结构、电子结构和表面能的差异,这些差异直接带来了金属催化性能的显著不同。相比于传统催化中对金属晶面催化差异性的广泛研究,该差异性在光解水制氢的研究中报道较少。直到最近,才有相关报道发表:在Pt-TiO2体系中Pt{111}晶面具有比{100}晶面更低(更正)的费米能级,这导致了光生电子由TiO2向Pt{111}晶面的转移比向{100}晶面的转移更容易、更快速,从而Pt{111}/TiO2展现出比Pt{100}/TiO2更高的光解水制氢性能[245]。
(3) 金属价态的影响。因制备方法的差异,可以得到不同价态的Pt助催化剂:光还原、化学还原(如NaBH4还原)或氢气热还原可以制备零价态的Pt(0);而浸渍-热分解过程往往得到氧化态的Pt(Ⅱ)、Pt(Ⅳ)或二者的混合物PtOx。最早在光催化降解污染物的研究中发现,Pt(0)比PtOx具有更好的助催化作用,这可能是因为存在变价的PtOx容易成为载流子复合中心。不过,在最近杨华桂等的研究中发现,在光解水制氢体系中,PtOx将有助于抑制H2的氧化逆反应[246]。而且,Domen等也发现PtOx比Pt(0)在WO3光解水制氧体系中具有更好的助催化作用[247]。
4.2.2.4 半导体类助催化剂
半导体类助催化剂指的是具有半导体性质的助催化剂,如金属氧化物和金属硫化物等。这类助催化剂负载在光催化剂上后,两者之间一般遵循4.2.1节中提到的半导体间的载流子转移机制,即能级匹配原则,如NiO由于带隙较大,只适合一些宽带隙的半导体材料。值得注意的是,在半导体类助催化剂的研究中,往往只考虑光催化剂向助催化剂的载流子转移,而没有考虑助催化剂向主催化剂的载流子转移。虽然半导体类助催化剂相对于光催化剂的比例是很少的,但助催化剂向主催化剂转移的载流子数量对参与光催化反应的载流子数量有多大的贡献至今还不是特别明确。
除了与光催化剂之间的能级匹配以外,助催化剂与光催化剂之间还存在物相匹配的特色,如金属硫化物助催化剂除了用于金属硫化物光催化剂以外,很难再用于其他半导体材料。例如,MoS2使CdS在可见光(λ>420 nm)和牺牲剂条件下的产氢速率提高36倍[233]。Pt和PdS共同负载CdS后[248],其在可见光(λ>420 nm)有牺牲剂条件下的产氢表观量子效率达到93%,这也是可见光下有牺牲剂存在的光解水产氢的最高表观量子效率。
负载的半导体类助催化剂的尺寸会非常小,由于纳米粒子的量子尺寸效应,其带隙会相应变大,这时在分析光催化剂与助催化剂之间的载流子转移时应将该变化考虑进去。譬如,谢英鹏等在WO3表面负载了大带隙的B2O3-xNx纳米团簇[249],按照两者之间的能级匹配关系(图 26),WO3上的光生载流子很难转移到B2O3-xNx上。但B2O3-xNx纳米团簇却可以大幅提高WO3的光解水产氧性能,他们分析这可能是因为电子隧穿的作用实现了WO3与B2O3-xNx之间的光生载流子有效转移。
此外,上面的讨论都是在光激发条件下载流子的转移机制,而没有考虑在光辐照前半导体间的相互作用。对半导体而言,有n型和p型两种类别。所以当半导体类的助催化剂(n型或p型)与半导体光催化剂(n型或p型)结合后,两者之间就会形成异质结构,组合可能有nn结,pp结或pn结。在这些异质结构中,会形成空间电荷区和内建电场,使半导体的能带在表面处发生弯曲。当他们被光激发后,这些表面弯曲的能带以及空间电荷区的内建电场自然会对载流子的转移过程产生影响。
4.2.2.5 复合类助催化剂
目前在光催化作用中应用的助催化剂还是主要以单相的金属或半导体为主,但复合相助催化剂的开发越来越受到人们的重视。正如复合材料间的协同作用能够大幅提高半导体材料的光催化活性一样,存在组分间协同作用的复合相助催化剂在对光催化材料性能的提高上也要优于单相助催化剂。除了具备促进光催化材料光生载流子转移这一基本功能,复合相助催化剂可实现单相助催化剂无法完成的特殊功能。
最近,Townsend等通过表面光伏测试等手段对NiOx-SrTiO3这一传统光催化材料体系进行了研究[241]。他们发现,在复合相助催化剂NiOx中,Ni和NiO存在组分间的协同作用:组分Ni主要捕获SrTiO3上的光生电子,而组分NiO则主要转移SrTiO3上的光生空穴。由此NiOx不但促进了SrTiO3上光生电子和空穴的双重转移,也实现了光催化氧化还原反应位的分离。
在助催化剂的发展过程中,日本的Domen研究小组发展了很多有创新性的复合类助催化剂。例如,他们在2006年发展的氧化铑和氧化铬的混合氧化物(RhxCr1-xO3)能够使几乎没有光催化能力的GaN-ZnO固溶体光催化剂具备了可观的全分解水能力[111]。此后,他们又发展的铑-氧化铬(Rh-Cr2O3)核-壳结构的助催化剂[123],如图 27a所示。在这样的核壳结构中,Rh-Cr2O3作为还原位而存在,氢离子可以穿过Cr2O3壳到达Rh核表面,并在Rh核表面被还原成H2,随后氢气分子可以通过Cr2O3壳而逸出。水分子则在GaN-ZnO固溶体光催化剂的表面被氧化成O2,但是氧气分子是不能通过Cr2O3壳的,从而也就抑制了H2和O2在金属Rh表面的逆反应(Cr2O3不能催化H2和O2的逆反应),提高了全光解水的效率。在此基础上,Domen小组又提出了在光催化剂上同时负载产氢和产氧助催化剂的概念,并基于此设计了Rh/Cr2O3作为产氢助催化剂和Mn3O4作为产氧助催化剂共同负载GaN-ZnO固溶体的新型光催化剂体系[232](如图 27b所示),尽管在可见光下(λ>420 nm)全分解水的表观量子效率不是很高(约为1%),但这是首次成功展示了在光催化剂上同时负载产氢和产氧助催化剂以实现全分解水的可行性,为助催化剂的设计和发展提供了新的思路。
 图27
(a) 以贵金属核-Cr2O3壳作为助催化剂的光催化体系全分解水过程示意图[123]; (b)以Rh/Cr2O3和Mn3O4双助催化剂负载的GaN-ZnO光催化剂进行全分解水过程示意图[232]
Figure27.
(a) Schematic model of H2 evolution reaction on core-shell noble-metal-Cr2O3 particulate system as a cocatalyst for photocatalytic overall water splitting[123]; (b) a proposed reaction mechanism for visible-light-driven overall water splitting on GaN-ZnO modified with Mn3O4 and Rh/Cr2O3 (core-shell) nanoparticles[232]
图27
(a) 以贵金属核-Cr2O3壳作为助催化剂的光催化体系全分解水过程示意图[123]; (b)以Rh/Cr2O3和Mn3O4双助催化剂负载的GaN-ZnO光催化剂进行全分解水过程示意图[232]
Figure27.
(a) Schematic model of H2 evolution reaction on core-shell noble-metal-Cr2O3 particulate system as a cocatalyst for photocatalytic overall water splitting[123]; (b) a proposed reaction mechanism for visible-light-driven overall water splitting on GaN-ZnO modified with Mn3O4 and Rh/Cr2O3 (core-shell) nanoparticles[232]
4.2.2.6 助催化剂的挑战
光解水制氢过程要想突破低效率的瓶颈问题,助催化剂的作用至关重要。但对助催化剂的研究存在很多的挑战,其中最大的问题是很难在实验上直接观测助催化剂的作用过程。特别是在粉体光解水体系中,光催化剂材料特性、助催化剂材料特性、溶液环境等诸多因素交织在一起,使得助催化剂的作用过程异常复杂。其中,最受关注的还是光催化剂与助催化剂之间的载流子转移过程。基于固体能带理论建立起来的载流子转移机制虽然还存在着一些疑问(如4.2.2.3和4.2.2.4节中论述),但其总体上可以很好的阐释相关的载流子转移行为。相比于对载流子转移作用的广泛研究,助催化剂的降低反应活化能作用受到的关注不是很多。现在被多数研究学者所接受的理解是光解水反应是一个上坡反应,助催化剂可以有效降低反应活化能,加速反应的进行。但不同助催化剂在降低光解水反应活化能上的差异,以及降低的具体数值都还没有相关的研究。因而,在助催化剂的核心作用中(促进载流子转移和降低反应活化能),究竟哪一个作用对光解水反应的影响更大?这个疑问的研究对助催化剂的发展具有重要意义,甚至会成为解决光解水反应瓶颈问题的关键突破口。
复合相助催化剂已展现出在光催化应用中的巨大潜力,是助催化剂发展的重要方向,但其发展仍然受到两个方面的限制:一是复合相助催化剂的制备,在光催化剂上进行纳米尺度(< 10 nm)的组分控制难度很大;二是组分间的协同作用机制目前只是针对特定案例的分析,尚不明确和完备,并不具有普适性。
4.2.3 复合其他导电材料
将半导体材料与一些导电聚合物[250]、碳纳米管[251]、石墨烯[252-255]等导电物质相复合也可促进半导体上的光生载流子分离,从而提高半导体材料的光催化活性。特别是自2004年以来被大量研究的石墨烯材料,由于其具有的高的载流子迁移率(20 000 cm2·V-1·s-1),高的比表面积(2 620 m2·g-1)和高的透光率(97.7%),被广泛用来构建石墨烯-半导体复合光催化材料。迄今为止,TiO2、ZnO、WO3、SnO2、Cu2O、Fe2O3、NiO、MnO2、ZrO2、C3N4、ZnS、CdS、CdSe、Bi2WO6、BiVO4、Sr2Ta2O7、ZnFe2O4、InNbO4等多种半导体与石墨烯复合后都可获得光催化性能的提高[194-197]。这主要得益于两个方面,一是石墨烯纳米材料的二维平面能够使半导体材料较好的分散,二是石墨烯优异的电子导电性能够有效的抑制光生电子和空穴的复合。例如,石墨烯含量为1.0%的石墨烯-CdS复合光催化剂在可见光(≥420 nm)照射下的光解水产氢速率可达1.12 mmol·h-1,比CdS的产氢速率提高了将近5倍,相应的在420 nm单色光下的表观量子效率为22.5%[256]。
在最近的研究中发现,还原石墨烯(PRGO)还可作为固态电子转移介质被应用于Z机制全分解水体系中(无氧化-还原电对参加的直接Z机制体系)[214]。譬如,在以Ru/SrTiO3:Rh为产氢光催化剂、以PRGO/BiVO4为产氧光催化剂的体系中(图 28a),PRGO可作为BiVO4上光生电子和Ru/SrTiO3:Rh上光生空穴的复合中心,从而触发Z机制全分解水过程(图 28b)。同样,在以金属硫化物为产氢光催化剂,TiO2为产氧光催化剂的体系中,还原石墨烯也起到电子转移介质的作用[257]。
 图28
(a) Ru/SrTiO3:Rh和PRGO/BiVO4在水中(pH= 3.5)的悬浮状态示意图; (b)可见光下Ru/SrTiO3:Rh和PRGO/BiVO4组成的Z机制全分解水过程示意图[214]
Figure28.
(a) Schematic image of a suspension of Ru/SrTiO3: Rh and PRGO/BiVO4 in water at pH 3.5;
(b) Mechanism of water splitting in a Z-scheme photocatalysis system consisting of Ru/SrTiO3:Rh and PRGO/BiVO4 under visible-light irradiation[214]
图28
(a) Ru/SrTiO3:Rh和PRGO/BiVO4在水中(pH= 3.5)的悬浮状态示意图; (b)可见光下Ru/SrTiO3:Rh和PRGO/BiVO4组成的Z机制全分解水过程示意图[214]
Figure28.
(a) Schematic image of a suspension of Ru/SrTiO3: Rh and PRGO/BiVO4 in water at pH 3.5;
(b) Mechanism of water splitting in a Z-scheme photocatalysis system consisting of Ru/SrTiO3:Rh and PRGO/BiVO4 under visible-light irradiation[214]
4.3 半导体的敏化
引入具有宽光谱响应的单元对半导体进行敏化也是一种扩展半导体光吸收范围和提高其光催化效率的有效手段。根据敏化剂的种类,可以将半导体的敏化分为有机分子敏化、量子点敏化和贵金属等离子共振敏化,其对应的敏化剂分别为有机配合物(多为染料分子),无机半导体和贵金属纳米颗粒。
4.3.1 有机分子敏化
有机分子敏化半导体提高其可见光吸收的方法起源于染料敏化太阳能电池的研究[258]。1981年Grätzel等利用联吡啶钌配合物敏化Nb掺杂的TiO2实现了可见光下的分解水制氢,这也被认为是染料敏化半导体光解水制氢的开山之作[259]。染料敏化半导体光解水制氢的反应过程如图 29所示[260],首先染料分子受光激发,电子由基态跃迁到激发态,即电子由染料分子中的最高占据轨道(HOMO)跃迁到最低未占据轨道(LUMO)上。如果染料分子的LUMO轨道高于半导体的导带,则电子会在能极差的驱动下由染料分子转移到半导体导带上,从而进行水的还原反应。在上述过程中,转移到半导体导带上的电子很容易与基态(氧化态)染料分子复合而不能去还原水分子,因而在染料敏化半导体光解水制氢体系中,电子给体的存在是必需的,电子给体要快速的还原掉氧化态的染料分子使之再生,从而保证体系中有足够的电子去参与水还原反应。除此之外,在半导体上负载助催化剂能够快速转移半导体导带上的电子去还原水分子,从而降低了电子与氧化态染料分子的复合,从而提高体系的光解水制氢能力。由此,染料敏化半导体光解水制氢过程包含4个步骤:(1)染料分子受光激发;(2)染料激发态电子转移到半导体导带上;(3)转移到半导体导带上的电子还原水分子;(4)染料分子的再生。
过去几十年以来研究人员在染料分子、半导体、电子给体和助催化剂上进行了广泛的研究[261-262]。用于敏化半导体进行光解水制氢的染料分子可以分为金属基和非金属基配合物两大类,其中金属基配合物主要为Ru基配合物、金属卟啉类化合物和金属酞菁类化合物,非金属基配合物主要有呫吨类染料(如曙红Y、罗丹明B、罗丹明6G、赤藓红、赤藓红B、伊红、玫瑰红、萤光素钠等)、阳离子有机染料以及给体-π桥-受体(D-π-A)类有机染料。TiO2是染料敏化半导体光解水制氢体系最常用的半导体材料,除此之外,ZnO、SnO2、TaON、CdS、g-C3N4、BiOCl、铌酸盐、钛酸盐等半导体材料也可用于染料敏化光解水制氢体系。在染料敏化半导体光解水制氢体系中,三乙醇胺(TEOA)和乙二胺四乙酸(EDTA)是最常用的电子给体,而当电子给体利用IO3-/I-或Fe2+/Fe3+氧化还原电对时,染料敏化半导体可以实现水的全分解[263]。助催化剂是染料敏化半导体光解水制氢体系中至关重要的部分,除了广谱助催化剂贵金属Pt之外,一些电解水产氢用催化剂(如Ni、Co、Cu(氢)氧化物、Mo硫化物等)、Ni(Co)类分子化合物以及氢化酶也可以起到助催化剂的作用。
利用染料分子敏化半导体进行光解水制氢的最大优势在于染料分子具有宽光谱响应,其响应范围甚至可以由可见光区扩展到红外光区。但该体系也存在着很大的弊端:(1)光生电子复合途径多,譬如染料分子中基态和激发态的复合,半导体导带上电子与基态(氧化态)染料分子的复合;(2)效率高的染料大多都是贵金属Ru基配合物,价格昂贵;(3)染料分子不稳定,容易被光生电子降解;(4)染料的过量吸附会阻碍光生电子的传输。
4.3.2 量子点敏化
在半导体敏化体系中,相比于利用染料敏化剂,量子点敏化剂具有稳定、价廉的优势。量子点敏化半导体的研究也是起源于敏化太阳能电池,在光解水领域,量子点敏化半导体更多的是应用于光电催化分解水。最常用的量子点是无机半导体材料,如Cd、Sn、Pd的硫化物、硒化物和碲化物等[264-266]。如在4.2.1.1节中所述,这些窄带隙半导体负载到宽带隙半导体后要形成Ⅱ型的异质结构,即敏化(窄带隙)半导体的导带低位置要高于宽带隙半导体的导带低位置,才能实现光生电子由敏化剂向宽带隙半导体的注入。
实际上,半导体敏化过程在任意一个含有可见光吸收组分的半导体异质结构中都可能存在。如余家国等在典型的Ⅱ型的半导体异质结构CdS/TiO2中发现[267]:当使用全光谱同时激发CdS和TiO2时,CdS中的光生电子向TiO2转移,而TiO2中的光生空穴则向CdS转移;当使用可见光只激发CdS时,则CdS中的光生电子会转移、注入到TiO2的导带,从而实现对TiO2的敏化。为了将敏化剂与半导体材料之间的接触最大化,以充分发挥敏化剂的敏化作用,将半导体敏化剂的颗粒尺寸减小到量子点尺寸,就成为了量子点敏化。有关Ⅱ型半导体异质结构的优势和缺点详见4.2.1.1节,这里需要说明的是:(1)由于量子尺寸效应,半导体量子点敏化剂的能带结构会发生较大的改变,通常表现为带隙扩大,导带低和价带顶的位置分别上移和下移;(2)相对于窄带隙半导体敏化体系,量子点半导体敏化体系的特点在于牺牲了部分可见光吸收能力,但增加了电子转移的驱动力(因量子点半导体和宽带隙半导体导带低之间的能极差增大)。
碳量子点因其优异的光学特性,近年来受到越来越多的重视。研究发现,因量子限域效应,使得氮掺杂石墨烯量子点具有了合适的带隙去全分解水[268]。而将碳量子点与半导体(如TiO2[269]、SiO2[269]、Ag3PO4[270]等)复合,也可实现量子点敏化过程。
4.3.3 金属等离子体敏化
当光照射贵金属纳米颗粒时(金属纳米颗粒的粒径要远小于光波长),如果入射光子频率与金属纳米颗粒(如Au、Ag和Cu)电子的振动频率相当,光子与金属表面的自由电子相互耦合,发生集体振荡产生表面等离子波,并且出现的表面波被束缚在很小的范围,即为局域表面等离子体共振。此时,与等离子体振荡频率相当的光就会被金属纳米颗粒吸收或者散射。金属纳米粒子的等离子体共振可以用来敏化半导体材料以提高其可见光下的光催化性能,研究人员针对该提高作用提出了3种可能的机理[271]:(1)金属共振电子转移到半导体上(图 30[7])。当金属的费米能级高于半导体的费米能级时,金属表面由等离子共振产生的热电子在能级差的作用下,由金属向半导体转移并在半导体表面参与光催化反应;(2)近场电磁场增强作用。局域表面等离子体共振效应可以在金属表面产生局域近场,并引起极大的近场增强。该电磁场会随着金属与半导体空间距离的拉大而衰减,衰减长度约为200 nm;(3)共振光子的散射作用。
金属对光的散射会随着金属粒子尺寸的增加而增强,从而增加光在金属-半导体之间的传输路径。表面等离子体吸收光的频率和范围一般在可见光区,且吸光峰位与金属纳米颗粒的尺寸、形貌、结构以及周围环境介质相关[272-274],譬如Au纳米颗粒的共振吸收峰可由可见光区延伸到红外光区[275-276]。等离子体敏化可以显著提高半导体材料的光解水性能,如Au-TiO2在可见光下的光电解水制氢性能相比于TiO2提高了66倍[277]。在Ag-AgCl等离子体敏化半导体体系中,可见光只能激发Ag纳米颗粒,不能激发宽带隙的AgCl(3.25 eV),从而避免了AgCl的光腐蚀[278]。
5 展望
光解水反应的最终目的是实现太阳能到氢能的转化,这是一个由热力学和动力学因素共同影响的过程。自Honda-Fujishima效应发现以来,光电催化和光催化分解水的研究快速发展,特别是对光电极材料和光催化材料的开发研究持续深入,很多关键基础科学问题也已获得重大突破。但光分解水过程仍受制于低能量转化效率这个瓶颈问题,距工业化应用还有很长的一段路要走。而突破瓶颈的关键是开发具有可见光响应的、高效的光催化材料体系。
本文论述了半导体光解水制氢的基本原理、扩展半导体可见光吸收和提高半导体光催化活性的方法策略、以及相关研究中所取得的重要进展。同时,也分析了该领域所面临的诸多问题:如全分解水的挑战、能带调控的困局、异质结构的弊端、助催化剂的瓶颈等。这些问题既相互矛盾又相互联系,一个瓶颈问题的制约因素有可能是解决另一个瓶颈问题的关键突破口。在以往的研究中,大多只针对某一个具体的问题进行研究,这虽然有助于加深对光解水反应影响因素的认识,但却远远不能解决实际应用问题。在对半导体光解水制氢的研究过程中,研究人员越来越深刻地认识到全面、宏观的理解整个过程,将诸多影响因素统一考虑、有所取舍是突破光解水制氢所面临的效率瓶颈问题的关键。我们认为未来半导体光解水制氢的研究应该着重关注以下几个问题:
(1) 如何在不影响光生载流子氧化还原能力的前提下引入半导体的可见光吸收?
(2) 直接Z机制的作用机理以及在全分解水中的应用;
(3) Ⅰ型半导体异质结构在光解水领域的开发和应用;
(4) 助催化剂作用机理的深入研究,并据此开发新型助催化剂。
-
-
[1]
Fujishima A, Honda K. Nature, 1972, 238(5358):37-38 doi: 10.1038/238037a0
-
[2]
Kudo A, Miseki Y. Chem. Soc. Rev., 2009, 38(1):253-278 doi: 10.1039/B800489G
-
[3]
Chen X B, Shen S H, Guo L J, et al. Chem. Rev., 2010, 110(11):6503-6570 doi: 10.1021/cr1001645
-
[4]
Kitano M, Hara M. J. Mater. Chem., 2010, 20(4):627-641 doi: 10.1039/B910180B
-
[5]
Chen X B, Li C, Gratzel M, et al. Chem. Soc. Rev., 2012, 41(23):7909-7937 doi: 10.1039/c2cs35230c
-
[6]
Yerga R M N, Galvan M C A, del Valle F, et al. Chemsus-chem, 2009, 2(6):471-485 doi: 10.1002/cssc.v2:6
-
[7]
Li X, Yu J G, Low J X, et al. J. Mater. Chem. A, 2015, 3(6): 2485-2534 doi: 10.1039/C4TA04461D
-
[8]
Osterloh F E. Chem. Mater., 2008, 20(1):35-54 doi: 10.1021/cm7024203
-
[9]
Artero V, Chavarot-Kerlidou M, Fontecave M. Angew. Chem. Int. Ed., 2011, 50(32):7238-7266 doi: 10.1002/anie.v50.32
-
[10]
Liu G, Yang H G, Pan J, et al. Chem. Rev., 2014, 114(19): 9559-9612 doi: 10.1021/cr400621z
-
[11]
Liu G, Yu J C, Lu G Q, et al. Chem. Commun., 2011, 47(24):6763-6783 doi: 10.1039/c1cc10665a
-
[12]
Zhang K, Guo L J. Catal. Sci. Technol., 2013, 3(7):1672-1690 doi: 10.1039/c3cy00018d
-
[13]
Hisatomi T, Kubota J, Domen K. Chem. Soc. Rev., 2014, 43(22):7520-7535 doi: 10.1039/C3CS60378D
-
[14]
Ekambaram S. J. Alloys Compd., 2008, 448(1/2):238-245
-
[15]
Liu G, Wang L Z, Yang H G, et al. J. Mater. Chem., 2010, 20(5):831-843 doi: 10.1039/B909930A
-
[16]
Ni M, Leung M K H, Leung D Y C, et al. Renewable Sustainable Energy Rev., 2007, 11(3):401-425 doi: 10.1016/j.rser.2005.01.009
-
[17]
Sayama K, Arakawa H. J. Phys. Chem., 1993, 97(3):531-533 doi: 10.1021/j100105a001
-
[18]
Chen X Y, Yu T, Fan X X, et al. Appl. Surf. Sci., 2007, 253(20):8500-8506 doi: 10.1016/j.apsusc.2007.04.035
-
[19]
Takahara Y, Kondo J N, Takata T, et al. Chem. Mater., 2001, 13(4):1194-1199 doi: 10.1021/cm000572i
-
[20]
Janaky C, Rajeshwar K, de Tacconi N R, et al. Catal. Today, 2013, 199:53-64 doi: 10.1016/j.cattod.2012.07.020
-
[21]
Domen K, Kudo A, Onishi T. J. Catal., 1986, 102(1):92-98 doi: 10.1016/0021-9517(86)90143-0
-
[22]
Ogura S, Kohno M, Sato K, et al. Appl. Surf. Sci., 1997, 121: 521-524
-
[23]
Inoue Y, Kubokawa T, Sato K. J. Phys. Chem., 1991, 95(10):4059-4063 doi: 10.1021/j100163a032
-
[24]
Inoue Y, Asai Y, Sato K. J. Chem. Soc. Faraday Trans., 1994, 90(5):797-802 doi: 10.1039/FT9949000797
-
[25]
Takata T, Furumi Y, Shinohara K, et al. Chem. Mater., 1997, 9(5):1063-1064 doi: 10.1021/cm960612b
-
[26]
Ogura S, Sato K, Inoue Y. Phys. Chem. Chem. Phys., 2000, 2(10):2449-2454 doi: 10.1039/b000187m
-
[27]
Kudo A, Sayama K, Tanaka A, et al. J. Catal., 1989, 120(2): 337-352 doi: 10.1016/0021-9517(89)90274-1
-
[28]
Kudo A, Kato H, Nakagawa S. J. Phys. Chem. B, 2000, 104(3):571-575 doi: 10.1021/jp9919056
-
[29]
Miseki Y, Kato H, Kudo A. Chem. Lett., 2005, 34(1):54-55 doi: 10.1246/cl.2005.54
-
[30]
Miseki Y, Kato H, Kudo A. Chem. Lett., 2006, 35(9):1052-1053 doi: 10.1246/cl.2006.1052
-
[31]
Kato H, Kudo A. Catal. Lett., 1999, 58(2/3):153-155 doi: 10.1023/A:1019082001809
-
[32]
Ishihara T, Nishiguchi H, Fukamachi K, et al. J. Phys. Chem. B, 1999, 103(1):1-3 doi: 10.1021/jp983590k
-
[33]
Kato H, Kudo A. Chem. Phys. Lett., 1998, 295(5/6):487-492
-
[34]
Machida M, Mitsuyama T, Ikeue K, et al. J. Phys. Chem. B, 2005, 109(16):7801-7806 doi: 10.1021/jp044833d
-
[35]
Shimizu K, Tsuji Y, Kawakami M, et al. Chem. Lett., 2002, 31(11):1158-1159 doi: 10.1246/cl.2002.1158
-
[36]
Kurihara T, Okutomi H, Miseki Y, et al. Chem. Lett., 2006, 35(3):274-275 doi: 10.1246/cl.2006.274
-
[37]
Otsuka H, Kim K Y, Kouzu A, et al. Chem. Lett., 2005, 34(6):822-823 doi: 10.1246/cl.2005.822
-
[38]
Kim D W, Cho I S, Lee S, et al. J. Am. Ceram. Soc., 2010, 93(11):3867-3872 doi: 10.1111/jace.2010.93.issue-11
-
[39]
Cheng H F, Huang B B, Liu Y Y, et al. Chem. Commun., 2012, 48(78):9729-9731 doi: 10.1039/c2cc35289c
-
[40]
Yourey J E, Bartlett B M. J. Mater. Chem., 2011, 21(21): 7651-7660 doi: 10.1039/c1jm11259g
-
[41]
Cho I S, Kwak C H, Kim D W, et al. J. Phys. Chem. C, 2009, 113(24):10647-10653 doi: 10.1021/jp901557z
-
[42]
Huang G L, Zhang C, Zhu Y F. J. Alloys Compd., 2007, 432(1/2):269-276
-
[43]
Ahmad M I, Mohanty G, Cambrea L R, et al. J. Cryst. Growth, 2012, 343(1):115-121 doi: 10.1016/j.jcrysgro.2011.12.081
-
[44]
Lee S, Teshima K, Fujisawa M, et al. Phys. Chem. Chem. Phys., 2009, 11(19):3628-3633 doi: 10.1039/b821209k
-
[45]
Konta R, Kato H, Kobayashi H, et al. Phys. Chem. Chem. Phys., 2003, 5(14):3061-3065 doi: 10.1039/b300179b
-
[46]
Tokunaga S, Kato H, Kudo A. Chem. Mater., 2001, 13(12): 4624-4628 doi: 10.1021/cm0103390
-
[47]
Hu Y W, Zhang W, Pan W. Mater. Res. Bull., 2013, 48(2): 668-671 doi: 10.1016/j.materresbull.2012.11.029
-
[48]
Girija K, Thirumalairajan S, Patra A K, et al. Curr. Appl. Phys., 2013, 13(4):652-658 doi: 10.1016/j.cap.2012.11.004
-
[49]
Apte S K, Garaje S N, Valant M, et al. Green Chem., 2012, 14(5):1455-1462 doi: 10.1039/c2gc16416g
-
[50]
Karunakaran C, Raadha S S, Gomathisankar P. J. Alloys Compd., 2013, 549:269-275 doi: 10.1016/j.jallcom.2012.09.035
-
[51]
Sato J, Saito N, Nishiyama H, et al. J. Phys. Chem. B, 2001, 105(26):6061-6063 doi: 10.1021/jp010794j
-
[52]
Sato J, Saito N, Nishiyama H, et al. Chem. Lett., 2001, 30(9): 868-869 doi: 10.1246/cl.2001.868
-
[53]
Sato J, Kobayashi H, Saito N, et al. J. Photochem. Photobiol. A, 2003, 158(2/3):139-144
-
[54]
Sato J, Kobayashi H, Inoue Y. J. Phys. Chem. B, 2003, 107(31):7970-7975 doi: 10.1021/jp030021q
-
[55]
Ikarashi K, Sato J, Kobayashi H, et al. J. Phys. Chem. B, 2002, 106(35):9048-9053 doi: 10.1021/jp020539e
-
[56]
Sato J, Kobayashi H, Ikarashi K, et al. J. Phys. Chem. B, 2004, 108(14):4369-4375 doi: 10.1021/jp0373189
-
[57]
Liu R M, Yin J Z, Du W, et al. Eur. J. Inorg. Chem., 2013, 2013(8):1358-1362 doi: 10.1002/ejic.201200975
-
[58]
Li B X, Liu T X, Hu L Y, et al. J. Phys. Chem. Solids, 2013, 74(4):635-640 doi: 10.1016/j.jpcs.2012.12.020
-
[59]
Zhu J X, Yin Z Y, Yang D, et al. Energy Environ. Sci., 2013, 6(3):987-993 doi: 10.1039/c2ee24148j
-
[60]
Abdulkarem A M, Elssfah E M, Yan N N, et al. J. Phys. Chem. Solids, 2013, 74(4):647-652 doi: 10.1016/j.jpcs.2012.12.027
-
[61]
Zhang J Y, Wang Y H, Zhang J, et al. ACS Appl. Mater. Interfaces, 2013, 5(3):1031-1037 doi: 10.1021/am302726y
-
[62]
Lei Z B, You W S, Liu M Y, et al. Chem. Commun., 2003(17):2142-2143 doi: 10.1039/b306813g
-
[63]
Tsuji I, Kato H, Kobayashi H, et al. J. Am. Chem. Soc., 2004, 126(41):13406-13413 doi: 10.1021/ja048296m
-
[64]
Jang J S, Borse P H, Lee J S, et al. J. Chem. Phys., 2008, 128(15):154717 doi: 10.1063/1.2900984
-
[65]
Tsuji I, Kato H, Kudo A. Angew. Chem. Int. Ed., 2005, 44(23):3565-3568 doi: 10.1002/(ISSN)1521-3773
-
[66]
Sato J, Saito N, Yamada Y, et al. J. Am. Chem. Soc., 2005, 127(12):4150-4151 doi: 10.1021/ja042973v
-
[67]
Ma S S K, Hisatomi T, Maeda K, et al. J. Am. Chem. Soc., 2012, 134(49):19993-19996 doi: 10.1021/ja3095747
-
[68]
Hara M, Hitoki G, Takata T, et al. Catal. Today, 2003, 78(1/2/3/4):555-560
-
[69]
Ishikawa A, Takata T, Kondo J N, et al. J. Am. Chem. Soc., 2002, 124(45):13547-13553 doi: 10.1021/ja0269643
-
[70]
Wang X C, Maeda K, Thomas A, et al. Nat. Mater., 2009, 8(1):76-80 doi: 10.1038/nmat2317
-
[71]
Zhang G G, Lan Z A, Lin L H, et al. Chem. Sci., 2016, 7(5): 3062-3066 doi: 10.1039/C5SC04572J
-
[72]
Liu J, Liu Y, Liu N Y, et al. Science, 2015, 347(6225):970-974 doi: 10.1126/science.aaa3145
-
[73]
Guo F, Chen J L, Zhang M W, et al. J. Mater. Chem. A, 2016, 4(28):10806-10809 doi: 10.1039/C6TA03424A
-
[74]
Martin D J, Reardon P J T, Moniz S J A, et al. J. Am. Chem. Soc., 2014, 136(36):12568-12571 doi: 10.1021/ja506386e
-
[75]
Zhao G X, Huang X B, Fina F, et al. Catal. Sci. Technol., 2015, 5(6):3416-3422 doi: 10.1039/C5CY00379B
-
[76]
Zhao Z W, Sun Y J, Dong F. Nanoscale, 2015, 7(1):15-37 doi: 10.1039/C4NR03008G
-
[77]
Zhang J S, Wang B, Wang X C. Prog. Chem., 2014, 26(1):19-29
-
[78]
Zheng Y, Lin L H, Wang B, et al. Angew. Chem. Int. Ed., 2015, 54(44):12868-12884 doi: 10.1002/anie.v54.44
-
[79]
Zhang J S, Chen Y, Wang X C. Energy Environ. Sci., 2015, 8(11):3092-3108 doi: 10.1039/C5EE01895A
-
[80]
Zheng Y, Lin L H, Ye X J, et al. Angew. Chem. Int. Ed., 2014, 53(44):11926-11930 doi: 10.1002/anie.201407319
-
[81]
Zhang J S, Zhang M W, Lin L H, et al. Angew. Chem. Int. Ed., 2015, 54(21):6297-6301 doi: 10.1002/anie.201501001
-
[82]
Niu P, Zhang L L, Liu G, et al. Adv. Funct. Mater., 2012, 22(22):4763-4770 doi: 10.1002/adfm.v22.22
-
[83]
Liu G, Niu P, Sun C H, et al. J. Am. Chem. Soc., 2010, 132(33):11642-11648 doi: 10.1021/ja103798k
-
[84]
Lin Z Z, Wang X C. Angew. Chem. Int. Ed., 2013, 52(6): 1735-1738 doi: 10.1002/anie.v52.6
-
[85]
Zhang G G, Zhang M W, Ye X X, et al. Adv. Mater., 2014, 26(5):805-809 doi: 10.1002/adma.201303611
-
[86]
Zhang J S, Chen X F, Takanabe K, et al. Angew. Chem. Int. Ed., 2010, 49(2):441-444 doi: 10.1002/anie.200903886
-
[87]
Zhang J S, Zhang G G, Chen X F, et al. Angew. Chem. Int. Ed., 2012, 51(13):3183-3187 doi: 10.1002/anie.v51.13
-
[88]
Zhang J S, Zhang M W, Lin S, et al. J. Catal., 2014, 310:24-30 doi: 10.1016/j.jcat.2013.01.008
-
[89]
郑华荣, 张金水, 王心晨, 等.物理化学学报, 2012, 28(10):2336-2342 doi: 10.3866/PKU.WHXB201209104ZHENG Hua-Rong, ZHANG Jin-Shui, WANG Xin-Chen, et al. Acta Phys-Chim. Sin., 2012, 28(10):2336-2342 doi: 10.3866/PKU.WHXB201209104
-
[90]
Sprick R S, Jiang J X, Bonillo B, et al. J. Am. Chem. Soc., 2015, 137(9):3265-3270 doi: 10.1021/ja511552k
-
[91]
Yang C, Ma B C, Zhang L Z, et al. Angew. Chem. Int. Ed., 2016, 55(32):9202-9206 doi: 10.1002/anie.201603532
-
[92]
Sprick R S, Bonillo B, Clowes R, et al. Angew. Chem. Int. Ed., 2016, 55(5):1824-1828
-
[93]
Schwab M G, Hamburger M, Feng X L, et al. Chem. Commun., 2010, 46(47):8932-8934 doi: 10.1039/c0cc04057f
-
[94]
Shao M W, Cheng L, Zhang X H, et al. J. Am. Chem. Soc., 2009, 131(49):17738-17739 doi: 10.1021/ja908085c
-
[95]
Wang F, Ng W K H, Yu J C, et al. Appl. Catal. B:Environ., 2012, 111:409-414
-
[96]
Liu G, Yin L C, Niu P, et al. Angew. Chem. Int. Ed., 2013, 52(24):6242-6245 doi: 10.1002/anie.201302238
-
[97]
Chiou Y D, Hsu Y J. Appl. Catal. B:Environ., 2011, 105(1/2):211-219
-
[98]
Liu G, Niu P, Yin L C, et al. J. Am. Chem. Soc., 2012, 134(22):9070-9073 doi: 10.1021/ja302897b
-
[99]
Takata T, Tanaka A, Hara M, et al. Catal. Today, 1998, 44(1/2/3/4):17-26
-
[100]
Ikeda S, Hara M, Kondo J N, et al. J. Mater. Res., 1998, 13(4):852-855 doi: 10.1557/JMR.1998.0113
-
[101]
Kato H, Asakura K, Kudo A. J. Am. Chem. Soc., 2003, 125(10):3082-3089 doi: 10.1021/ja027751g
-
[102]
Soldat J, Marschall R, Wark M. Chem. Sci., 2014, 5(10): 3746-3752 doi: 10.1039/C4SC01127A
-
[103]
Liu H, Yuan J, Jiang Z, et al. J. Mater. Chem., 2011, 21(41):16535-16543 doi: 10.1039/c1jm11809a
-
[104]
Mu L C, Zhao Y, Li A L, et al. Energy Environ. Sci., 2016, 9(7):2463-2469 doi: 10.1039/C6EE00526H
-
[105]
Maeda K, Lu D L, Domen K. Chem. Eur. J., 2013, 19(16): 4986-4891 doi: 10.1002/chem.201300158
-
[106]
Pan C S, Takata T, Nakabayashi M, et al. Angew. Chem. Int. Ed., 2015, 54(10):2955-2959 doi: 10.1002/anie.201410961
-
[107]
Zou Z G, Ye J H, Sayama K, et al. Nature, 2001, 414(6864): 625-627 doi: 10.1038/414625a
-
[108]
Xu J S, Pan C S, Takata T, et al. Chem. Commun., 2015, 51(33):7191-7194 doi: 10.1039/C5CC01728A
-
[109]
Zhang P, Zhang J J, Gong J L. Chem. Soc. Rev., 2014, 43(13):4395-4422 doi: 10.1039/C3CS60438A
-
[110]
Hara M, Kondo T, Komoda M, et al. Chem. Commun., 1998(3):357-358 doi: 10.1039/a707440i
-
[111]
Maeda K, Teramura K, Lu D L, et al. Nature, 2006, 440(7082):295 doi: 10.1038/440295a
-
[112]
Maeda K, Teramura K, Domen K. J. Catal., 2008, 254(2): 198-204 doi: 10.1016/j.jcat.2007.12.009
-
[113]
Li J, Liu B D, Yang W J, et al. Nanoscale, 2016, 8(6):3694-3703 doi: 10.1039/C5NR08663A
-
[114]
Takanabe K, Uzawa T, Wang X C, et al. Dalton Trans., 2009(45):10055-10062 doi: 10.1039/b910318j
-
[115]
Maeda K, Saito N, Inoue Y, et al. Chem. Mater., 2007, 19(16):4092-4097 doi: 10.1021/cm0709828
-
[116]
Ni L, Tanabe M, Irie H. et al. Chem. Commun., 2013, 49(86):10094-10096 doi: 10.1039/c3cc45222k
-
[117]
Zhang N, Shi J W, Mao S S. Chem. Commun., 2014, 50(16): 2002-2004 doi: 10.1039/c3cc48026g
-
[118]
Maeda K. Catal. Sci. Technol., 2014, 4(7):1949-1953 doi: 10.1039/C4CY00251B
-
[119]
Li R G, Weng Y X, Zhou X, et al. Energy Environ. Sci., 2015, 8(8):2377-2382 doi: 10.1039/C5EE01398D
-
[120]
Maeda K, Lu D L, Teramura K, et al. Energy Environ. Sci., 2010, 3(4):471-478 doi: 10.1039/B915064A
-
[121]
Zhang K, Kim W, Ma M, et al. J. Mater. Chem. A, 2015, 3(9):4803-4810 doi: 10.1039/C4TA05571C
-
[122]
Kudo A. MRS Bull., 2011, 36(1):32-38 doi: 10.1557/mrs.2010.3
-
[123]
Maeda K, Domen K. J. Phys. Chem. Lett., 2010, 1(18):2655-2661 doi: 10.1021/jz1007966
-
[124]
Maeda K. ACS Catal., 2013, 3(7):1486-1503 doi: 10.1021/cs4002089
-
[125]
Maeda K, Higashi M, Lu D L, et al. J. Am. Chem. Soc., 2010, 132(16):5858-5868 doi: 10.1021/ja1009025
-
[126]
Chen S, Qi Y, Hisatomi T, et al. Angew. Chem. Int. Ed., 2015, 54(29):8498-8501 doi: 10.1002/anie.201502686
-
[127]
Sasaki Y, Iwase A, Kato H, et al. J. Catal., 2008, 259(1): 133-137 doi: 10.1016/j.jcat.2008.07.017
-
[128]
Kato H, Sasaki Y, Iwase A, et al. Bull. Chem. Soc. Jpn., 2007, 80(12):2457-2464 doi: 10.1246/bcsj.80.2457
-
[129]
Sasaki Y, Nemoto H, Saito K, et al. J. Phys. Chem. C, 2009, 113(40):17536-17542 doi: 10.1021/jp907128k
-
[130]
Yang L L, Zhou H, Fan T X, et al. Phys. Chem. Chem. Phys., 2014, 16(15):6810-6826 doi: 10.1039/c4cp00246f
-
[131]
Walsh A, Yan Y, Huda M N, et al. Chem. Mater., 2009, 21(3):547-551 doi: 10.1021/cm802894z
-
[132]
Wang D F, Tang J W, Zou Z G, et al. Chem. Mater., 2005, 17(20):5177-5182 doi: 10.1021/cm051016x
-
[133]
Yi Z G, Ye J H, Kikugawa N, et al. Nat. Mater., 2010, 9(7): 559-564 doi: 10.1038/nmat2780
-
[134]
Tang J W, Zou Z G, Ye J H. Catal. Lett., 2004, 92(1/2):53-56 doi: 10.1023/B:CATL.0000011086.20412.aa
-
[135]
Xie Y P, Liu G, Yin L C, et al. J. Mater. Chem., 2012, 22(14):6746-6751 doi: 10.1039/c2jm16178h
-
[136]
Wang D E, Jiang H F, Zong X, et al. Chem. Eur. J., 2011, 17(4):1275-1282 doi: 10.1002/chem.v17.4
-
[137]
Zhang G G, Huang C J, Wang X C. Small, 2015, 11(9/10): 1215-1221
-
[138]
Zhang G G, Zang S H, Lin L H, et al. ACS Appl. Mater. Interfaces, 2016, 8(3):2287-2296 doi: 10.1021/acsami.5b11167
-
[139]
Zhang G G, Zang S H, Lan Z A, et al. J. Mater. Chem. A, 2015, 3(35):17946-17950 doi: 10.1039/C5TA04767F
-
[140]
Zhang G G, Zang S H, Wang X C. ACS Catal., 2015, 5(2): 941-947 doi: 10.1021/cs502002u
-
[141]
Pan H, Zhu S, Lou X, et al. RSC Adv., 2015, 5(9):6543-6552 doi: 10.1039/C4RA09546D
-
[142]
Zhang H, Lü X J, Li Y M, et al. ACS Nano, 2010, 4(1):380-386 doi: 10.1021/nn901221k
-
[143]
Kim I Y, Lee J M, Kim T W, et al. Small, 2012, 8(7):1038-1048 doi: 10.1002/smll.201101703
-
[144]
Lee J S, You K H, Park C B. Adv. Mater., 2012, 24(8):1084-1088 doi: 10.1002/adma.201104110
-
[145]
Guo J J, Li Y, Zhu S M, et al. RSC Adv., 2012, 2(4):1356-1363 doi: 10.1039/C1RA00621E
-
[146]
Sun Z H, Guo J J, Zhu S M, et al. Nanoscale, 2014, 6(4): 2186-2193 doi: 10.1039/C3NR05249D
-
[147]
Meng F K, Li J T, Cushing S K, et al. ACS Catal., 2013, 3(4):746-751 doi: 10.1021/cs300740e
-
[148]
Sun Z H, Guo J J, Zhu S M, et al. RSC Adv., 2014, 4(53): 27963-27970 doi: 10.1039/C4RA03533J
-
[149]
Huang H, Yue Z K, Li G, et al. J. Mater. Chem. A, 2013, 1(47):15110-15116 doi: 10.1039/c3ta13433d
-
[150]
Bai S, Shen X P, Lü H W, et al. J. Colloid Interface Sci., 2013, 405:1-9 doi: 10.1016/j.jcis.2013.05.023
-
[151]
Liu L, Liu J C, Sun D D. Catal. Sci. Technol., 2012, 2(12): 2525-2532 doi: 10.1039/c2cy20483e
-
[152]
Borgarello E, Kiwi J, Gratzel M, et al. J. Am. Chem. Soc., 1982, 104(11):2996-3002 doi: 10.1021/ja00375a010
-
[153]
Asahi R, Morikawa T, Ohwaki T, et al. Science, 2001, 293(5528):269-271 doi: 10.1126/science.1061051
-
[154]
Irie H, Watanabe Y, Hashimoto K. J. Phys. Chem. B, 2003, 107(23):5483-5486 doi: 10.1021/jp030133h
-
[155]
Ihara T, Miyoshi M, Iriyama Y, et al. Appl. Catal. B: Environ., 2003, 42(4):403-409 doi: 10.1016/S0926-3373(02)00269-2
-
[156]
Liu G, Pan J, Yin L C, et al. Adv. Funct. Mater., 2012, 22(15):3233-3238 doi: 10.1002/adfm.v22.15
-
[157]
Liu G, Wang L Z, Sun C H, et al. Chem. Mater., 2009, 21(7):1266-1274 doi: 10.1021/cm802986r
-
[158]
Liu G, Yin L C, Wang J Q, et al. Energy Environ. Sci, 2012, 5(11):9603-9610 doi: 10.1039/c2ee22930g
-
[159]
Pan X Y, Yang M Q, Fu X Z, et al. Nanoscale, 2013, 5(9): 3601-3614 doi: 10.1039/c3nr00476g
-
[160]
Lü Y H, Yao W Q, Ma X G, et al. Catal. Sci. Technol., 2013, 3(12):3136-3146 doi: 10.1039/c3cy00369h
-
[161]
Niu P, Liu G, Cheng H M. J. Phys. Chem. C, 2012, 116(20):11013-11018 doi: 10.1021/jp301026y
-
[162]
Niu P, Yin L C, Yang Y Q, et al. Adv. Mater., 2014, 26(47): 8046-8052 doi: 10.1002/adma.v26.47
-
[163]
Li R G, Wang X L, Jin S Q, et al. Sci. Rep., 2015, 5:13475 doi: 10.1038/srep13475
-
[164]
Chen X B, Liu L, Yu P Y, et al. Science, 2011, 331(6018): 746-750 doi: 10.1126/science.1200448
-
[165]
Kang Y Y, Yang Y Q, Yin L C, et al. Adv. Mater., 2015, 27(31):4572-4577 doi: 10.1002/adma.v27.31
-
[166]
Wang Z Q, Wen B, Hao Q Q, et al. J. Am. Chem. Soc., 2015, 137(28):9146-9152 doi: 10.1021/jacs.5b04483
-
[167]
Kakuta N, Park K H, Finlayson M F, et al. J. Phys. Chem., 1985, 89(5):732-734 doi: 10.1021/j100251a002
-
[168]
Xing C J, Zhang Y J, Yan W, et al. Int. J. Hydrogen Energy, 2006, 31(14):2018-2024 doi: 10.1016/j.ijhydene.2006.02.003
-
[169]
Tsuji I, Kato H, Kobayashi H, et al. J. Phys. Chem. B, 2005, 109(15):7323-7329 doi: 10.1021/jp044722e
-
[170]
Torimoto T, Adachi T, Okazaki K, et al. J. Am. Chem. Soc., 2007, 129(41):12388-12389 doi: 10.1021/ja0750470
-
[171]
Tsuji I, Kato H, Kudo A. Chem. Mater., 2006, 18(7):1969-1975 doi: 10.1021/cm0527017
-
[172]
Liu H, Yuan J, Shangguan W F, et al. J. Phys. Chem. C, 2008, 112(23):8521-8523 doi: 10.1021/jp802537u
-
[173]
Wang Q Z, Liu H, Jiang L, et al. Catal. Lett., 2009, 131(1/2):160-163
-
[174]
Maeda K, Takata T, Hara M, et al. J. Am. Chem. Soc., 2005, 127(23):8286-8287 doi: 10.1021/ja0518777
-
[175]
Lee Y, Terashima H, Shimodaira Y, et al. J. Phys. Chem. C, 2007, 111(2):1042-1048 doi: 10.1021/jp0656532
-
[176]
Maeda K, Domen K. Chem. Mater., 2010, 22(3):612-623 doi: 10.1021/cm901917a
-
[177]
Yoshida M, Hirai T, Maeda K, et al. J. Phys. Chem. C, 2010, 114(36):15510-15515 doi: 10.1021/jp100106y
-
[178]
Liu G, Yang H G, Wang X W, et al. J. Am. Chem. Soc., 2009, 131(36):12868-12869 doi: 10.1021/ja903463q
-
[179]
Liu G, Sun C H, Yang H G, et al. Chem. Commun., 2010, 46(5):755-757 doi: 10.1039/B919895D
-
[180]
Pan J, Liu G, Lu G M, et al. Angew. Chem. Int. Ed., 2011, 50(9):2133-2137 doi: 10.1002/anie.v50.9
-
[181]
Pan J, Wu X, Wang L Z, et al. Chem. Commun., 2011, 47(29):8361-8363 doi: 10.1039/c1cc13034j
-
[182]
Jiao W, Xie Y P, Chen R Z, et al. Chem. Commun., 2013, 49(100):11770-11772 doi: 10.1039/c3cc46527f
-
[183]
Bi Y, Ouyang S, Umezawa N, et al. J. Am. Chem. Soc., 2011, 133(17):6490-6492 doi: 10.1021/ja2002132
-
[184]
Ohno T, Sarukawa K, Matsumura M. New J. Chem., 2002, 26(9):1167-1170 doi: 10.1039/b202140d
-
[185]
Li R G, Zhang F X, Wang D G, et al. Nat. Commun., 2013, 4: 1432 doi: 10.1038/ncomms2401
-
[186]
Zhu J, Fan F T, Chen R T, et al. Angew. Chem. Int. Ed., 2015, 54(31):9111-9114 doi: 10.1002/anie.201504135
-
[187]
Yang H G, Sun C H, Qiao S Z, et al. Nature, 2008, 453(7195):638-641 doi: 10.1038/nature06964
-
[188]
Song H S, Zhang W J, Cheng C, et al. Cryst. Growth. Des., 2011, 11(1):147-153 doi: 10.1021/cg101062e
-
[189]
Kim H N, Kim T W, Kim I Y, et al. Adv. Funct. Mater., 2011, 21(16):3111-3118 doi: 10.1002/adfm.201100453
-
[190]
Lin C J, Chen S Y, Liou Y H. Electrochem. Commun., 2010, 12(11):1513-1516 doi: 10.1016/j.elecom.2010.08.021
-
[191]
Sun Y H, Zhao Q, Gao J Y, et al. Nanoscale, 2011, 3(10): 4418-4426 doi: 10.1039/c1nr10922g
-
[192]
Wei S Q, Chen Y Y, Ma Y Y, et al. J. Mol. Catal. A:Chem., 2010, 331(1/2):112-116
-
[193]
Cui Y F, Wang C, Liu G, et al. Mater. Lett., 2011, 65(14): 2284-2286 doi: 10.1016/j.matlet.2011.04.041
-
[194]
Yan W, Fan H Q, Yang C. Mater. Lett., 2011, 65(11):1595-1597 doi: 10.1016/j.matlet.2011.03.026
-
[195]
Agrawal M, Gupta S, Pich A, et al. Chem. Mater., 2009, 21(21):5343-5348 doi: 10.1021/cm9028098
-
[196]
Lin D D, Wu H, Zhang R, et al. J. Am. Ceram. Soc., 2010, 93(10):3384-3389 doi: 10.1111/jace.2010.93.issue-10
-
[197]
Cun W, Wang X M, Xu B Q, et al. J. Photochem. Photobiol. A, 2004, 168(1/2):47-52
-
[198]
Lü K Z, Li J, Qing X X, et al. J. Hazard. Mater., 2011, 189(1/2):329-335
-
[199]
Yan H J, Zhang X J, Zhou SQ, et al. J. Alloys Compd., 2011, 509(24):L232-L235 doi: 10.1016/j.jallcom.2011.03.181
-
[200]
Kim H I, Kim J, Kim W, et al. J. Phys. Chem. C, 2011, 115(19):9797-9805 doi: 10.1021/jp1122823
-
[201]
Wang Z Y, Huang B B, Dai Y, et al. J. Phys. Chem. C, 2009, 113(11):4612-4617 doi: 10.1021/jp8107683
-
[202]
Chen S F, Zhao W, Liu W, et al. Chem. Eng. J., 2009, 155(1/2):466-473
-
[203]
Chen S F, Zhao W, Liu W, et al. J. Sol-Gel Sci. Technol., 2009, 50(3):387-396 doi: 10.1007/s10971-009-1908-3
-
[204]
Chen S F, Zhao W, Liu W, et al. Appl. Surf. Sci., 2008, 255(5):2478-2484 doi: 10.1016/j.apsusc.2008.07.115
-
[205]
Wang X W, Liu G, Chen Z G, et al. Chem. Commun., 2009(23):3452-3454 doi: 10.1039/b904668b
-
[206]
Miyauchi M, Nukui Y, Atarashi D, et al. ACS Appl. Mater. Interfaces, 2013, 5(19):9770-9776 doi: 10.1021/am402929d
-
[207]
Zhang L J, Li S, Liu B K, et al. ACS Catal., 2014, 4(10): 3724-3729 doi: 10.1021/cs500794j
-
[208]
Wang J C, Yao H C, Fan Z Y, et al. ACS Appl. Mater. Interfaces, 2016, 8(6):3765-3775 doi: 10.1021/acsami.5b09901
-
[209]
Tada H, Mitsui T, Kiyonaga T, et al. Nat. Mater., 2006, 5(10):782-786 doi: 10.1038/nmat1734
-
[210]
Yu Z B, Xie Y P, Liu G, et al. J. Mater. Chem. A, 2013, 1(8):2773-2776 doi: 10.1039/c3ta01476b
-
[211]
Yun H J, Lee H, Kim N D, et al. ACS Nano, 2011, 5(5): 4084-4090 doi: 10.1021/nn2006738
-
[212]
Kobayashi R, Tanigawa S, Takashima T, et al. J. Phys. Chem. C, 2014, 118(39):22450-22456 doi: 10.1021/jp5069973
-
[213]
Wang Q, Hisatomi T, Ma S S K, et al. Chem. Mater., 2014, 26(14):4144-4150 doi: 10.1021/cm5011983
-
[214]
Iwase A, Ng Y H, Ishiguro Y, et al. J. Am. Chem. Soc., 2011, 133(29):11054-11057 doi: 10.1021/ja203296z
-
[215]
Jo W K, Natarajan T S. ACS Appl. Mater. Interfaces, 2015, 7(31):17138-17154 doi: 10.1021/acsami.5b03935
-
[216]
Xian J J, Li D Z, Chen J, et al. ACS Appl. Mater. Interfaces, 2014, 6(15):13157-13166 doi: 10.1021/am5029999
-
[217]
Wang X W, Yin L C, Liu G. Chem. Commun., 2014, 50(26): 3460-3463 doi: 10.1039/c4cc00044g
-
[218]
Wang Q, Hisatomi T, Jia Q, et al. Nat. Mater., 2016, 15: 611-615 doi: 10.1038/nmat4589
-
[219]
Huang J, Mulfort K L, Du P W, et al. J. Am. Chem. Soc., 2012, 134(40):16472-16475 doi: 10.1021/ja3062584
-
[220]
Zhu H M, Song N H, Lü H J, et al. J. Am. Chem. Soc., 2012, 134(28):11701-11708 doi: 10.1021/ja303698e
-
[221]
Xie Y P, Yu Z B, Liu G, et al. Energy Environ. Sci., 2014, 7(6):1895-1901 doi: 10.1039/c3ee43750g
-
[222]
Zhang J, Xu Q, Feng Z, et al. Angew. Chem. Int. Ed., 2008, 47(9):1766-1769 doi: 10.1002/(ISSN)1521-3773
-
[223]
Wang X, Xu Q, Li M R, et al. Angew. Chem. Int. Ed., 2012, 51(52):13089-13092 doi: 10.1002/anie.201207554
-
[224]
Wang D E, Li R G, Zhu J, et al. J. Phys. Chem. C, 2012, 116(8):5082-5089 doi: 10.1021/jp210584b
-
[225]
Lee J S, Choi W Y. J. Phys. Chem. B, 2005, 109(15):7399-7406 doi: 10.1021/jp044425+
-
[226]
Zhang F X, Maeda K, Takata T, et al. Catal. Today, 2012, 185(1):253-258 doi: 10.1016/j.cattod.2011.09.025
-
[227]
Inoue Y. Energy Environ. Sci., 2009, 2(4):364-386 doi: 10.1039/b816677n
-
[228]
Domen K, Kudo A, Onishi T, et al. J. Phys. Chem., 1986, 90(2):292-295 doi: 10.1021/j100274a018
-
[229]
Lalitha K, Sadanandam G, Kumari V D, et al. J. Phys. Chem. C, 2010, 114(50):22181-22189 doi: 10.1021/jp107405u
-
[230]
Sayama K, Hayashi H, Arai T, et al. Appl. Catal. B, 2010, 94(1/2):150-157
-
[231]
Arai T, Horiguchi M, Yanagida M, et al. J. Phys. Chem. C, 2009, 113(16):6602-6609 doi: 10.1021/jp8111342
-
[232]
Maeda K, Xiong A K, Yoshinaga T, et al. Angew. Chem. Int. Ed., 2010, 49(24):4096-4099 doi: 10.1002/anie.201001259
-
[233]
Zong X, Yan H J, Wu G P, et al. J. Am. Chem. Soc., 2008, 130(23):7176-7177 doi: 10.1021/ja8007825
-
[234]
Zong X, Han J F, Ma G J, et al. J. Phys. Chem. C, 2011, 115(24):12202-12208 doi: 10.1021/jp2006777
-
[235]
Zhang W, Wang Y B, Wang Z, et al. Chem. Commun., 2010, 46(40):7631-7633 doi: 10.1039/c0cc01562h
-
[236]
Tabata M, Maeda K, Ishihara T, et al. J. Phys. Chem. C, 2010, 114(25):11215-11220 doi: 10.1021/jp103158f
-
[237]
Zhang F X, Maeda K, Takata T, et al. Chem. Commun., 2010, 46(39):7313-7315 doi: 10.1039/c0cc02425b
-
[238]
Wang J, Li B, Chen J Z, et al. Appl. Surf. Sci., 2012, 259: 118-123 doi: 10.1016/j.apsusc.2012.07.003
-
[239]
Shen S H, Chen X B, Ren F, et al. Nanoscale Res. Lett., 2011, 6:290 doi: 10.1186/1556-276X-6-290
-
[240]
Yang J H, Yan H J, Wang X L, et al. J. Catal., 2012, 290: 151-157 doi: 10.1016/j.jcat.2012.03.008
-
[241]
Townsend T K, Browning N D, Osterloh F E. Energy Environ. Sci., 2012, 5(11):9543-9550 doi: 10.1039/c2ee22665k
-
[242]
Maeda K, Sakamoto N, Ikeda T, et al. Chem. Eur. J., 2010, 16(26):7750-7759 doi: 10.1002/chem.201000616
-
[243]
刘恩科, 朱秉升, 罗晋生.半导体物理学.7版. Beijing:Electronic Industry Press, 2011: 216-219LIU En-Ke, ZHU Bing-Sheng, LUO Jin-Sheng. Physics of Semiconductor. 7th Ed. Beijing:Electronic Industry Press, 2011: 216-219
-
[244]
Luo M H, Yao W F, Huang C P, et al. J. Mater. Chem. A, 2015, 3(26):13884-13891 doi: 10.1039/C5TA00218D
-
[245]
Cui E T, Lu G X. J. Phys. Chem. C, 2013, 117(50):26415-26425 doi: 10.1021/jp4104933
-
[246]
Li Y H, Xing J, Chen Z J, et al. Nat. Commun., 2013, 4:2500
-
[247]
Ma S S K, Maeda K, Abe R, et al. Energy Environ. Sci., 2012, 5(8):8390-8397 doi: 10.1039/c2ee21801a
-
[248]
Yan H J, Yang J H, Ma G J, et al. J. Catal., 2009, 266(2): 165-168 doi: 10.1016/j.jcat.2009.06.024
-
[249]
Xie Y P, Liu G, Lu G Q, et al. Nanoscale, 2012, 4(4):1267-1270 doi: 10.1039/c2nr11846g
-
[250]
Zhao X, Lü L, Pan B C, et al. Chem. Eng. J., 2011, 170(2/3):381-394
-
[251]
Zhu L W, Zhou L K, Li H X, et al. Mater. Lett., 2013, 95: 13-16 doi: 10.1016/j.matlet.2013.01.004
-
[252]
Xiang Q J, Yu J G, Jaroniec M. Chem. Soc. Rev., 2012, 41(2):782-796 doi: 10.1039/C1CS15172J
-
[253]
Xie X Q, Kretschmer K, Wang G X. Nanoscale, 2015, 7(32):13278-13292 doi: 10.1039/C5NR03338A
-
[254]
Upadhyay R K, Soin N, Roy S S. RSC Adv., 2014, 4(8): 3823-3851 doi: 10.1039/C3RA45013A
-
[255]
An X Q, Yu J C. RSC Adv., 2011, 1(8):1426-1434 doi: 10.1039/c1ra00382h
-
[256]
Li Q, Guo B D, Yu J G, et al. J. Am. Chem. Soc., 2011, 133(28):10878-10884 doi: 10.1021/ja2025454
-
[257]
Iwashina K, Iwase A, Ng Y H, et al. J. Am. Chem. Soc., 2015, 137(2):604-607 doi: 10.1021/ja511615s
-
[258]
Yu Z, Lia F, Sun L C. Energy Environ. Sci., 2015, 8(3):760-775 doi: 10.1039/C4EE03565H
-
[259]
Borgarello E, Kiwi J, Pelizzetti E, et al. Nature, 1981, 289(5794):158-160 doi: 10.1038/289158a0
-
[260]
Maeda K, Eguchi M, Youngblood W J, et al. Chem. Mater., 2008, 20(21):6770-6778 doi: 10.1021/cm801807b
-
[261]
Zhang X, Peng T Y, Song S. J. Mater. Chem. A, 2016, 4(7): 2365-2402 doi: 10.1039/C5TA08939E
-
[262]
Willkomm J, Orchard K L, Reynal A. Chem. Soc. Rev., 2016, 45(1):9-23 doi: 10.1039/C5CS00733J
-
[263]
Abe R, Shinmei K, Hara K. Chem. Commun., 2009(24): 3577-3579 doi: 10.1039/b905935k
-
[264]
Tada H, Fujishima M, Kobayashi H. Chem. Soc. Rev., 2011, 40(7):4232-4243 doi: 10.1039/c0cs00211a
-
[265]
Wang G, Yang X, Qian F, et al. Nano Lett., 2010, 10(3): 1088-1092 doi: 10.1021/nl100250z
-
[266]
Sheng W, Sun B, Shi T, et al. ACS Nano, 2014, 8(7):7163-7169 doi: 10.1021/nn502121t
-
[267]
Qi L, Yu J, Jaroniec M. Phys. Chem. Chem. Phys., 2011, 13(19):8915-8923 doi: 10.1039/c1cp20079h
-
[268]
Yeh T F, Teng C Y, Chen S J, et al. Adv. Mater., 2014, 26(20):3297-3303 doi: 10.1002/adma.v26.20
-
[269]
Li H T, He X D, Kang Z H, et al. Angew. Chem. Int. Ed., 2010, 49(26):4430-4434 doi: 10.1002/anie.200906154
-
[270]
Zhang H, Huang H, Ming H, et al. J. Mater. Chem., 2012, 22(21):10501-10506 doi: 10.1039/c2jm30703k
-
[271]
Zhou N, López-Puente V, Wang Q, et al. RSC Adv., 2015, 5(37):29076-29097 doi: 10.1039/C5RA01819F
-
[272]
Warren S C, Thimsen E. Energy Environ. Sci., 2012, 5(1): 5133-5146 doi: 10.1039/C1EE02875H
-
[273]
Wang P, Huang B, Dai Y, et al. Phys. Chem. Chem. Phys., 2012, 14(28):9813-9825 doi: 10.1039/c2cp40823f
-
[274]
Kelly K L, Coronado E, Zhao L L, et al. J. Phys. Chem. B, 2003, 107(3):668-677 doi: 10.1021/jp026731y
-
[275]
Murphy C J, Gole A M, Stone J W, et al. Acc. Chem. Res., 2008, 41(12):1721-1730 doi: 10.1021/ar800035u
-
[276]
Biesso A, Qian W, Huang X H, et al. J. Am. Chem. Soc., 2009, 131(7):2442-2443 doi: 10.1021/ja8088873
-
[277]
Liu Z W, Hou W B, Pavaskar P, et al. Nano Lett., 2011, 11(3):1111-1116 doi: 10.1021/nl104005n
-
[278]
Wang P, Huang B B, Qin X Y, et al. Angew. Chem., Int. Ed., 2008, 47(41):7931-7933 doi: 10.1002/anie.v47:41
-
[1]
-

图 1 半导体光解水制氢基本过程示意图
Figure 1 Scheme of the basic processes in photocatalytic water splitting using semiconductors




图 7 金属掺杂对半导体能带结构的影响: (a)掺杂形成浅施主能级; (b)掺杂形成浅受主能级; (c)掺杂形成深施主能级; (d)掺杂形成深受主能级
Figure 7 Effects of metal doping on band structure of semiconductors: (a) shallow donor level, (b) shallow acceptor level, (c) deep donor level and (d) deep acceptor level formed by metal doping

图 8 (a) 纤铁矿型钛酸盐的晶体结构示意图; (b)不同样品的吸收光谱: (ⅰ) Cs0.68Ti1.83O4; (ⅱ) Cs0.68Ti1.83O4-xNx; (ⅲ) H0.68Ti1.83O4; (ⅳ) H0.68Ti1.83O4-xNx; (ⅴ)商业P25型二氧化钛; (ⅵ)氮掺杂P25; (b)中的插图是(ⅰ), (ⅱ), (ⅲ)和(ⅳ) 4个样品的光学照片[157]
Figure 8 (a) Crystal structure model of typical lepidocrocite-type titanate; (b) UV-visible light absorption spectra of (ⅰ) Cs0.68Ti1.83O4, (ⅱ) Cs0.68Ti1.83O4-xNx, (ⅲ) H0.68Ti1.83O4, (ⅳ) H0.68Ti1.83O4-xNx, (ⅴ) commercial P25 titania, and (ⅵ) nitrogen-doped P25; Inset is the photos of samples (ⅰ), (ⅱ), (ⅲ) and (ⅳ)[157]

图 9 (a) 红色TiO2微米球的扫描电镜照片; (b)红色TiO2样品的光学照片; (c)白色和红色TiO2的吸收光谱(d)红色TiO2的梯度能带结构示意图[158]
Figure 9 (a) SEM image of a red TiO2 microsphere; (b) optical photograph of the prepared red TiO2 sample; (c) UV-visible absorption of the white TiO2 and red TiO2; (d) schematic of band structures of the red TiO2 with a bandgap gradient[158]

图 10 (a) 氢气处理前(白色TiO2)和(b)氢气处理后(黑色TiO2)TiO2纳米晶体的高分辨透射电镜照片; (c)黑色和白色TiO2的XPS价带谱; (d)表面无序化处理后的黑色TiO2与未处理的白色TiO2的态密度示意图[164]
Figure 10 (a) and (b) HRTEM images of TiO2 nanocrystals before and after hydrogenation, respectively; (c) Valence-band XPS spectra of the white and black TiO2 nanocrystals; (d) Schematic illustration of the DOS of disorder-engineered black TiO2 nanocrystals, as compared to that of unmodified TiO2 nanocrystals[164]


图 12 (a) (AgIn)xZn2(1-x)S2固溶体(ZnS-AgInS2)的紫外-可见吸收光谱, 其中x的值分别为: (ⅰ) 0, (ⅱ) 0.17, (ⅲ) 0.22, (ⅳ) 0.29, (ⅴ) 0.33, (ⅵ) 0.40, (ⅶ) 0.5和(ⅷ) 1; (b) (AgIn)xZn2(1-x)S2固溶体、ZnS和AgInS2的能带结构示意图[63]
Figure 12 (a) UV-vis light absorption spectra of (AgIn)xZn2(1-x)S2 solid solutions; the values of x are (ⅰ) 0, (ⅱ) 0.17, (ⅲ) 0.22, (ⅳ) 0.29, (ⅴ) 0.33, (ⅵ) 0.40, (ⅶ) 0.5, and (ⅷ) 1; (b) Band structures of (AgIn)xZn2(1-x)S2 solid solutions, ZnS, and AgInS2[63]

图 13 (a) GaN、ZnO和GaN-ZnO固溶体的能带结构示意图[176]; (b) GaN、Zn掺杂GaN、和GaN-ZnO固溶体(GaN为主)的杂质能级结构示意图[177]
Figure 13 (a) Schematic illustration of band structures of GaN, ZnO, and their solid solution[176]; (b) Expected energy level diagram for impurity levels in undoped GaN, Zn-doped GaN, and GaN-rich (Ga1-xZnx)(N1-xOx) solid solutions[177]

图 14 (a) 纳米尺寸{101}晶面暴露比例为82%和微米尺寸{001}晶面暴露比例为72%的锐钛矿TiO2晶体的吸收光谱[179]; (b) {001}、{101}和{010}晶面分别占优、微米尺寸的锐钛矿TiO2晶体的吸收光谱[180]; (c)亚微米尺寸的类立方体和长方体WO3的紫外-可见吸收光谱[135]; (d)亚微米尺寸的菱形十二面体和立方体形貌的Ag3PO4晶体的吸收光谱[176]
Figure 14 (a) UV-visible absorption spectra of nanosized anatase crystals with 82% {101} facets and micron-sized anatase crystals with 72% {001} facets[179]; (b) UV-visible absorption spectra of T001, T101 and T010[180]; (c) UV-visible absorption spectra of quasi-cubic-like WO3 crystals and rectangular sheet-like WO3 crystals[135]; (d) UV-vis diffusive reflectance spectra of Ag3PO4 sub-microcrystals with rhombic dodecahedrons and cubes morphologies[176]


图 16 (a) 光沉积Pt-MnOx-BiVO4颗粒的扫描电镜图片; (b) BiVO4系列样品的光解水产氧性能, 其中助催化剂的负载量为0.1%, imp为浸渍法负载, P.D.为光沉积法负载[185]
Figure 16 (a) SEM images of Pt and MnOx deposited rutile BiVO4 particle; (b) photocatalytic water oxidation performance of BiVO4 (imp, impregnation method; P.D., photo-deposition method; the contents of the deposited cocatalysts are all 0.1%)[185]

图 17 (a) 敏化过程, (b) Ⅱ型半导体异质结构, (c) p-n结和(d)矢量Z机制中的载流子转移过程
Figure 17 Charge-carrier transfer process in sensitization (a), traditional (b), p-n junction (c) and vectorial Z-scheme (d) mechanisms

图 18 (a) CdS/ZnO中的矢量Z机制和(b) CdS/Au/ZnO中的矢量Z机制载流子转移过程[210]; (c) Ru修饰的SrTiO3:La, Rh/Au/BiVO4:Mo全分解水示意图[218]
Figure 18 Schematic of (a) the vectorial Z-scheme charge-carrier transfer process in the CdS/ZnO heterostructure and (b) the vectorial Z-scheme charge-carrier transfer process in the CdS/Au/ZnO heterostructure[210]; (c) Schematic of overall water splitting on the Ru-modified SrTiO3:La, Rh/Au/BiVO4:Mo sheet[218]

图 19 (a) Ⅰ型半导体异质结构和(b) Ⅰ型半导体核-壳结构的能带结构示意图
Figure 19 Band alignments of (a) type-Ⅰ and (b) core-shell type-Ⅰ semiconductor heterostructures








图 27 (a) 以贵金属核-Cr2O3壳作为助催化剂的光催化体系全分解水过程示意图[123]; (b)以Rh/Cr2O3和Mn3O4双助催化剂负载的GaN-ZnO光催化剂进行全分解水过程示意图[232]
Figure 27 (a) Schematic model of H2 evolution reaction on core-shell noble-metal-Cr2O3 particulate system as a cocatalyst for photocatalytic overall water splitting[123]; (b) a proposed reaction mechanism for visible-light-driven overall water splitting on GaN-ZnO modified with Mn3O4 and Rh/Cr2O3 (core-shell) nanoparticles[232]

图 28 (a) Ru/SrTiO3:Rh和PRGO/BiVO4在水中(pH= 3.5)的悬浮状态示意图; (b)可见光下Ru/SrTiO3:Rh和PRGO/BiVO4组成的Z机制全分解水过程示意图[214]
Figure 28 (a) Schematic image of a suspension of Ru/SrTiO3: Rh and PRGO/BiVO4 in water at pH 3.5; (b) Mechanism of water splitting in a Z-scheme photocatalysis system consisting of Ru/SrTiO3:Rh and PRGO/BiVO4 under visible-light irradiation[214]

-





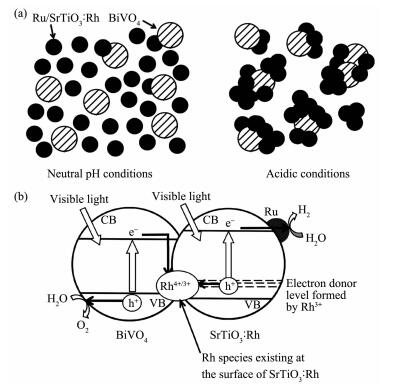


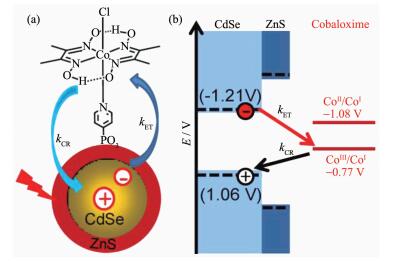









 下载:
下载:





























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 786
- 文章访问数: 31847
- HTML全文浏览量: 7882
 下载:
下载:
