 图 1
配合物的合成路线
Figure 1.
Synthetic routes of complexes 1~4
图 1
配合物的合成路线
Figure 1.
Synthetic routes of complexes 1~4
引用本文:
骆开均, 耿浩, 张晨阳, 倪海亮, 李权. 以2, 3-二苯基吡嗪或2, 3-二苯基喹喔啉为配体的单核和双核金属铂配合物的晶体结构与光物理性质[J]. 无机化学学报,
2017, 33(3): 405-414.
doi:
10.11862/CJIC.2017.026

Citation: LUO Kai-Jun, GENG Hao, ZHANG Cheng-Yang, NI Hai-Liang, LI Quan. Mononuclear and Dinuclear Platinum (Ⅱ) Complexes Based on Ligands 2, 3-Diphenylpyrazine or 2, 3-Diphenylquinoxaline: Crystal Structures and Photophysical Properties[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(3): 405-414. doi: 10.11862/CJIC.2017.026
Citation: LUO Kai-Jun, GENG Hao, ZHANG Cheng-Yang, NI Hai-Liang, LI Quan. Mononuclear and Dinuclear Platinum (Ⅱ) Complexes Based on Ligands 2, 3-Diphenylpyrazine or 2, 3-Diphenylquinoxaline: Crystal Structures and Photophysical Properties[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(3): 405-414. doi: 10.11862/CJIC.2017.026
以2, 3-二苯基吡嗪或2, 3-二苯基喹喔啉为配体的单核和双核金属铂配合物的晶体结构与光物理性质
摘要:
以2,3-二苯基吡嗪(H2dpp)、5-甲基-2,3-二苯基吡嗪(H2mdpp)和2,3-二苯基喹喔啉(H2dpq)为配体,乙酰丙酮(Hacac)为辅助配体,合成了一类单核和双核金属铂配合物[Pt(Hdpp)(acac)](1)、[Pt2(dpp)(acac)2](2)、[Pt(Hmdpp)(acac)](3)和[Pt(Hdpq)(acac)](4),并且得到了配合物2、3和4的晶体结构数据。通过对单核配合物1的类似物配合物3和双核配合物2的配位平面、分子扭曲程度等的晶体结构分析,我们合理地推断以2,3-二苯基吡嗪为配体的双核配合物2具有比相应的单核配合物1更加扭曲的分子平面。通过对配合物1和2的紫外-可见吸收光谱和激发光谱的比较,发现由于双核配合物2在激发态的构型变化造成了激发光谱中最低能带比相应的最低能量吸收带光谱红移了18 nm。因此,尽管双核配合物2具有与单核配合物1类似的紫外-可见吸收光谱,最低能吸收带仅比单核配合物1红移5 nm,但是双核配合物2的最大发射峰值λmax为609 nm,比单核配合物1(λmax=546 nm)红移了63 nm。双核配合物2的发射光谱红移现象与配合物的分子构型直接相关。分子扭曲程度更大的双核配合物2在激发态可能发生了一个向平面性更好的构型转变过程,从而进一步降低了激发态能量,造成了发射光谱的红移。
-
关键词:
- 合成
- / 单核/双核金属铂 (Ⅱ) 配合物
- / 晶体结构
- / 光谱红移
English
Mononuclear and Dinuclear Platinum (Ⅱ) Complexes Based on Ligands 2, 3-Diphenylpyrazine or 2, 3-Diphenylquinoxaline: Crystal Structures and Photophysical Properties
Abstract:
Luminescent mononuclear platinum (Ⅱ) complexes, [Pt (Hdpp)(acac)] (1), [Pt (Hmdpp)(acac)] (3) and [Pt (Hdpq)(acac)] (4) and dinuclearnum (Ⅱ) complex, [Pt2(dpp)(acac)2] (2) were obtained by using 2, 3-diphenylp-yrazine (H2dpp), 5-methyl-2, 3-diphenylpyrazine (H2mdpp) and 2, 3-diphenylquinoxaline (H2dpq) as ligands. It is noted that the maximal emission peak value for dinuclear complex 2 is at 609 nm, which has red-shift of about 63 nm compared with its analogue mononuclear complexes 1 and 3, although they seem somewhat analogue in lowest energy absorption band. To examine the cause of anomalous lower emission for dinuclear complex 2, the single crystals for complexes 2, 3 and 4 were grown from dichloromethane and methanol solution. It is found that dinuclear platinum (Ⅱ) complexes 2 incorporating 2, 3-diphenylpyrazine-based bridging ligand has more twisted conformation in the ground than its mononuclear analogue, complexes 3, which has similar structure with complex 1. Furthermore, lowest energy band for dinuclear complex 2 in exciting spectrum shows red-shift of 18 nm compared with its absorption spectrum. The twisted geometry in ground and red-shifted exciting spectrum make us infer reasonably that dinuclear complex 2 may undergo a more profound change from twisted geometry to planar conformation before emission, which led a lower triplet excited sate, resulting in the red-shifted emission of the dinuclear complex 2.
-
0 引言
红色和近红外金属铂磷光材料在有机发光二级管 (OLEDs)[1-11],生物传感器[12-17]等领域具有潜在的应用价值。因此,如何通过分子设计将光谱移到红色和/或近红外区域一直是人们研究的热点。对于环金属铂配合物而言,有2种常见的方式可以使光谱红移,其中之一是通过对配体的适当修饰,如引入一些能够增大配体共轭程度的稠环芳烃。已有的研究表明,配体的共轭程度越大,配合物的最高占有轨道 (HOMO) 和最低空轨道 (LUMO) 之间的能隙越小,吸收和发射光谱向长波方向红移。但是随着能隙的降低,尤其是在红色和近红外区域,往往会伴随着非辐射衰减速度的增减,导致发光量子产率的降低。近年来,人们发现在分子中引入一个以上的金属铂,形成双核或多核铂配合物能够有效的将光谱红移,同时又不至于降低发光量子效率[1-2, 18-23]。例如,以3, 5-二取代吡唑为桥联体,2-苯基吡啶、苯并喹啉等为主配体的双核金属铂配合物,通过对吡唑取代基的选择,可以调节配合物中2个铂原子间的距离,当Pt-Pt距离小于0.35 nm时,产生所谓金属-金属到配体的电荷跃迁 (MMLCT),MMLCT跃迁比金属到配体的电荷跃迁 (MLCT) 能量低,因此能够产生吸收和发射光谱的红移。最近,Williams等[1]利用具有多个配位点的吡嗪衍生物配体合成了一系列单核和双核金属铂配合物,它们发现与单核配合物相比较,双核金属铂配合物的吸收和发射光谱都产生较大的红移现象,因为随着第二个铂原子的引入,配合物的平面性和共轭程度增强,其吸收和发射光谱红移。但是对于以2,3-二苯基吡嗪和2,3-二苯基喹喔啉为配体的双核配合物Pt2L3和Pt2L5是一个例外,在吸收光谱中,Pt2L3和Pt2L5在低能吸收峰仅比相应的单核配合物红移了几个纳米。但是令人惊讶的是在光致发光 (PL) 光谱中,双核配合物的最大发射峰却比单核配合物分别红移了约60和80 nm。对于这种反常的现象,作者推测与双核配合物在基态和激发态的几何构型变化有关,但没有给出相应的单核和双核配合物的晶体学证据。本文以2,3-二苯基吡嗪 (H2dpp)、5-甲基-2,3-二苯基吡嗪 (H2mdpp) 和2,3-二苯基喹喔啉 (H2dpq) 为配体,合成了3个单核和1个双核配合物,并得到了它们的单晶结构。通过对单核和双核配合物的单晶结构分析,结合紫外-可见吸收光谱、PL光谱以及激发光谱进一步详细阐述配合物晶体结构与其吸收和发射光谱的关系。
1 实验部分
1.1 试剂、仪器和测试
所用试剂均为分析纯。反应所用溶剂无水乙醇、三乙胺等使用前经无水处理。表征所用仪器由核磁共振仪 (400和600 MHz, Varian Unity公司);MS质谱仪 (FINNIN LCQDECA质谱仪);紫外-可见分光光度计 (Perkin Elmer Lambda 950,美国);荧光光谱:HORIBA FlouroMax-4稳态/瞬态荧光光谱仪。寿命采用FlouroMax-4测试,NANOLED370为激发光源,光子收集使用TCSPC模式;元素分析采用CARLO ERBA 1106元素分析仪;量子效率在除水、脱气四氢呋喃 (THF) 溶液中测定,参比物为Ru[(bpy)3]Cl2·6H2O (水溶液量子效率为4.2%),配合物的浓度在10~5.0 μmol·L-1;溶剂THF的脱气采用冷冻-脱气-溶解循环脱气方法。
1.2 配合物的合成
5,6-二苯基-2, 3-二氢吡嗪 (dpdhp):将乙二胺 (0.6 g,10 mmol) 和二苯乙二酮 (2.1 g,10 mmol) 放入100 mL双颈瓶中,加入30 mL乙醇,回流5 h后停止反应。冷却至室温,有淡黄色晶体析出,抽滤后的固体用乙醇洗涤3次,把固体放入真空干燥箱烘干,得淡黄色固体2.18 g,产率81%。1H NMR (CDCl3,400 MHz):δ 7.41~7.39(m, 4H), 7.34~7.30(m, 2H),7.27~7.23(m,4H),3.71(s,1H)。13C NMR (DMSO-d6,400 MHz):δ 160.30,137.67,129.69,128.14,127.90,45.77。
2-甲基-5,6-二苯基-2,3-二氢吡嗪 (mdpdhp):将1,2-丙二胺 (0.74 g,10 mmol) 和二苯乙二酮 (2.1 g,10 mmol) 放入100 mL双颈瓶中,加入30 mL乙醇,回流5 h后停止反应。冷却至室温,有淡黄色晶体析出,抽滤后的固体用乙醇洗涤3次,把固体放入真空干燥箱烘干,得淡黄色固体1.85 g,产率75%。1H NMR (CDCl3,400 MHz):δ 7.40~7.30(m, 4H),7.29~7.22(m,6H),4.05~4.00(m,1H),3.53~3.48(m,1H),3.19~3.11(m,1H),1.48~1.47(d,J=2.5 Hz,3H)
2,3-二苯基喹喔啉 (H2mdpp):将邻苯二胺 (1.08 g,10 mmol) 和二苯乙二酮 (2.1 g,10 mmol) 放入100 mL双颈瓶中,加入36 mL乙醇和4 mL乙酸,回流5 h后停止反应。冷却至室温,有白色晶体析出,抽滤后的固体用乙醇洗涤3次,把固体放入真空干燥箱烘干,得白黄色固体2.45 g,产率77%。1H NMR (CDCl3,400 MHz):δ 8.23~8.20(m,1H),7.81~7.78(m,1H),7.53~7.51(d,J=5 Hz,2H),7.38~7.32(m,3H)。
配合物的合成路线如图 1所示:
图 1
配合物的合成路线
Figure 1.
Synthetic routes of complexes 1~4
配合物1的合成:称取dpdhp (0.35 g,1.5 mmol), K2PtCl4(0.65 g,1.56 mmol) 加入100 mL双颈瓶中,加入5 mL水和15 mL乙二醇乙醚,在90 ℃下反应24 h。反应结束后,减压蒸去溶剂,抽滤得到的固体分别用水和乙醇洗涤,干燥后得到0.71 g血红色固体A,产率71%。将A和碳酸铯 (0.05 g,0.15 mmol),新蒸乙酰丙酮 (0.06 g,0.60 mmol) 和15 mL乙二醇乙醚加入100 mL双颈瓶中,氩气保护下回流反应10 h。停止反应,减压蒸去溶剂,用二氯甲烷萃取,有机层用蒸馏水洗涤3次,加入适量无水硫酸镁干燥过夜,抽滤旋蒸并用硅胶柱分离提纯,淋洗剂为石油醚+乙酸乙酯 (5:1,V/V),得血红色固体0.14 g,产率35%。1H NMR (CDCl3,400 MHz):δ 9.12~9.11(d,J=2.5 Hz,1H),8.32~8.31(d,J=2.5 Hz,1H),7.66~7.64(d,J=5 Hz,1H),7.57~7.52(m,5H),7.15~7.10(t,J=5 Hz,1H),6.75~5.70(t,J=5 Hz,1H),6.66~6.64(d,J=5 Hz,1H),5.52(s,1H),2.06~2.05(s,6H)。13C NMR (DMSO-d6,100 MHz):δ 186.35,184.25,163.15, 161.24, 142.35, 139.04, 138.52, 136.97,132.31,130.25,130.34,129.35, 129.25, 129.05, 128.71, 126.40,125.32,123.17,102.71,28.60,27.11。MS (FAB) m/z:526.102 6[M+1]+。元素分析,C21H18N2O2Pt计算值 (%):C 48.00,H 3.45,N 5.33;实验值 (%):C 48.02,H 3.47,N 5.53。
配合物2的合成:反应条件和实验过程同1的合成。只是在制备氯桥联中间体时,配体dpdhp与氯亚铂酸钾的物质的量之比为1:2,产率22%。1H NMR (CDCl3,400 MHz):δ 8.72(s,1H),8.04~8.02(d,J=8 Hz,1H),7.69~7.67(d,J=8 Hz,1H),7.02~6.99(t,J=8 Hz,1H),5.50(s,1H),5.30(s,1H),2.06(s,3H),2.03(s,3H)。13C NMR (DMSO-d6,400 MHz):δ 186.50,184.20, 162.32,142.28,141.64,137.48,131.07, 130.55, 127.30, 122.80,102.68,28.22,27.05。MS (FAB) m/z:819.11[M+1]+。元素分析,C26H24N2O4Pt2计算值 (%):C 38.15,H 2.96,N 3.42;实验值 (%):C 38.17,H 3.01,N 3.62。
配合物3的合成:反应条件和实验过程同1的合成。产率为32%。1H NMR (CDCl3,400 MHz):δ 8.60(s,1H),7.64~7.62(m,5H),7.11~7.07(t,J=5 Hz,1H),6.72~6.68(t,J=5 Hz,1H),6.56~6.54(d,J=5 Hz,1H),5.51(s, 1H), 2.69(s, 3H), 2.07(3H), 2.04(s,3H)。13C NMR (DMSO-d6,400 MHz):δ 186.49, 184.44, 152.51, 150.86,140.22, 138.47, 138.27,130.28,129.51,129.09,128.96, 128.72, 127.00, 123.18, 102.69, 29.71,28.40,21.36。MS (FAB) m/z:540.118 9[M+1]+。元素分析,C22H20N2O2Pt计算值 (%):C 48.98,H 3.74,N 5.19;实验值 (%):C 48.99,H 3.73,N 5.28。
配合物4的合成:反应条件和实验过程同1的合成。产率36.5%。1H NMR (CDCl3,400 MHz):δ 9.33~9.31(d,J=5 Hz,1H),8.11~8.09(d,J=5 Hz,1H),7.75~7.73(m,4H),7.55(m,4H),7.14(s,1H),6.75(s,1H),5.61(s,1H),2.09~2.05(m,6H)。13C NMR (DMSO-d6,400 MHz):δ 186.09, 184.47, 142.58, 141.53, 139.16, 130.51, 129.86, 129.85, 129.48,129.32,129.22,129.04,128.90, 126.01, 123.37,102.01,29.72,28.35。MS (FAB) m/z:576.138 9[M+1]+。元素分析,C25H20N2O2Pt计算值 (%):C 52.17, H 3.50, N 4.87;实验值 (%):C 52.19,H 3.43,N 4.98。
单晶的制备:将配合物溶解在装有三氯甲烷溶液的10 mL小瓶中,然后将小瓶放在装有甲醇的100 mL棕色试剂瓶中,盖好瓶塞后静置直到晶体出现。通过这种方式得到了单核配合物3和4的单晶和双核配合物2的单晶。
1.3 单晶的测试
单晶X射线衍射数据由Xcalibur Eos单晶衍射仪收集 (石墨单色Mo Kα辐射源,λ=0.071 073 nm)。数据还原和吸收校正采用CrysAlisPro (Agilent Technologies) 程序,结构解析和精修采用OLEX2[24]和SHELXS-97[25-26]程序,采用理论加氢获得氢原子。主要的单晶数据和键长、键角列在表 1和表 2中。
 表 1
配合物2、3、4的主要晶体参数
Table 1.
Main crystal parameters of complexes 2, 3 and 4
表 1
配合物2、3、4的主要晶体参数
Table 1.
Main crystal parameters of complexes 2, 3 and 4
Complex 2 3 4 Empirical formula C26H24N2O4Pt2 C22H2ON2O2Pt C25H2ON2O2Pt Formula weight 818.65 1 078.98 575.52 Crystal system Monoclinic Triclinic Triclinic Space group P21/c P1 P1 a/nm 0.908 92(2) 1.230 43(6) 1.004 44(6) b/nm 2.009 26(4) 1.326 89(5) 1.026 63(5) c/nm 1.346 89(3) 1.379 40(7) 1.036 48(6) α/(º) 90 108.903(4) 99.712(4) β/(º) 108.492(3) 113.138(5) 108.526(5) γ/(º) 90 93.025(3) 95.070(4) V/nm3 2.332(10) 1.916(17) 0.987 3(10) Z 4 2 2 Dc/(g·cm-3) 2.331 1.869 1.936 F(000) 1 528 1 040 556 Crystal size / mm 0.3×0.25×0.2 0.3×0.3×0.2 0.3×0.2×0.2 2θ range/(º) 6.228~52.742 5.826~52.744 6.246~52.744 Reflections collected 10 346 15 993 8 276 Independent reflections 4 765 7 833 4 035 Data, restraints, parameters 4 765, 0, 311 7 833, 0, 493 4 035, 0, 273 Goodness-of-fit F2 1.05 1.014 1.006 Final R indexes [I≥2σ(I)] R1=0.028 3, wR2=0.048 3 R1=0.030 8, wR2=0.0514 R1=0.028 9, wR2=0.0500 Final R indexes (all data) R1=0.036 6, wR2=0.050 8 R1=0.042 5, wR2=0.055 6 R1=0.033 3, wR2=0.052 1 Largest diff. peak and hole / (e·nm-3) 930 and-930 950 and-1 190 810 and-1 040 表 1 配合物2、3、4的主要晶体参数
Table 1. Main crystal parameters of complexes 2, 3 and 4
表 2
配合物2、3、4的主要键长和键角
Table 2.
Selected bond lengths (nm) and bond angels (°) for 2, 3 and 4
2 Pt1-O1 0.208 7(4) Pt1-O2 0.199 7(3) Pt1-N1 0.197 9(4) Pt1-C1 0.195 1(5) Pt2-O3 0.209 4(3) Pt2-O4 0.200 3(3) Pt2-N2 0.197 8(4) Pt2-C14 0.196 2(5) C6-C7 0.147 2(7) C8-C9 0.146 6(6) 02-Pt1-O1 91.80(14) N1-Pt1-O1 93.42(15) N1-Pt1-O2 174.44(15) C1-Pt1-O1 170.86(17) C1-Pt1-O2 93.85(18) C1-Pt1-N1 81.21(19) 04-Pt2-O3 92.29(13) N2-Pt2-O3 93.03(15) N2-Pt2-O4 174.05(16) C14-Pt2-O3 172.64(16) C14-Pt2-O4 93.41(17) C14-Pt2-N2 81.52(18) 3 N3-Pt2 0.198 1(4) 03-Pt2 0.199 6(3) 04-Pt2 0.208 5(3) C7-Pt1 0.196 2(5) C18-O1 0.126 6(6) C20-O2 0.126 9(6) N1-Pt1 0.197 4(4) 01-Pt1 0.199 4(3) O2-Pt1 0.208 6(3) C29-Pt2 0.195 3(5) C25-C34 0.148 8(6) C26-C28 0.145 9(7) C24-C27 0.150 8(7) C3-C12 0.147 8(7) C4-C6 0.146 8(6) C29-Pt2-N3 81.45(19) C29-Pt2-O3 92.63(18) C29-Pt2-O4 173.79(17) N3-Pt2-O3 174.05(15) N3-Pt2-O4 93.47(15) 03-Pt2-O4 92.48(13) 4 N3-Pt2 0.198 1(4) 03-Pt2 0.199 6(3) 04-Pt2 0.208 5(3) C7-Pt1 0.196 2(5) C20-O2 0.126 9(6) 01-Pt1 0.199 4(3) C18-O1 0.126 6(6) N1-Pt1 0.197 4(4) 02-Pt1 0.208 6(3) C29-Pt2 0.195 3(5) C26-C28 0.145 9(7) C3-C12 0.147 8(7) C25-C34 0.148 8(6) C24-C27 0.150 8(7) C4-C6 0.146 8(6) C29-Pt2-N3 81.45(19) C29-Pt2-O4 173.79(17) N3-Pt2-O4 93.47(15) C29-Pt2-O3 92.63(18) N3-Pt2-O3 174.05(15) O3-Pt2-O4 92.48(13) 表 2 配合物2、3、4的主要键长和键角
Table 2. Selected bond lengths (nm) and bond angels (°) for 2, 3 and 4CCDC:1517500,2;1517502,3;1517501,4。
2 结果与讨论
2.1 配合物的合成
配体2,3-二苯基吡嗪和5-甲基2,3-二苯基吡嗪的合成是十分有趣的,一般而言,二胺与二苯基乙二酮反应得到5,6-二苯基-2,3-二氢吡嗪和2-甲基5,6-二苯基-2,3-二氢吡嗪,需要在三氯化铁等催化剂下氧化得到2,3-二苯基吡嗪和5-甲基2,3-二苯基吡嗪。我们直接将未氧化的配体与氯亚铂酸钾反应,在生成氯桥联二聚体的同时吡嗪上的2个碳原子也脱氢氧化成共轭的吡嗪环。我们分析二价金属铂离子可能在反应中起到了催化氧化的作用。采用三氯化铱也得到了同样的结果。说明金属离子在形成金属桥联配合物过程中也同时参与了吡嗪环的氧化。另外,由于2,3-二苯基吡嗪和2,3-二苯基喹喔啉有2个与金属离子配位的中心,所以可以形成单核和双核配合物。当配体与金属铂等物质的量反应时主要形成单核配合物;当配体与金属离子比例为1:2时,主要形成双核配合物。但对于5-甲基-2,3-二苯基吡嗪而言,配体与氯亚铂酸钾的物质的量之比从1:2增加到1:5时,仍然主要得到单核产物,仅发现痕量的双核配合物。这可能是吡嗪环的N原子旁边的甲基对第二个金属铂的配位产生了阻碍。
2.2 吸收和发射光谱
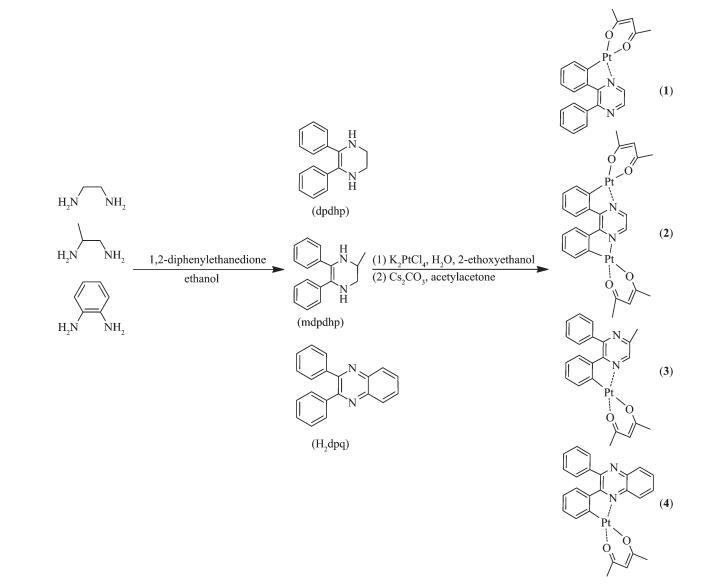
图 5是常温下配合物1~4在四氢呋喃 (THF) 溶液中的紫外-可见吸收光谱。主要的吸收数据见表 3。从图 5可见,4个配合物具有与一般环金属铂配合物类似的吸收特征,高能带 ( < 350 nm) 的强吸收,摩尔吸光系数 (ε) 在5.4×104~3.45×104 L·mol-1·cm-1之间,归属于环金属配合物中配体2,3-二苯基吡嗪或2,3-二苯基喹喔啉的自旋允许的1π→π*跃迁。在低能 (>350 nm) 的弱吸收带,ε值在1.38×104~2.5×103 L·mol-1·cm-1之间,归属于金属增强的自旋-轨道偶合而产生3LC和金属到配体的电荷转移跃迁 (MLCT)[27-28]。比较配合物1和2的吸收光谱可知,配合物2在高能带和低能带的吸收强度都比相应的单核配合物1强。尤其在低能吸收带,ε值大约是配合物1的2倍,这是因为双核分子中有2个金属铂配位中心。另外,尽管配合物2有2个金属铂配合物单元,但是配合物2具有与配合物1相似的吸收光谱。在最低能带,配合物1和2的吸收带分别为444 nm和449 nm。配合物2在最低能带的吸收仅比配合物1红移了约5 nm。在3.2节的讨论中,双核配合物具有比单核配合物更加扭曲的分子构型。我们知道扭曲的分子平面并不能有效地增加分子的共轭程度,因此双核和单核配合物具有基本相同的吸收光谱也是正常的。以5-甲基-2,3-二苯基吡嗪为配体的单核配合物3,在化学结构上只是比配合物1的配体在吡嗪环上多了1个甲基,由于环金属配合物的电子跃迁主要与配体的共轭程度有关,吡嗪环上的甲基对电子跃迁的贡献较小[29-31],因此它们具有类似的吸收光谱,配合物3在低能带的吸收仅比配合物1红移了约5 nm。对于以2,3-二苯基喹喔啉为配体的配合物4,由于喹喔啉环的共轭程度要大于吡嗪环,所以配合物4在最低能带的吸收红移到481 nm。
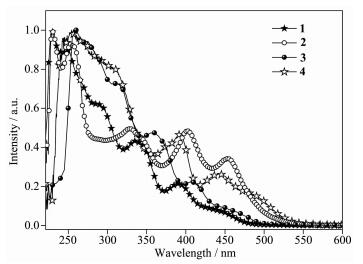 图 5
配合物1~4在THF溶液中的紫外-可见吸收光谱
Figure 5.
UV-Vis absorption spectra of the complexes 1~4 in diluted DCM solution
表 3
配合物1~4的紫外吸收数据
Table 3.
UV-Vis absorption of the complexes 1~4 in diluted DCM solution
图 5
配合物1~4在THF溶液中的紫外-可见吸收光谱
Figure 5.
UV-Vis absorption spectra of the complexes 1~4 in diluted DCM solution
表 3
配合物1~4的紫外吸收数据
Table 3.
UV-Vis absorption of the complexes 1~4 in diluted DCM solution
Complex λabs / nm (ε/ (L·mol-1·cm-1)) 1 228(3.71×104), 248(3.4×104), 290(2.14×104), 340(1.52×104), 394(7.3×103), 444(2.5×103) 2 2229(5.47×104), 256(3.65×104), 327(1.64×104), 401(1.54×104), 449 (6.4×103) 3 3259(3.14×104), 274(2.81×104), 315(1.64×104), 356(1.1×104), 399 (6.2×103), 448(3.3×103) 4 4242(3.16×104), 258(3.43×104), 306(3.21×104), 389(1.74×104), 441(1.15×104), 481 (4.0×103) 表 3 配合物1~4的紫外吸收数据
Table 3. UV-Vis absorption of the complexes 1~4 in diluted DCM solution图 6和图 7是在常温和低温77 K时,配合物1~4在脱氧THF中的光致发光 (PL) 光谱。配合物1~4在常温下和77 K下都具有强的发射光谱。采用瞬态荧光光谱仪对配合物1~4在常温和低温77 K的寿命进行了测试,常温下配合物的寿命在0.1~0.3 μs之间,微秒级的寿命说明这些配合物的发光来源于重金属增强的自旋-轨道偶合而产生的3MLCT激发态,属于典型的磷光发射。低温77 K时,由于温度和溶剂引起的淬灭被抑制,使配合物的寿命增加到1.3~2.6 μs范围。77 K下,PL光谱也变得更具有精细结构,同时PL光谱产生一定程度的蓝移。如配合物1,最大强度发射峰 (λmax) 从546 nm蓝移到536 nm。配合物在常温和77 K下的寿命、常温下溶液量子效率、计算的辐射和非辐射常数[32]见表 4。
 图 6
常温下配合物1~4在THF溶液中的PL光谱
Figure 6.
PL spectra of complexes 1~4 in THF solution at 298 K
图 6
常温下配合物1~4在THF溶液中的PL光谱
Figure 6.
PL spectra of complexes 1~4 in THF solution at 298 K
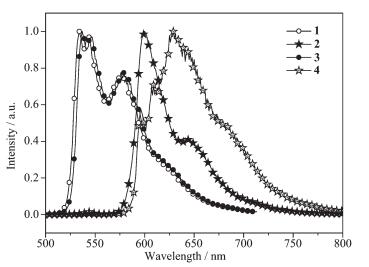 图 7
77 K下配合物1~4在THF溶液中的PL光谱
Figure 7.
PL spectra of complexes 1~4 in THF solution at 77 K
表 4
配合物1~4的PL发射、量子效率、辐射常数、非辐射常数和寿命
Table 4.
PL emission, quantum yield (φem), radiation constant, non-radiation constant and lifetime of complexes 1~4
图 7
77 K下配合物1~4在THF溶液中的PL光谱
Figure 7.
PL spectra of complexes 1~4 in THF solution at 77 K
表 4
配合物1~4的PL发射、量子效率、辐射常数、非辐射常数和寿命
Table 4.
PL emission, quantum yield (φem), radiation constant, non-radiation constant and lifetime of complexes 1~4
Complex λ / nm (298 K) λ / nm (77 K) φem Kr / s-1 Knr / s-1 τ/μs (77 K) 1 540, 565(sh) 535, 545, 579 0.31 2.58×106 5.75×2.58×106 0.12(1.3) 2 609 601, 644 0.12 6.6×105 4.89×2.58×106 0.18(1.6) 3 544, 575(sh) 534, 543, 579 0.33 1.43×2.58×106 2.92×2.58×106 0.23(2.3) 4 642 597, 607, 631 0.18 6.4×105 2.93×2.58×106 0.28(2.6) 表 4 配合物1~4的PL发射、量子效率、辐射常数、非辐射常数和寿命
Table 4. PL emission, quantum yield (φem), radiation constant, non-radiation constant and lifetime of complexes 1~4在常温下的配合物1和3具有基本一致的PL光谱,配合物1的λmax位于546 nm,在578 nm处伴随着一个强度较弱的肩峰。配合物3的λmax位于550 nm,仅仅比配合1红移了4 nm。与相应的吸收光谱类似,吡嗪环上甲基的引入对于发射光谱的影响是很小的。对于以2,3-二苯基喹喔啉为配体的配合物4,由于喹喔啉环的共轭度比吡嗪环大,与2,3-二苯基吡嗪为配体的配合物1相比,λmax从546 nm红移到了632 nm。而在配合物1~4的PL光谱中,配合物1和配合物2的PL光谱最为特别,在对它们的紫外-可见吸收光谱的讨论中,2个配合物具有近似的吸收光谱,配合物2在最低能带的吸收仅比配合物1红移了5 nm,但是在配合物2的PL光谱中,λmax为609 nm,与配合物1(λmax=546 nm) 相比较红移63 nm。是什么原因造成配合物2的光谱产生如此大的红移呢?在许多双核金属铂配合物中,往往由于配合物中Pt-Pt之间的距离小于0.38 nm,从而产生分子内的Pt-Pt相互作用,产生所谓金属-金属到配体的电荷跃迁 (MMLCT)[18, 21-22],使得双核配合物的PL光谱发生较大的红移。对配合物2的晶体中Pt-Pt距离的计算表明,分子内2个铂原子间的距离为0.669 1 nm,金属间距离显然远大于产生金属间相互作用的距离。因此在配合物2中,金属铂-铂之间的相互作用显然不是PL光谱红移的原因。而从配合物1和2的紫外-可见吸收光谱知道,它们的基态跃迁基本相同。在3.2节的讨论中,通过配合物1与2的晶体结构比较,证实了配合物2的确具有比配合物1具有更加扭曲的分子构型。因此,可以合理的推测配合物2的PL光谱红移与激发态构型变化有关,即在辐射衰减之前,配合物2发生了扭曲的构型向平面构型的转变,进而降低了激发态能级,导致配合物2的PL光谱红移。为了进一步证实这种推测是合理的,我们对配合物1和2的激发光谱进行了比较 (图 8),将图 8与图 5相比较,单核配合物1在低能带的吸收和激发光谱基本一致,说明配合物1的发光的确来源于MLCT跃迁。而在配合物2的激发光谱中,最低能带位于467 nm,比相应的最低能带的吸收光谱 (449 nm) 红移了18 nm。结合配合物2的扭曲平面构型,其PL光谱的红移是由于配合物2在激发态发生的构型转变造成的 (图 9)。
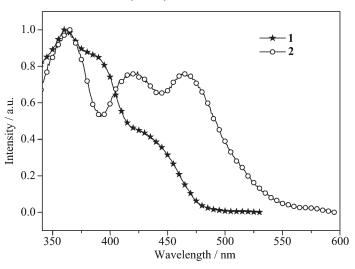 图 8
常温下配合物1和配合物2在THF溶液中的激发光谱
Figure 8.
Excitation spectra of complexes 1 and 2 in THF solution at 298 K
图 8
常温下配合物1和配合物2在THF溶液中的激发光谱
Figure 8.
Excitation spectra of complexes 1 and 2 in THF solution at 298 K
 图 9
配合物1和配合物2电荷跃迁示意图
Figure 9.
Charge transition diagrammatic sketch of complexes 1 and 2
图 9
配合物1和配合物2电荷跃迁示意图
Figure 9.
Charge transition diagrammatic sketch of complexes 1 and 2
2.2 晶体结构的解析
图 2是配合物2的分子结构图,其结构属于单斜晶系,空间群为P21/c。在配合物2中, 吡嗪环上2、3位碳原子相连接的2个苯环都参与了配位,形成双核配合物分子中以2个铂离子为中心的2个扭曲的平面正方形,配位键角O1-Pt1-O2,O3-Pt1-O4分别为910.80(14)°和92.29(13)°;C1-Pt1-N1和C14-Pt2-N2分别为81.21(19)°和81.52(18)°。配位面C-Pt-N和O-Pt-O之间的对向角 (subtend angle) 分别为7.79°(对Pt1配合物) 和5.76°(对Pt2配合物)。与5-甲基-2,3-二苯基吡嗪形成的配合物3相比较,配合物2中的2个配位平面发生了较大的扭曲,配位面C-Pt-N和O-Pt-O之间的二面角比配合物3大了一倍以上。配位键Pt-C、Pt-N和Pt-O键长分别在0.195 1(5)~0.196 2(5) nm,0.197 8(4)~0.197 9(4) nm和0.199 7(3)~0.209 4(3) nm范围。另一方面,吡嗪环与苯环之间的夹角分别为24.07°(相对于Pt1配合物) 和22.76°(相对于Pt2配合物),比相似的配合物3增大了近10°。由于吡嗪环2,3位碳原子上连接的2个苯环相互拥挤,在第二个金属铂原子进入时,另一个配位面必然采取一种更加扭曲的形式来避免苯环之间的拥挤程度。这样形成的双核配合物平面性受到很大的破坏。这种扭曲的结构降低了配合物的共轭程度。
 图 2
配合物2的晶体结构
Figure 2.
Crystal structure of complex 2
图 2
配合物2的晶体结构
Figure 2.
Crystal structure of complex 2
图 3是配合物3的晶体结构图,配合物3属于三斜晶体,空间群为P1,在一个不对称单元中有2个独立的分子。晶体中每个分子均为二聚体方式,交替堆积形成晶体的三维结构。二聚体Pt1和二聚体Pt2面间距分别为0.331 9和0.318 9 nm,说明晶体中仅有弱的π-π相互作用。二聚体中Pt-Pt距离分别为0.500 5和0.502 0 nm,说明晶体中没有金属-金属间相互作用。每一个分子中,Pt (Ⅱ) 分别与1个苯环上的C原子,吡嗪环的1个N原子以及乙酰丙酮的2个O原子配位。配位面C-Pt-N和O-Pt-O之间的对向角仅为2.59°(对Pt1配合物) 和3.60°(对Pt2配合物)。配位键角C-Pt-N和O-Pt-O分别为80.96(18)°,92.16(13)°(对Pt1配合物) 和81.45(19)°,92.48(13)°(对Pt2配合物),可见Pt与配体形成稍微扭曲的平面正方形构型。Pt-C距离在0.195 3(5)~0.196 2(5) nm之间;Pt-N距离在0.197 4(4)~0.198 1(4) nm之间;Pt-O距离在0.199 4(3)~0.208 6(3) nm之间。这些配位键之间的距离都在一般的环金属铂配合物的范围内。此外,分子中参与配位的苯环与吡嗪环平面之间的夹角分别为13.43°(对Pt1配合物) 和15.29°(对Pt2配合物),2个苯环之间的夹角分别为63.97°和58.58°。由于配合物3的化学结构与配合物1近似,仅在吡嗪环的5位上多了一个甲基,因此我们推测配合物1有与配合物3相似的晶体结构。
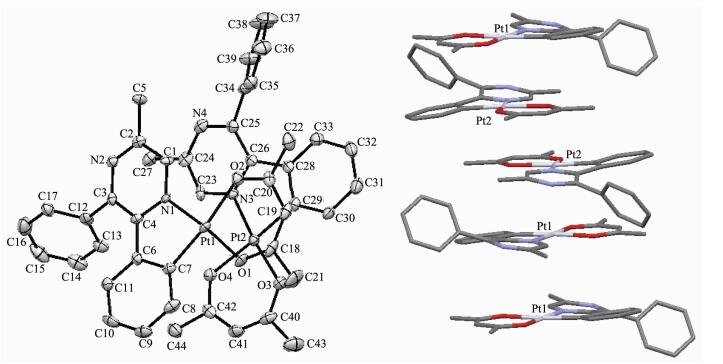 图 3
配合物3的晶体结构 (左图) 和晶胞堆积图 (右图)
Figure 3.
Crystal structure of complex 3 (left) and packing diagram (right)
图 3
配合物3的晶体结构 (左图) 和晶胞堆积图 (右图)
Figure 3.
Crystal structure of complex 3 (left) and packing diagram (right)
配合物4的晶体结构如图 4所示,属于三斜晶系,空间群为P1。配位平面与配合物3相似,金属铂与配体形成扭曲的平面正方形。配位键角C1-Pt1-N1和O1-Pt1-O2分别为80.60(17)°和89.27(11)°。配位面C-Pt-N和O-Pt-O之间的夹角 (7.62°) 大于配合物3(2.59°),相关的配位键长Pt1-C1(0.196 7(4) nm),Pt1-N1(0.202 0(4) nm),Pt1-O1(0.200 4(3) nm) 和Pt1-O2(0.209 2(3) nm) 均比配合物3长,反映了配合物4没有配合物3稳定。喹喔啉环与参与配位的苯环之间的夹角为20.97°,苯环之间夹角为61.42°。
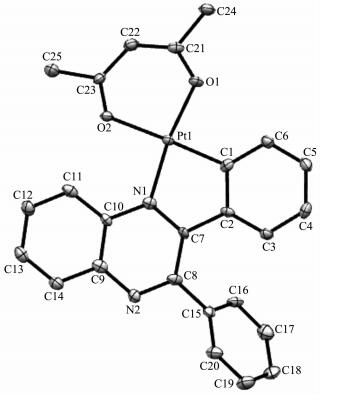 图 4
配合物4的晶体结构
Figure 4.
Crystal structure of complex 4
图 4
配合物4的晶体结构
Figure 4.
Crystal structure of complex 4
3 结论
以2,3-二苯基吡嗪 (H2dpp)、5-甲基-2,3-二苯基吡嗪 (H2mdpp) 和2,3-二苯基喹喔啉 (H2mdpp) 为配体,合成了单核配合物1、3、4以及双核配合物2。通过对配合物晶体结构的分析,发现配合物2具有比配合物3更加扭曲的平面构型。配合物2在激发态产生了从扭曲构型向平面构型的转变,从而降低了激发态能量,导致配合物2最大发射峰λmax(609 nm) 比相应的配合物1(λmax=546 nm) 红移了63 nm。而由于配体2,3-二苯基喹喔啉的共轭体系比2,3-二苯基吡嗪大,使配合物4较配合物3的PL光谱呈现出更大的红移。
Supporting information is available at http://www.wjhxxb.cn
-
-
[1]
Stacey C, Pierre H L, Victoria L, et al. Inorg. Chem., 2013, 52(19):10992-11003 doi: 10.1021/ic401131x
-
[2]
Valery N K, Marcus C D, Gareth W J A. Inorg. Chem., 2011, 50(13):6304-6313 doi: 10.1021/ic200706e
-
[3]
Cui L S, Liu Y, Liu X, et al. ACS Appl. Mater. Interfaces, 2015, 7(21):11007-11014
-
[4]
Tao R, Qiao J, Zhang G L, et al. J. Phys. Chem. C, 2012, 116(21):11658-11664 doi: 10.1021/jp301740c
-
[5]
Davide D C, Simona F P, de Angelis C, et al. Inorg. Chem., 2008, 47(3):980-989 doi: 10.1021/ic701814h
-
[6]
Tamayo A B, Garon S, Sajoto T, et al. Inorg. Chem., 2005, 44(24):8723-8732 doi: 10.1021/ic050970t
-
[7]
Xiao L X, Chen Z J, Qu B, et al. Adv. Mater., 2011, 23(8): 926-952 doi: 10.1002/adma.v23.8
-
[8]
王玲霞, 梅群波, 颜芳, 等.物理化学学报, 2012, 28(7):1556-1569 http://www.cnki.com.cn/Article/CJFDTotal-WLHX201207003.htmWANG Lin-Xia, MEI Qun-Bo, YAN Fang, et al. Acta Phys.-Chim. Sin., 2012, 28(7):1556-1569 http://www.cnki.com.cn/Article/CJFDTotal-WLHX201207003.htm
-
[9]
Tsuboyama A, Iwawaki H, Furugori M, et al. J. Am. Chem. Soc., 2003, 125(42):12971-12979 doi: 10.1021/ja034732d
-
[10]
Baranoff E, Yum J H, Gratzel M, et al. J. Organomet. Chem., 2009, 694(17):2661-2670 doi: 10.1016/j.jorganchem.2009.02.033
-
[11]
Li C L, Su Y J, Tao Y T, et al. Adv. Funct. Mater., 2005, 15(3):387-395 doi: 10.1002/(ISSN)1616-3028
-
[12]
Zhao Q, Li F Y, Huang C H. Chem. Soc. Rev., 2010, 39: 3007-3030 doi: 10.1039/b915340c
-
[13]
Kenneth K W L, Steve P Y L, Kenneth Y Z. New J. Chem., 2011, 35:265-287 doi: 10.1039/C0NJ00478B
-
[14]
Yu M X, Zhao Q, Shi L X, et al. Chem. Commun., 2008: 2115-2117 http://europepmc.org/abstract/med/18438486
-
[15]
Koo C K, Wong K L, Man C W Y, et al. Inorg. Chem., 2009, 48(3):872-878 doi: 10.1021/ic801261h
-
[16]
Zhao Q, Huang C H, Li F Y. Chem. Soc. Rev., 2011, 40: 2508-2524 doi: 10.1039/c0cs00114g
-
[17]
Ma B W, Li J, Peter I D, et al. J. Am. Chem. Soc., 2005, 127(1):28-29 doi: 10.1021/ja044313w
-
[18]
Violeta S, Juan F, Jose M, et al. Inorg. Chem., 2012, 51(6): 3427-3435 doi: 10.1021/ic201910t
-
[19]
Mitsuya U, Tomona Y, Masaki M, et al. Inorg. Chem., 2007, 46(26):11291-11296 doi: 10.1021/ic700953k
-
[20]
Tzeng B C, Chiu T H, Lin S Y, et al. Cryst. Growth Des., 2009, 9(12):5356-5362 doi: 10.1021/cg901100z
-
[21]
Arnab C, Joseph C D, Alexandre H, et al. Organometallics, 2013, 32(14):3819-3829 doi: 10.1021/om400276v
-
[22]
Aaron A R, Felix N C. Inorg. Chem., 2009, 48(23):10865-10867 doi: 10.1021/ic901156z
-
[23]
Masako K, Ai O, Asami T, et al. Angew. Chem. Int. Ed., 2002, 41(17):3183-3185 doi: 10.1002/1521-3773(20020902)41:17<>1.0.CO;2-C
-
[24]
Dolomanov O V, Bourhis L J, Gildea R J, et al. J. Appl. Cryst., 2009, 42(2):339-341 doi: 10.1107/S0021889808042726
-
[25]
Sheldrick G M. SHELXS-97, Program for the Solution of Crystal Structures, University of Göttingen, Germany, 1997.
-
[26]
Sheldrick G M. SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1997.
-
[27]
Ge G P, Zhang G L, Guo H Q, et al. Inorg. Chim. Acta, 2009, 362(7):2231-2236 doi: 10.1016/j.ica.2008.10.001
-
[28]
Ying L, Zou J H, Zhang A Q, et al. J. Organomet. Chem., 2009, 694(17):2727-2734 doi: 10.1016/j.jorganchem.2009.05.007
-
[29]
Brooks J, Babayan Y, Lamansky S, et al. Inorg. Chem., 2002,41(12):3055-3066 doi: 10.1021/ic0255508
-
[30]
Lamansky S, Djurovich P, Murphy D, et al. Inorg. Chem., 2001, 40(7):1704-1711 doi: 10.1021/ic0008969
-
[31]
Lamansky S, Djurovich P, Murphy D, et al. J. Am. Chem. Soc., 2001, 123(18):4304-4312 doi: 10.1021/ja003693s
-
[32]
Daniel V, Martin N, Jana F, et al. Langmuir, 2013, 29(9):3034-3044 doi: 10.1021/la3039522
-
[1]
-

图 2 配合物2的晶体结构
Figure 2 Crystal structure of complex 2
Ellipsoid probability level: 50%; Hydrogen atoms in the molecule are omitted for clarity

图 3 配合物3的晶体结构 (左图) 和晶胞堆积图 (右图)
Figure 3 Crystal structure of complex 3 (left) and packing diagram (right)
Ellipsoid probability level: 50%; Hydrogen atoms in the molecule are omitted for clarity

图 4 配合物4的晶体结构
Figure 4 Crystal structure of complex 4
Ellipsoid probability level: 50%; Hydrogen atoms in the molecule are omitted for clarity

图 5 配合物1~4在THF溶液中的紫外-可见吸收光谱
Figure 5 UV-Vis absorption spectra of the complexes 1~4 in diluted DCM solution

图 6 常温下配合物1~4在THF溶液中的PL光谱
Figure 6 PL spectra of complexes 1~4 in THF solution at 298 K

图 7 77 K下配合物1~4在THF溶液中的PL光谱
Figure 7 PL spectra of complexes 1~4 in THF solution at 77 K

图 8 常温下配合物1和配合物2在THF溶液中的激发光谱
Figure 8 Excitation spectra of complexes 1 and 2 in THF solution at 298 K

图 9 配合物1和配合物2电荷跃迁示意图
Figure 9 Charge transition diagrammatic sketch of complexes 1 and 2
表 1 配合物2、3、4的主要晶体参数
Table 1. Main crystal parameters of complexes 2, 3 and 4
Complex 2 3 4 Empirical formula C26H24N2O4Pt2 C22H2ON2O2Pt C25H2ON2O2Pt Formula weight 818.65 1 078.98 575.52 Crystal system Monoclinic Triclinic Triclinic Space group P21/c P1 P1 a/nm 0.908 92(2) 1.230 43(6) 1.004 44(6) b/nm 2.009 26(4) 1.326 89(5) 1.026 63(5) c/nm 1.346 89(3) 1.379 40(7) 1.036 48(6) α/(º) 90 108.903(4) 99.712(4) β/(º) 108.492(3) 113.138(5) 108.526(5) γ/(º) 90 93.025(3) 95.070(4) V/nm3 2.332(10) 1.916(17) 0.987 3(10) Z 4 2 2 Dc/(g·cm-3) 2.331 1.869 1.936 F(000) 1 528 1 040 556 Crystal size / mm 0.3×0.25×0.2 0.3×0.3×0.2 0.3×0.2×0.2 2θ range/(º) 6.228~52.742 5.826~52.744 6.246~52.744 Reflections collected 10 346 15 993 8 276 Independent reflections 4 765 7 833 4 035 Data, restraints, parameters 4 765, 0, 311 7 833, 0, 493 4 035, 0, 273 Goodness-of-fit F2 1.05 1.014 1.006 Final R indexes [I≥2σ(I)] R1=0.028 3, wR2=0.048 3 R1=0.030 8, wR2=0.0514 R1=0.028 9, wR2=0.0500 Final R indexes (all data) R1=0.036 6, wR2=0.050 8 R1=0.042 5, wR2=0.055 6 R1=0.033 3, wR2=0.052 1 Largest diff. peak and hole / (e·nm-3) 930 and-930 950 and-1 190 810 and-1 040  下载: 导出CSV
下载: 导出CSV
表 2 配合物2、3、4的主要键长和键角
Table 2. Selected bond lengths (nm) and bond angels (°) for 2, 3 and 4
2 Pt1-O1 0.208 7(4) Pt1-O2 0.199 7(3) Pt1-N1 0.197 9(4) Pt1-C1 0.195 1(5) Pt2-O3 0.209 4(3) Pt2-O4 0.200 3(3) Pt2-N2 0.197 8(4) Pt2-C14 0.196 2(5) C6-C7 0.147 2(7) C8-C9 0.146 6(6) 02-Pt1-O1 91.80(14) N1-Pt1-O1 93.42(15) N1-Pt1-O2 174.44(15) C1-Pt1-O1 170.86(17) C1-Pt1-O2 93.85(18) C1-Pt1-N1 81.21(19) 04-Pt2-O3 92.29(13) N2-Pt2-O3 93.03(15) N2-Pt2-O4 174.05(16) C14-Pt2-O3 172.64(16) C14-Pt2-O4 93.41(17) C14-Pt2-N2 81.52(18) 3 N3-Pt2 0.198 1(4) 03-Pt2 0.199 6(3) 04-Pt2 0.208 5(3) C7-Pt1 0.196 2(5) C18-O1 0.126 6(6) C20-O2 0.126 9(6) N1-Pt1 0.197 4(4) 01-Pt1 0.199 4(3) O2-Pt1 0.208 6(3) C29-Pt2 0.195 3(5) C25-C34 0.148 8(6) C26-C28 0.145 9(7) C24-C27 0.150 8(7) C3-C12 0.147 8(7) C4-C6 0.146 8(6) C29-Pt2-N3 81.45(19) C29-Pt2-O3 92.63(18) C29-Pt2-O4 173.79(17) N3-Pt2-O3 174.05(15) N3-Pt2-O4 93.47(15) 03-Pt2-O4 92.48(13) 4 N3-Pt2 0.198 1(4) 03-Pt2 0.199 6(3) 04-Pt2 0.208 5(3) C7-Pt1 0.196 2(5) C20-O2 0.126 9(6) 01-Pt1 0.199 4(3) C18-O1 0.126 6(6) N1-Pt1 0.197 4(4) 02-Pt1 0.208 6(3) C29-Pt2 0.195 3(5) C26-C28 0.145 9(7) C3-C12 0.147 8(7) C25-C34 0.148 8(6) C24-C27 0.150 8(7) C4-C6 0.146 8(6) C29-Pt2-N3 81.45(19) C29-Pt2-O4 173.79(17) N3-Pt2-O4 93.47(15) C29-Pt2-O3 92.63(18) N3-Pt2-O3 174.05(15) O3-Pt2-O4 92.48(13)
下载: 导出CSV
表 3 配合物1~4的紫外吸收数据
Table 3. UV-Vis absorption of the complexes 1~4 in diluted DCM solution
Complex λabs / nm (ε/ (L·mol-1·cm-1)) 1 228(3.71×104), 248(3.4×104), 290(2.14×104), 340(1.52×104), 394(7.3×103), 444(2.5×103) 2 2229(5.47×104), 256(3.65×104), 327(1.64×104), 401(1.54×104), 449 (6.4×103) 3 3259(3.14×104), 274(2.81×104), 315(1.64×104), 356(1.1×104), 399 (6.2×103), 448(3.3×103) 4 4242(3.16×104), 258(3.43×104), 306(3.21×104), 389(1.74×104), 441(1.15×104), 481 (4.0×103)
下载: 导出CSV
表 4 配合物1~4的PL发射、量子效率、辐射常数、非辐射常数和寿命
Table 4. PL emission, quantum yield (φem), radiation constant, non-radiation constant and lifetime of complexes 1~4
Complex λ / nm (298 K) λ / nm (77 K) φem Kr / s-1 Knr / s-1 τ/μs (77 K) 1 540, 565(sh) 535, 545, 579 0.31 2.58×106 5.75×2.58×106 0.12(1.3) 2 609 601, 644 0.12 6.6×105 4.89×2.58×106 0.18(1.6) 3 544, 575(sh) 534, 543, 579 0.33 1.43×2.58×106 2.92×2.58×106 0.23(2.3) 4 642 597, 607, 631 0.18 6.4×105 2.93×2.58×106 0.28(2.6)
下载: 导出CSV
-










 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 9
- 文章访问数: 2070
- HTML全文浏览量: 339
 下载:
下载:
