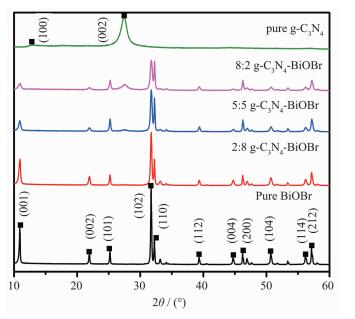
 图 1
g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的XRD图
Figure 1.
patterns of pure g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts
图 1
g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的XRD图
Figure 1.
patterns of pure g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts
引用本文:
李娜, 王茗, 赵北平, 曹雪丽. g-C3N4-BiOBr复合材料制备及可见光催化性能[J]. 无机化学学报,
2016, 32(6): 1033-1040.
doi:
10.11862/CJIC.2016.134

Citation: LI Na, WANG Ming, ZHAO Bei-Ping, CAO Xue-Li. g-C3N4-BiOBr Composites: Synthesis and High Photocatalytic Performance under Visible-Light Irradiation[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(6): 1033-1040. doi: 10.11862/CJIC.2016.134
Citation: LI Na, WANG Ming, ZHAO Bei-Ping, CAO Xue-Li. g-C3N4-BiOBr Composites: Synthesis and High Photocatalytic Performance under Visible-Light Irradiation[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(6): 1033-1040. doi: 10.11862/CJIC.2016.134
g-C3N4-BiOBr复合材料制备及可见光催化性能
摘要:
利用原位沉积法将BiOBr纳米片生长到g-C3N4表面,制得g-C3N4-BiOBr p-n型异质结复合光催化剂。采用X射线衍射 (XRD)、红外光谱 (FTIR)、场发射扫描电子显微镜 (FE-SEM)、透射电子显微镜 (TEM)、紫外可见漫反射 (UV-Vis-DRS) 和荧光光谱 (PL) 等测试对光催化剂结构和性能进行表征。通过可见光辐照降解甲基橙水溶液检测评估复合光催化剂光催化活性。研究结果表明:复合光催化剂由BiOBr和g-C3N4两相组成,BiOBr纳米片在片状g-C3N4表面快速形核生长形成面-面复合结构。相比于纯相g-C3N4和BiOBr,g-C3N4-BiOBr复合材料具有更强可见光吸收能力,吸收带边红移。在可见光辐照100 min后,性能最佳的2:8 g-C3N4-BiOBr复合光催化剂光催化活性分别是纯相g-C3N4和BiOBr的1.8和1.2倍,经过4次循环实验后,其降解率仍达84%,说明复合结构光催化剂催化性能和稳定性增强。复合光催化剂的荧光强度显著降低,说明光生载流子复合得到了有效抑制。复合光催化剂催化性能的提高归因于p-n型异质结促进电荷有效分离、抑制电子-空穴复合和吸收光波长范围的扩展,相比单一成分材料具有更好的催化活性和稳定性。自由基捕获实验证明,可见光降解甲基橙光催化过程中的主要活性成分为空穴, 并据此提出了可能的光催化机理。
English
g-C3N4-BiOBr Composites: Synthesis and High Photocatalytic Performance under Visible-Light Irradiation
Abstract:
A novel p-n heterojunction composite photocatalyst of graphitic carbon nitride-BiOBr (g-C3N4-BiOBr) fabricated by deposition of BiOBr nanoflakes on g-C3N4 surface were presented. The structures and properties of as-synthesized samples were characterized by X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), field emission scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM), UV-Vis diffuse reflection spectroscopy (DRS) and photoluminescence (PL). The photocatalytic activity was evaluated by degradation of methyl orange (MO) aqueous solution under visible-light irradiation. The study results show that the composite photocatalysts were consisted of two components of g-C3N4 and BiOBr, and the BiOBr nanoflakes can be rapidly deposited on g-C3N4 sheet surface. In comparison with pure BiOBr and g-C3N4, the g-C3N4-BiOBr composite photocatalysts shows more absorption intensity within the visible light range and the sorption edge shifts to lower energy direction. The optimum photocatalytic activity of the 2:8 g-C3N4-BiOBr composite sample was 1.8 and 1.2 times as high as those of individual g-C3N4 and BiOBr after 100 minutes irradiation with visible light. After reusing 4 cycles, the photodecomposition rate of MO still remains 84%, which proves the enhancement of photocatalytic activity and stability of the composite photocatalyst. The PL emission intensity of the composite photocatalyst decreased remarkably due to the suppression of photogenerated charges recombination. The enhancement in both photocatalytic performance and stability was induced by a synergistic effect, including the improved charge separation efficiency of the photoinduced electron-hole pair at the interface of g-C3N4 and BiOBr, the inhibition of photoinduced charge recombination and the extension of the absorption bands comparing with the sole component. A series of radical trapping experiments demonstrate that the holes should be the main active species in MO photodegradation and a possible photocatalytic mechanism is proposed.
-
Key words:
- g-C3N4
- / BiOBr
- / composite materials
- / visible light responded photocatalysts
-
0 引言
自1972年Honda Fujishima发现TiO2半导体电极光催化分解水现象以来,半导体光催化材料得到了广泛关注和飞速发展[1]。到目前为止,大多数的研究聚焦于TiO2,这主要是由于TiO2具有抗光腐蚀性强、化学稳定性好、无毒廉价等优点。然而,TiO2禁带宽度为3.2 eV,属宽带隙半导体,只能吸收占太阳光谱3%~5%、波长小于390 nm的紫外光,这一缺点限制了TiO2的广泛应用[2]。因此,探索可见光应用的新型光催化材料逐渐成为研究者关注的重点。
最近,石墨相氮化碳 (g-C3N4) 材料逐渐引起研究者的广泛关注。g-C3N4是一种带宽约为2.7 eV的非金属聚合物型光催化剂,对可见光有较好的吸收能力,并且具有较高的热稳定性、化学稳定性和廉价无污染等优势[3]。这使得g-C3N4在光解水制氢和降解有机污染物等方面具有广泛的应用前景。然而,单体g-C3N4材料由于表面缺陷较多,其光生电子空穴的复合率较高,导致光催化性能较差[4]。为了解决上述问题,研究者们采取了一些策略,例如过渡金属元素掺杂[5],制备多孔结构g-C3N4[6],以及与半导体复合[7]等。将2种半导体材料复合形成异质结结构,是减少光生载流子复合率的有效策略。到目前为止,已经报道了一些g-C3N4基异质结复合光催化剂,例如g-C3N4-TiO2[8],g-C3N4-ZnO[9],g-C3N4-CdS[10],g-C3N4-Bi2WO6[11]和g-C3N4-TaOH[12]等。以上复合体系光催化剂的催化活性比单体材料都有不同程度的提高,这归因于光生载流子的有效分离和迁移。根据2种半导体的能带结构,通常会形成3种结构异质结,其中Ⅱ型异质结被认为是促进电荷有效分离的最佳能级匹配结构。在Ⅱ型异质结中,电子从一种半导体转移到另一种半导体,同时,空穴也向相反的方向迁移[13]。g-C3N4的导带 (CB) 和价带 (VB) 位置分别为-1.12 eV和1.58 eV[14],而BiOBr的导带 (CB) 和价带 (VB) 分别0.30 eV和3.06 eV[15],二者的能带结构非常匹配,即BiOBr被认为是最适合与g-C3N4形成Ⅱ型异质结的材料之一。
本论文采用原位沉积法将BiOBr纳米片生长到g-C3N4表面制得g-C3N4-BiOBr复合光催化剂。对合成的光催化剂进行了结构、形貌和光学性能表征,并以甲基橙为模拟污染物评估了复合光催化剂的光催化降解能力。实验结果表明g-C3N4和BiOBr两相成分共存并且具有紧密接触界面,形成了g-C3N4-BiOBr面面接触的异质结结构。除此之外,g-C3N4-BiOBr异质结较好的能带匹配结构确保了复合物的光催化性能,比单一的g-C3N4或BiOBr有较大提高,并且具有良好的光化学稳定性。结合捕获实验和分析,提出了g-C3N4-BiOBr异质结复合光催化剂的光催化机理。
1 实验部分
1.1 原料与试剂
三聚氰胺 (C3N6H6, CP)、五水硝酸铋 (Bi (NO3)3·5H2O)、溴化钾 (KBr)、冰醋酸 (C2H4O2)、甲基橙 (C14H14N3NaO3S)、指示剂 (IND) 购自国药集团化学试剂有限公司; 乙二胺四乙酸二钠 (C10H14N2Na2O8·2H2O)、叔丁醇 (C4H10O)、对苯醌 (C6H4O2)、无水乙醇 (C2H6O) 均为分析纯 (AR),购自西陇化工股份有限公司。
1.2 样品合成与制备
1.3 样品的表征
用X射线粉末衍射仪 (XPert Pro,荷兰Panalytical公司) 表征粉末颗粒的晶体物相,X射线源为Cu Kα射线,λ=0.154 059 nm,扫描范围为10°~80°,工作电压为40 kV。用傅里叶变换红外光谱仪 (Thermo Nexus 470,美国NICOLET公司) 测定样品的红外谱图,KBr压片。扫描电子显微镜 (FE-SEM-S-4800,日本日立,测试电压10 kV) 表征样品的微观结构形貌。透射电镜 (JEM-2100F,日本电子,测试电压200 kV) 观察粉末颗粒的形貌、分散状态。用紫外-可见光谱仪 (UV-3600,日本岛津公司) 测定样品的紫外-可见漫反射吸收光谱 (DRS),以BaSO4作为参比。用荧光分光光度计 (VARIAN,美国安捷伦公司) 测试样品的发光性能,激发波长选为365 nm。用电化学工作站 (PARSTAT-4000,美国普林斯顿应用研究) 测试样品的光电响应。
1.4 光催化实验
通过可见光辐照降解甲基橙 (MO) 溶液来测定催化剂的光催化活性。具体测试过程如下:将50 mg样品分散于50 mL的5 mg·L-1的MO溶液中,黑暗条件下搅拌120 min使其达到吸附平衡。可见光光源选用450 W的氙灯,用滤光片去除波长λ<420 nm的光。反应开始后,每隔一定时间取3 mL反应液,转移至离心管中,将离心管中的样品离心分离,收集离心后的上清液,转移至石英比色皿中,在甲基橙最大吸收波长463 nm处测试溶液吸光度。根据朗伯-缪尔公式计算MO的降解率:η=(A0-At)/A0×100%,其中:η为降解率;A0为吸附平衡后MO溶液的吸光度;At为光照不同时间后MO溶液的吸光度。
1.5 电化学性能测试
g-C3N4、BiOBr和g-C3N4-BiOBr复合材料的电化学性能是在PARSTAT-4000型电化学工作站上进行测量的,采用三电极体系,其中以WO3薄膜/FTO玻璃为工作电极,Ag/AgCl为参比电极,铂片作为对电极,电解质溶液为0.5 mol·L-1的稀硫酸水溶液,入射光强度为100 mW·cm-2,光照面积为3 cm2。
1.2.1 g-C3N4的制备
采用高温热解三聚氰胺的方法制备g-C3N4[16]。称量4 g三聚氰胺置于带盖的刚玉坩埚中,将坩埚放入马弗炉内,在空气气氛下以2 ℃·min-1的升温速率加热到550 ℃,保温4 h,自然冷却后将所得的黄色粉末研磨备用。
1.2.2 g-C3N4-BiOBr复合光催化剂的制备
采用原位沉积法将BiOBr纳米片生长到g-C3N4表面。首先,将一定量g-C3N4粉末加入到50 mL去离子水中,超声30 min后制得均匀悬浮液。将10 mL一定浓度的KBr溶液滴加到g-C3N4悬浮液中。然后,将一定比例的Bi (NO3)3·5H2O溶于3 mL冰醋酸中,滴加到上述悬浮液中,连续搅拌12 h,静置3 h,离心收集沉淀。最后用去离子水充分洗涤,60 ℃下烘干12 h。按照这种方法制得不同质量比的g-C3N4-BiOBr复合光催化剂,分别命名为2:8 g-C3N4-BiOBr, 5:5 g-C3N4-BiOBr和8:2 g-C3N4-BiOBr。同样方法,在不加g-C3N4条件下制得纯的BiOBr样品。
2 结果与讨论
2.1 XRD分析
图 1为g-C3N4、BiOBr及g-C3N4-BiOBr复合体系样品的XRD图。从图中可见,纯相g-C3N4样品有2个衍射峰,其中2θ位于12.86°的衍射峰对应于g-C3N4的 (100) 晶面衍射,是由石墨相氮化碳层内基本结构单元均三嗪环周期性排列而产生的,其衍射强度较弱。2θ位于27.46°的衍射峰对应于具有层状结构特征的g-C3N4(002) 晶面衍射,是由环状芳香物的层间堆积形成的[17],故其衍射强度较强。BiOBr样品展示了一系列窄而尖的衍射峰,说明其结晶性较好,所有衍射峰均与衍射数据库卡片 (PDF No. 73-2061) 的四方相BiOBr衍射峰相一致,说明合成的样品为纯相BiOBr。将两种材料复合之后,它们的衍射峰在复合光催化剂中全部出现,说明复合光催化剂确实是由g-C3N4和BiOBr两相组成的,同时各相含量比例与其对应衍射峰强度成正比。
图 1
g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的XRD图
Figure 1.
patterns of pure g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts
2.2 FTIR分析
图 2为g-C3N4、BiOBr和g-C3N4-BiOBr复合材料的FTIR图谱。图 2A中位于3 160 cm-1的峰是N-H键伸缩振动峰[16-18]。3 460和1 636 cm-1的吸收峰分别对应于O-H键的伸缩振动和弯曲振动[19]。图 2B中,纯BiOBr的特征峰位于512 cm-1,是由Bi-O键的伸缩振动形成的[20]。在纯g-C3N4样品中,主要的吸收峰依次位于1 568、1 414、1 326和1 243 cm-1,对应于碳氮六元杂环典型的伸缩振动[21]。807 cm-1处的吸收峰对应均三嗪环弯曲振动[22]。g-C3N4和BiOBr所有的特征吸收峰在g-C3N4-BiOBr复合光催化剂中基本都能观察到,说明g-C3N4-BiOBr复合光催化剂是由g-C3N4和BiOBr两相组成的,与XRD测试结果一致。
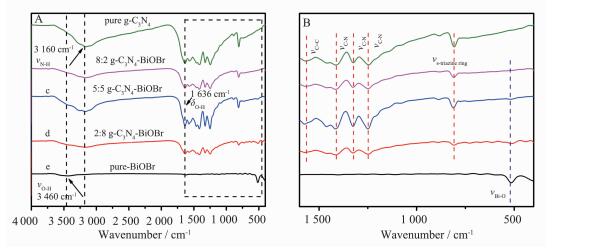 图 2
纯相g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的傅里叶变换红外光谱
Figure 2.
FTIR spectra of pure g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts
图 2
纯相g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的傅里叶变换红外光谱
Figure 2.
FTIR spectra of pure g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts
2.3 SEM和EDS分析
用SEM观察g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的形貌。如图 3A所示,刚合成出来的g-C3N4样品为聚集状的块体,通过进一步超声分散处理后,块状g-C3N4进一步分散成较小尺寸片状纳米片结构 (图 3C)。如图 3B所示,纯BiOBr样品为典型的层状纳米薄片结构,与文献报道的一致,薄片的厚度小于50 nm,宽度约为几百纳米。将BiOBr纳米片沉积到g-C3N4表面后,BiOBr沿g-C3N4面内形核生长形成g-C3N4-BiOBr异质结 (图 3C)。复合光催化剂的EDS图谱可以检测到样品只含有C、N、Bi、O和Br元素 (图 3D)。该结果进一步证实所制备的样品为g-C3N4-BiOBr复合材料。
 图 3
g-C3N4(A)、BiOBr (B) 和2:8 g-C3N4-BiOBr (C) 样品的扫描电子显微镜图像;(D) 2:8 g-C3N4-BiOBr能谱图
Figure 3.
FE-SEM images of g-C3N4(A), BiOBr (B) and 2:8 g-C3N4-BiOBr samples (C); (D) EDS of 2:8 g-C3N4-BiOBr sample
图 3
g-C3N4(A)、BiOBr (B) 和2:8 g-C3N4-BiOBr (C) 样品的扫描电子显微镜图像;(D) 2:8 g-C3N4-BiOBr能谱图
Figure 3.
FE-SEM images of g-C3N4(A), BiOBr (B) and 2:8 g-C3N4-BiOBr samples (C); (D) EDS of 2:8 g-C3N4-BiOBr sample
2.4 TEM形貌观察
图 4为2:8 g-C3N4-BiOBr复合光催化剂的透射电镜照片。从图 4A中可以看出,BiOBr优先在片层状的g-C3N4平面内形核生长,形成面与面结合的g-C3N4-BiOBr复合结构,与扫描电镜照片结果相一致。图 4B为方框内复合区域的高分辨照片,其中d值为0.352 nm的晶格条纹属于四方相BiOBr的 (101) 晶面,这与XRD分析结果是一致的。此外,从图上我们还可以看到,这种通过原位异质形核生长形成的g-C3N4-BiOBr面-面结合界面非常紧密,有利于光生电子和空穴在g-C3N4和BiOBr之间传输和转移,抑制光生电子-空穴对的复合,从而提高光催化剂的光催化活性。
 图 4
2:8 g-C3N4-BiOBr复合光催化剂的TEM (A) 和HR-TEM (B) 图像分析
Figure 4.
TEM (A) and HR-TEM (B) images of 2:8 g-C3N4-BiOBr composite photocatalyst
图 4
2:8 g-C3N4-BiOBr复合光催化剂的TEM (A) 和HR-TEM (B) 图像分析
Figure 4.
TEM (A) and HR-TEM (B) images of 2:8 g-C3N4-BiOBr composite photocatalyst
2.5 UV-Vis吸收光谱分析
图 5为g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的紫外可见漫反射图谱。由图 5A可知,纯g-C3N和BiOBr的吸收边分别位于460和442 nm处,而复合材料的吸收边主要位于纯g-C3N和BiOBr之间,说明g-C3N4-BiOBr复合光催化剂是由g-C3N4和BiOBr两相组成, 并且将2种半导体复合后,复合光催化剂比纯的g-C3N4和BiOBr在可见光区拥有了更强的吸收,而随着g-C3N4含量的提高,其吸收强度也在增加。对于半导体材料,其禁带宽度Eg可以由公式αhν=(hν-Eg)n/2估算[23]。其中n值由半导体光跃迁的类型决定:直接跃迁半导体n=1,间接跃迁半导体n=4。纯g-C3N4的n值为1,纯BiOBr和2:8 g-C3N4-BiOBr的n值为4。由图 5B可知,纯g-C3N4和BiOBr对应的禁带宽度分别为2.70和2.80 eV。2:8 g-C3N4-BiOBr的禁带宽度为2.60 eV,低于纯的g-C3N4和BiOBr。能带估算的结果表明制备的复合光催化剂具有较好的可见光响应。
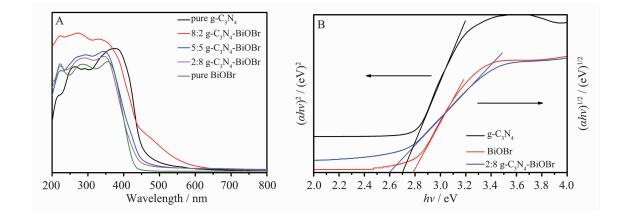 图 5
纯g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的紫外可见漫反射光谱 (A) 及 (αhν)1/2-h或 (αhν)2-h曲线 (B)
Figure 5.
UV-Vis diffuse reflectance spectra (A) and (αhν)1/2-h or (αhν)2-h curves (B) of pure g-C3N4、BiOBr and g-C3N4-BiOBr composite photocatalysts
图 5
纯g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的紫外可见漫反射光谱 (A) 及 (αhν)1/2-h或 (αhν)2-h曲线 (B)
Figure 5.
UV-Vis diffuse reflectance spectra (A) and (αhν)1/2-h or (αhν)2-h curves (B) of pure g-C3N4、BiOBr and g-C3N4-BiOBr composite photocatalysts
2.6 光催化性能的研究
基于以上结论,我们通过在可见光下降解甲基橙来评估g-C3N4-BiOBr复合光催化剂的光催化活性。光照之前,悬浮液在黑暗环境中搅拌120 min以确保达到吸附平衡。从图 6可以看到,在不加光催化剂的对照实验中,MO几乎没有降解,这表明MO相当稳定,排除了MO自降解过程发生的可能。当可见光照100 min后,g-C3N4和BiOBr对MO的降解率分别为51.16%和75.86%。可以看出g-C3N4和BiOBr均具有可见光光催化活性。当g-C3N4和BiOBr复合后,对MO的降解活性显著提高,随着复合物中BiOBr含量的增加,其可见光催化活性提高。其中2:8 g-C3N4-BiOBr复合物光催化活性最佳。在可见光照100 min后,对MO的降解率达到93.27%,为纯g-C3N4和BiOBr的1.8和1.2倍。而5:5 g-C3N4-BiOBr和8:2 g-C3N4-BiOBr复合物对MO的降解率分别为83.62%和74.71%。可以看出,通过g-C3N4-BiOBr异质结的协同作用效应,其光催化性能得到明显的提高。
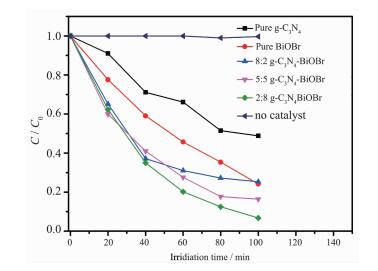 图 6
g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂在可见光下光催化降解甲基橙
Figure 6.
photocatalytic degradation of MO over g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts under visible light irradiation
图 6
g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂在可见光下光催化降解甲基橙
Figure 6.
photocatalytic degradation of MO over g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts under visible light irradiation
光催化降解甲基橙的动力学反映了光催化剂对反应速率常数的影响,甲基橙浓度随时间的的改变可通过一级动力学方程-ln (C/C0)=kt拟合得出相应的表观降解速率常数 (k)[24]。如图 7A所示。所有照射时间t对ln (C0/C) 的拟合曲线都近似一条直线,其中回归系数均大于0.967, 说明该反应较好的符合一级动力学模型[25]。如图 7B所示,经拟合后,2:8 g-C3N4-BiOBr光催化降解MO的速率常数为0.027 min-1,分别是纯g-C3N4和BiOBr的3.5和1.9倍。上述结果表明,该g-C3N4-BiOBr复合光催化剂具有较好的可见光催化性能。
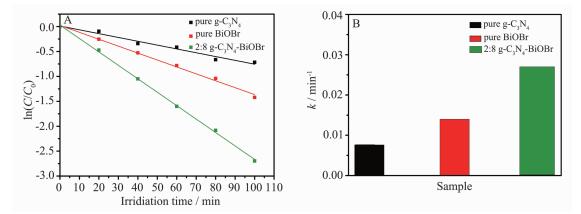 图 7
g-C3N4、BiOBr和2:8 g-C3N4-BiOBr复合光催化剂在可见光下光催化降解甲基橙的一级动力学曲线 (A) 和表观降解速率常数 (B)
Figure 7.
First-order kinetics plot (A) and the kinetic constants (B) for the photodegradation of MO under visible light irradiation by g-C3N4, BiOBr and 2:8 g-C3N4-BiOBr composite photocatalyst
图 7
g-C3N4、BiOBr和2:8 g-C3N4-BiOBr复合光催化剂在可见光下光催化降解甲基橙的一级动力学曲线 (A) 和表观降解速率常数 (B)
Figure 7.
First-order kinetics plot (A) and the kinetic constants (B) for the photodegradation of MO under visible light irradiation by g-C3N4, BiOBr and 2:8 g-C3N4-BiOBr composite photocatalyst
2.7 可见光催化活性提高的研究
2.8 光催化剂稳定性的研究
除了光催化性能,光催化剂的稳定性和重复性在实际应用中也具有十分重要的意义。因此,我们还研究了g-C3N4-BiOBr复合光催化剂的稳定性。将2:8 g-C3N4-BiOBr样品在同样的实验条件下循环4次,每次循环后,将样品用乙醇和去离子水充分洗涤,60 ℃下烘干,待用。如图 10所示,经过4次循环之后,催化剂对MO的降解率为84%,显示其具有较好的稳定性[30]。
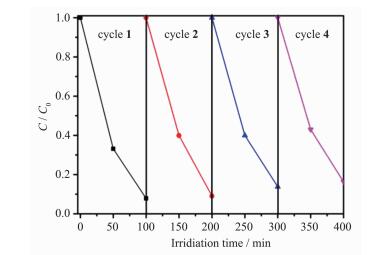 图 10
2:8 g-C3N4-BiOBr复合光催化剂在可见光下降解甲基橙的4次循环实验过程
Figure 10.
Four consecutive reaction processes for the degradation of MO over 2:8 g-C3N4-BiOBr composite photocatalyst under visible light irradiation
图 10
2:8 g-C3N4-BiOBr复合光催化剂在可见光下降解甲基橙的4次循环实验过程
Figure 10.
Four consecutive reaction processes for the degradation of MO over 2:8 g-C3N4-BiOBr composite photocatalyst under visible light irradiation
2.9 g-C3N4-BiOBr复合物光催化反应机理
由光催化实验可知,g-C3N4-BiOBr复合光催化剂在可见光下降解MO已经显示了极好的光催化效果。为了进一步增强其光催化性能,我们研究了g-C3N4-BiOBr复合光催化剂在可见光下降解MO的可能光催化机理。
我们通过自由基捕获实验揭示了g-C3N4-BiOBr复合光催化剂在可见光下降解甲基橙的机理。如图 11所示,当加入羟基自由基捕获剂叔丁醇 (TBA, 6 mmol·L-1) 和超氧自由基捕获剂苯醌 (BQ, 0.5 mmol·L-1) 时,光降解能力没有受到明显影响,说明羟基自由基和超氧自由基都不是光催化过程中的主要活性物种。而当加入空穴捕获剂乙二胺四乙酸二钠 (EDTA, 6 mmol·L-1) 时,甲基橙的降解被显著地抑制了,光照100 min只有不到30%的降解率,表明空穴就是光催化降解甲基橙的主要活性物种[31-32]。基于上述实验结果,我们提出了可能的光催化机理 (图 12):由于g-C3N4-BiOBr复合光催化剂交错的能带结构,当其在可见光照射下,g-C3N4导带上的电子就会自发的转移到BiOBr的导带,同时,BiOBr价带的空穴也会自发的转移到g-C3N4的价带。通过转移,可以有效地抑制光生电子空穴对的复合,增强光催化活性。
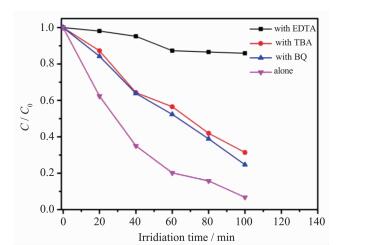 图 11
8 g-C3N4-BiOBr复合光催化剂的捕获实验
Figure 11.
Trapping experiments of 2:8 g-C3N4-BiOBr composite photocatalyst
图 11
8 g-C3N4-BiOBr复合光催化剂的捕获实验
Figure 11.
Trapping experiments of 2:8 g-C3N4-BiOBr composite photocatalyst
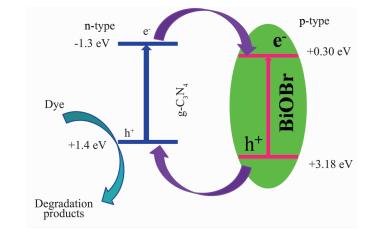 图 12
g-C3N4-BiOBr复合光催化剂的光催化机理图
Figure 12.
Proposed photocatalytic mechanism for g-C3N4-BiOBr composite photocatalyst
图 12
g-C3N4-BiOBr复合光催化剂的光催化机理图
Figure 12.
Proposed photocatalytic mechanism for g-C3N4-BiOBr composite photocatalyst
2.7.1 PL分析
对于光催化剂而言,光照时材料的电子-空穴分离效率是影响其光催化能力强弱的重要因素之一[26-27],因为只有分离的光生电子和空穴才能参与后续的氧化还原反应。我们采用荧光光致发光谱对光催化剂的光生电子行为进行了研究,由图 8的PL光谱图可见,纯g-C3N4在435 nm处有一强发光峰,说明其光生电子-空穴复合率较高[28],而g-C3N4与BiOBr形成复合材料后该发光峰强度明显降低,表明复合材料中的电子-空穴得到有效分离,复合率进一步降低,有利于提高材料的光催化活性[29]。
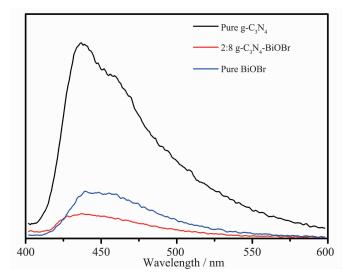 图 8
纯g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的PL图谱
Figure 8.
PL spectra of g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts
图 8
纯g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的PL图谱
Figure 8.
PL spectra of g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts
2.7.2 光电流分析
为进一步研究光催化剂的光生电荷行为,我们通过光电流测试研究光催化过程中光生电子和空穴的分离以及迁移情况。图 9是g-C3N4、BiOBr和g-C3N4-BiOBr复合材料在可见光 (λ>420 nm) 照射下光电流响应性能的对比图。从开关可见光灯后光电流的变化情况来判断,纯g-C3N4的光电流响应很低,g-C3N4-BiOBr复合光催化剂的光电流较纯g-C3N4和BiOBr都高,产生了强烈的光电流响应。可见光电流响应性能的提高说明g-C3N4与BiOBr形成复合材料后,有效的提高了光生电子空穴的分离效率,有利于提高材料的光催化活性[8]。
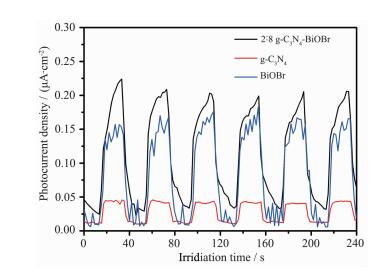 图 9
纯g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的光电流响应曲线
Figure 9.
Photoresponses spectra of g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts
图 9
纯g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的光电流响应曲线
Figure 9.
Photoresponses spectra of g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts
3 结论
通过原位沉积法将BiOBr纳米片负载到g-C3N4表面,制得了g-C3N4-BiOBr p-n型异质结复合光催化剂。光催化结果表明,通过g-C3N4-BiOBr异质结的协同效应,提高了光催化活性。其中,2:8 g-C3N4-BiOBr复合光催化剂具有最好的光催化活性,在可见光照射下,100 min后降解MO达93.27%,而g-C3N4和BiOBr分别为51.63%和75.86%。g-C3N4-BiOBr复合光催化剂存在下MO的表观降解速率常数分别是g-C3N4和BiOBr催化时的3.5和1.9倍。稳定性测试表明g-C3N4-BiOBr复合光催化剂在光催化反应中拥有优异的稳定性和可重复性。本工作可为制备高效的复合光催化剂提供帮助。
-
-
[1]
Fujishima A. Nature, 1972, 238:37-38 doi: 10.1038/238037a0
-
[2]
OReagan B, Gräetel M. Nature, 1991, 353:737-746 doi: 10.1038/353737a0
-
[3]
桂明生, 王鹏飞, 袁东, 等.无机化学学报, 2013, 29(10):2057-2064 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20131006&journal_id=wjhxxbcnGUI Ming-Sheng, WANG Peng-Fei, YUAN Dong, et al. Chinese J. Inorg. Chem., 2013, 29(10):2057-2064 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20131006&journal_id=wjhxxbcn
-
[4]
Niu P, Zhang L, Liu G, et al. Adv. Funct. Mater., 2012, 22: 4763-4770 doi: 10.1002/adfm.v22.22
-
[5]
Liu G, Niu P, Sun C, et al. J. Am. Chem. Soc., 2010, 132: 11642-11648 doi: 10.1021/ja103798k
-
[6]
Zheng Y, Jiao Y, Chen J, et al. J. Am. Chem. Soc., 2011, 133:20116-20119 doi: 10.1021/ja209206c
-
[7]
楚增勇, 原博, 颜廷楠.无机材料学报, 2014, 29(8):785-794 http://kns.cnki.net/KCMS/detail/detail.aspx?filename=wgcl201408001&dbname=CJFD&dbcode=CJFQCU Zeng-Yong, YUAN Bo, YAN Ting-Nan. J. Inorg. Mater., 2014, 29(8):785-794 http://kns.cnki.net/KCMS/detail/detail.aspx?filename=wgcl201408001&dbname=CJFD&dbcode=CJFQ
-
[8]
Zhu H, Chen D, Yue D, et al. J. Nanopart. Res., 2014, 16:1-10 doi: 10.1007/s11051-014-2632-7
-
[9]
Sun J X, Yuan Y P, Qiu L G, et al. Dalton Trans., 2012, 41: 6756-6763 doi: 10.1039/c2dt12474b
-
[10]
Yu H, Chen F, Chen F, et al. Appl. Surf. Sci., 2015, 358: 385-392 doi: 10.1016/j.apsusc.2015.06.074
-
[11]
Wang Y, Bai X, Pan C, et al. J. Mater. Chem., 2012, 22: 11568-11573 doi: 10.1039/c2jm16873a
-
[12]
Yan S C, Lü S B, Li Z S, et al. Dalton Trans., 2010, 39:1488-1491 doi: 10.1039/B914110C
-
[13]
Reiss P, Protiere M, Li L. Small, 2009, 5:154-168 doi: 10.1002/smll.200800841
-
[14]
Dai K, Lu L, Liang C, et al. Dalton Trans., 2015, 44:7903-7910 doi: 10.1039/C5DT00475F
-
[15]
Kong L, Jiang Z, Lai H H, et al. J. Catal., 2012, 293:116-125 doi: 10.1016/j.jcat.2012.06.011
-
[16]
Yan S C, Li Z S, Zou Z G. Langmuir, 2009, 25:10397-10401 doi: 10.1021/la900923z
-
[17]
Cao S, Low J, Yu J, et al. Adv. Mater., 2015, 27:2150-2176 doi: 10.1002/adma.201500033
-
[18]
Bojdys M J, Müller J O, Antonietti M, et al. Chem. Eur. J., 2008, 14:8177-8182 doi: 10.1002/chem.v14:27
-
[19]
Chen Z J, Lin B Z, Xu B H, et al. J. Porous Mater., 2011, 18:185-193 doi: 10.1007/s10934-010-9369-1
-
[20]
Song J M, Mao C J, Niu H L, et al. CrystEngComm, 2010, 12:3875-3881 doi: 10.1039/c003497p
-
[21]
Xu H, Yan J, Xu Y, et al. Appl. Catal. B, 2013, 129:182-193 doi: 10.1016/j.apcatb.2012.08.015
-
[22]
Li D, Wu Z, Xing C, et al. J. Mol. Catal. A: Chem., 2014, 395:261-268 doi: 10.1016/j.molcata.2014.08.036
-
[23]
Chang F, Li C, Chen J, et al. Superlattices Microstruct., 2014, 76:90-104 doi: 10.1016/j.spmi.2014.10.002
-
[24]
Li W, Li C, Chen B, et al. RSC Adv., 2015, 5:34281-34291 doi: 10.1039/C5RA04100G
-
[25]
Ge L, Han C, Liu J. Appl. Catal. B, 2011, 108:100-107 http://www.sciencedirect.com/science/article/pii/S0926337311003833
-
[26]
Chen J S, Wang Z, Dong X C, et al. Nanoscale, 2011, 3: 2158-2161 doi: 10.1039/c1nr10162e
-
[27]
Long M, Cai W, Cai J, et al. J. Phys. Chem. B, 2006, 110: 20211-20216 doi: 10.1021/jp063441z
-
[28]
Yang S, Gong Y, Zhang J, et al. Adv. Mater., 2013, 25:2452-2456 doi: 10.1002/adma.v25.17
-
[29]
Zhang S, Li J, Zeng M, et al. ACS Appl. Mater. Interface, 2013, 5:12735-12743 doi: 10.1021/am404123z
-
[30]
Ye S, Qiu L G, Yuan Y P, et al. J. Mater. Chem. A, 2013, 1: 3008-3015 doi: 10.1039/c2ta01069k
-
[31]
Cao J, Luo B, Lin H, et al. J. Hazard. Mater., 2012, 217: 107-115 http://www.sciencedirect.com/science/article/pii/S0304389412002701
-
[32]
Cao J, Xu B, Lin H, et al. Chem. Eng. J., 2012, 185:91-99 http://www.sciencedirect.com/science/article/pii/S1385894712000381
-
[1]
-

图 1 g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的XRD图
Figure 1 patterns of pure g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts

图 2 纯相g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的傅里叶变换红外光谱
Figure 2 FTIR spectra of pure g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts

图 3 g-C3N4(A)、BiOBr (B) 和2:8 g-C3N4-BiOBr (C) 样品的扫描电子显微镜图像;(D) 2:8 g-C3N4-BiOBr能谱图
Figure 3 FE-SEM images of g-C3N4(A), BiOBr (B) and 2:8 g-C3N4-BiOBr samples (C); (D) EDS of 2:8 g-C3N4-BiOBr sample

图 4 2:8 g-C3N4-BiOBr复合光催化剂的TEM (A) 和HR-TEM (B) 图像分析
Figure 4 TEM (A) and HR-TEM (B) images of 2:8 g-C3N4-BiOBr composite photocatalyst

图 5 纯g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的紫外可见漫反射光谱 (A) 及 (αhν)1/2-h或 (αhν)2-h曲线 (B)
Figure 5 UV-Vis diffuse reflectance spectra (A) and (αhν)1/2-h or (αhν)2-h curves (B) of pure g-C3N4、BiOBr and g-C3N4-BiOBr composite photocatalysts

图 6 g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂在可见光下光催化降解甲基橙
Figure 6 photocatalytic degradation of MO over g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts under visible light irradiation

图 7 g-C3N4、BiOBr和2:8 g-C3N4-BiOBr复合光催化剂在可见光下光催化降解甲基橙的一级动力学曲线 (A) 和表观降解速率常数 (B)
Figure 7 First-order kinetics plot (A) and the kinetic constants (B) for the photodegradation of MO under visible light irradiation by g-C3N4, BiOBr and 2:8 g-C3N4-BiOBr composite photocatalyst

图 8 纯g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的PL图谱
Figure 8 PL spectra of g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts

图 9 纯g-C3N4、BiOBr和g-C3N4-BiOBr复合光催化剂的光电流响应曲线
Figure 9 Photoresponses spectra of g-C3N4, BiOBr and g-C3N4-BiOBr composite photocatalysts

图 10 2:8 g-C3N4-BiOBr复合光催化剂在可见光下降解甲基橙的4次循环实验过程
Figure 10 Four consecutive reaction processes for the degradation of MO over 2:8 g-C3N4-BiOBr composite photocatalyst under visible light irradiation

图 11 8 g-C3N4-BiOBr复合光催化剂的捕获实验
Figure 11 Trapping experiments of 2:8 g-C3N4-BiOBr composite photocatalyst
-


 下载:
下载:











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 944
- HTML全文浏览量: 192
 下载:
下载:
