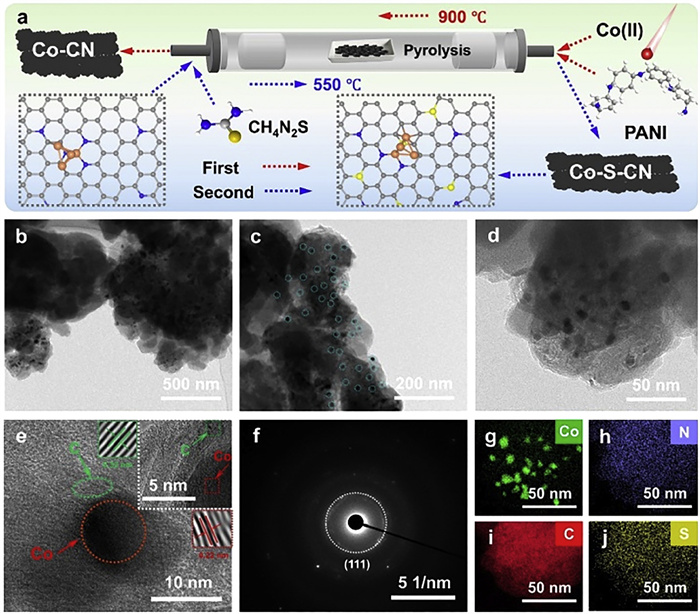
Figure 1.
(a) The flowchart of the preparation process of Co-S-CN and Co-CN. (b–d) TEM images of Co-S-CN. (e) HRTEM images of Co-S-CN. (f) SAED image of Co-S-CN. (g–j) EDS mapping images of Co-S-CN.
Sulfur-nitrogen synergy anchored and modulated cobalt nanoparticles to construct dual micro-reduction environments for efficient peroxymonosulfate activation: Nonradical-dominated mechanism
Fuming Miao , Zexiang Gao , Ting Cheng , Cibin Wang , Youzhi Liu , Xingyue Wei , Xinping Duan , Weizhou Jiao

The presence of recalcitrant organic pollutants in aquatic environments poses severe threats to both ecological systems and human health [1,2]. In recent years, advanced oxidation processes (AOPs) have been extensively utilized for the remediation of aqueous contaminants [3]. Among these, peroxymonosulfate (PMS) AOPs (PMS-AOPs) have garnered significant research attention owing to their potent oxidative capability, operational simplicity, and broad pH applicability [4,5]. The efficacy of PMS-AOPs in pollutant degradation fundamentally relies on the design of efficient catalysts to activate PMS, generating radical species (e.g., sulfate radical (SO4•−) [6], hydroxyl radical (•OH) [7], superoxide radical (O2•−) [8]) and nonradical species (singlet oxygen (1O2)) [9]. Nonradical species have the following characteristics [10]: (Ⅰ) Long half-life. (Ⅱ) Low sensitivity to interference from background anions. (Ⅲ) High selectivity towards electron-rich organic pollutants. Consequently, nonradical species demonstrate significant superiority in contaminant purification processes [11].
However, efficient generation of nonradical species requires effective PMS activation. Transition metal catalysts have been extensively validated for superior PMS activation performance [12], with cobalt-based systems exhibiting exceptional capability due to their distinctive valence electron configuration [13,14]. Although cobalt-based heterogeneous catalysts have been extensively explored for persulfate activation, their practical application is often hampered by inadequate stability and significant metal leaching. While cobalt single-atom catalysts can enhance atomic utilization efficiency and stability [15], their synthesis remains challenging and metal loading is typically low. In contrast, cobalt nanoparticles (Co NPs) offer greater practical applicability potential but are hindered by aggregation tendencies [16,17]. More critically, leaching of toxic cobalt ions causes significant secondary pollution, severely constraining their practical implementation in heterogeneous catalysis. Furthermore, efficient redox cycling between different cobalt oxidation states (e.g., Co2+/Co3+) is crucial for sustainable operational lifetime [18].
Researchers have established that incorporating metal species and nitrogen heteroatoms into carbon matrices forms metal-nitrogen-carbon (M-N-C) composites, where electron-rich pyridinic and pyrrolic N sites form strong metal-nitrogen coordination structures that effectively inhibit aggregation of metal active centers [19,20]. Compared to carbon atoms, nitrogen possesses higher electronegativity, inducing electron accumulation around N and effectively reducing electron cloud density on adjacent carbon atoms, thereby facilitating PMS adsorption and activation [21]. Xu et al. [22] demonstrated that N atoms bonding with metallic Co to form CoNX sites not only enable uniform distribution of active metal components on porous carbon nitride but also enhance catalyst stability and activity through N-mediated confinement of Co atoms. Similarly, studies show that bimetallic species can be anchored by N heteroatoms, reducing leaching losses and contamination during application [23]. Although N heteroatom coordination effectively anchors active components and delivers good catalytic activity, its limited electron-donating capacity and insufficient modulation depth of metal d-orbitals constrain further catalytic enhancement. To achieve deeper electronic structure modulation of metal centers, introducing foreign electrons into the unoccupied 3d orbitals of central metal atoms is considered. This means that substituting heteroatoms for nitrogen can directly regulate the electronic structure of active metal centers [24]. Sulfur, possessing an additional p-orbital electron and lower electronegativity (χ = 2.58) than nitrogen, serves as an ideal heteroatom substitute [25,26]. Furthermore, thiophene sulfur (C–S–C) formed by sulfur incorporation in carbon matrices can itself function as an active site for PMS activation [27]. Building upon this foundation, this study proposes the synergistic anchoring of Co-NPs by S and N to profoundly modulate the 3d orbital electronic structure of Co atoms, aiming to significantly enhance catalytic activity. More crucially, to establish a “dual micro-reductive environment” through the combined reducing properties of low-valent sulfur (S2-) and zero-valent cobalt (Co0), which will more effectively promote redox cycling (Co2+/Co3+) at the metal active center, thereby improving catalytic efficiency and stability. Two critical issues urgently require resolution: (Ⅰ) The mechanism of S, N synergistic regulation on the electronic distribution of metal nanoparticles and its impact on PMS activation performance remains unclear, hindering further performance optimization and mechanistic understanding. (Ⅱ) Difficulty in achieving efficient redox cycling between different metal valence states. Notably, the mechanistic role of constructed “S2--Co0” dual micro-reductive environment in promoting redox cycling has rarely been explored.
In conclusion, this study proposes the synergistic anchoring and modulation of metallic Co-NPs by sulfur and nitrogen, with in-depth investigation of their structure-activity relationships and activation mechanisms. The main research objectives encompass six key aspects: (Ⅰ) Construction of a cobalt nanoparticle catalyst synergistically anchored by sulfur and nitrogen (Co-S-CN), with multi-technique characterization employed to validate the S, N-anchoring mechanism. (Ⅱ) Investigation of the chemical states and surface microenvironment in Co-S-CN. (Ⅲ) Comprehensive evaluation performance of Co-S-CN in PMS activation, including efficiency, stability, and anti-interference capability. (Ⅳ) Identification of reactive oxygen species and elucidation of the PMS activation mechanism. (Ⅴ) Elucidation of sulfur’s modulation mechanism on metallic Co nanoparticles and the S, N-synergistic mechanism through density functional theory (DFT) calculations.
Comprehensive experimental details, including chemicals and reagents, catalyst characterization, experimental procedures, analytical techniques, kinetic study, electrochemical characterization and density functional theory were systematically documented in Texts S1-S7 (Supporting information). The catalyst was synthesized via a two-step protocol: (1) Polyaniline (PANI, 0.5 g) and cobalt chloride hexahydrate (CoCl2·6H2O, 0.2 × 10–3 mol) were mixed and ground in an agate mortar for 10 min, and calcined in an N2 atmosphere at 900 ℃ (5 ℃/min, 4 h) to obtain the catalyst Co-CN; (2) Co-CN (0.1 g) and a certain quality of thiourea (CH4N2S, 0.05–0.25 g) were mixed and ground for 15 min, calcined in an N2 atmosphere at 550 ℃ (5 ℃/min, 4 h) [28,29]. The resulting catalysts were named Co-S-CN-1, Co-S-CN-2, Co-S-CN-3, Co-S-CN-4, and Co-S-CN-5, respectively. Comparative phenol degradation tests (Fig. S1 in Supporting information) identified Co-S-CN-4 as optimal, hereafter designated Co-S-CN. Control materials included Co-CN and CN (direct PANI pyrolysis at 900 ℃ under identical conditions).
The catalyst preparation process is illustrated in Fig. 1a, where the first calcination step yielded Co-CN, followed by a second calcination step producing Co-S-CN. Scanning electron microscopy (SEM) characterization of CN (Fig. S2 in Supporting information), Co-CN (Fig. S3 in Supporting information), and Co-S-CN (Fig. S4 in Supporting information) revealed that the introduction of Co and S did not alter the spherical interconnected structure of the CN material, corresponding energy dispersive spectroscopy (EDS) mapping confirmed the successful incorporation and homogeneous dispersion of Co and S. Further analysis of the geometric structure and morphology of Co-S-CN by transmission electron microscopy (TEM) clearly revealed well-dispersed Co NPs within the carbon matrix (Figs. 1b–d), demonstrating the synergistic anchoring effect of S and N on the Co NPs [30]. High-resolution transmission electron microscopy (HRTEM) indicated that the Co NPs on the carbon support ranged from 5 nm to 10 nm in size (Fig. 1e), with observed lattice fringes of 0.22 nm and 0.32 nm corresponding to the Co0 (111) and graphitic carbon (002) crystal planes, respectively [31,32]. The corresponding selected area electron diffraction (SAED) image (Fig. 1f) exhibited the diffraction rings corresponding to Co (111) crystal planes. EDS elemental mapping confirmed the uniform distribution of C, N, and S and further verified the existence of Co NPs form (Figs. 1g–j). Collectively, the TEM results substantiated that the Co NPs in Co-S-CN were synergistically anchored within the carbon support by S and N, exhibiting excellent dispersion. Further analysis of the specific surface area and pore structure of catalyst via automated physisorption revealed N2 adsorption-desorption isotherms and pore size distributions for CN, Co-CN, and Co-S-CN as shown in Figs. S5a–c (Supporting information). Co-S-CN exhibited a specific surface area of 41.1 m2/g, slightly lower than that of Co-CN (53.8 m2/g), indicating that sulfur incorporation induced structural rearrangement within the carbon material. The average pore size increased from 12.9 nm for Co-CN to 18.6 nm for Co-S-CN. This larger pore size is highly advantageous for catalytic reactions, as it facilitates enhanced mass transport of reactants and products, thereby improving the accessibility of active sites. This textural modification, combined with the electronic structure modulation, collectively contributes to the superior performance of Co-S-CN despite its reduced surface area.
As shown in Fig. 2a, X-ray diffraction (XRD) analysis revealed two broad diffraction peaks at 23.5° and 44.3° for CN, Co-CN, and Co-S-CN, corresponding to the (002) and (100) planes of graphitic carbon [32], demonstrating the structural disruption of PANI during high-temperature calcination and its transformation into low-crystallinity disordered carbon intermediate between graphitic and amorphous carbon [33]. Additionally, three distinct characteristic peaks at 44.2°, 51.4°, and 75.7° were observed for Co-CN and Co-S-CN, assignable to the (111), (200), and (220) planes of Co0 (PDF #15–0806), confirming the presence of Co0 in these catalysts [34]. Notably, the characteristic peaks of Co0 intensified with increasing sulfur content (Fig. S6 in Supporting information). The presence of Co° can establish a reductive microenvironment, facilitating the reduction of Co3+ to Co2+, which is more conducive to catalytic reactions [35]. The defect characteristics of the catalysts were assessed by Raman spectroscopy, where the ID/IG ratio typically evaluates the defect density/graphitization level [36]. As shown in Fig. 2b, the ID/IG values followed the order Co-S-CN (1.03) > Co-CN (1.02) > CN (0.98), indicating that Co-S-CN has more defects. This is primarily attributed to the larger atomic radius of sulfur (1.03 Å) compared to carbon (0.75 Å) and nitrogen (0.71 Å). Therefore, sulfur incorporation induced local lattice distortion, hindering the maintenance of a planar configuration, and consequently generating more defects, which also provides superior active sites for PMS activation. Fourier transform infrared (FT-IR) spectroscopy detected functional groups and chemical bonds in CN, Co-CN, and Co-S-CN, showing several common characteristic peaks for all three catalysts (Fig. 2c), such as O–H (3418 cm−1), C–H (2975, 2910, 2879 cm−1), C–N/C=N (1600–1200 cm−1), C–O (1100 cm−1) [37,38]. Significantly, unlike CN, new vibration peaks appeared in the 500−800 cm−1 region for Co-CN and Co-S-CN, attributed to the coordination of Co with N or S within the material, forming Co-N and Co-S bonds [39], providing further evidence for the potential synergistic anchoring of Co by S and N.
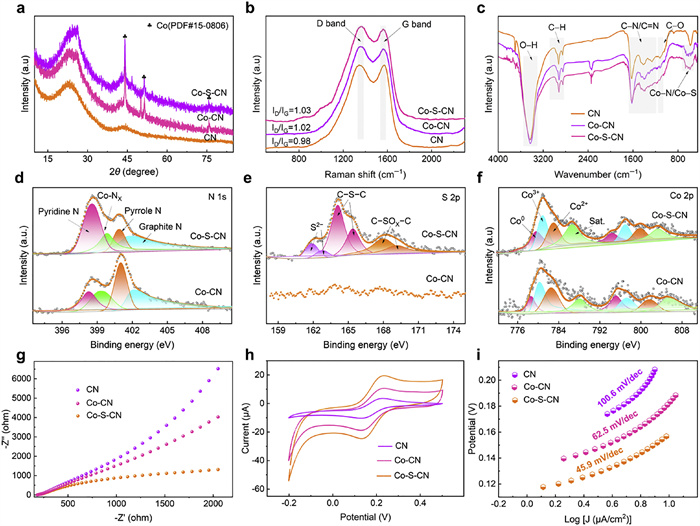
X-ray photoelectron spectroscopy (XPS) was employed to analyze the elemental composition and surface chemical states of the materials, as shown in Figs. 2d-f and Fig. S7 (Supporting information), with corresponding contents listed in Table S1 (Supporting information). The N 1s XPS spectrum of Co-S-CN (Fig. 2d) was deconvoluted into four characteristic peaks assigned to pyridinic N (398.5 eV), Co−NX (399.8 eV), pyrrolic N (400.8 eV), and graphitic N (402.3 eV) [40]. The pyridinic N content in Co-S-CN (40.4%) was more than double that in Co-CN (18.2%). Since pyridinic N readily bonds with metals, this suggests that sulfur introduction may enhance the anchoring effect of nitrogen. The Co−NX content in Co-S-CN (17.9%) was slightly lower compared to Co-CN (20.5%), possibly due to partial substitution of N in Co-N bonds by S to form Co-S bonds. As shown in Fig. 2e, sulfur species in Co-S-CN were identified as S2− (161.5 and 162.6 eV), C–S–C (164.0 and 165.4 eV), and oxidized sulfur (C–SOX–C) (167.3 and 169.1 eV) [41,42]. It is worth noting that the presence of S2− can promote the Co3+/Co2+ redox cycle [43]. C–S–C constituted the dominant form at 55.5%, importantly serving not only to effectively bond with and anchor metals but also to provide excellent active sites for PMS activation. The Co 2p XPS spectrum (Fig. 2f) revealed that the Co° content in Co-S-CN (26.7%) increased compared to Co-CN (18.7%), indicating that the introduction of S increased the content of Co0, and this result is consistent with that of XRD. Therefore, the dual micro-reduction environment “S2--Co0” constructed on the catalyst surface can effectively promote the reduction of Co3+ to Co2+, which is beneficial for achieving efficient redox cycling during catalytic reactions [34].
To further elucidate the electron transfer capability of the catalysts, electrochemical analyses were performed. The electrochemical impedance spectroscopy (EIS) results show that Co-S-CN exhibits a smaller diameter of the Nyquist arc, indicating the lowest charge transfer resistance and the most efficient electron transfer ability (Fig. 2g) [44]. From the cyclic voltammetry (CV) curves (Fig. 2h), it can be observed that Co-S-CN possesses a stronger reduction capability compared to Co-CN and CN, which can be attributed to the dual reducing environment provided by “S2--Co0”. Generally, a smaller Tafel slope suggests a faster electron transfer rate [45]. As shown in Fig. 2i, the Tafel slope of Co-S-CN is 45.9 mV/dec, which is lower than those of Co-CN (62.5 mV/dec) and CN (100.6 mV/dec). In conclusion, Co-S-CN has the potential for faster electron transfer rate.
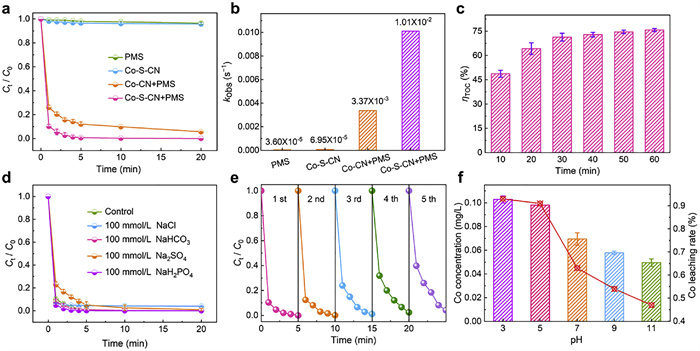
The catalytic performance of Co-S-CN was evaluated by activating PMS for phenol degradation. Parameter optimization investigated the effects of catalyst dosage (10, 15, 20, 25, 30, 35 mg) (Fig. S8 in Supporting information), PMS concentration (0.75, 1.00, 1.25, 1.50 mmol/L) (Fig. S9 in Supporting information), pH (3, 5, 7, 9, 11) (Fig. S10 in Supporting information), and phenol concentration (25, 50, 75, 100 mg/L) (Fig. S11 in Supporting information). Optimal conditions were established as pH 7, catalyst dosage 30 mg, PMS concentration 1.25 mmol/L, and phenol concentration 50 mg/L, subsequent experiments employed these parameters. Notably, Co-S-CN exhibited broad pH applicability (3–11) for PMS activation. Fig. 3a compares phenol degradation across different processes, with corresponding pseudo-first-order kinetic constants derived (Fig. 3b and Fig. S12 in Supporting information). Results demonstrated near-complete phenol removal (~100%) within 5 min in the Co-S-CN+PMS system, whereas degradation remained incomplete after 20 min in the Co-CN+PMS system. The reaction rate constant for Co-S-CN+PMS was 3.0-fold higher than for Co-CN+PMS. As shown in Fig. 3c, total organic carbon (TOC) analysis further revealed 71.3% mineralization of phenol within 30 min, indicating abundant reactive species in the Co-S-CN+PMS system enabling both highly selective degradation and deep mineralization. To elucidate environmental adaptability, the influence of various salts (NaCl, NaHCO3, Na2SO4, NaH2PO4) on phenol removal was investigated (Fig. 3d and Figs. S13–S16 in Supporting information). The results show that different concentrations of salt have almost no inhibitory effect on the removal of phenol, which also proves the universality of Co-S-CN. Furthermore, Co-S-CN maintained high removal efficiency after five consecutive reuse cycles (Fig. 3e), demonstrating excellent stability. As shown in Fig. 3f, cobalt leaching concentration and leaching rate in the Co-S-CN+PMS system exhibited pH dependence, with active metallic Co being more susceptible to leaching under acidic conditions, while leaching diminished progressively as pH increased. However, overall leaching remained minimal (0.05–0.10 mg/L), corresponding to leaching rates of merely 0.47% – 0.93%, which not only verifies the excellent stability of Co-S-CN but also demonstrates the synergistic anchoring effect of S and N on the Co NPs. Furthermore, as shown in Fig. S17 (Supporting information), the homogeneous catalytic contribution from the leached metal ions at this concentration was negligible, confirming that the PMS activation was predominantly governed by the heterogeneous catalyst.
Meanwhile, degradation experiments were conducted on various pollutants including phenol, sulfamethoxazole (SMX), bisphenol A (BPA), and benzoic acid (BA) (Fig. S18 in Supporting information). The results demonstrated that all pollutants except BA achieved 100% degradation within 20 min of reaction, confirming the broad applicability of this system. Comparative analysis with reported cobalt-based catalysts for PMS activation and phenol degradation (Table S2 in Supporting information) revealed that the Co-S-CN+PMS system developed in this study achieved phenol degradation within shorter timeframes, utilizing lower catalyst and oxidant dosages compared to most reported systems; this demonstrates its superior degradation efficiency, with the notably enhanced mineralization capability being of particular significance. Consequently, considering both comprehensive performance and cost-effectiveness, the proposed Co-S-CN catalyst exhibits a distinct advantage for PMS-activated phenol degradation.
The preceding findings confirmed that Co-S-CN effectively activated PMS to generate highly oxidizing reactive species. As reported [46], potential reactive oxygen species (ROS) in cobalt-based catalyst/PMS systems include SO4•−, •OH, O2•−, and 1O2. Therefore, electron paramagnetic resonance (EPR) spectroscopy and quenching experiments were conducted to identify the specific reactive species and their contributions. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) served as the spin-trapping agent for SO4•−, •OH, and O2•−, while 2,2,6,6-tetramethylpiperidine (TEMP) was used for 1O2 [10]. In quenching experiments, tert-butyl alcohol (TBA), methanol (MeOH), p-benzoquinone (PBQ), and furfuryl alcohol (FFA) were employed as quenchers to probe the existence and contribution of different ROS. TBA is a typical •OH quencher because its reaction rate constant with •OH (k = 3.8 − 7.6 × 108 L mol-1 s-1) is approximately 1000-fold higher than with SO4•− (k = 4.0 − 9.1 × 105 L mol-1 s-1) [47]. MeOH can quench both •OH (k = 1.2 − 2.8 × 108 L mol-1 s-1) and SO4•− (k = 1.6 − 7.8 × 107 L mol-1 s-1) [48]. PBQ is a common quencher for O2•− (k = 9.6 × 108 L mol-1 s-1) [49], and FFA is a specific quencher for the nonradical species 1O2 (k = 1.2 × 108 L mol-1 s-1) [50].
As shown in Fig. 4a, neither PMS alone nor the Co-S-CN system exhibited characteristic signals for •OH or SO4•−, only weak signals corresponding to SO4•− and •OH were detected in the Co-S-CN+PMS system after 5 min, suggesting these radicals were unlikely the primary reactive species. However, distinct characteristic signals for both O2•− (Fig. 4b) and 1O2 (Fig. 4c) were detected in the Co-S-CN+PMS system, indicating their potential as the main species controlling pollutant degradation. Quenching experiments further elucidated the roles of these species. The addition of TBA and MeOH had almost no inhibitory effect on phenol degradation (Fig. 4d, Figs. S19 and S20 in Supporting information), confirming that SO4•− and •OH were not the dominant active species. Conversely, adding PBQ reduced phenol removal from 100% to 56.6% after 5 min (Fig. 4d and Fig. S21 in Supporting information), while adding FFA (10 times the PMS concentration) reduced it to 22.0% (Fig. 4d and Fig. S22 in Supporting information). Comparison of pseudo-first-order rate constants with different quenchers (Fig. 4e and Fig. S23 in Supporting information): FFA (3.08 × 10−4 s−1) < PBQ (1.37 × 10−3 s−1) < MeOH (8.90 × 10−3 s−1) < TBA (9.92 × 10−3 s−1) < no quencher (1.01 × 10−2 s−1), it can be concluded that O2•− and 1O2 contributed significantly to phenol degradation. Given that O2•− readily converts to 1O2 [51], further investigation was required. Fig. 4f showed that PBQ addition markedly weakened the O2•− signal, with the weakening effect intensifying with increasing PBQ concentration, confirming PBQ inhibited O2•− generation. Crucially, inhibiting O2•− also led to a concurrent decrease in the 1O2 signal intensity (Fig. 4g). This strongly demonstrated that 1O2 originated from O2•− within the Co-S-CN+PMS system and played the predominant role in the reaction pathway.
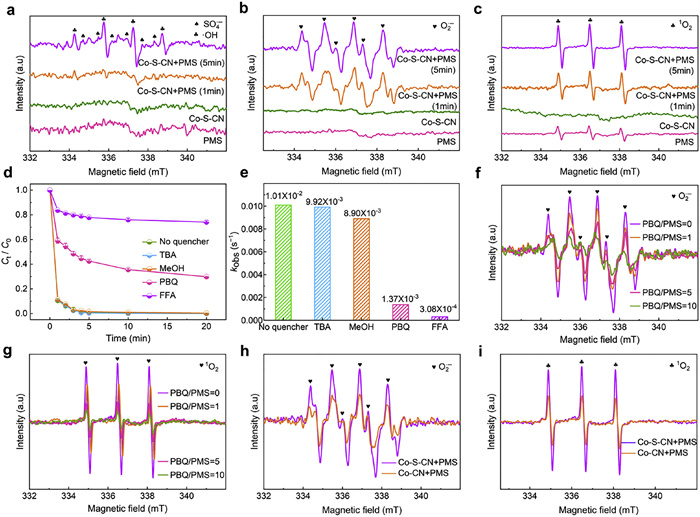
To elucidate the influence of sulfur introduction, combined with nitrogen, on the synergistic modulation of Co active sites and its impact on reactive species generation. EPR spectroscopy was used to compare the signals of reactive species in the Co-CN+PMS and Co-S-CN+PMS systems. Results demonstrated that both the Co-CN and Co-S-CN systems exhibited weak signal intensities for SO4•− and •OH (Fig. S24 in Supporting information), confirming that neither system was governed by these radical species. Significantly, the EPR signal intensities for both O2•− and 1O2 were substantially higher in the Co-S-CN system compared to the Co-CN system (Figs. 4h and i). This unequivocally proves that the introduction of sulfur effectively synergizes with nitrogen to modulate the Co active sites, promoting the generation of the key reactive species O2•− and consequently leading to a surge in 1O2 production.
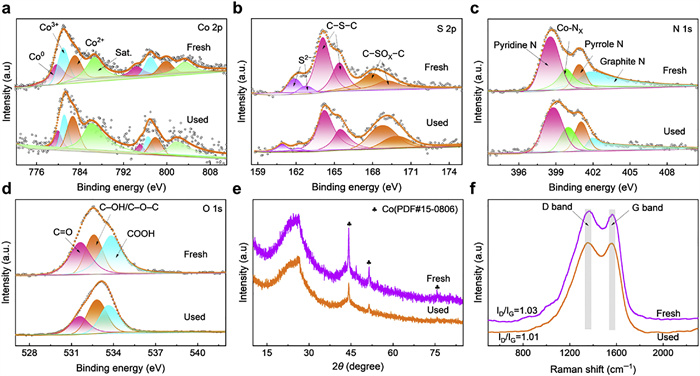
Comparative analysis of Co-S-CN before and after use was conducted to elucidate the PMS activation mechanism. XPS analysis of fresh and used Co-S-CN as shown in Figs. 5a−d and Fig. S25 (Supporting information), and the corresponding contents are presented in Table S3 (Supporting information). The surface chemical state of Co before and after using Co-S-CN is shown in Fig. 5a. Compared with the fresh Co-S-CN, the content of Co0 decreased from 26.7% to 17.4%, accompanied by increases in Co2+ (36.0% to 41.9%) and Co3+ (37.3% to 40.7%), which indicates that while PMS oxidizes Co2+ to Co3+, there is also a reduction of Co3+ to Co2+. Based on the standard redox potentials of Co3+/Co2+ (1.82 V) and Co2+/Co0 (−0.28 V), Co0 can thermodynamically reduce Co3+ to Co2+ [34]. Concurrently, the S 2p spectrum (Fig. 5b) showed a decrease in S2− content from 13.7% to 7.3%, as the reducing S2− species also facilitates Co3+ reduction to Co2+ while itself being oxidized to S0 [52]. To underscore the synergy of this dual micro-reduction environment, we compared it with the Co0-only system (Co-CN). As shown in Fig. S26 (Supporting information), the Co2+ content in used Co-CN increased only marginally from 34.7% to 36.5%, a significantly smaller increase compared to the Co-S-CN. This indicates that the concurrent electron donation from both S2− and Co0 in Co-S-CN creates a more efficient redox cycle, leading to a markedly higher regeneration rate of active Co3+ sites. A schematic illustration detailing this synergistic electron flow is provided in Fig. S27 (Supporting information). The content of C–S–C decreased from 55.5% to 50.6%, suggesting its involvement not only in anchoring Co nanoparticles but also in activating PMS due to its electron-rich nature [53]. As shown in Fig. 5c, the N 1s spectrum indicated a decrease in graphitic N content, implying its participation in the reaction. However, the content of Co-NX showed significant changes, indicating that it can effectively anchor the Co NPs. Notably, O 1s analysis (Fig. 5d) showed a decrease in C=O content (31.2% to 23.3%), suggesting its role where C=O groups are oxidized by persulfate to form epoxy groups, subsequent oxidation of these epoxies by SO52− induces 1O2 generation [54]. XRD (Fig. 5e) and FT-IR (Fig. S28 in Supporting information) spectra showed negligible structural changes before and after use, demonstrating the effective anchoring of active components by S and N and consequent catalyst stability. Raman spectroscopy indicated a decrease in the ID/IG ratio for the used catalyst, suggesting the involvement of defects in the reaction (Fig. 5f).
In summary, this study proposes a possible mechanism of Co-S-CN activating PMS. (ⅰ) O2•−-mediated 1O2 generation: Metallic species in Co-S-CN transfer electrons to PMS, inducing O2•− formation (Eq. 1), while graphitic N also donates electrons to PMS to produce O2•− (Eqs. 2–4) [55,56]. Subsequently, O2•− reacts with HO2• or H+ to generate 1O2 ((5), (6)) [56]. (ⅱ) 1O2 generation via •OH and O2•− interaction: Oxidation of Co2+ by PMS produces •OH (Eq. 7), and •OH reacts with O2•− to form 1O2 (Eq. 8) [50]. (ⅲ) SO5•− decomposition to 1O2: Co3+ may be reduced by PMS, yielding SO5•−, which undergoes self-decomposition to produce 1O2 ((9), (10)) [57]. (ⅳ) C=O induced 1O2 pathway: C=O groups on Co-S-CN are oxidized by persulfate to form epoxy structures, which are further oxidized by SO52− to induce 1O2 generation ((11), (12)) [54]. Critically, the synergistic “S2−-Co0” micro-reductive environment facilitates the reduction of Co3+ back to Co2+ ((13), (14)) [35,43], thereby significantly promoting the cobalt redox cycling essential for sustained catalytic activity.
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
|
|
(6) |
|
|
(7) |
|
|
(8) |
|
|
(9) |
|
|
(10) |
|
|
(11) |
|
|
(12) |
|
|
(13) |
|
|
(14) |
To elucidate the synergistic mechanism of S and N in PMS activation, DFT calculations were performed. Models for CN, Co-CN, and Co-S-CN were successfully constructed based on prior characterization results as shown in Fig. S29 (Supporting information). Calculation of the adsorption energies (Eads) for PMS on these models revealed that CN required the most energy (Eads = −1.95 eV) for PMS adsorption compared to Co-CN and Co-S-CN (Fig. 6a), indicating that introducing metallic Co enhances the affinity of catalyst for PMS. Significantly, Co-S-CN exhibited a lower Eads for PMS (−3.77 eV) than Co-CN (−3.51 eV) (Figs. 6b and c), demonstrating a synergistic effect between S and N that promotes PMS adsorption. Analysis of the differential charge density for adsorbed PMS on CN, Co-CN, and Co-S-CN (Figs. 6d–f) showed that S introduction effectively modulates the electronic structure and charge distribution of the carbon nitride material [26,58]. Bader charge analysis confirmed a higher number of electrons transferred from Co-S-CN to PMS (0.80 e) compared to Co-CN (0.75 e) and CN (0.73 e), further corroborating that S incorporation synergizes with N to induce electronic redistribution within the catalytic material, thereby enhancing PMS activation [24]. Changes in bond lengths upon adsorption support this conclusion: the O–H bond in Co-S-CN-PMS (0.987 Å) lengthened compared to that in Co-CN-PMS (0.983 Å), facilitating its cleavage and favoring SO5•− generation, which promotes 1O2 formation. Similarly, the elongation of the S–O bond (1.520 Å → 1.527 Å, indicating bond weakening) is conducive to O2•− generation and its subsequent conversion to 1O2. These computational results are fully consistent with the preceding quenching experiments and proposed activation mechanism.
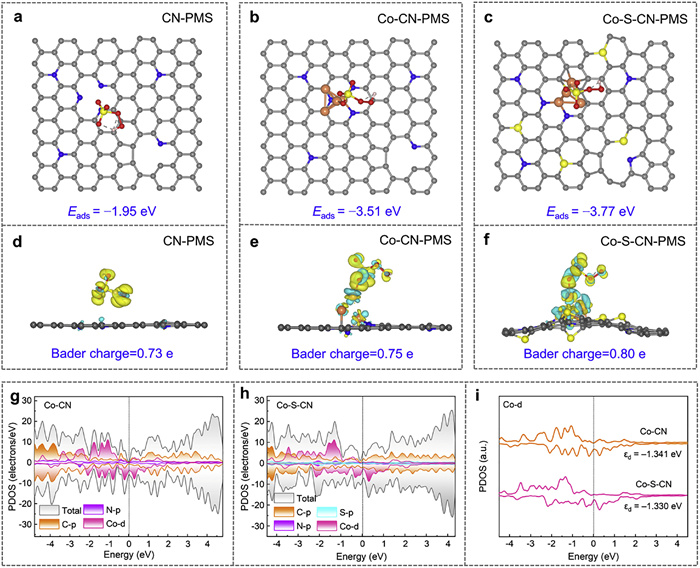
Projected density of states (PDOS) calculations was performed for individual atoms in Co-CN and Co-S-CN. As shown in Fig. 6g, Co-CN exhibited relatively weak electronic states near the Fermi level. Upon sulfur introduction (Co-S-CN, Fig. 6h), pronounced electronic states emerged near the Fermi level, indicating that the synergistic effect of S and N enhances electrical conductivity. The increased electron occupancy in these states implies greater availability of electrons for PMS activation, consistent with the preceding Bader charge analysis and further confirming that S and N synergistically optimize orbital configurations to facilitate interatomic electron transfer. The partial density of states for Co atoms (Fig. 6i) revealed that the d-band center in Co-S-CN (−1.330 eV) lies closer to the Fermi level than in Co-CN (−1.341 eV), demonstrating that S and N cooperatively modulate the electronic distribution of the Co 3d orbitals, leading to enhanced reactivity between Co-S-CN and PMS.
In summary, this work designed and fabricated a sulfur-nitrogen co-anchored cobalt nanoparticle catalyst, effectively addressing the critical issue of active metal leaching. By constructing a “S2--Co0” dual micro-reduction environment within the catalyst, it significantly accelerated the Co3+/Co2+ redox cycle. Experimental validation, characterization techniques, and theoretical calculations collectively elucidated the strong metal anchoring effect and modulation mechanism enabled by S-N synergy. Results demonstrated that the Co-S-CN+PMS system achieved 100% phenol degradation within 5 min, exhibiting exceptional resistance to salt interference (SO42−, Cl−, HCO3−, H2PO4−). Minimal cobalt leaching (<1%) across various pH values confirmed the strong anchoring effect and structural stability imparted by S-N synergy. Structural characterizations verify the existence of S2- and Co0, and co-optimizing cobalt valence cycling. EPR and radical quenching experiments indicated the reaction proceeds primarily via a nonradical pathway, with 1O2 identified as the key reactive species. DFT calculations revealed S-N synergy enhanced PMS adsorption strength, increased charge density and optimized the electronic structure of Co 3d orbitals. This study provides a strategy for designing high-performance and stable catalysts, offering a practical paradigm for applying persulfate-based advanced oxidation processes in wastewater purification.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Fuming Miao: Writing – original draft, Methodology, Investigation, Formal analysis. Zexiang Gao: Visualization, Investigation. Ting Cheng: Validation, Supervision. Cibin Wang: Data curation. Youzhi Liu: Supervision, Conceptualization. Xingyue Wei: Writing – review & editing, Supervision. Xinping Duan: Supervision, Software. Weizhou Jiao: Writing – review & editing, Supervision, Project administration.
This research was supported by Research Project Supported by the National Natural Science Foundation of China (No. U23A20676), Key Research & Development Plan of Shanxi Province (No. 202502090302004), Shanxi Scholarship Council of China (No. 2023–128), Innovative Projects for Postgraduate Education in Shanxi (No. 2024KY578).
Supplementary material associated with this article can be found, in the online version, at doi:
S. Zhang, H. Zheng, P.G. Tratnyek, Nat. Water 1 (2023) 666–681. doi: 10.1038/s44221-023-00098-1
C.J. Li, W.C. Liu, X.L. Chen, et al., Chin. Chem. Lett. 35 (2024) 109652. doi: 10.1016/j.cclet.2024.109652
X. Li, M. Long, Nat. Water 3 (2025) 140–141. doi: 10.1038/s44221-024-00383-7
M. Yang, W.Y. Liu, Q. Liu, et al., Angew. Chem. Int. Ed. 64 (2025) e202421797. doi: 10.1002/anie.202421797
S.Y. Xue, C. Cheng, J.Q. Kang, et al., Chin. Chem. Lett. 36 (2025) 110776. doi: 10.1016/j.cclet.2024.110776
Z.Y. Zhao, P.F. Wang, C.L. Song, et al., Angew. Chem. Int. Ed. 62 (2023) e202216403. doi: 10.1002/anie.202216403
Q.L. Liu, H. Qie, Z.Q. Sun, et al., Chin. Chem. Lett. 34 (2023) 108397. doi: 10.1016/j.cclet.2023.108397
Z.Y. Zhao, H.B. Tan, P. Zhang, et al., Angew. Chem. Int. Ed. 62 (2023) e202219178. doi: 10.1002/anie.202219178
C.H. Gu, S. Wang, A.Y. Zhang, et al., Nat. Commun. 15 (2024) 5771. doi: 10.1038/s41467-024-50240-0
F. Miao, J. Song, Y. Liu, et al., Chem. Eng. J. 522 (2025) 168002. doi: 10.1016/j.cej.2025.168002
T. Liu, S. Xiao, N. Li, et al., Nat. Commun. 14 (2023) 2881. doi: 10.1038/s41467-023-38677-1
Z.D. Zhao, M.Z. Hu, T.T. Nie, et al., Environ. Sci. Technol. 57 (2023) 4556–4567. doi: 10.1021/acs.est.2c09336
Y.L. Cao, Z. Wang, S.X. He, et al., Environ. Sci. Technol. 58 (2024) 3564–3575.
Q.Y. Wu, Z.W. Yang, Z.W. Wang, et al., Proc. Natl. Acad. Sci. U. S. A. 120 (2023) 2219923120. doi: 10.1073/pnas.2219923120
X. Li, X. Wen, J.Y. Lang, et al., Angew. Chem. Int. Ed. 62 (2023) e202303267. doi: 10.1002/anie.202303267
H.Y. Deng, N. Zhao, J.J. You, et al., Chin. Chem. Lett. 36 (2025) 110650. doi: 10.1016/j.cclet.2024.110650
Y.Y. Ahn, H. Bae, H.I. Kim, et al., Appl. Catal. B: Environ. 241 (2019) 561–569. doi: 10.1016/j.apcatb.2018.09.056
X.B. Dong, B.X. Ren, X.W. Zhang, et al., Appl. Catal. B: Environ. 272 (2020) 118971. doi: 10.1016/j.apcatb.2020.118971
B.H. Liu, W.Q. Guo, Q.S. Si, et al., Chem. Eng. J. 446 (2022) 137277. doi: 10.1016/j.cej.2022.137277
L.L. Chen, X.L. Xu, W.X. Yang, et al., Chin. Chem. Lett. 31 (2020) 626–634. doi: 10.1016/j.cclet.2019.08.008
Z.Y. Choong, K.Y.A. Lin, G. Lisak, et al., J. Hazard. Mater. 426 (2022) 128077. doi: 10.1016/j.jhazmat.2021.128077
B.K. Xu, X. Zhang, Y. Zhang, et al., Chem. Eng. J. 466 (2023) 143155. doi: 10.1016/j.cej.2023.143155
X.B. Jin, J.J. Lu, J. Li, et al., J. Clean. Prod. 471 (2024) 143392. doi: 10.1016/j.jclepro.2024.143392
X. Liu, Y. Pei, X.R. Mao, et al., Chem. Eng. J. 505 (2025) 159513. doi: 10.1016/j.cej.2025.159513
W. Du, Q. Zhang, Y. Shang, et al., Appl. Catal. B: Environ. 262 (2020) 118302. doi: 10.1016/j.apcatb.2019.118302
A. Mohammad, P. Chandra, M.E. Khan, et al., Adv. Colloid. Interface Sci. 322 (2023) 103048. doi: 10.1016/j.cis.2023.103048
M. Huang, X.L. Wang, C. Liu, et al., J. Environ. Chem. Eng. 9 (2021) 106536. doi: 10.1016/j.jece.2021.106536
S. Li, B. Feng, X. Zhang, et al., Appl. Catal. B: Environ. 335 (2023) 122879. doi: 10.1016/j.apcatb.2023.122879
S.Q. Liu, Z.C. Zhang, F. Huang, et al., Appl. Catal. B: Environ. 286 (2021) 119921. doi: 10.1016/j.apcatb.2021.119921
Z.W. Chen, S.W. Yang, J. Yang, et al., ACS Catal. 14 (2024) 18256–18267. doi: 10.1021/acscatal.4c05569
J.W. Lin, L.C. Li, L.S. Ma, et al., Chem. Eng. J. 445 (2022) 136668. doi: 10.1016/j.cej.2022.136668
K. Silambarasan, A.V.N. Kumar, W.S. Shin, Chem. Eng. J. 474 (2023) 145922. doi: 10.1016/j.cej.2023.145922
C. Tian, Y. Du, P. Xu, et al., ACS Appl. Mater. Interfaces 7 (2015) 20090–20099. doi: 10.1021/acsami.5b05259
Y.B. Wang, D. Li, X.L. Ge, et al., Adv. Mater. 36 (2024) 2402935. doi: 10.1002/adma.202402935
Y. Zhou, X. Hu, Y. Zhang, et al., Appl. Surf. Sci. 566 (2021) 150657. doi: 10.1016/j.apsusc.2021.150657
S. Chen, J.X. Li, W. Zhou, et al., Coordin. Chem. Rev. 507 (2024) 215749. doi: 10.1016/j.ccr.2024.215749
Y. Wei, Y. Liu, T. Wang, et al., Sep. Purif. Technol. 328 (2024) 125080. doi: 10.1016/j.seppur.2023.125080
H. Zeng, Z.P. Zhou, W.B. Li, et al., Sci. Total. Environ. 915 (2024) 170191. doi: 10.1016/j.scitotenv.2024.170191
Z.Y. Xu, Y.M. Zhang, F. Wang, et al., Chem. Eng. J. 452 (2023) 139229. doi: 10.1016/j.cej.2022.139229
M. Cao, J.Y. Lei, J.L. Zhang, et al., J. Clean. Prod. 375 (2022) 134114. doi: 10.1016/j.jclepro.2022.134114
Y. Guo, Z. Zeng, Y. Zhu, et al., Appl. Catal. B: Environ. 220 (2018) 635–644. doi: 10.1016/j.apcatb.2017.08.073
G.S. Zhang, J.Y. Gao, J. Wang, et al., J. Environ. Chem. Eng. 11 (2023) 109355. doi: 10.1016/j.jece.2023.109355
K. Hou, Z. Pi, F. Chen, et al., J. Hazard. Mater. 435 (2022) 128970. doi: 10.1016/j.jhazmat.2022.128970
X. Liu, Y. Xiang, L. Yi, et al., Sep. Purif. Technol. 348 (2024) 127719. doi: 10.1016/j.seppur.2024.127719
R. Wu, Z. Zhang, B. Yu, et al., Chem. Eng. J. 502 (2024) 158122. doi: 10.1016/j.cej.2024.158122
W.J. Zhao, Q.W. Shen, T.T. Nan, et al., J. Alloys. Compd. 958 (2023) 170370. doi: 10.1016/j.jallcom.2023.170370
L.Y. Wu, P.P. Guo, X. Wang, et al., Chemosphere 288 (2022) 132646. doi: 10.1016/j.chemosphere.2021.132646
D.Y. Chen, Q. Bai, T.T. Ma, et al., Chem. Eng. J. 435 (2022) 134916. doi: 10.1016/j.cej.2022.134916
W.B. Xu, D.L. Huang, G.F. Wang, et al., Chem. Eng. J. 499 (2024) 156245. doi: 10.1016/j.cej.2024.156245
X.Q. Zhou, Q.D. Zhao, J. Wang, et al., Chem. Eng. J. 410 (2021) 128312. doi: 10.1016/j.cej.2020.128312
S. Li, J.C. Tang, C. Yu, et al., J. Hazard. Mater. 431 (2022) 128581. doi: 10.1016/j.jhazmat.2022.128581
C.Y. He, Z.Y. Liu, J. Chen, et al., Chem. Eng. J. 507 (2025) 160768. doi: 10.1016/j.cej.2025.160768
Q.Z. Zhao, D. Liu, Y.Z. Wu, et al., Sep. Purif. Technol. 352 (2025) 128089. doi: 10.1016/j.seppur.2024.128089
Y. Ding, X. Wang, L. Fu, et al., Sci. Total. Environ. 765 (2021) 142794. doi: 10.1016/j.scitotenv.2020.142794
H.Y. Luo, H.Y. Fu, H. Yin, et al., J. Hazard. Mater. 426 (2022) 128044. doi: 10.1016/j.jhazmat.2021.128044
X.X. Zhang, Y.H. Tian, L. Zhou, et al., Chemosphere 314 (2023) 137684. doi: 10.1016/j.chemosphere.2022.137684
C.B. Wang, H.X. Dai, L. Liang, et al., J. Hazard. Mater. 458 (2023) 132002. doi: 10.1016/j.jhazmat.2023.132002
Y.B. Xie, Y. Liu, Y.J. Yao, et al., Chin. Chem. Lett. 33 (2022) 1298–1302. doi: 10.1016/j.cclet.2021.07.055

Figure 1 (a) The flowchart of the preparation process of Co-S-CN and Co-CN. (b–d) TEM images of Co-S-CN. (e) HRTEM images of Co-S-CN. (f) SAED image of Co-S-CN. (g–j) EDS mapping images of Co-S-CN.

Figure 2 (a) XRD patterns, (b) Raman spectra, and (c) FT-IR spectra of CN, Co-CN, and Co-S-CN. (d) N 1s, (e) S 2p, and (f) Co 2p XPS spectra of Co-CN, and Co-S-CN. (g) EIS, (h) CV, and (i) Tafel slope of CN, Co-CN, and Co-S-CN.

Figure 3 (a) Degradation curves and (b) kinetic constants of phenol in PMS, Co-S-CN, Co-CN+PMS, and Co-S-CN+PMS reaction systems. (c) Phenol mineralization rate at Co-S-CN+PMS reaction system. (d) Effect of phenol degradation with different salts in Co-S-CN+PMS reaction system. (e) Repeated use experiment of Co-S-CN. (f) Co concentration and leaching rate at different pH in Co-S-CN+PMS system. Conditions: [pH] = 7, [Catalyst] = 30 mg, [PMS] = 1.25 mmol/L, [Phenol] = 50 mg/L, normal temperature.

Figure 4 EPR spectra of •OH/SO4•− (a), O2•− (b), and 1O2 (c) for PMS, Co-S-CN, and Co-S-CN+PMS systems. (d) Effects of different quenching agents (TBA, MeOH, PBQ, and FFA) on phenol degradation and (e) kinetic constants for Co-S-CN+PMS system. Influence of PBQ on the signal strength of O2•− (f), and 1O2 (g) in the Co-S-CN+PMS system. (h) O2•− and (i) 1O2 for Co-CN+PMS and Co-S-CN+PMS systems.

Figure 5 (a) Co 2p, (b) S 2p, (c) N 1s, and (d) O 1s XPS spectra of fresh and used Co-S-CN. (e) XRD patterns and (f) Raman spectra of fresh and used Co-S-CN.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: