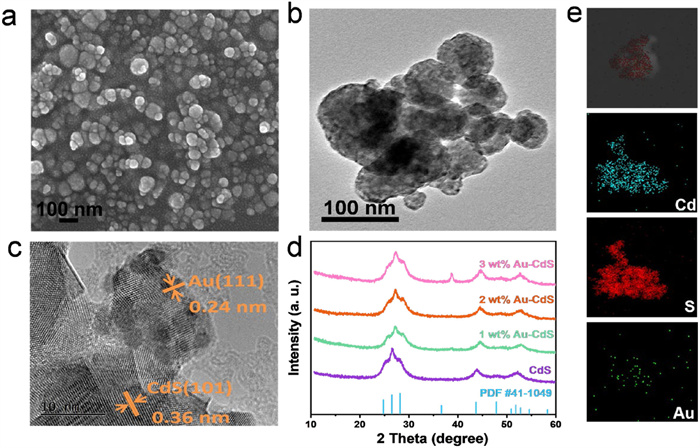
Figure 1.
(a) SEM image, (b) TEM image, and (c) HRTEM image of 2 wt% Au-CdS. (d) XRD patterns of CdS, 1 wt% Au-CdS, 2 wt% Au-CdS and 3 wt% Au-CdS. (e) EDS elemental mappings of Cd, S, and Au elements in 2 wt% Au-CdS.
Boosting α-C bond activation to enable photocatalytic upcycling of waste polylactic acid plastics into value-added pyruvic acid over Au-CdS
Yuqing Chen , Xinxin Liang , Detlef W. Bahnemann , Chuanyi Wang

Plastics, as vital necessities, play an indispensable role in human societal production and daily life [1,2]. However, the problem of plastic waste generated thereby has posed a serious threat to the environment [3,4]. Global plastic production climbed to 413.8 million tons in 2023, while the current situation of plastic waste treatment is a cause for concern: as much as 95% of plastics are discarded after only one use [5,6]. Among discarded plastics, post-consumer plastics that are mechanically recycled accounts for only 8.7%, while the proportion of chemical recycling is even as low as 0.1% [7,8]. The stability of plastics makes it difficult for them to degrade in the natural environment [9,10]. This characteristic causes them to continuously pollute the ecosystem through various channels such as land, the atmosphere, and the ocean [11-13]. Conventional methods for treating waste plastics, such as landfilling and incineration, generally suffer from problems of low efficiency and environmental pollution [14-16]. The mechanical recycling method also has its limitations. It often downgrades waste plastics into low-quality products, resulting in a serious waste of carbon resources [17].
Polylactic acid (PLA) is currently the biodegradable plastic with the highest production volume on an industrial scale and the largest consumption globally [18]. However, some studies have shown that PLA cannot be completely degraded under natural water and soil conditions [19,20]. Its continuous accumulation in the environment will also lead to more severe pollution from PLA microplastics [21]. At present, the products of most of the methods for degrading PLA contain CO2, and the massive emission of CO2 will exacerbate the greenhouse effect [22,23]. The inefficient recycling of carbon resources will severely restrict the achievement of carbon peaking and carbon neutrality goals. Consequently, there is an urgent need to develop greener and more economical technologies to enable the recycling of PLA and the production of high-value-added products.
Pyruvic acid (PA) exhibits versatile applications in pharmaceuticals, food, cosmetics, and chemical engineering-particularly in the synthesis of bio-based materials, preparation of pharmaceutical intermediates, and development of green chemical processes [24,25]. Therefore, exploring a catalytic system that can effectively achieve the selective conversion of waste PLA into PA is particularly crucial for addressing the pollution caused by waste PLA, realizing efficient resource recycling, and expanding the green preparation routes of PA. The photocatalytic oxidation strategy harnesses inexhaustible solar energy as the primary power source, using waste PLA as the reaction substrate which consumes photogenerated holes to be oxidized into small organic molecules. This approach offers energy-efficient, economical, and sustainable advantages, positioning it as an ideal waste plastic remediation solution [26,27]. Current PLA pretreatment technologies predominantly rely on strongly alkaline catalytic hydrolysis systems. However, the use of highly corrosive reagents brings about issues such as safety risks and increased costs [28]. Moreover, complex side reactions are likely to occur during hydrolysis, posing difficulties for the separation and purification of products [29]. Miao et al. proposed a strategy for the direct hydrolytic photoreforming of PLA and verified its feasibility in sustaining the efficient production of LA [30].
CdS, due to its suitable bandgap and excellent visible-light harvesting properties, is regarded as one of the most promising photocatalysts in the field of photocatalytic reactions [31]. However, in practical applications, CdS photocatalysts are hampered by significant photogenerated charge carrier recombination phenomena and insufficient capability in activating chemical bonds, thereby resulting in difficulties in regulating product selectivity [32-34]. These limitations collectively hinder the enhancement of its photocatalytic activity. Efficient adsorption and activation of C—H bonds by photocatalysts constitute an essential prerequisite for enabling the formation of value-added chemical compounds [35]. Noble metal nanoparticles as cocatalysts represent an efficacious strategy for augmenting the photocatalytic performance of CdS [36]. Among them, Au nanoparticles can act as photoelectron reservoirs, optimizing the efficiency of charge separation [37,38]. Furthermore, Au nanoparticles exhibit exceptional ability in activating Cα-H bonds during the photocatalytic oxidation of methane and ethane, holding the potential to achieve the selective synthesis of target products [39,40].
Inspired by the above considerations, we developed an Au-loaded CdS nanosphere catalyst. It was demonstrated that Au-CdS can selectively promote the continuous dehydrogenation of α-C in LA, thus enabling its conversion into value-added PA. Au-CdS simultaneously enhances the effectiveness of separating photogenerated charge carriers, catalyzes the efficient conversion of O2•− to •OH radicals, and reduces the Cα-H bond homolysis energy barrier in LA molecules from 1.02 eV to 0.49 eV, thereby significantly promoting the generation of carbon-centered radical intermediates. In the hydrolysis-pretreated PLA system, when the Au loading is optimized to 2 wt%, 2 wt% Au-CdS exhibits optimal performance during 5 h photocatalytic reaction, achieving a PA production rate of 1340 ± 25 μmol g-1 h-1 and showing an 8.9-fold superiority over pristine CdS. In-situ EPR characterizations reveal the critical role of •OH radicals in the successive dehydrogenation reactions occurring at the α-C of LA. This work establishes a green and mild pathway for resource recovery, enabling the value-added upcycling of plastic waste.
For the purpose of clarifying the morphology, structure, and Au distribution in 2 wt% Au-CdS, TEM and SEM analyses were utilised to explore these characteristics. SEM analysis reveals that 2 wt% Au-CdS catalyst maintains an identical homogeneous nanospherical architecture to pristine CdS, with no discernible Au nanoparticles observed (Fig. 1a and Fig. S1 in Supporting information). TEM images (Fig. 1b) further reveal that the black spots, averaging 7.96 nm in particle size, correspond to Au nanoparticles loaded onto CdS surfaces. Combined with these characterization results, it can be confirmed that the Au nanoparticles achieve uniform dispersion on the CdS substrate. HRTEM images (Fig. 1c) of 2 wt% Au-CdS show two kinds of lattice fringes: Those with an interplanar spacing of 0.24 nm point to the (111) plane of cubic Au, while those at 0.36 nm correspond to the (100) plane of hexagonal CdS [41,42]. EDS elemental mappings further evidence the high dispersion of Cd, S and Au within 2 wt% Au-CdS (Fig. 1e).
Verification of sample structural composition was achieved through XRD characterization. The characteristic diffraction peaks for CdS at 2theta of 25.1°, 26.5°, 28.1°, 43.8°, 47.9° and 52.3° are indexed as (100), (002), (101), (110), (103), (112) planes of hexagonal CdS (PDF No 41–1049) respectively (Fig. 1d) [30]. Further analysis reveals that with the incremental elevation of Au loading content, the characteristic diffraction peak corresponding to the (111) crystallographic plane of Au progressively emerges at 2theta of 38.2°, whereas the peak positions and relative intensities of all other diffraction profiles remain virtually unaltered in comparison to pristine CdS.
Fig. S2a (Supporting information) presents the N2 adsorption-desorption isotherms of CdS photocatalysts with varying Au loadings. All synthesized catalysts present type Ⅳ isotherms accompanied by H3 hysteresis loops, which confirms that a mesoporous structure is present [43]. With all catalysts displaying strikingly similar specific surface areas and average pore sizes (Fig. S2b in Supporting information), it can be inferred that the photocatalytic activity of 2 wt% Au-CdS correlates weakly with either specific surface area or pore size distribution.
X-ray photoelectron spectroscopy (XPS) was utilized to ascertain the surface elemental composition and chemical state of the samples. The survey spectrum of 2 wt% Au-CdS confirms the presence of constituent elements Cd, S, and Au within the composite material (Fig. S3 in Supporting information). Through XPS analysis, it is observed that the Cd 3d orbital binding energies in 2 wt% Au-CdS undergo significant positive shifts relative to pristine CdS, with the characteristic Cd 3d5/2 and Cd 3d3/2 peaks positioned at 405.10 eV and 411.84 eV, respectively (Fig. 2a). Analogous positive shifts are observed in relation to the S 2p orbitals, with the S 2p3/2 and S 2p1/2 characteristic peaks appearing at 161.37 eV and 162.38 eV, respectively (Fig. 2b). The positive shifts in binding energies serve as evidence that photogenerated electrons are efficiently transferred from the CdS to Au [44]. Furthermore, the characteristic peaks corresponding to Au 4f7/2 and Au 4f5/2 are found at 84.17 eV and 87.85 eV, respectively (Fig. 2c), which provide definitive evidence for the existence of Au0.
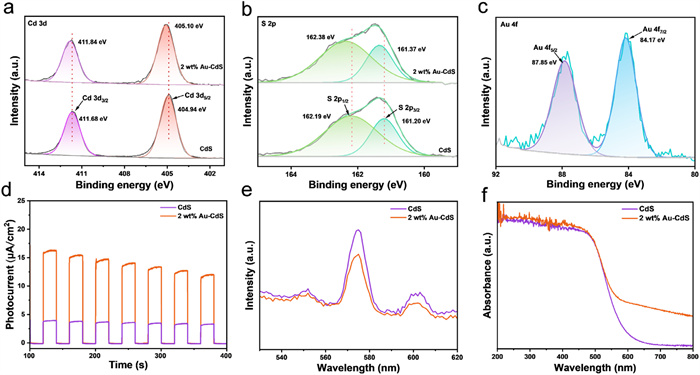
Photoelectrochemical properties serve as pivotal indicators for evaluating catalytic performance. Transient photocurrent measurements reveal that all samples exhibit pronounced photoresponse characteristics, with the 2 wt% Au-CdS demonstrating substantially enhanced photocurrent density compared to pristine CdS (Fig. 2d). Detailed analysis of electrochemical impedance spectroscopy (EIS) Nyquist plots indicates that 2 wt% Au-CdS has a smaller semicircle radius than CdS (Fig. S4a in Supporting information). This characteristic reflects reduced charge transfer resistance and more effective facilitation of electron-hole pair separation. These results corroborate that the introduction of Au facilitates the efficient transfer of charge carriers and simultaneously suppresses the recombination process of photogenerated carriers to a significant extent [45].
The charge separation and transfer dynamics were further investigated using photoluminescence (PL) spectra and time-resolved fluorescence spectroscopy. As illustrated in Fig. 2e, the most pronounced PL intensity quenching is observed in the 2 wt% Au-CdS. These findings clearly demonstrate the efficient charge transfer from the CdS matrix to Au nanoparticles [46]. The average fluorescence lifetime of 2 wt% Au-CdS is 17.30 ns, which is longer than the average lifetime of CdS (Fig. S4b in Supporting information). The extended lifetime of photogenerated charges also means that their probability of recombination is greatly reduced [47]. Derived from linear sweep voltammetry (LSV) measurements, a critical observation unveils that when Au nanoparticles are decorated onto CdS, the resulting 2 wt% Au-CdS exhibits a distinct positive shift in its onset oxidation potential (Fig. S4c in Supporting information). The cause of this occurrence is the co-catalytic action of Au [48].
The optical absorption characteristics of both CdS and 2 wt% Au-CdS were examined using solid-state UV–vis diffuse reflectance spectroscopy (DRS). The bandgap and absorption edge of these materials can be estimated by extrapolating the intersection of the slope and the baseline in the spectrum [49]. As illustrated in Fig. 2f, CdS and 2 wt% Au-CdS exhibit a comparable characteristic absorption edge at 602 nm, while 2 wt% Au-CdS displays substantially intensified absorbance across the visible spectrum (500–800 nm), indicative of augmented light-harvesting capacity imparted by Au incorporation [50]. Furthermore, the bandgap energy of 2 wt% Au-CdS (2.22 eV) remains virtually unchanged relative to pristine CdS (2.25 eV) (Fig. S4d in Supporting information). These optimized optoelectronic properties signify an expanded visible-light response range in 2 wt% Au-CdS catalysts, which facilitates the generation of electron-hole pairs upon photoexcitation, thereby significantly improving its photocatalytic activity. Through meticulous analysis of the intercepts derived from Fig. S4e (Supporting information), the valence band (EVB) position of CdS is measured as 1.42 eV. Applying the fundamental relationship of semiconductor band theory, ECB = EVB - Eg, the conduction band (ECB) potential of CdS is calculated as −0.83 eV relative to the normal hydrogen electrode (NHE) [51]. Under this favorable conduction band potential, 2 wt% Au-CdS endows photogenerated electrons with a robust reductive driving force to react with O2 to form O2•− radicals, while also facilitating the further conversion of O2•−into •OH radicals [52,53].
The catalytic activity in the PLA plastics photocatalytic oxidation can be evaluated by detecting the generation of products of economic and application value. In view of environmental friendliness, the hydrothermal hydrolysis pretreatment was chosen for PLA, which avoids the use of large amounts of chemical reagents and reduces the negative impact on the environment while safeguarding the material properties. The liquid products of PLA photocatalytic oxidation in pretreated PLA samples were verified via proton nuclear magnetic resonance (1H NMR) spectroscopy, with 2 mmol/L maleic acid as an internal standard (Figs. S5a-c in Supporting information). Spectral analysis reveals that PLA undergoes hydrolysis subsequent to pretreatment, yielding LA and trace amounts of ethyl lactate, with 86% of the PLA being hydrolyzed into LA (Table S1 in Supporting information). After 5 h of photocatalysis, a marked attenuation in the characteristic peak intensity corresponding to LA is observed, with its concentration declining from 113 mmol/L to 101 mmol/L. Concurrently, emerging peaks attributable to PA, its derivatives and acetate (AA) are observed in the spectrum (Fig. 3a). High-performance liquid chromatography (HPLC) was used to conduct quantitative analysis of liquid products after 5 h of PLA photocatalysis (Figs. S6a-c in Supporting information). As illustrated in Fig. 3b, 2 wt% Au-CdS catalyst attains a PA production rate of 1340 ± 25 μmol g-1 h-1, achieving 80.4% selectivity and 9.9% conversion. In comparison to pristine CdS, which yields a mere 150 ± 4 μmol g-1 h-1, this rate is 8.9-fold higher and surpasses all control samples. AA is generated at a rate of 326 ± 34 μmol g-1 h-1, exhibiting selectivity of 19.6%. The calculated carbon balance yields a value of 91.1%, whereas TOC analysis gives a value of 90.1%, a difference that can ascribed to trace side products undetected under the employed 1H NMR and HPLC (Table S2 in Supporting information). These experimental data demonstrate that 2 wt% Au-CdS exhibits superior performance in PLA photocatalytic reactions, showcasing efficacy superior to previously reported photocatalytic systems (Table S3 in Supporting information). Findings derived from control experiments reveal that in the absence of any one of the critical conditions (illumination, catalyst, or PLA substrate), no formation of PA product is detected (Fig. S7 in Supporting information). These observations thus underscore that the photocatalytic process is indispensable for enabling the conversion of PLA to PA. To pinpoint active species in PLA photocatalytic oxidation, a suite of scavengers was systematically introduced into the reaction system. These included Na2C2O4, p-benzoquinone (PBQ), isopropyl alcohol (IPA), and furfuryl alcohol (FFA), which were employed to selectively quench holes, O2•−radicals, •OH radicals, and 1O2 radicals, respectively. Fig. 3c illustrates that the inclusion of 0.05 mol/L Na2C2O4 and IPA gives rise to a marked decline in PA yield, while the addition of PBQ scavenger induces a comparatively negligible effect on PA yield. These results establish that photogenerated holes and •OH radicals constitute the predominant reactive species dictating the oxidation pathway of LA. Furthermore, by investigating the differential effects of air and Ar atmospheres on the photocatalytic activity of PLA, it is observed that the PA yield under an Ar atmosphere is markedly diminished (Fig. 3d). These comparative experimental findings strongly corroborate that the •OH radicals generated under O2-containing conditions exert a pivotal role in the formation process of PA.
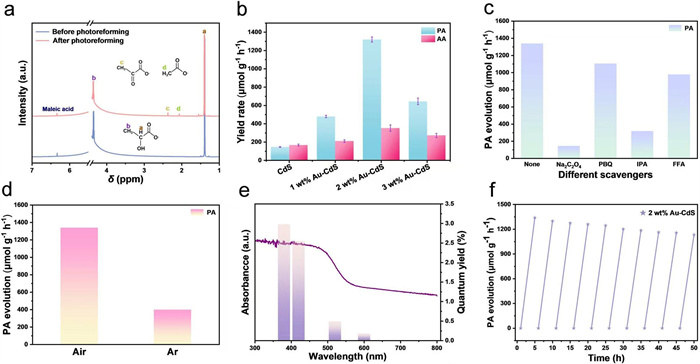
To elucidate the origin of photocatalytic oxidation performance, systematic catalytic activity tests were conducted on 2 wt% Au-CdS under various monochromatic light irradiations. As depicted in Fig. 3e, the catalytic performance exhibits pronounced wavelength dependency, demonstrating excellent correspondence with the ultraviolet-visible diffuse reflectance spectroscopy (UV–vis DRS) profile of 2 wt% Au-CdS. Under light irradiation at a wavelength of 420 nm, 2 wt% Au-CdS exhibits a quantum yield (QY) of 2.55%, accompanied by a relatively high PA yield. When the irradiation wavelength extends beyond 520 nm, the QY of PA decreases remarkably. Under 600 nm light irradiation, the PA generation capability of 2 wt% Au-CdS is extremely limited. This indicates that photons with energy lower than the band gap of CdS cannot be effectively absorbed and utilized, resulting in the failure of effective excitation of photogenerated electron-hole pairs. Consequently, the insufficient photoinduced electrons fail to activate O2 into reactive species (O2•− and •OH radicals), ultimately limiting the photocatalytic oxidation of PLA. These collective findings establish that the localized surface plasmon resonance (LSPR) characteristics of Au nanoparticles contribute negligibly to the LA-to-PA oxidation pathway [54]. Following five consecutive illumination cycles cumulating 25 h of photoirradiation, the PA production rate maintained remarkable stability (Fig. 3f). ICP-OES analysis indicates that the Cd2+ concentration remains undetectable in post-reaction solutions. Moreover, XRD and TEM analyses collectively demonstrate that the recycled 2 wt% Au-CdS catalyst undergoes no significant alterations in either morphology or crystalline structure following the reaction (Figs. S8a and b in Supporting information). HRTEM image further reveals that the characteristics of its lattice fringes remain consistent (Fig. S8c in Supporting information). These findings collectively validate that the 2 wt% Au-CdS catalyst with a relatively low Au loading exhibits excellent photocatalytic stability and environmental safety.
For a more profound understanding of the reaction mechanism behind the photocatalytic oxidation of PLA, the application of in situ electron paramagnetic resonance (EPR) spectroscopy and in situ Fourier-transform infrared spectroscopy (FTIR) for detecting reaction intermediates in pretreated PLA solutions is of significant importance. As shown in Figs. 4a and b, with DMPO used as a spin-trapping agent under light irradiation, the signals of O2•− and •OH radicals generated by 2 wt% Au-CdS were captured in methanol and aqueous solutions respectively, confirming the presence of these two radicals. Further comparison reveals that the signal intensities of O2•− and •OH radicals over 2 wt% Au-CdS are significantly enhanced compared to pristine CdS, implying that 2 wt% Au-CdS more effectively improves the utilization of photogenerated electrons to promote photocatalytic reactions. Furthermore, with TEMP employed as the trapping agent, the characteristic signal of 1O2 radicals in both cases of CdS and 2 wt% Au-CdS could be detected, but the signal intensity for the latter is higher (Fig. S9a in Supporting information). However, combined with the analysis of the energy band structure, the valence band potential of 2 wt% Au-CdS measures 1.42 V vs. RHE, a value that falls short of the 2.40 V vs. RHE required for •OH generation [55]. This conclusion is experimentally validated: No •OH signals are detected over 2 wt% Au-CdS during light irradiation under Ar atmosphere, indicating that holes in 2 wt% Au-CdS cannot directly produce •OH, thus inferring that •OH originates from the conversion of O2•−[56,57]. Notably, as shown in Fig. 4c, in the Ar-pretreated PLA solution, 2 wt% Au-CdS only exhibits a sextet characteristic peak corresponding to carbon-centered radicals (intensity ratio of 1:1:1:1:1:1) (•C center AN = 15.51 G, AH = 22.02 G, and g = 2.0057), with no •OH signals detected, suggesting that holes (h+) are involved in PLA oxidation. Additionally, in the light irradiation experiment under air atmosphere, the signal intensity of •OH radicals over 2 wt% Au-CdS significantly decreases in the presence of PLA compared to that in pure water, while the signal of carbon-centered radicals increases remarkably, confirming that •OH radicals perform a vital function in the dehydrogenation of PLA. As shown in Figs. 4d and e and Fig. S9b (Supporting information), during the photocatalytic reaction of PLA, 2 wt% Au-CdS exhibits stronger carbon-centered radical signals and weaker •OH signals than CdS, as revealed by spectral fitting, indicating that 2 wt% Au-CdS promotes the attack of •OH radicals on the Cα-H bond in LA. To investigate O2 adsorption behavior on photocatalyst surfaces, in situ Fourier-transform infrared spectroscopy (FTIR) was employed. As illustrated in Fig. 4f, within the vibrational frequency range of 850–1250 cm-1, CdS exhibits no distinct absorption bands, whereas 2 wt% Au-CdS displays more intense characteristic vibrational peaks of O2 adsorption at 1022 cm-1 and 960 cm-1 [58,59]. The observed spectral enhancement correlates with Au nanoparticle-promoted O2 chemisorption on CdS surfaces, promoting •OH production to enhance PLA photocatalytic efficiency.
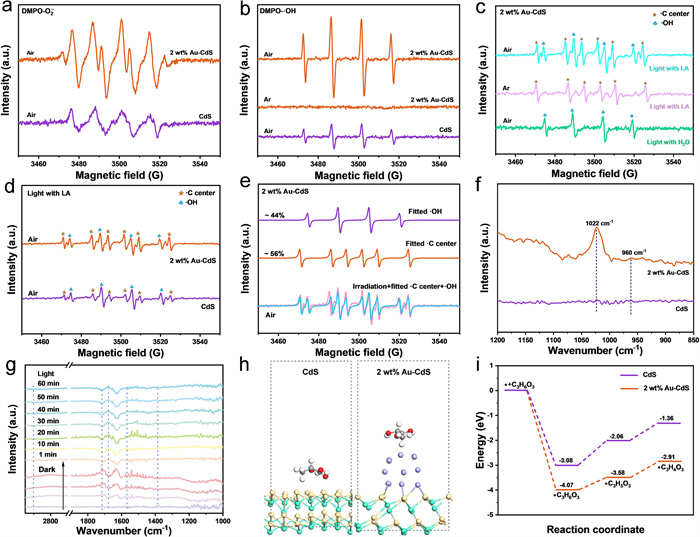
In-situ diffuse reflectance infrared Fourier transform spectroscopy (in-situ DRIFTS) indicates that the characteristic peaks observed at 1710 and 2900 cm-1 are respectively ascribed to the stretching vibrations of COO− groups and the Cα-H bond in LA (Fig. 4g). As the photocatalytic reaction progresses, the signal of the characteristic peak assigned to the Cα-H bond gradually diminishes and eventually disappears. This observation indicates the cleavage of the Cα-H bond within LA. Concomitantly, a substantial enhancement in absorption intensity emerges at 1670 cm-1, corresponding to the C=O stretching vibration of PA. These spectral changes over time clearly demonstrate that 2 wt% Au-CdS facilitates the cleavage of the Cα-H bond in LA and the concomitant formation of the C=O bond in PA. Furthermore, newly emerged characteristic peaks at 1385 and 1570 cm-1 can be respectively assigned to the stretching vibrations of -CH3 groups and COO− groups in PA [60]. Collectively, these in-situ infrared spectroscopic analyses provide compelling experimental evidence for the progressive consumption of LA and the formation of PA.
Computational investigations based on density functional theory (DFT) further elucidate the catalytic behavior of 2 wt% Au-CdS and CdS catalysts during LA conversion reactions (Figs. 4h and i and Fig. S10). As shown in Fig. 4i, theoretical computations reveal that CdS and 2 wt% Au-CdS exhibit substantial discrepancies with respect to the dissociation energy barriers of the Cα-H bond within lactic acid molecules: 2 wt% Au-CdS (0.49 eV) < CdS (1.02 eV). This theoretical prediction aligns remarkably well with electron paramagnetic resonance (EPR) experimental results, which confirmed that a greater number of carbon-centered radicals are generated in the 2 wt% Au-CdS catalyst system. Collectively, these findings substantiate that the incorporation of Au sites effectively diminishes the activation energy barrier for Cα-H bond cleavage while concomitantly promoting the generation of carbon-centered radicals.
From the characterization results presented above, the photocatalytic oxidation mechanism of pretreated PLA catalyzed by 2 wt% Au-CdS can be speculated as follows (Fig. 5): Under light irradiation, Au nanoparticles on CdS surfaces act as electron traps to capture photogenerated conduction band (CB) electrons. There, the photogenerated electrons react with the dissolved O2 in water to generate O2•− radicals, and the enhanced reducing ability of these electrons further drives the conversion of O2•− into more •OH. The •OH radicals attack the Cα-H bonds of LA, abstracting a hydrogen atom to form carbon-centered radical intermediates. Subsequently, the •OH radicals further react with the α-OH bonds of the carbon-centered radical to generate a C=O bond, thereby achieving the continuous dehydrogenation of LA and effectively driving the selective generation of PA. In summary, the proposed mechanism for photocatalytic oxidation can be possibly inferred as follows:
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
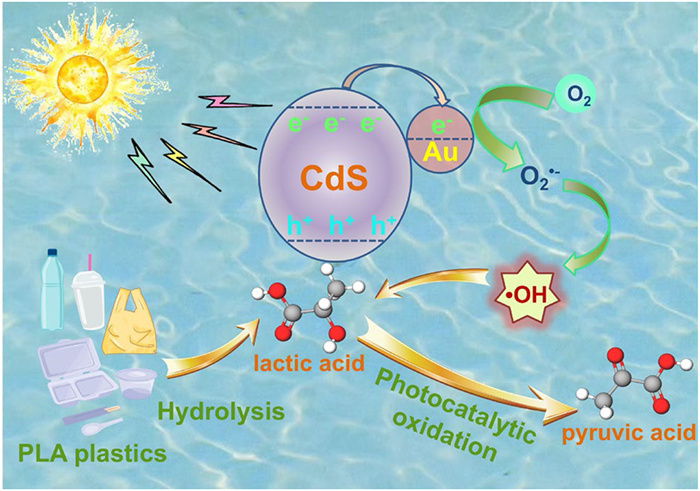
In summary, we fabricated an Au-loaded CdS nanosphere photocatalyst wherein the incorporation of Au active sites facilitates the activation of the Cα-H bond in LA, aiming to achieve the photocatalytic oxidation of waste PLA into high-value-added PA. In-situ X-ray photoelectron spectroscopy (XPS) and electron paramagnetic resonance (EPR) characterizations reveal that 2 wt% Au-CdS not only significantly enhances the separation efficiency of photogenerated charge carriers but also efficiently catalyzes the activation of O2 into O2•− and further drives the conversion of O2•− to •OH radicals, compared to pristine CdS. Theoretical calculations corroborate that 2 wt% Au-CdS reduces the Cα-H bond cleavage energy barrier from 1.02 eV to 0.49 eV, markedly boosting the generation of carbon-centered radical intermediates. The production of •OH radicals facilitates the successive dehydrogenation reactions by interacting with the Cα-H and α-OH bonds on the α-C of LA, thereby promoting the selective conversion of LA to PA. As a result, 2 wt% Au-CdS exhibits optimal catalytic performance: the PA production rate reaches 1340 ± 25 μmol g-1 h-1, surpassing that of pristine CdS by 8.9-fold. In general, this work provides a new solution for the value-added upcycling of plastic waste.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yuqing Chen: Writing – review & editing, Writing – original draft, Investigation, Data curation. Xinxin Liang: Validation, Data curation. Detlef W. Bahnemann: Validation, Conceptualization. Chuanyi Wang: Visualization, Validation, Methodology, Funding acquisition.
This work was supported by the National Natural Science Foundation of China (Nos. 52161145409, 21976116), “Belt and Road” Innovative Talent Exchange Foreign Expert Project (No. 2023041004L), High-end Foreign Expert Project (No. G2023041021L), and Alexander-von-Humboldt Foundation of Germany (Group-Linkage Program). D. W. Bahnemann gratefully acknowledges the support of this work by St. Petersburg University, with the research project (No. 125021902439-8).
Supplementary material associated with this article can be found, in the online version, at doi:
P.G.C.N.T. Pilapitiya, A.S. Ratnayake, Clean. Mater. 11 (2024) 100220. doi: 10.1016/j.clema.2024.100220
A.D. Macheca, B. Mutuma, J.L. Adalima, et al., Recycling 9 (2024) 77. doi: 10.3390/recycling9050077
A.S. Pottinger, R. Geyer, N. Biyani, et al., Science 386 (2024) 1168–1173. doi: 10.1126/science.adr3837
M. MacLeod, H.P.H. Arp, M.B. Tekman, et al., Science 373 (2021) 61–65. doi: 10.1126/science.abg5433
Plastics Europe, Plastics-the fast facts 2024,
OECDGlobal Plastics Outlook: Economic Drivers, Environmental Impacts and Policy Options, OECD publishing, 2022.
X. Zhang, M. Jun, W. Zu, et al., Small 20 (2024) 2403347. doi: 10.1002/smll.202403347
S. Zhang, J. Wang, D. Su, et al., Nat. Commun. 16 (2025) 4188. doi: 10.1038/s41467-025-59540-5
D. Gibovic, A. Bikfalvi, Recycling 6 (2021) 29. doi: 10.3390/recycling6020029
M. Shams, I. Alam, I. Chowdhury, Water Res. 171 (2020) 115401. doi: 10.1016/j.watres.2019.115401
G. Bonifazi, L. Fiore, C. Pelosi, et al., Polym. Degrad. Stab. 207 (2023) 110215. doi: 10.1016/j.polymdegradstab.2022.110215
I.E. Gilani, S. Sayadi, N. Zouari, et al., Bioresour. Technol. Rep. 24 (2023) 101606. doi: 10.1016/j.biteb.2023.101606
M.G. Kibria, N.I. Masuk, R. Safayet, et al., Int. J. Environ. Res. 17 (2023) 20. doi: 10.1007/s41742-023-00507-z
M.Q. Zhang, M. Wang, B. Sun, et al., Chem 8 (2022) 2912–2923. doi: 10.1016/j.chempr.2022.08.004
T. Uekert, C.M. Pichler, T. Schubert, et al., Nat. Sustain. 4 (2021) 383–391.
J. Meng, Y. Zhou, D. Li, et al., Sci. Bull. 68 (2023) 1522–1530. doi: 10.1016/j.scib.2023.06.024
I. Vollmer, M.J.F. Jenks, M.C.P. Roelands, et al., Angew. Chem. Int. Ed. 59 (2020) 15402–15423. doi: 10.1002/anie.201915651
Y. Li, S. Wang, S. Qian, et al., ACS Omega 9 (2024) 13509–13521. doi: 10.1021/acsomega.3c08674
P. McKeown, M.D. Jones, Sustain. Chem. 1 (2020) 1–22. doi: 10.3390/suschem1010001
Y. Niu, F. Pan, K. Shen, et al., Sustainability-Basel 16 (2024) 9395. doi: 10.3390/su16219395
M.S. Nasir, I. Tahir, A. Ali, et al., Heliyon 10 (2024) e25883. doi: 10.1016/j.heliyon.2024.e25883
C.X. Liu, K. Liu, Y. Xu, et al., Angew. Chem. Int. Ed. 63 (2024) e202401255. doi: 10.1002/anie.202401255
A. Garratt, K. Nguyen, A. Brooke, et al., ACS Environ. Au 3 (2023) 342–347. doi: 10.1021/acsenvironau.3c00040
Q. Luo, N. Ding, Y. Liu, et al., Molecules 28 (2023) 1418. doi: 10.3390/molecules28031418
A. Corma, S. Iborra, A. Velty, Chem. Rev. 107 (2007) 2411–2502. doi: 10.1021/cr050989d
T.K.A. Nguyen, T. Trần-Phú, R. Daiyan, et al., Angew. Chem. Int. Ed. 63 (2024) e202401746. doi: 10.1002/anie.202401746
R. Cao, D. Xiao, M. Wang, et al., Appl. Catal. B 341 (2024) 123357. doi: 10.1016/j.apcatb.2023.123357
C. Peng, G. Zhang, J. Yue, et al., Fuel Process. Technol. 124 (2014) 212–221. doi: 10.1016/j.fuproc.2014.02.025
S. Zhang, H. Li, L. Wang, et al., J. Am. Chem. Soc. 145 (2023) 6410–6419. doi: 10.1021/jacs.2c13590
Y. Miao, Y. Zhao, J. Gao, et al., J. Am. Chem. Soc. 146 (2024) 4842–4850. doi: 10.1021/jacs.3c13000
I. Ahmad, S. Ben Ahmed, M. Shabir, et al., J. Ind. Eng. Chem. 130 (2024) 105–124. doi: 10.1016/j.jiec.2023.10.001
Y. Li, L.H. Gan, Int. J. Hydrog. Energy 60 (2024) 1500–1508. doi: 10.1016/j.ijhydene.2024.02.113
W. Li, X. Zheng, B.B. Xu, et al., Angew. Chem. Int. Ed. 63 (2024) e202320014. doi: 10.1002/anie.202320014
Y. You, P. Han, S. Song, et al., Angew. Chem. Int. Ed. 62 (2024) e202306452.
Y. Shi, P. Li, H. Chen, et al., Nat. Commun. 15 (2024) 4641. doi: 10.1038/s41467-024-49005-6
N. Xiao, S. Li, X. Li, et al., Chin. J. Catal. 41 (2020) 642–671. doi: 10.1016/S1872-2067(19)63469-8
B. He, Y. Cao, K. Lin, et al., Angew. Chem. Int. Ed. 63 (2024) e202402435. doi: 10.1002/anie.202402435
Q. Zhang, B. Fan, Y. Wang, et al., Chem. Synth. 5 (2025) 25.
Y. Jiang, S. Li, S. Wang, et al., J. Am. Chem. Soc. 145 (2023) 2698–2707. doi: 10.1021/jacs.2c13313
K. Xu, Y. Xiao, S. Liu, et al., ACS Catal. 15 (2025) 9962–9975. doi: 10.1021/acscatal.5c01446
C. Zhang, Z. Ai, X. Xu, et al., Small 22 (2025) 2411128.
J. Jiang, S. Chu, Y. Zhang, et al., Sci. Adv. 10 (2024) eadn5946. doi: 10.1126/sciadv.adn5946
Q. Zhao, J. Hu, Z. Gui, et al., ChemSusChem 18 (2024) e202401727.
Z. Xu, W. Yue, C. Li, et al., ACS Nano 17 (2023) 11655–11664. doi: 10.1021/acsnano.3c02092
W. Xu, J. Jia, T. Wang, et al., Angew. Chem. Int. Ed. 132 (2020) 22430–22435. doi: 10.1002/ange.202010613
W. Su, Y. Zhang, A. Kuklin, et al., ACS Catal. 14 (2024) 13927–13939. doi: 10.1021/acscatal.4c02269
Y. Wang, R. Zhao, Y. Xu, et al., Nano Energy 141 (2025) 111103. doi: 10.1016/j.nanoen.2025.111103
X. Liang, T. Gao, Y. Cui, et al., Appl. Catal. B 357 (2024) 124326. doi: 10.1016/j.apcatb.2024.124326
P. Makuła, M. Pacia, W. Macyk, et al., J. Phys. Chem. Lett. 9 (2018) 6814–6817. doi: 10.1021/acs.jpclett.8b02892
F. Ge, Y. Zhao, C. Feng, et al., ACS Appl. Mater. Interfaces 16 (2024) 32847–32856. doi: 10.1021/acsami.4c04195
X. Li, B. Kang, F. Dong, et al., Nano Energy 81 (2021) 105671. doi: 10.1016/j.nanoen.2020.105671
H. Wang, Y. Shi, J. Guo, et al., Chin. Chem. Lett. 36 (2025) 110779. doi: 10.1016/j.cclet.2024.110779
M. Yang, H. Li, F. Liu, et al., Appl. Catal. B 354 (2024) 124071. doi: 10.1016/j.apcatb.2024.124071
N. Ghorai, Z. Yang, S.T. Gebre, et al., Nano Lett. 25 (2025) 3253–3258. doi: 10.1021/acs.nanolett.4c06154
Y. Wang, X. Li, S. Liu, et al., ACS Catal. 12 (2022) 2770–2780. doi: 10.1021/acscatal.1c05447
A. Sampath, T. Ricciardulli, P. Priyadarshini, et al., ACS Catal. 12 (2022) 9549–9558. doi: 10.1021/acscatal.2c02076
X. Li, W. Yang, J. Yue, et al., Nat. Commun. 16 (2025) 3590. doi: 10.1038/s41467-025-58840-0
J. Li, Y. Deng, Y. Shi, et al., Chem. Eng. J. 520 (2025) 166375. doi: 10.1016/j.cej.2025.166375
J. Wang, S. Shen, X. Li, et al., Chem. Rev. 125 (2025) 7811–7917. doi: 10.1021/acs.chemrev.5c00074
X. Liang, Y. Cui, Q. Tian, et al., Chin. Chem. Lett. 37 (2025) 111097.

Figure 1 (a) SEM image, (b) TEM image, and (c) HRTEM image of 2 wt% Au-CdS. (d) XRD patterns of CdS, 1 wt% Au-CdS, 2 wt% Au-CdS and 3 wt% Au-CdS. (e) EDS elemental mappings of Cd, S, and Au elements in 2 wt% Au-CdS.

Figure 2 High-resolution XPS spectra of (a) Cd 3d, (b) S 2p and (c) Au 4f for CdS and 2 wt% Au-CdS. (d) Photocurrents, (e) PL spectra, (f) UV–vis DRS spectra of CdS and 2 wt% Au-CdS.

Figure 3 (a) 1H NMR spectra of the pretreated PLA before and after photocatalytic oxidation. (b) PA evolution rate over Au-CdS with different Au content following 5 h of photocatalytic oxidation. (c) The control experiments with different scavengers over 2 wt% Au-CdS. (d) Different atmospheres on photocatalytic oxidation of PLA over 2 wt% Au-CdS. (e) Quantum yield values at varied monochromatic wavelengths along with UV–vis DRS of 2 wt% Au-CdS. (f) PA evolution rate for 50 h over 2 wt% Au-CdS.

Figure 4 In-situ EPR signals of (a) DMPO-O2•-, and (b) DMPO-•OH in presence of CdS and 2 wt% Au-CdS under visible light irradiation. (c) In-situ EPR signals of 2 wt% Au-CdS in the photocatalytic system under different atmospheres. (d) In-situ EPR signals in the CdS and 2 wt% Au-CdS photocatalytic system. (e) In-situ EPR spectra of 2 wt% Au-CdS during the photocatalytic system. (f) In-situ FTIR spectra after O2 adsorption on CdS and 2 wt% Au-CdS. (g) Time-resolved in-situ DRlFTS of pretreated PLA on 2 wt% Au-CdS under dark and irradiated conditions. (h) Optimized models of adsorbed LA on the CdS and 2 wt% Au-CdS. (i) Energy profile of the LA oxidation process.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: