Citation:
Zhihao Li, Zhiruo Zhou, Yuan Qin, Dan Huang, Meizhen Wang. Density functional theory assists in promoting advanced oxidation processes: Toward efficient antibiotic degradation in waters[J]. Chinese Chemical Letters,
2026, 37(6): 112318.
doi:
10.1016/j.cclet.2025.112318

Density functional theory assists in promoting advanced oxidation processes: Toward efficient antibiotic degradation in waters
English
Density functional theory assists in promoting advanced oxidation processes: Toward efficient antibiotic degradation in waters
Abstract:
Antibiotic contamination in aquatic environments poses a serious threat to ecological safety and public health. However, traditional advanced oxidation processes (AOPs) face critical bottlenecks due to unclear microscopic reaction mechanisms, including ambiguous reactive species generation pathways and a lack of theoretical guidance for catalyst design. This review systematically elucidates the pivotal role of density functional theory (DFT) in antibiotic degradation via AOPs: (1) By accurately simulating catalyst electronic structures, adsorption energies, and reaction energy barriers, DFT reveals the evolution rules of active sites (e.g., multi-element doping reduces the O–O bond cleavage energy barrier by 36%), thereby optimizing reaction pathways across photocatalysis, electrochemical oxidation, and persulfate activation systems; (2) Combined with Fukui index and molecular orbital analyses, DFT enables precise identification of vulnerable sites in antibiotic molecules (e.g., C8/C13 of ofloxacin and O23/N15 of ciprofloxacin), and predicts the thermodynamics and kinetics of reactive species (e.g., 1O2, SO4•‒) formation; (3) Under a closed-loop "computational guidance — experimental validation" framework, DFT drives catalyst structure optimization and reaction pathway regulation, significantly enhancing AOP mineralization efficiency (e.g., 2.5-fold increase in tetracycline removal rate). Future directions should focus on integrating non-adiabatic molecular dynamics, machine learning-assisted screening, and toxicity prediction of degradation products to promote the intelligent design and green engineering of AOPs, thereby building an efficient and precise antibiotic pollution control system.
-
1. Introduction
Emerging antibiotic-related pollutants, characterized by high biological toxicity and their potential to facilitate the dissemination of antibiotic resistance genes (ARGs), have intensified concerns regarding water pollution and ecological safety. These contaminants pose a substantial threat to the already vulnerable "water-energy-health" nexus [1-4]. The World Health Organization (WHO) has recently assessed that infections caused by antimicrobial resistance (AMR) result in over 1.27 million deaths annually. Given the role of aquatic environments as critical reservoirs for ARG propagation, their associated risks must not be overlooked. Recent studies have revealed alarming concentrations of antibiotics in both engineered wastewater treatment systems and natural water bodies [5]. For example, average concentrations of quinolones, sulfonamides, and tetracyclines in influents of wastewater treatment plants have been reported at 2004.76, 3863.63, and 476.29 ng/L, respectively [6]. In natural environments, the average total antibiotic concentration in the upper reaches of the Yangtze River in China reaches 525.40 ng/L [7], while nearshore waters in China exhibit average concentrations of approximately 185.00 ng/L [8,9]. Furthermore, the peak concentration of sulfamethoxazole in the Ganges River in India exceeds 390 ng/L, and the average concentration of fluoroquinolone antibiotics in the Mississippi River in the United States is approximately 220 ng/L [10]. Currently, conventional methods for antibiotic removal from water bodies include the activated sludge process, phytoremediation, and advanced oxidation processes. However, both activated sludge and phytoremediation techniques exhibit limited removal efficiency for persistent antibiotics [11], especially when antibiotic concentrations in water are in the µg/L range, and face challenges related to degradation or metabolic kinetics [12]. In contrast, Advanced Oxidation Process (AOPs) can overcome kinetic limitations by generating reactive species to achieve efficient pollutant mineralization, emerging as a core research direction in environmental remediation [13-17].
In recent years, AOPs systems based on photocatalysis (PC), electrochemical (EC) oxidation, and persulfate (PS) activation have demonstrated significant advantages in antibiotic removal [18-22], for example, Jiang et al. achieved 99% degradation of 25 mg/L tetracycline (TC) within 30 min using photocatalytic activation of xylitol-modified bismuth nitrate [23]; while Liu et al. developed CoFe2O4 magnetic nanoparticles to activate peroxymonosulfate (PMS), efficiently removing 99.8% of 5 mg/L moxifloxacin in the same timeframe [24]. However, the core mechanism of antibiotic degradation by such AOPs remains unclear: existing experimental methods face dual challenges in analyzing dynamic processes and capturing microscopic mechanisms. Although traditional characterization tools such as liquid chromatography-mass spectrometry (LC-MS) and vibrational spectroscopy provide certain analytical capabilities [25], they are limited by their inability to capture transient intermediates and rapid reaction steps [26], hindering comprehensive mechanistic analysis. Taking the TC degradation process by Fe-g-C3N4 as an example [27], conventional X-ray absorption spectroscopy (XAS) can reveal the ligand environment and oxidation state of Fe [28], but its averaged signals fail to distinguish the specific contributions of different Fe sites in TC degradation [29], thereby limiting the understanding of active site structure-activity relationships. Furthermore, in studying key reaction steps, traditional static characterization methods can detect active species generation in Co3O4/PMS systems [30-32], but struggle to identify intermediates, potentially leading to misinterpretation of reaction pathways [33,34]. These limitations not only fragment the mechanistic understanding of AOPs but also hinder the rational design of high-performance catalysts, trapping research in a trial-and-error cycle [35]. The gap between mechanism and technology impedes the development of AOPs systems that are both efficient and practical for antibiotic degradation.
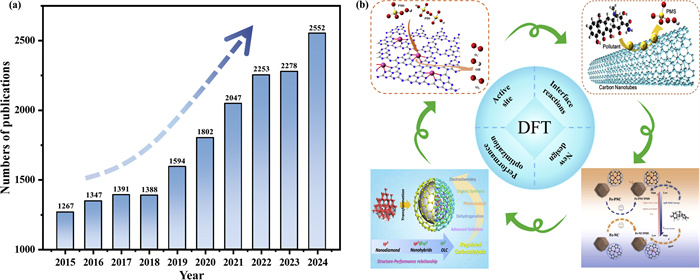
Density functional theory (DFT) calculations can predict microscopic mechanisms (e.g., electronic state distributions, reaction transition states) by simulating material properties such as electronic structure [36-38], bridging the disconnect between mechanistic insights and technological applications. Over the past decade, publications on DFT-mediated AOPs reactions have surged from 1267 in 2014 to 2552 in 2024 (Fig. 1a). As a cornerstone of theoretical simulations, DFT enables deep analysis of AOPs reaction processes, including mechanistic pathways [39,40], kinetics [41] and product formation [42,43] through molecular simulation and computational chemistry, and has played a pivotal role in: (1) Identifying catalyst active sites [44], (2) elucidating reaction mechanisms at catalyst-pollutant interfaces [45], (3) optimizing catalyst performance [46,47], and (4) designing novel catalysts (Fig. 1b) [48-50]. This computational approach establishes a revolutionary paradigm for uncovering AOPs’ microscopic mechanisms and advancing their performance [51-53].
Figure 1

However, the current application of DFT in AOPs research still faces methodological gaps. Most studies focus on validating isolated mechanisms and lack a closed-loop "computational guidance — experimental validation" framework, resulting in mismatches between theoretical predictions and real-world performance. In this review, we propose a novel perspective to explore how DFT computations can transform the understanding of advanced oxidation mechanisms. We establish a multi-scale synergistic research paradigm centered on two core objectives: (1) Elucidating pollutant degradation mechanisms and (2) rational catalyst design. This approach aims to provide theoretical foundations for developing precise and intelligent AOPs technologies, while maximizing the practical value of DFT in this field.
2. Urgent technical bottlenecks in advanced oxidative degradation of antibiotics
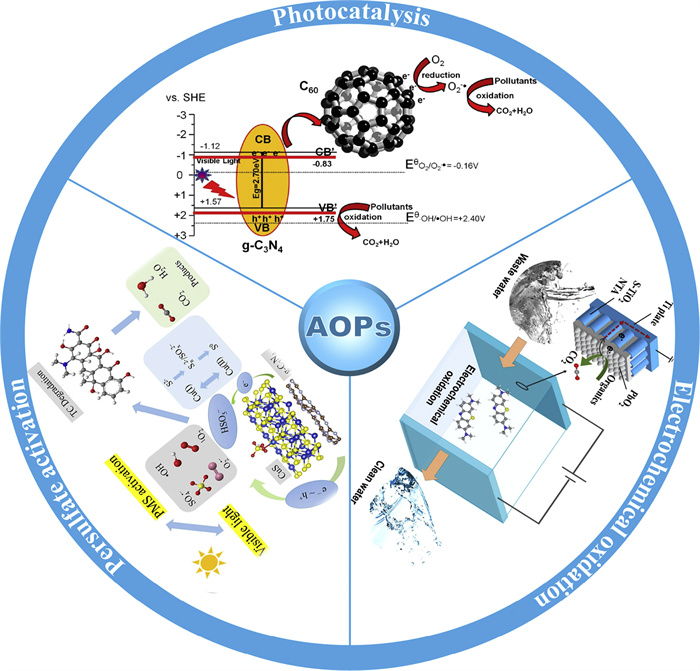
AOPs achieve oxidative degradation of antibiotics by generating reactive species (e.g., SO4•‒, •OH, O2•−, and 1O2), resulting in pollutant mineralization (conversion to CO2, H2O, and inorganic ions) [54]. The core mechanisms involve: (1) Reactive species generation, (2) critical reaction pathways targeting antibiotic structures, and (3) regulation of environmental factors’ impact on reaction efficiency [55]. Currently, AOPs subfields such as photocatalytic AOPs (PC-AOPs) [56], electrochemical oxidation AOPs (EO-AOPs) [57], and persulfate-activated AOPs (PS-AOPs) [58] are widely recognized for their high efficiency, environmental compatibility, and broad applicability (Fig. 2). Table S1 (Supporting information) further compares the characteristics of these three types of AOPs in practical applications, demonstrating their respective advantages and applicable scenarios.
Figure 2

To systematically examine the advancements and challenges, this section analyzes the key bottlenecks of these three representative AOP pathways, and highlights the pivotal role of DFT in elucidating microscopic mechanisms and guiding catalyst design.
2.1 Technical bottlenecks in photocatalytic degradation of pollutants
With the in-depth development and application of catalysts in PC-AOPs, despite significant advancements in extending the photoresponse range and enhancing activity of photocatalytic materials, their large-scale application in PC-AOPs remains constrained by high energy consumption and costs. The fundamental challenge lies in the rapid recombination of photogenerated electron-hole (e--h+) pairs, resulting in low energy conversion efficiency. Although strategies such as morphology control and crystal facet engineering have been widely adopted to improve charge separation [59]. the underlying mechanisms remain poorly understood. For instance, the photocatalytic activity disparity in BiOBr nanosheets is not primarily governed by highly exposed crystal facets but by band bending-induced spatial charge separation between coexisting facets [60]. However, conventional experimental techniques cannot quantitatively resolve surface-specific band structures or oxygen vacancy formation energies, leaving catalyst design without robust theoretical guidance. In contrast, DFT simulations enable precise determination of facet-dependent properties such as band structures, oxygen vacancy energetics, and charge migration pathways. For example, the (100) facet of CeO2 exhibits an exceptionally low energy barrier (−0.59 eV) for PMS adsorption and subsequent SO4•‒ generation [61], providing atomic-level insights for structure optimization and targeted synthesis.
Furthermore, the uncontrolled formation of toxic byproducts in PC-AOPs poses major practical limitations [62]. While studies like Ling et al. [63] demonstrated that carbon-nitrogen materials can achieve 97.6% selectivity in active species generation via cyanide-sodium synergy, experimental methods struggle to correlate e--h+ separation pathways with intermediate evolution, leading to unpredictable accumulation of hazardous byproducts. DFT-based modeling addresses this gap by simulating pollutant-catalyst adsorption configurations, radical attack sites, and reaction energy barriers. This approach successfully predicts degradation pathways for antibiotics like norfloxacin [64], enabling strategic suppression of toxic intermediates. Such advancements are critical for advancing PC-AOPs toward real-world wastewater treatment containing complex refractory organics (e.g., antibiotics, dyes and perfluoroalkyl and polyfluoroalkyl substances) [65].
2.2 Technical bottlenecks in electrochemical degradation of pollutants
EO-AOPs demonstrate remarkable potential in antibiotic wastewater treatment due to their high efficiency and operational simplicity; however, their scalability remains hindered by critical bottlenecks. First, the selectivity and stability of electrode materials require substantial improvement. For instance, low active site density on electrode surfaces and suboptimal interfacial reaction pathways lead to competition between antibiotic degradation and the oxygen evolution reaction (OER) under high overpotentials, severely compromising energy efficiency [66]. To address this, researchers often employ multi-element doping strategies to enhance bifunctional ORR/OER performance [67]. Yet, such approaches obscure the identification of dominant active sites and reaction pathways. Breaking this impasse, Ma et al. [68] leveraged DFT simulations to analyze binary NiFe-CN materials, revealing that Fe sites dominate both ORR and OER, while adjacent Ni sites reduce overpotentials to 0.436 V (ORR) and 0.420 V (OER) by modulating Fe’s electronic structure, which significantly promoted the generation, binding and dissociation of OOH* intermediates.
Atomic-scale mechanisms, such as antibiotic molecule adsorption-dissociation dynamics and intermediate evolution pathways, remain elusive to experimental observation. DFT bridges this gap by precisely calculating molecular adsorption energies to determine the most stable antibiotic-catalyst configurations [69], and identifying high-reactivity sites via Fukui index analysis [70], enabling prediction of degradation pathways and intermediate trajectories [71]. These insights not only establish a theoretical foundation for catalyst design and reaction optimization but also propel EO-AOPs toward scalable applications, achieving near-complete pollutant mineralization while minimizing secondary pollution risks.
2.3 Technical bottlenecks in persulfates degradation of pollutants
To address the limitations of single-mode PS activation in AOPs, including low energy efficiency, catalyst deactivation, and environmental risks, strategies such as physicochemical coupling [72], multi-component composites [73], and energy-field synergy [74] have emerged to synergistically enhance performance through multi-mechanistic pathways. However, two major challenges persist in elucidating reaction pathways and electron-transfer mechanisms: First, experimental limitations in dynamically tracking short-lived reactive species (e.g., SO4•‒, •OH) and intermediates. For instance, the 1O2 in non-radical pathways remains contentious, as traditional characterization methods (e.g., EPR, quenching assays) only permit indirect speculation, leading to divergent mechanistic interpretations; second, Unresolved regulatory mechanisms of reaction pathways by reactive center size in composite systems. For example, the charge polarization effect induced by multi-element doping in straw biochar-supported Co3O4 systems [52] cannot be experimentally quantified.
In this context, DFT emerges as a pivotal tool for mechanistic elucidation, with its advantages manifested in three key aspects: (1) Dynamic pathway resolution: Huang et al. [75] combined DFT with in situ spectroscopy to confirm that surface-generated phenoxyl radicals on CuO are critical for peroxydisulfate (PDS) activation; (2) Electronic structure quantification: B-doping in Fe-N2B4 six-coordinated catalysts induces selective adsorption of terminal O atoms in PMS, lowering the 1O2 generation energy barrier and achieving 98.7% selectivity [76]. (3) Synergistic mechanism decoding: Li et al. [77] demonstrated that Fe-based N-doped carbon catalysts enhance PMS activation for tetracycline (TC) removal via DFT-guided analysis, revealing that interfacial built-in electric fields between Fe single atoms and N-doped carbon shift TC degradation from radical- to non-radical-dominated pathways, boosting removal efficiency from 48% to 100%. These breakthroughs underscore DFT’s irreplaceable role in unraveling microscopic and synergistic mechanisms, providing a theoretical blueprint for precision-engineered PS-AOPs systems.
In summary, although the three mainstream AOP systems differ in terms of reaction pathways, energy input forms, and interface structures, they all face the common bottleneck of uncertainty in the microscopic mechanisms involved in antibiotic degradation. DFT serves not only as a "microscope" for revealing the essence of reactions but also as a ‘connector’ bridging the common issues and technical heterogeneity across the three categories of AOPs. By establishing a closed-loop research framework integrating "theoretical calculations and experimental validation", DFT holds the potential to overcome the current developmental bottlenecks in AOPs, driving a shift from experience-driven technology to precision-designed solutions. This approach provides a unified and robust theoretical foundation for achieving efficient and safe antibiotic removal in complex aquatic environments (as illustrated in Fig. S1 in Supporting information).
3. Theoretical framework and methodological choices for DFT
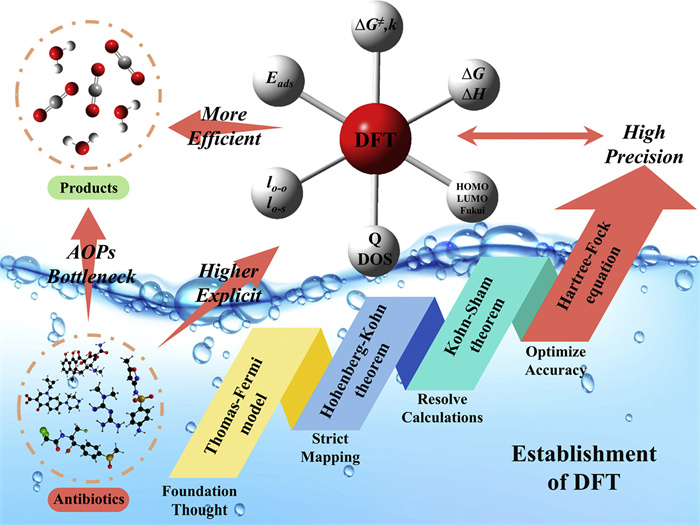
The foundation of DFT stems from the evolution of the Thomas-Fermi model [78]. Its uniqueness lies in mapping all physical properties of a system to electron density distributions via the Hohenberg-Kohn theorem [79], while simplifying many-body problems into single-particle approximations through the Kohn-Sham theorem by introducing a hypothetical non-interacting particle system [80]. This framework not only extends quantum mechanical principles to multiparticle systems (e.g., molecules, clusters, and periodic crystals), but also accurately computes system potentials and total energies through electron density functions. Here, the Kohn-Sham equations directly yield charge density distributions and kinetic energy profiles [81]. To overcome the accuracy limitations of traditional exchange-correlation functionals, the scientific community developed hybrid functionals that linearly combine Hartree-Fock exact exchange energy with DFT exchange-correlation terms. This advancement significantly improves descriptions of strongly correlated electron systems, establishing a robust theoretical foundation for atomic-level design in AOPs (Fig. 3).
Figure 3
 Figure 3. Development and role of DFT’s in AOPs.
Figure 3. Development and role of DFT’s in AOPs.3.1 Selection of generalized functions and basis groups
In practical applications, the selection of exchange-correlation functionals and basis sets must align with the physicochemical properties of the target system. For molecular-scale reactive species (e.g., •OH, O2•−), the hybrid functional B3LYP, which combines Hartree-Fock exact exchange energy with DFT correlation energy [82,83], can accurately captures energy changes in strongly correlated electron systems. The inclusion of dispersion correction (B3LYP-D3) further improves the accuracy of interfacial adsorption energies and solvation effect calculations [84,85]. In recent years, machine learning functionals based on neural networks (such as DM21 proposed by DeepMind) have demonstrated excellent performance in describing the potential energy surface, reaction barriers, and electron density distribution of open-shell free radicals, especially showing stronger generalization capabilities when dealing with multi-reference electronic states [86]. Additionally, models like NeuralXC can be used as correctors for traditional functionals like B3LYP to improve the energy prediction accuracy of functionals in specific reaction pathways or small molecule dynamics simulations [87]. For periodic systems (e.g., WO3 photocatalyst surfaces), the generalized gradient approximation (GGA) functional PBE is widely adopted for band structure calculations due to its computational efficiency and stability [88]. However, strong electron correlation effects in transition metal oxides necessitate the Hubbard U correction (PBE+U) to address electronic localization errors [89]. Building on this foundation, some studies have further attempted to extend machine learning functionals to periodic material systems by training high-precision benchmark datasets (such as QMC or CCSD(T)) to fit energy functionals or Kohn-Sham potential energy functions, thereby improving the accuracy of describing the band structure and defect state energy levels of transition metal oxides [90]. It is worth noting that, with the growing demand for large-scale reaction process modeling, some studies have explored the construction of orbital-free machine learning functionals to efficiently simulate the evolution of electron density and energy changes in complex AOP systems, thereby further reducing computational costs while maintaining high accuracy [91].
Regarding the choice of basis sets, the moderately accurate 6–31G* basis set enhances the description of molecular orbital hybridization through polarization functions and is commonly used for the initial exploration of radical reaction pathways. When higher accuracy is required and computational resources permit, cc-pVDZ or 6–311G* may be used, while considering whether dispersion functions are needed to deal with a specific chemical environment. The advantages and disadvantages of some commonly used basis sets are summarized in this section, as shown in Table 1.
Table 1
 Table 1. Comparison of the application of different base groups.
Table 1. Comparison of the application of different base groups. DownLoad:
CSV
DownLoad:
CSV
Basis set Accuracy level Core advantages Application scenarios in AOPs 6–31G* Moderate Fast and broadly applicable Rapid screening of active species generation pathways; overcoming the experimental bottleneck in capturing transient reactive species; optimizing radical yield on catalyst surfaces. 6–31+G* Moderate (+ long range) Improved description of anions/adsorption Accurately simulation of solvation shell structures of antibiotic anions; correction of adsorption energy deviations; guidance for the design of acidic sites on catalysts to enhance adsorption. 6–311G* Medium-High More precise valence electron distribution Precise calculation of transition state energy barriers for antibiotic molecule cleavage; overcoming predictive bias for C—N/C-S bond dissociation energies in conventional basis sets; enabling targeted design of low-activation-energy active sites. cc-pVDZ Medium-High Optimized electron correlation energy Accurate description of multi-electron cooperative transfer in metal-antibiotic complexes; resolution of prediction inaccuracies of redox potentials in Fe/Cu based catalysts; enhancement of electron transfer efficiency. LANL2DZ Moderate (for heavy elements) Efficient handling of metal centers Balancing efficiency and accuracy in computing d-orbital electron structures of transition metals (e.g., Co, Fe); overcoming bottlenecks in elucidating metal-support synergistic mechanisms in supported catalysts. 3.2 Software tools comparison
In the application of DFT software tools, different platforms are suited to specific scenarios in the study of AOPs due to their functional characteristics. Gaussian, which supports a wide range of exchange-correlation functionals (e.g., B3LYP, M06–2X) and basis sets (e.g., 6–311++G**), excels at molecular-scale reaction mechanism analysis, such as Fenton-like reactions involving SO4•‒ attack on organic pollutants and bond dissociation energy calculations [92,93]. VASP, based on a plane-wave pseudopotential approach, efficiently handles the analysis of optoelectronic properties in periodic systems. Its advanced features, such as hybrid functionals (e.g., HSE06) and van der Waals corrections (e.g., DFT-D3) — make it particularly effective in studying the effects of oxygen doping in TiO2 on conduction band potentials [94], or adsorption activation mechanisms on g-C3N4 surfaces [95]. Materials Studio, a commercial software with an integrated graphical interface, supports both molecular and periodic calculations. Through the integration of its DMol3 and CASTEP modules, it enables streamlined workflows from model construction and structural optimization to reaction pathway visualization. This paper systematically summarizes the core features of these three mainstream DFT platforms in terms of functionality, accuracy, and application domains (Table S2 in Supporting information), providing a useful reference for researchers selecting appropriate computational tools in the AOPs field.
4. Application of DFT in advanced oxidation
The application of DFT in AOPs primarily focuses on three interrelated dimensions: (1) Revealing the principles of catalysts, (2) elucidating pollutant degradation, and (3) combining theoretical simulation with experimental verification to enhance prediction reliability. The following sections will systematically elaborate on these three aspects, outlining the multidimensional role of DFT in promoting advanced oxidation processes.
4.1 Applications in catalyst design
4.1.1 Catalyst active site identification
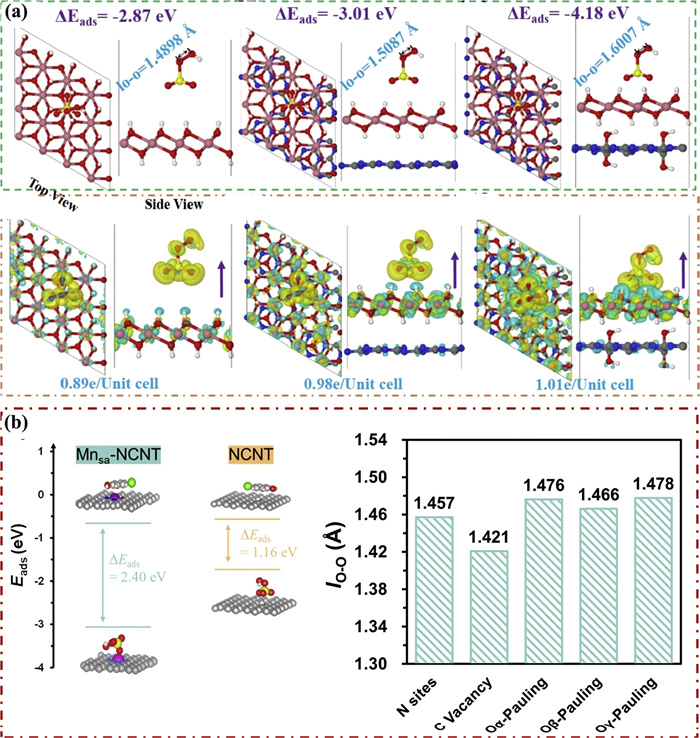
In catalyst active site identification studies, the integration of DFT with multiscale modeling enables atomic-scale resolution of active site evolution dynamics, elucidating interfacial interaction mechanisms governed by chemical bonding and revealing structure-activity relationships and catalytic behaviors of active centers. This synergy establishes a theoretical foundation for designing high-performance catalysts with tailored efficiency and stability. For instance, Mao et al. [96] systematically decoupled the active sites and mechanisms of hydroxylated g-C3N4@Co(OH)2 catalysts using DFT. Their calculations revealed that hydroxylation shifts the hybridization state of C atoms in g-C3N4 from sp2 to sp3, significantly reducing electron binding energy and enhancing charge transfer from the catalyst to PMS (Fig. 4a). This electronic modulation promotes O—O bond cleavage, evidenced by bond elongation from 1.4494 Å to 1.6007 Å. Furthermore, the novel ≡CoOH+ active center, formed via coordination between surface -OH groups and Co2+ ions, exhibits strong PMS adsorption (−4.18 eV adsorption energy) and achieves 50% higher charge transfer efficiency. Consequently, the PMS activation rate reaches 64.7% within 30 min — 3.1-fold higher than the CNH system alone (20.8%) — driving pollutant removal efficiency from 43% to 96%.
Figure 4

While Duan et al. similarly [97] systematically revealed the interfacial interaction mechanism of Mn-N4 single-atom catalysts for the activation of PMS in Fenton-like catalysis by DFT and combined with multiscale modeling. It was found that the atomically dispersed Mn-N4 sites form chemical bonds with PMS through inner-sphere interactions (Mn-O bond length of about 2.08 Å), while the nitrogen-doped carbon substrate (NCNT) adsorbs PMS through van der Waals force outer-sphere interactions. Further DFT calculations revealed that the electronic structural properties of the Mn-N4 sites were revealed to enable the adsorption energy of Mn-N4 sites to PMS up to about −2.9 eV. This strong inner-sphere interaction significantly reduces the O—O bond breaking energy (0.291 eV vs. 1.393 eV for NCNT), which leads to an increase in the slope of the linear relationship of the activation rate of PMS from 0.0541 to 0.1129 (109% increase) for the NCNT/PMS system (Fig. 4b). This optimization of the interfacial interaction mechanism led to a significant increase in PMS utilization, with the Mn-NCNT catalyst achieving 3.57 mmol/g of 2.0 mmol/L PMS digestion, which was 58% higher than that of the NCNT reference material (2.26 mmol/g). The theoretically constructed interfacial model is in high agreement with the experimental results, confirming the key role of the inner-sphere-dominated activation mechanism in enhancing the oxidation performance.
4.1.2 Catalyst surface reaction mechanisms
Oxidant adsorption on catalysts serves as the critical initiation step for both radical and non-radical pathways, where rapid adsorption kinetics dictate subsequent chemical interactions [98]. Quantitative DFT analysis of adsorption energy (Eads) between oxidants and catalyst surfaces provides atomic-level insights into surface reaction mechanisms [53,99,100]. The adsorption energy is defined as:
(1) where EC, EOX, and EOX+C represent the total energies of the catalyst, free oxidizer molecules, and catalyst oxidizer energy, respectively. Negative Eads values indicate thermodynamically spontaneous processes. A representative case by Song et al. [101] demonstrated a dual-path electron transfer strategy to overcome mass transfer and activity limitations in PMS-based Fenton-like systems. By confining reactions to catalyst surfaces through synergistic adsorption-oxidation, they created localized high-concentration reactive zones, boosting apparent reaction rates by 54-fold. DFT calculations revealed that N/P co-doping optimized electron density distribution in Mn d orbitals, establishing efficient electron transport channels that simultaneously enhanced PMS activation efficiency and accelerated Mn3+/Mn4+ valence cycling. This overcame kinetic limitations of traditional Mn-based catalysts while alleviating mass transfer resistance through surface confinement effects.
DFT further deciphers oxidant activation mechanisms by resolving catalyst-oxidant interfacial dynamics. For example, Le et al. [22] revealed the activation mechanism of multiphase MoS2 (1T/2H phase)/PMS system by DFT calculations: The elevated 1T phase ratio (2.0% → 49.3%) increased the PMS adsorption energy from −0.93 eV to −1.40 eV, and the O—O bond length was stretched from 1.45 Å to 1.64 Å, which promoted the generation of SO4•‒ and •OH. In addition, the elevated free energy of desorption (−0.52 eV → −0.65 eV) of active species such as SO4•‒ and •OH generated on the MoS2 surface made them more easily bound to the material surface, enhancing the surface-dominated inhomogeneous oxidation pathway.
4.1.3 Catalyst performance optimization
DFT provides a robust theoretical framework for catalyst performance optimization through systematic analysis of electronic band structures, density of states (DOS), and optoelectronic properties. For instance, Halder et al. [102] employed the DFT+U method to investigate the electronic and optical characteristics of bicalcite Ln2AlMnO6 (Ln = La, Pr, Nd), revealing direct/indirect bandgap transitions, optical anisotropy, and enhanced visible-light absorption capabilities. Calculations showed narrow bandgaps (1.28–1.77 eV) ideal for visible-light harvesting, with indirect bandgap features in NAM effectively suppressing electron-hole (e--h+) recombination and boosting photocatalytic activity. Strong agreement between theoretical predictions and experimental validation confirmed these materials’ exceptional solar energy conversion efficiency, positioning them as promising candidates for photocatalysis and photovoltaic applications.
In a complementary study, Xie et al. [103] leveraged DFT to demonstrate that Eu doping introduces impurity states near the valence band maximum of BiOCl, reducing bandgap energy and promoting photogenerated charge separation. This electronic engineering enhanced visible-light absorption by 38% compared to pristine BiOCl. Guided by these insights, Eu-doped BiOCl catalysts were synthesized and exhibited superior photocatalytic performance under visible light, validating DFT’s predictive power in designing efficient visible-light-driven catalysts.
Recent studies demonstrate that enhancing H2O2 production via metal single-atom photocatalysts (M-SACs) can be achieved by tailoring electronic configurations and excitation dynamics [17,104-107]. A representative example by Teng et al. [108] integrated DFT with experimental validation to show that atomically dispersed indium (In) sites on polymer carbon nitride (PCN) optimize oxygen adsorption geometry and steer the oxygen reduction reaction (ORR) toward a 2-electron pathway. This electronic modulation tripled the H2O2 yield from 2.5 mg/L to 7.5 mg/L within 30 min compared to pristine PCN. The amplified H2O2 generation critically enhances pollutant degradation by serving as both a potent oxidant and a precursor for secondary reactive species (e.g., •OH). Such studies emphasize the critical role of DFT in deciphering electronic structures, optimizing doping strategies, and supporting rational material design. Moreover, DFT offers multi-scale theoretical insights that enable precise regulation of key catalytic properties such as light absorption, charge carrier dynamics, and reaction selectivity.
4.2 Applications in pollutant degradation
4.2.1 Electronic structure analysis
DFT can accurately identify the reactive sites of antibiotic molecules by analyzing electronic structure information such as molecular orbitals (HOMO/LUMO) and Fukui indices [109]. The HOMO (highest occupied molecular orbital) is electron-rich and thus prone to electrophilic attack by reactive species such as SO4•‒, •OH, and 1O2. In contrast, the LUMO (lowest unoccupied molecular orbital) is electron-deficient and more susceptible to nucleophilic attack. Furthermore, quantitative analysis using Fukui functions provides a clearer understanding of the reactivity tendencies at different molecular sites.
(2) where qNA is the number of electrons of atom A in the molecule, and N is the total number of electrons. Regions with high f+ values tend to react with nucleophilic reagents, while regions with high f- values are more susceptible to attack by electrophilic reagents. High f0 value regions, on the other hand, are more active against reactive species. For example, Yang et al. [110] calculated the f0 value of the Fukui index using DFT to characterize the active sites for radical attack and showed that C27, C26, C30, C22, O5, and O3 are susceptible to •OH and SO4•‒ attack. They further derived the π* orbitals by analyzing the energies of the LUMO (0.079 eV) and HOMO (0.209 eV); the anti-bonding orbitals and π-bonding orbitals are located on the benzene ring D and the -N(CH6)7 group, respectively, thereby further verifying the validity of the f 0 calculations. Similarly, Su et al. [111] first analyzed the susceptible sites of ofloxacin (OFL) using its HOMO and LUMO and further verified them with electrostatic potential (ESP) diagrams, which concluded that the tetrahydropyridine ring, the benzene ring binding site, and the carboxylic acid group may be more susceptible to attack by negatively charged nucleophilic radicals such as O2•−. Based on the Hirshfeld charge, the f- and f+ values of OFL were calculated, further supporting the above conclusions. In addition, Qi et al. [112] similarly utilized the Fukui function to identify the most vulnerable active sites on OFL and CIP molecules. The computational results showed that C8, O14, C13, N20, C16, and C5 of OFL, and O23, N15, C12, C4, C22, and C11 of CIP were the most vulnerable active sites (Fig. S2 in Supporting information).
Altogether, the analysis of Fukui indices and molecular orbital distributions provides a robust theoretical foundation for qualitatively identifying the reactive sites of antibiotic molecules, and offering targeted input for subsequent simulations of reaction energy barriers and kinetics.
4.2.2 Prediction of adsorption behavior of pollutants
DFT has also been employed to quantify the adsorption energy and conformational differences of pollutants on catalyst surfaces, providing a foundation for optimizing material surface design. For example, Aline et al. [115] used DFT to predict the adsorption behavior of antibiotic molecules — such as levofloxacin (LV), tetracycline (TC), and benzylpenicillin (BP) — on the surface of silver nanoparticles (AgNPs) by constructing theoretical models of the antibiotic molecules and analyzing their molecular vibrational properties. The results showed that LV molecules were adsorbed through interactions between their carboxylic acid groups and the silver surface; TC interacted with silver atoms via the carbonyl group on the intermediate ring; and BP was partially adsorbed through both the carboxylate and carbonyl groups on the acyclic amide. By analyzing and predicting the adsorption behavior of antibiotics, researchers can tailor catalyst surface designs to improve antibiotic degradation performance. Notably, the adsorption configurations predicted by DFT were validated by surface-enhanced Raman spectroscopy (SERS) experiments, and the observed spectral pattern changes demonstrated a high level of agreement between the theoretical models and experimental results.
4.2.3 Analysis of the generation mechanism of active species
In AOPs, the efficiency and selectivity of reactive species generation critically influence pollutant degradation. Due to their short lifetimes and low concentrations, traditional experiments struggle to capture their formation mechanisms. DFT, especially when combined with TDDFT, offers powerful atomic-level insights into these processes. For instance, in PS-based systems, DFT can calculate the O–O bond dissociation energy under various adsorption configurations, compare the activation performance of different catalytic sites (e.g., metals, non-metal dopants, vacancies), and elucidate the underlying factors governing reactivity and selectivity.
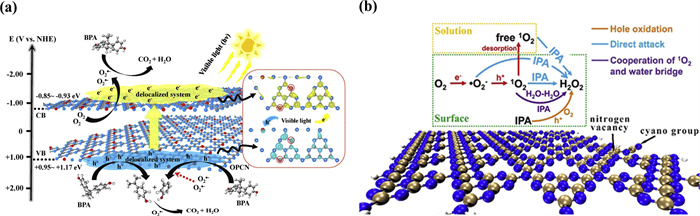
For example, Zhang et al. [113] experimentally found that double-oxygen-doped g-C3N4 selectively generates O2•− (Fig. 5a). To elucidate the theoretical basis of this phenomenon, the researchers performed a systematic analysis using DFT calculations. First, formation energy calculations confirmed that the most stable doping mode involves para-substitution of oxygen atoms in the melam unit, which narrows the band gap to 1.22 eV and significantly enhances visible light absorption. Charge distribution analysis further revealed that oxygen doping effectively induces an out-of-domain conjugated system of e--h+ pairs, thereby promoting charge separation. Additionally, the conduction band potential derived from DFT results confirmed that dioxygen-doped g-C3N4 can efficiently generate O2•− from reduced O2, while the valence band potential is insufficient to produce •OH, consistent with the experimental observations. Furthermore, Luo et al. also applied DFT to investigate the multipath mechanism of H2O2 generation by photocatalytic g-C3N4 [114]. Through model screening, the researchers concluded that nitrogen vacancies enhance O2 adsorption and serve as oxidation/reduction centers. TDDFT analysis showed that NV sites and their neighboring regions dominate the generation of h+ and radical reaction intermediates (RFR), respectively. Five feasible pathways were identified using DFT, among which the water-molecule-bridge-mediated 1O2 pathway (with an energy barrier of 0.66 eV) was found to be optimal (Fig. 5b), outperforming the conventional h⁺-dominated pathway (energy barrier of 0.82 eV). The central role of 1O2 was further validated experimentally, with EPR and RRDE results indicating that O2•− acts as an intermediate, while 1O2 contributes to 64% of H2O2 generation, revealing the multifacetedroles of active species in photocatalysis.
Figure 5
 Figure 5. (a) Intrinsic mechanism of selective O2•− generation by doped g-C3N4 with double oxygen. Reproduced with permission [113]. Copyright 2019, Elsevier. (b) Multi-path mechanism of photocatalytic g-C3N4 generation of H2O2. Reproduced with permission [114]. Copyright 2021, American Chemical Society.
Figure 5. (a) Intrinsic mechanism of selective O2•− generation by doped g-C3N4 with double oxygen. Reproduced with permission [113]. Copyright 2019, Elsevier. (b) Multi-path mechanism of photocatalytic g-C3N4 generation of H2O2. Reproduced with permission [114]. Copyright 2021, American Chemical Society.These findings collectively demonstrate that DFT and TDDFT are powerful tools to elucidate the formation mechanisms of various reactive species by correlating electronic structure with catalytic performance. This approach not only rationalizes experimental observations but also enables targeted optimization of catalysts for selective activation of desired species in AOPs.
4.2.4 Reaction energy barriers and thermodynamic analysis
In PS-AOPs, the low generation efficiency and selectivity of 1O2 are key factors limiting the effective degradation of antibiotics. To improve the selectivity of PMS activation and promote non-radical pathways, Shen et al. [116] explored the regulatory mechanisms of non-metal doping on the reaction energy barriers and thermodynamic processes of PMS activation using DFT. The authors employed VASP software to calculate the energy evolution of HSO5- adsorption configurations and activation pathways in different doped systems (B/S/P-Fe1/GLCNs). They found that B doping significantly reduced the activation energy barrier for PMS decomposition (ΔE = 0.78 eV), whereas the energy barriers for S/P-doped systems increased to 1.24 eV and 1.32 eV, respectively. Thermodynamic analysis showed that the B-FeN3B sites in Fe1/GLCNs exhibited a much lower HSO5- adsorption energy (−2.08 eV) compared to the undoped system (−1.45 eV), and the Gibbs free energy of the reaction (ΔG) was reduced to −1.92 eV, confirming its thermodynamic spontaneity and high efficiency. A transition state (TS) search revealed that B-doping induces PMS adsorption in a Type Ⅱ configuration, resulting in a 36% reduction in the O—O bond cleavage energy barrier compared to the Type Ⅰ configuration, along with a decrease in electron transfer potential along the reaction pathway, thereby accelerating the kinetics of 1O2 generation and increased its yield by 62%. These calculations aligned well with XPS and EPR experimental results, revealing the atomic-scale mechanism by which non-metal doping modulates the electronic structure to reduce the reaction energy barrier and optimize the thermodynamic driving force, thus facilitating the preferential formation of non-radical pathways.
DFT can also elucidate the atomic-scale mechanisms underlying the selective modulation of activation pathways by different metals through analysis of the reaction energy barriers and thermodynamic processes in PMS activation catalyzed by single-atom catalysts (MeN@C). Hu et al. [53] investigated PMS adsorption configurations and energy evolution of activation pathways in metal-doped systems including Mn, Fe, Co, Ni, and Cu. They found that high-valent metal-oxygen species (e.g., FeN=O, MnN=O) have significantly lower formation energy barriers compared to other reactive species. Specifically, the energy barriers for forming high-valent species in MnN@C and FeN@C were only 0.25 eV and 0.48 eV, respectively, while those for CoN@C, NiN@C, and CuN@C ranged from 1.66 eV to 2.79 eV, suggesting that Mn and Fe systems are thermodynamically more favorable for forming reactive intermediates through heterolytic pathways. Furthermore, analysis of ΔG revealed that the electron transfer-mediated surface-activated PMS* pathway required no energy input, and the system’s work function significantly increased after PMS adsorption, for instance, NiN@C increased from 4.12 eV to 5.33 eV, further confirming its thermodynamic spontaneity advantage.
4.3 Mechanism deepening and model optimization in theory-experiment synergy
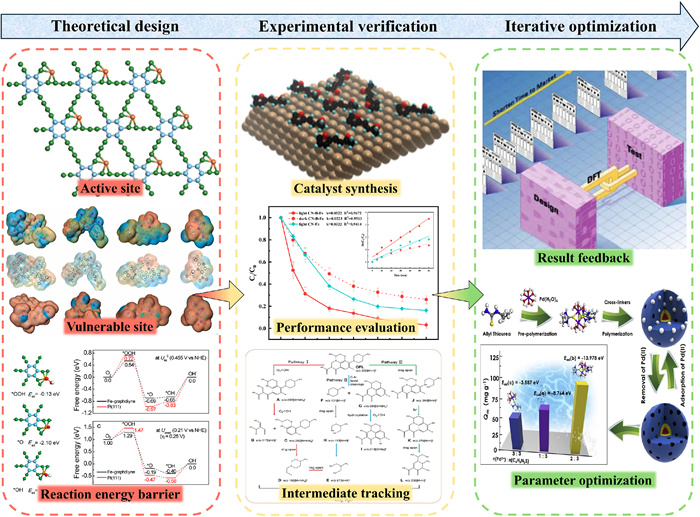
Although DFT plays a crucial role in rational catalyst design and the analysis of pollutant degradation mechanisms, its true value can only be fully realized through deep integration with experimental observations [117], forming a dynamic closed-loop system of "theoretical design — experimental verification — iterative optimization" (Fig. 6) [111,118-120]. In real-world systems, theoretical models often exhibit prediction errors due to environmental complexity (e.g., pH/ion strength fluctuations), transient intermediates, and material synthesis deviations, necessitating continuous calibration using experimental data [121]. This synergistic mechanism not only validates the reliability of computational models but also deepens mechanistic understanding and enhances prediction accuracy through iterative optimization, ultimately enabling precise control of catalytic systems.
Figure 6
 Figure 6. “Computational guidance — experimental validation” research flowchart. Reproduced with permission [111]. Copyright 2022, Elsevier. Reproduced with permission [117]. Copyright 2018, American Chemical Society. Reproduced with permission [118]. Copyright 2019, Elsevier. Reproduced with permission [119]. Copyright 2024, Elsevier. Reproduced with permission [120]. Copyright 2025, American Chemical Society.
Figure 6. “Computational guidance — experimental validation” research flowchart. Reproduced with permission [111]. Copyright 2022, Elsevier. Reproduced with permission [117]. Copyright 2018, American Chemical Society. Reproduced with permission [118]. Copyright 2019, Elsevier. Reproduced with permission [119]. Copyright 2024, Elsevier. Reproduced with permission [120]. Copyright 2025, American Chemical Society.4.3.1 Novel catalyst design
Selectively generating active species and balancing activity with stability remain fundamental challenges in developing advanced catalytic materials. Overcoming these limitations requires a deep understanding of the atomic-scale relationship between catalytic site structure and function, along with the design of tunable synergistic mechanisms to simultaneously enhance both activity and stability.
DFT enables rational active site design through multiscale modeling, offering a transformative approach. For instance, guided by DFT, Zhang et al. [122] constructed a core-shell catalyst featuring a highly graphitized shell layer and short C-Fe bonds bridging two reaction centers. This design achieved synergistic optimization of electron transfer and reaction kinetics. Compared to unoptimized samples, the catalyst significantly enhanced norfloxacin degradation performance: degradation efficiency increased from 68.9% to 93.8%, and the reaction rate constant nearly doubled (0.0275 vs. 0.0109 min-1). Additionally, MORANKAR et al. [123] used DFT to systematically compare Fe-N-C catalyst structures — including zigzag edges, bulk, and aggregated FeN4 sites. DFT guidance led them to propose an atomic-level modulation strategy: optimizing active site exposure by adjusting catalyst morphology (e.g., extending edge length), and balancing redox kinetics by regulating the operating potential window. This "computational guidance-experimental validation" approach establishes an atomic-resolution framework for designing selective and stable catalysts, highlighting DFT’s indispensable role in next-generation catalyst innovation.
4.3.2 Solvent effects and kinetic simulations
The multi-scale integration of DFT and kinetics not only quantifies the influence of solvent effects on reaction pathways, but also dynamically calibrates theoretical models with experimental data, achieving mechanism closure from electronic structure to macroscopic kinetics. This provides accurate theoretical support for the design of highly efficient oxidants. For example, Wang et al. [124] simulated the Fe(Ⅱ)-peroxyacid (POA) reaction mechanism in aqueous solution by incorporating solvent effects and kinetic simulations via DFT. The authors calculated ligand coordination modes, O—O bond dissociation energy barriers, and kinetic parameters for the hydrogen atom transfer steps between Fe(Ⅱ) and various POAs (e.g., PFA, PAA, and PPA), as well as H2O2, with a focus on the solvent effect in aqueous media. The DFT-calculated O—O bond dissociation energies for POAs (40–40.5 kcal/mol) were significantly lower than that of H2O2 (46 kcal/mol), resulting in a much lower reaction energy barrier (8–10 kcal/mol vs. 13.6 kcal/mol), thereby explaining the higher reactivity of POAs. Further stepwise thermodynamic analysis indicated that O—O bond cleavage is the rate-determining step. Moreover, the bidentate coordination pathway — featuring synergistic binding of the carbonyl oxygen and peroxy oxygen — was identified as the key mechanism underlying the high selectivity of POAs due to its lower energy barrier. Combined with experimental evidence that Fe(Ⅳ) is the dominant reactive species, these findings elucidate the molecular basis for the minimal contribution of reactive species in the POA system.
5. Results and outlook
This review systematically demonstrates the central role of DFT in overcoming the technical bottlenecks of advanced oxidation degradation technology for antibiotics. DFT overcomes the limitations of traditional experimental methods by precisely analyzing the structure-property relationships of catalysts, the mechanisms of active species formation, and the degradation pathways of pollutants at the atomic scale. In AOPs such as PC, EO, and PS, DFT can precisely predict active site configurations, Eads, and interfacial electron transfer pathways. Further integration with the Fukui index and molecular orbital analysis enables qualitative identification of reactive sites on antibiotic molecules, predicting degradation pathways and the evolution of intermediates. Most importantly, DFT and experimental validation are deeply synergistic, establishing a closed-loop research paradigm of "computational guidance — experimental validation", which can effectively guide catalyst design and reaction system optimization, significantly enhancing the efficiency of AOPs in degrading antibiotics.
Despite the remarkable progress DFT has made in AOPs, several challenges hinder its broader application. Most existing studies still focus on single-radical degradation pathways, whereas actual reaction processes often involve non-radical mechanisms and synergistic interactions among multiple reactive species, whose microscopic mechanisms remain insufficiently understood. Additionally, simulating dynamic electron transfer in heterogeneous catalytic systems remains difficult, as traditional DFT methods based on adiabatic approximations struggle to capture electron behavior in excited or transition states, and the treatment of non-adiabatic effects and solvent interactions remains underdeveloped. Furthermore, current DFT calculations often rely on idealized models, limiting their ability to accurately reflect the complex and dynamic nature of aqueous environments, such as fluctuations in pH, temperature, ionic strength, and competitive adsorption — thereby reducing their effectiveness in guiding experimental research. To address these limitations, future research should focus on: (1) Deepening multi-scale mechanism analysis by coupling non-adiabatic molecular dynamics with TDDFT to dynamically simulate electron excitation, migration, and reactive species generation; (2) enabling data-driven intelligent catalyst design through the integration of machine learning and high-throughput computing to build a "catalyst structure–electronic properties–degradation performance" database; and (3) expanding the dimensions of sustainability assessment by extending DFT applications to predict the toxicity of degradation products, and incorporating life cycle assessment to evaluate the environmental risks and carbon footprint of AOP technologies — ultimately promoting their transformation toward green and practical engineering applications.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Zhihao Li: Writing – original draft. Zhiruo Zhou: Writing – review & editing, Writing – original draft, Supervision, Funding acquisition. Yuan Qin: Writing – review & editing. Dan Huang: Writing – review & editing, Funding acquisition. Meizhen Wang: Supervision, Funding acquisition.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 22306163, 42477032, 82404680), Natural Science Foundation of Zhejiang Province (No. LQ23B070001), Basic Research Funds for Zhejiang Gongshang University (No. FR24002Z).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:
10.1016/j.cclet.2025.112318 .
-
-
[1]
L.H. Santos, A.N. Araujo, A. Fachini, et al., J. Hazard. Mater. 175 (2010) 45–95. doi: 10.1016/j.jhazmat.2009.10.100
-
[2]
M. Naddaf, The world faces a water crisis — 4 powerful charts show how, Nature Publishing Group, 2023.
-
[3]
N. Editorial, Nature 613 (2023) 611–612. doi: 10.1038/d41586-023-00182-2
-
[4]
S. Dickin, M. Bayoumi, R. Giné, et al., NPJ. Clean. Water 3 (2020) 24. doi: 10.1038/s41545-020-0072-8
-
[5]
S. Li, Y. Liu, Y. Wu, et al., Natl. Sci. Open 1 (2022) 20220029. doi: 10.1360/nso/20220029
-
[6]
B. Wang, Z. Xu, B. Dong, J. Hazard. Mater. 469 (2024) 133925. doi: 10.1016/j.jhazmat.2024.133925
-
[7]
D.L. Fan, Y. Zhang, S. Sun, et al., Emerging Contam 11 (2025) 100437. doi: 10.1016/j.emcon.2024.100437
-
[8]
L.X. He, L.Y. He, F.Z. Gao, et al., J. Hazard. Mater. 452 (2023) 131208. doi: 10.1016/j.jhazmat.2023.131208
-
[9]
Z. Maghsodian, A.M. Sanati, T. Mashifana, et al., Antibiotics 11 (2022) 1461. doi: 10.3390/antibiotics11111461
-
[10]
L. Yao, Y. Wang, L. Tong, et al., Ecotoxicol. Environ. Saf. 135 (2017) 236–242. doi: 10.1016/j.ecoenv.2016.10.006
-
[11]
P. Sahay, D. Mohite, S. Arya, et al., Emergent. Mater. 6 (2023) 373–404. doi: 10.1007/s42247-023-00460-9
-
[12]
F. Mahmud, T.S. Banhi, H. Roy, et al., Groundwater Sust. Dev. 25 (2024) 101181. doi: 10.1016/j.gsd.2024.101181
-
[13]
A. Joss, S. Zabczynski, A. Gobel, et al., Water Res. 40 (2006) 1686–1696. doi: 10.1016/j.watres.2006.02.014
-
[14]
P.K. Mutiyar, A.K. Mittal, Desalin. Water Treat. 51 (2013) 6158–6164. doi: 10.1080/19443994.2013.770199
-
[15]
S.J. Zhang, H.C. Zheng, P.G. Tratnyek, Nat. Water 1 (2023) 666–681. doi: 10.1038/s44221-023-00098-1
-
[16]
Z. Zhou, X. Liu, K. Sun, et al., Chem. Eng. J. 372 (2019) 836–851. doi: 10.1016/j.cej.2019.04.213
-
[17]
W. Ren, C. Cheng, P. Shao, et al., Environ. Sci. Technol. 56 (2022) 78–97. doi: 10.1021/acs.est.1c05374
-
[18]
B. Liu, Y.M. Li, Y.S. Wu, et al., Chem. Eng. J. 417 (2021) 127972. doi: 10.1016/j.cej.2020.127972
-
[19]
H. Li, Y. Liu, F. Jiang, et al., Sci. Total Environ. 806 (2022) 150968. doi: 10.1016/j.scitotenv.2021.150968
-
[20]
Y.M. Li, B. Liu, S.T. Xing, Desalin. Water Treat. 206 (2020) 349–357. doi: 10.5004/dwt.2020.26354
-
[21]
Y. Tang, J. Kang, M. Wang, et al., J. Environ. Chem. Eng. 9 (2021) 105864. doi: 10.1016/j.jece.2021.105864
-
[22]
L. Wang, X.Y. Zheng, L.G. Yan, et al., Sep. Purif. Technol. 317 (2023) 123907. doi: 10.1016/j.seppur.2023.123907
-
[23]
R. Jiang, G. Lu, L. Zhang, et al., J. Hazard. Mater. 463 (2024) 132887. doi: 10.1016/j.jhazmat.2023.132887
-
[24]
L. Liu, H. Mi, M. Zhang, et al., Chem. Eng. J. 407 (2021) 127201. doi: 10.1016/j.cej.2020.127201
-
[25]
Z. Zhao, S. Ma, B. Gao, et al., Sep. Purif. Technol. 314 (2023) 123510. doi: 10.1016/j.seppur.2023.123510
-
[26]
Y. Li, L. Zhu, X. Wu, et al., Angew. Chem. Int. Ed. 63 (2024) e202318169. doi: 10.1002/anie.202318169
-
[27]
X. Peng, J. Wu, Z. Zhao, et al., Chem. Eng. J. 427 (2022) 130803. doi: 10.1016/j.cej.2021.130803
-
[28]
B.E. Snyder, P. Vanelderen, M.L. Bols, et al., Nature 536 (2016) 317–321. doi: 10.1038/nature19059
-
[29]
Y. Li, A.G. Walsh, P. Zhang, et al., CCS Chem. 6 (2024) 1110–1129. doi: 10.31635/ccschem.024.202303690
-
[30]
Q. Wang, Z. Xu, Y. Cao, et al., Chem. Eng. J. 427 (2022) 131953. doi: 10.1016/j.cej.2021.131953
-
[31]
L. Liu, Y. Li, W. Li, et al., Environ. Res. 187 (2020) 109665. doi: 10.1016/j.envres.2020.109665
-
[32]
Q. Yi, X. Li, Y. Li, et al., ACS ES&T Eng 2 (2022) 1836–1846. doi: 10.1021/acsestengg.2c00087
-
[33]
W. Liu, Y.R. Lu, Y.B. Dong, et al., Chem. Eng. J. 466 (2023) 143161. doi: 10.1016/j.cej.2023.143161
-
[34]
X. Cheng, Y. Zhang, Q. Fan, et al., Chem. Eng. J. 454 (2023) 140450. doi: 10.1016/j.cej.2022.140450
-
[35]
J. Xie, S.H. Wu, C.Y. Luo, et al., Appl. Catal. B: Environ. 354 (2024) 124138. doi: 10.1016/j.apcatb.2024.124138
-
[36]
J. Chen, Y. Ren, Y. Fu, et al., ACS Nano 18 (2024) 13035–13048. doi: 10.1021/acsnano.4c01637
-
[37]
Y. Ren, Y. Fu, N. Li, et al., Nat. Commun. 15 (2024) 4675. doi: 10.1038/s41467-024-49003-8
-
[38]
Z. Tang, H. Li, P. Lin, et al., Nat. Commun. 15 (2024) 8815. doi: 10.1038/s41467-024-53028-4
-
[39]
Z. Xiao, B. Yang, X. Feng, et al., Environ. Sci. Technol. 57 (2023) 3951–3961. doi: 10.1021/acs.est.2c09034
-
[40]
H.X. Li, X.R. Miao, J. Zhang, et al., Chem. Eng. J. 381 (2020) 122680. doi: 10.1016/j.cej.2019.122680
-
[41]
R.L. Yin, W.Q. Guo, H.Z. Wang, et al., Chem. Eng. J. 335 (2018) 145–153. doi: 10.1016/j.cej.2017.10.063
-
[42]
B. Jing, J. Li, C. Nie, et al., Crit. Rev. Environ. Sci. Technol. 53 (2023) 483–503. doi: 10.1080/10643389.2022.2070404
-
[43]
E.V. Rokhina, R.P. Suri, Sci. Total Environ. 417-418 (2012) 280–290. doi: 10.1016/j.scitotenv.2011.12.008
-
[44]
H. Tian, K. Cui, X. Chen, et al., J. Hazard. Mater. 461 (2024) 132647. doi: 10.1016/j.jhazmat.2023.132647
-
[45]
J. Zhang, S.Y. Li, H.T. Xiang, et al., J. Environ. Chem. Eng. 12 (2024) 112495. doi: 10.1016/j.jece.2024.112495
-
[46]
X. Duan, W. Tian, H. Zhang, et al., ACS Catal. 9 (2019) 7494–7519. doi: 10.1021/acscatal.9b01565
-
[47]
H. Shi, S. Gao, X. Liu, et al., Small 20 (2024) 2309557. doi: 10.1002/smll.202309557
-
[48]
Y. Chai, H. Dai, X. Duan, et al., Appl. Catal. B: Environ. 341 (2024) 123289. doi: 10.1016/j.apcatb.2023.123289
-
[49]
B. Li, C. Feng, T. Wang, et al., Sep. Purif. Technol. 354 (2025) 128955. doi: 10.1016/j.seppur.2024.128955
-
[50]
M. Li, C. Guo, X. Yuan, et al., Appl. Catal. B 370 (2025) 125175. doi: 10.1016/j.apcatb.2025.125175
-
[51]
L. He, X. Sun, F. Zhu, et al., Sci. Total Environ. 592 (2017) 33–40. doi: 10.1016/j.scitotenv.2017.03.041
-
[52]
P.P. Zhang, Y.Y. Yang, X.G. Duan, et al., ACS Catal. 11 (2021) 11129–11159. doi: 10.1021/acscatal.1c03099
-
[53]
J. Hu, Y. Li, Y. Zou, et al., Chem. Eng. J. 437 (2022) 135428. doi: 10.1016/j.cej.2022.135428
-
[54]
Y. Deng, R.Z. Zhao, Curr. Pollut. Rep. 1 (2015) 167–176. doi: 10.1007/s40726-015-0015-z
-
[55]
M. Salimi, A. Esrafili, M. Gholami, et al., Environ. Monit. Assess. 189 (2017) 414. doi: 10.1007/s10661-017-6097-x
-
[56]
X. Bai, L. Wang, Y. Wang, et al., Appl. Catal. B 152-153 (2014) 262–270. doi: 10.1016/j.apcatb.2014.01.046
-
[57]
C. Yang, S. Shang, X.Y. Li, Sep. Purif. Technol. 258 (2021) 118035. doi: 10.1016/j.seppur.2020.118035
-
[58]
G. Cao, Z. Shen, J. Cui, et al., Chem. Eng. J. 483 (2024) 149082. doi: 10.1016/j.cej.2024.149082
-
[59]
J. Qu, Y. Wang, X. Mu, et al., Adv. Mater. 34 (2022) e2203320. doi: 10.1002/adma.202203320
-
[60]
M. Shi, G. Li, J. Li, et al., Angew. Chem. Int. Ed. 59 (2020) 6590–6595. doi: 10.1002/anie.201916510
-
[61]
V.A. Le, H.T. Nguyen, T.D.H. Vo, et al., J. Environ. Chem. Eng. 13 (2025) 115699. doi: 10.1016/j.jece.2025.115699
-
[62]
M. Malayeri, C.S. Lee, J. Niu, et al., J. Hazard. Mater. 419 (2021) 126411. doi: 10.1016/j.jhazmat.2021.126411
-
[63]
M. Xu, R. Wang, H. Fu, et al., Proc. Natl. Acad. Sci. U. S. A. 121 (2024) e2318787121. doi: 10.1073/pnas.2318787121
-
[64]
T. Wang, A. Kumar, G. Sharma, et al., NPJ Clean. Water 7 (2024) 97. doi: 10.1038/s41545-024-00393-8
-
[65]
H.L. Ran, Q.L. Xu, Y. Yang, et al., ACS Catal. 14 (2024) 11675–11704. doi: 10.1021/acscatal.4c02738
-
[66]
Y.L. Xu, X.Y. Zhang, Y.Y. Zhao, et al., Int. J. Hydrogen Energy 48 (2023) 33746–33762. doi: 10.1016/j.ijhydene.2023.05.167
-
[67]
Z. Zhang, Y. Li, Z. Zhang, et al., Chem. Sci. 13 (2022) 8876–8884. doi: 10.1039/d2sc02845j
-
[68]
M. Ma, A. Kumar, D.N. Wang, et al., Appl. Catal. B: Environ. 274 (2020) 119091. doi: 10.1016/j.apcatb.2020.119091
-
[69]
J. Li, S. Ma, Z. Qi, et al., Appl. Catal. B 322 (2023) 122076. doi: 10.1016/j.apcatb.2022.122076
-
[70]
H. Hai, X. Xing, S. Li, et al., Sci. Total Environ. 738 (2020) 139909. doi: 10.1016/j.scitotenv.2020.139909
-
[71]
H.L. Zhao, Z.X. Zheng, S.K. Zhu, et al., Chem. Eng. J. 477 (2023) 147094. doi: 10.1016/j.cej.2023.147094
-
[72]
Y. Feng, Y. Tao, Q.Q. Meng, et al., Chem. Eng. J. 441 (2022) 135924. doi: 10.1016/j.cej.2022.135924
-
[73]
J. Lu, Y. Zhou, L. Ling, et al., Chem. Eng. J. 446 (2022) 137067. doi: 10.1016/j.cej.2022.137067
-
[74]
B. Dutta, S. Datta, M.H. Mir, Chem. Commun. 60 (2024) 9149–9162. doi: 10.1039/d4cc02093f
-
[75]
M. Huang, Y. Han, W. Xiang, et al., Environ. Sci. Technol. 55 (2021) 15361–15370. doi: 10.1021/acs.est.1c03758
-
[76]
Y. Long, Z. Cao, W. Wu, et al., Appl. Catal. B 344 (2024) 123643. doi: 10.1016/j.apcatb.2023.123643
-
[77]
H. Li, N. Wang, H. Li, et al., Appl. Catal. B 341 (2024) 123323. doi: 10.1016/j.apcatb.2023.123323
-
[78]
E.H. Lieb, Rev. Mod. Phys. 53 (1981) 603. doi: 10.1103/RevModPhys.53.603
-
[79]
T.L. Gilbert, Phys. Rev. B 12 (1975) 2111. doi: 10.1103/PhysRevB.12.2111
-
[80]
F.M. Bickelhaupt, E.J. Baerends, Rev. Comput. Chem. 15 (2000) 1–86. doi: 10.1002/9780470125922.ch1
-
[81]
W. Kohn, L. Sham, Density functional theory, Conference Proceedings-Italian Physical Society, Editrice Compositori. (1996) 561–572.
-
[82]
H.Y. Yao, Y. Huang, X. Li, et al., Environ. Sci.: Nano 7 (2020) 782–792. doi: 10.1039/c9en01295h
-
[83]
W.X. Jiang, J.G. Han, H. Guo, Sep. Purif. Technol. 330 (2024) 125309. doi: 10.1016/j.seppur.2023.125309
-
[84]
G. Yu, Y. Wu, H. Cao, et al., Environ. Sci. Technol. 56 (2022) 7853–7863. doi: 10.1021/acs.est.1c08666
-
[85]
H.G. Zhang, P.T. Cen, J.S. Li, et al., Appl. Catal. B: Environ. 359 (2024) 124484. doi: 10.1016/j.apcatb.2024.124484
-
[86]
J. Kirkpatrick, B. McMorrow, D.H.P. Turban, et al., Science 374 (2021) 1385–1389. doi: 10.1126/science.abj6511
-
[87]
S. Dick, M. Fernandez-Serra, Nat. Commun. 11 (2020) 3509. doi: 10.1038/s41467-020-17265-7
-
[88]
M. Talukdar, S.K. Behera, S. Jana, et al., Adv. Mater. Interfaces 9 (2022) 2101943. doi: 10.1002/admi.202101943
-
[89]
E.M. Flores, M.L. Moreira, M.J. Piotrowski, J. Phys. Chem. A 124 (2020) 3778–3785. doi: 10.1021/acs.jpca.9b11415
-
[90]
R. Nagai, R. Akashi, O. Sugino, npj Comput. Mater. 6 (2020) 43. doi: 10.1038/s41524-020-0310-0
-
[91]
Y. Chen, L. Zhang, H. Wang, et al., J. Chem. Theory Comput. 17 (2021) 170–181. doi: 10.1021/acs.jctc.0c00872
-
[92]
L. Chen, H.D. Ji, J.J. Qi, et al., Chem. Eng. J. 406 (2021) 126877. doi: 10.1016/j.cej.2020.126877
-
[93]
X. Peng, J. Li, S. Liu, et al., Mol. Catal. 572 (2025) 114751.
-
[94]
T. Vazhappilly, D.A. Micha, Optical properties of the TiO2 (110) surface with adsorbed Ag atoms relevant to photocatalysis and photovoltaics, in: D. Kilin, S. Kilina, Y. Han (Eds.), Computational Photocatalysis: Modeling of Photophysics and Photochemistry at Interfaces, Chapter 3, 2019, pp. 47–66. doi:
10.1021/bk-2019-1331.ch003 . -
[95]
H. Li, X. Liu, H. Hu, et al., Sep. Purif. Technol. 335 (2024) 126103. doi: 10.1016/j.seppur.2023.126103
-
[96]
S. Mao, P. Zhao, Y. Wu, et al., Chem. Eng. J. 451 (2023) 138503. doi: 10.1016/j.cej.2022.138503
-
[97]
P. Duan, M. Li, X. Xu, et al., Appl. Catal. B 344 (2024) 123619. doi: 10.1016/j.apcatb.2023.123619
-
[98]
L.Z. Wang, D.H. Ding, Z. Qian, et al., Chem. Eng. J. 450 (2022) 138353. doi: 10.1016/j.cej.2022.138353
-
[99]
Z. Jiang, Z. Zhao, X. Li, et al., Appl. Surf. Sci. 607 (2023) 154997. doi: 10.1016/j.apsusc.2022.154997
-
[100]
P. Chen, Z.L. Cheng, X.Z. Zhang, et al., J. Cleaner Prod. 445 (2024) 141365. doi: 10.1016/j.jclepro.2024.141365
-
[101]
Y. Song, Y. Feng, T. Wu, et al., Sep. Purif. Technol. 354 (2025) 128587. doi: 10.1016/j.seppur.2024.128587
-
[102]
S. Halder, T.K. Bhowmik, A. Dutta, et al., Ceram. Int. 46 (2020) 21021–21032. doi: 10.1016/j.ceramint.2020.05.170
-
[103]
K. Xie, W. Hao, K. Xu, et al., Ultrason. Sonochem. 99 (2023) 106543. doi: 10.1016/j.ultsonch.2023.106543
-
[104]
K. He, Z. Huang, C. Chen, et al., Nano-Micro Lett. 16 (2023) 23.
-
[105]
J. Gao, H.b. Yang, X. Huang, et al., Chem 6 (2020) 658–674. doi: 10.1016/j.chempr.2019.12.008
-
[106]
S.Y. Ma, W.G. Han, W.L. Han, et al., J. Mater. Chem. A 11 (2023) 3315–3363. doi: 10.1039/d2ta08735a
-
[107]
Y. Bu, R. Ma, Y. Wang, et al., Adv. Mater. 36 (2024) 2412670. doi: 10.1002/adma.202412670
-
[108]
Z. Teng, W. Cai, W. Sim, et al., Appl. Catal. B 282 (2021) 119589. doi: 10.1016/j.apcatb.2020.119589
-
[109]
C. Qin, Y. Yang, X. Wu, et al., Nat. Commun. 14 (2023) 6740. doi: 10.1038/s41467-023-42513-x
-
[110]
B. Yang, X. Cheng, Y. Zhang, et al., Environ. Sci. Ecotechnol. 8 (2021) 100127. doi: 10.1016/j.ese.2021.100127
-
[111]
Q. Su, J. Li, H. Yuan, et al., Chem. Eng. J. 427 (2022) 131594. doi: 10.1016/j.cej.2021.131594
-
[112]
Q. Su, J. Li, B. Wang, et al., Appl. Catal. B 318 (2022) 121820. doi: 10.1016/j.apcatb.2022.121820
-
[113]
S. Zhang, Y. Liu, P. Gu, et al., Appl. Catal. B: Environ. 248 (2019) 1–10.
-
[114]
J. Luo, Y. Liu, C. Fan, et al., ACS Catal. 11 (2021) 11440–11450. doi: 10.1021/acscatal.1c03103
-
[115]
A.L. Filgueiras, D. Paschoal, H.F. Dos Santos, et al., Spectrochim. Acta Part A 136 (2015) 979–985. doi: 10.1016/j.saa.2014.09.120
-
[116]
Y. Shen, M. Yang, C. Zhu, et al., ACS ES&T Eng 4 (2024) 2839–2851. doi: 10.1021/acsestengg.4c00400
-
[117]
Y. Gao, Z. Cai, X. Wu, et al., ACS Catal. 8 (2018) 10364–10374. doi: 10.1021/acscatal.8b02360
-
[118]
W. Lai, K. Zhang, P. Shao, et al., J. Hazard. Mater. 379 (2019) 120791. doi: 10.1016/j.jhazmat.2019.120791
-
[119]
I. Salahshoori, M. Namayandeh Jorabchi, A. Mazaheri, et al., Environ. Res. 252 (2024) 118856. doi: 10.1016/j.envres.2024.118856
-
[120]
M. Rebarchik, E. Smith, H. Chang, et al., ACS Catal. 15 (2025) 6662–6672. doi: 10.1021/acscatal.4c07231
-
[121]
G. Liu, S. Li, C. Shi, et al., Sep. Purif. Technol. 352 (2025) 128141. doi: 10.1016/j.seppur.2024.128141
-
[122]
X. Zhang, Z. Yao, J. Wang, et al., Appl. Catal. B 307 (2022) 121205. doi: 10.1016/j.apcatb.2022.121205
-
[123]
A. Morankar, S. Deshpande, Z. Zeng, et al., Proc. Natl. Acad. Sci. U. S. A. 120 (2023) e2308458120. doi: 10.1073/pnas.2308458120
-
[124]
J. Wang, J. Kim, J. Li, et al., Environ. Sci. Technol. 58 (2024) 17157–17167.
-
[1]
-
Figure 5 (a) Intrinsic mechanism of selective O2•− generation by doped g-C3N4 with double oxygen. Reproduced with permission [113]. Copyright 2019, Elsevier. (b) Multi-path mechanism of photocatalytic g-C3N4 generation of H2O2. Reproduced with permission [114]. Copyright 2021, American Chemical Society.
Figure 6 “Computational guidance — experimental validation” research flowchart. Reproduced with permission [111]. Copyright 2022, Elsevier. Reproduced with permission [117]. Copyright 2018, American Chemical Society. Reproduced with permission [118]. Copyright 2019, Elsevier. Reproduced with permission [119]. Copyright 2024, Elsevier. Reproduced with permission [120]. Copyright 2025, American Chemical Society.
Table 1. Comparison of the application of different base groups.
Basis set Accuracy level Core advantages Application scenarios in AOPs 6–31G* Moderate Fast and broadly applicable Rapid screening of active species generation pathways; overcoming the experimental bottleneck in capturing transient reactive species; optimizing radical yield on catalyst surfaces. 6–31+G* Moderate (+ long range) Improved description of anions/adsorption Accurately simulation of solvation shell structures of antibiotic anions; correction of adsorption energy deviations; guidance for the design of acidic sites on catalysts to enhance adsorption. 6–311G* Medium-High More precise valence electron distribution Precise calculation of transition state energy barriers for antibiotic molecule cleavage; overcoming predictive bias for C—N/C-S bond dissociation energies in conventional basis sets; enabling targeted design of low-activation-energy active sites. cc-pVDZ Medium-High Optimized electron correlation energy Accurate description of multi-electron cooperative transfer in metal-antibiotic complexes; resolution of prediction inaccuracies of redox potentials in Fe/Cu based catalysts; enhancement of electron transfer efficiency. LANL2DZ Moderate (for heavy elements) Efficient handling of metal centers Balancing efficiency and accuracy in computing d-orbital electron structures of transition metals (e.g., Co, Fe); overcoming bottlenecks in elucidating metal-support synergistic mechanisms in supported catalysts.  下载: 导出CSV
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 9
- HTML全文浏览量: 1
 下载:
下载:
