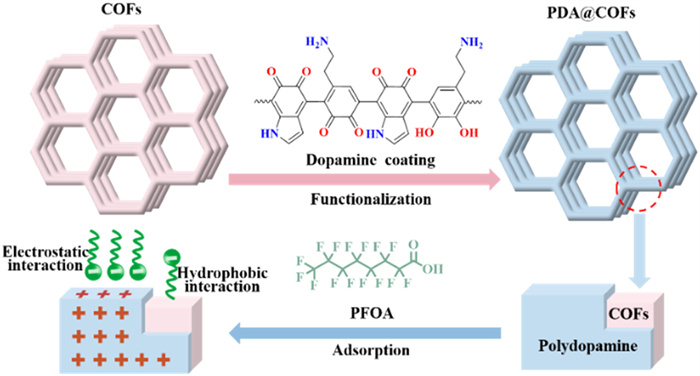
Scheme 1.
Synthetic scheme of highly efficient and selective adsorbents.
Amine/imine-functionalized covalent organic frameworks with high selective capacity of perfluoroalkyl substances: Mechanism and potential application as anode in capacitive deionization
Taiwen Chen , Baohua Wang , Lu Zhang , Wenbo Bi , Cong Lyu

Per- and poly-fluoroalkyl substances (PFASs), categorized as emerging contaminants, were typically termed “forever chemical” due to their hydrophobic fluorinated carbon chains stabilized by exceptionally strong C–F bonds (450 kJ/mol) [1]. Prolonged exposure to these compounds can lead to severe health issues, including liver damage, ulcerative colitis, and even cancers [2,3]. In response to growing concerns, the U.S. Environmental Protection Agency has recently established mandatory standards for PFASs in drinking water [4], necessitating the development of remediation technologies that are economical, efficient, and sustainable.
Adsorption is the most prevalent method for PFASs removal, owing to its ease of implementation and notable economic viability in water treatment [5]. Current adsorbents that have been investigated for PFASs adsorption include activated carbon [6], ion-exchange resins [7], and surfactants [8]. However, these adsorbents often suffer from insufficient adsorption capacity, poor selectivity, slow adsorption kinetics, and challenge in reusability [9], which hindered their practical application. In addition, for porous adsorbents, PFASs aggregation due to the hydrophobic interactions hindered the mass transfer diffusion process, whereas the adsorption efficiency of these compounds can be enhanced via cooperative adsorption [10], so the construction of adsorbents with unobstructed channels can help to realize the balance between hydrophobic aggregation of PFASs and mass transfer and diffusion.
Covalent organic frameworks (COFs) with highly ordered pore architectures, hydrophobicity of carbon skeleton, tunable pore sizes [11], robust chemical stability [12], and surface functionalization [13] are novel advanced materials with appealing adsorption properties. However, COFs still lack specific binding sites for PFASs, and the post-synthetic modification of COFs is complicated and expensive, which limit their practical application. Polydopamine (PDA), a biocompatible polymer, features numerous functional amine and imine moieties that generate positive charges, enabling the electrostatic interaction with anionic PFASs during adsorption [14-16]. Herein, this study introduces a novel and simple strategy, constructing PFASs adsorbents with multi-functionalized sites by coating PDA onto COFs (PDA@COFs), which can selectivity and efficiently adsorb negatively charged PFASs molecules (Scheme 1), and further improving the adsorption performance of PFASs adsorbents by utilizing the aggregation of PFASs. However, to our knowledge, the adsorption of PFASs by PDA-coated COFs has not been reported previously.
COFs typically exist as powder due to their inherent hyper-crosslinked and insolubility of the framework, which poses challenges for practical application and recycling in adsorption processes. To explore the practical application potential and reusability of PDA@COFs adsorbent, PDA@COFs was evaluated as a promising anode in capacitive deionization (CDI), improving the desorption of the adsorbent and the recovery of PFASs through polarity reversal of the electrochemical cell.
Perfluorooctanoic acid (PFOA) was selected as a representative PFASs because of its wide availability and resistance to conventional adsorption methods. This study provided valuable guidance on constructing adsorbents for efficient and selective removal of PFASs, as well as a promising strategy for exploring the practicality of adsorbents via CDI.
Supporting information included preparation process, characterization, experimental methods, and analytical methods. COFs was first synthesized via polycondensation of TAPT and TFTA under Schiff base reaction conditions. Subsequently, PDA was coated onto COFs using a modified solvothermal method to construct the PDA@COFs adsorbent (Fig. 1a). A peak at 1578 cm−1 was observed in the FT-IR spectra of the COFs, which assigned to the newly formed C=N bond [17], while the bands corresponding to C=O (1697 cm−1) from TFTA and –NH2 (3150–3430 cm−1) from TAPT almost vanished, indicating completion of the condensation reaction (Fig. S1 in Supporting information). Meanwhile, the aromatic C=C vibration at around 1600 cm−1 [18] persists in PDA@COFs, confirming that the intrinsic hydrophobic regions remain unshielded. The optimal conditions (Fig. S2 in Supporting information) for the preparation of PDA@COFs were found: 40 mg dopamine hydrochloride and 40 mg potassium carbonate, with a reaction temperature of 100 ℃ for one day. Surface SEM images revealed that COFs exhibited a coral-like morphology consisting of irregular nanoparticles (Fig. 1b and Fig. S3 in Supporting information), whereas PDA@COFs displayed larger and rougher particles with rounded edges, which was attributed to PDA coating-induced surface modification. In addition, TEM (Figs. S4b and d in Supporting information) confirmed that PDA@COFs retain intact crystallinity and ordered pores without collapse.
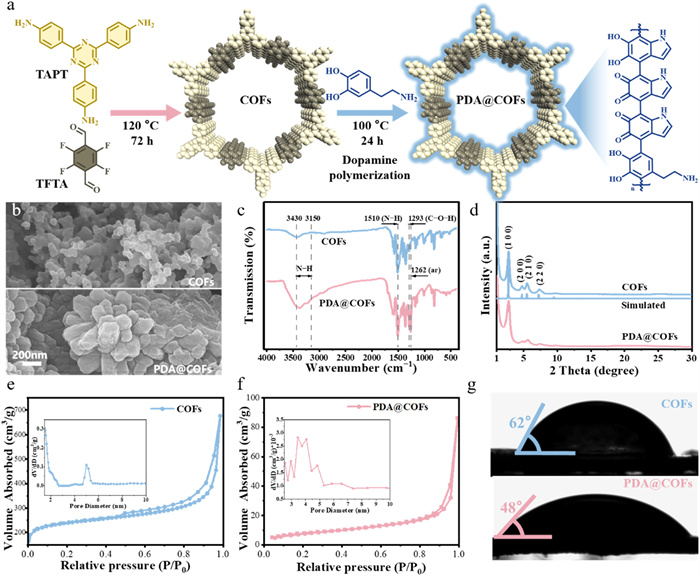
The FT-IR spectra of PDA@COFs exhibited enhanced stretching vibrations peaks at 3150–3430 cm−1 compared to COFs (Fig. 1c), indicating that PDA conferred additional amine functional groups to the COFs [19]. Furthermore, the peaks at 1293 and 1262 cm−1 corresponded to C–O–H bending and aromatic ring stretching vibration in PDA, respectively [20,21]. PDA and COFs were connected via non-covalent interactions, and the rightward shift (Fig. S5 in Supporting information) of the hydroxyl stretching vibration peak confirms the enhanced hydrogen bonding interaction [22]. XPS analysis showed that the PDA@COFs contained C, N, O, and F (Fig. S6 in Supporting information). The O 1s spectra revealed that peaks at 531.5 eV and 532.9 eV corresponded to the C=O and C–O structures, respectively [23], which were associated with the ketone and hydroxyl groups in polydopamine.
The XRD patterns of COFs and PDA@COFs showed distinct diffraction peaks at approximately 2θ = 2.75°, 4.89°, 5.65°, and 7.39° (Fig. 1d), corresponding to the (100), (200), (210), and (220) crystallographic planes, respectively [24]. The N2 adsorption-desorption isotherms revealed the porous properties of COFs and PDA@COFs, with the specific surface area decreasing from 617.306 m2/g (COFs) to 28.361 m2/g (PDA@COFs) (Figs. 1e and f), suggesting that the specific surface area of PDA and PDA@COFs decreased significantly during the synthesis process, which was attributed to pore blockage and surface coverage by PDA. Both adsorbents displayed type Ⅳ adsorption isotherms accompanied by H3 hysteresis loops [25], characterized by mesoporous structures, with average pore diameters decreasing from 5.2 nm (COFs) to 3.8 nm (PDA@COFs) (Table S1 in Supporting information). As shown in Fig. S7 (Supporting information), these pores were sufficiently large to trap PFOA [26]. Notably, the PDA coating endows the COFs with amine and imine functional groups. The corresponding increase in surface hydrophilicity (Fig. 1g) directly demonstrates the enhanced number of these amine/imine groups, which strengthens electrostatic interactions with PFOA.
To assess the PDA@COFs adsorption performance, adsorption kinetics were investigated on its parent materials COFs and PDA. The adsorption of PFOA by PDA@COFs was rapid and approached equilibrium within 5 min, adsorbing about 80% of PFOA (Fig. 2a). In contrast, pure COFs, PDA, and physical mixtures of COFs and PDA adsorbents were only able to adsorb small amounts of PFOA. This phenomenon can be ascribed to the fact that COFs possess unobstructed channels, but lack adsorption affinity for PFOA. Meanwhile, pure PDA have the necessary adsorption sites for PFOA but lack regular pores, resulting in poor mass transfer and limited contact area with PFOA. The pseudo-second-order model demonstrated superior fitting performance (Fig. S8 in Supporting information), evidenced by a higher correlation coefficient (R2 = 0.99) and a rate constant k2 of 9.6 g mg−1 h−1 (Table S2 in Supporting information), demonstrated that the adsorption process involves a variety of mechanisms and not just changes in adsorbent concentration [27].
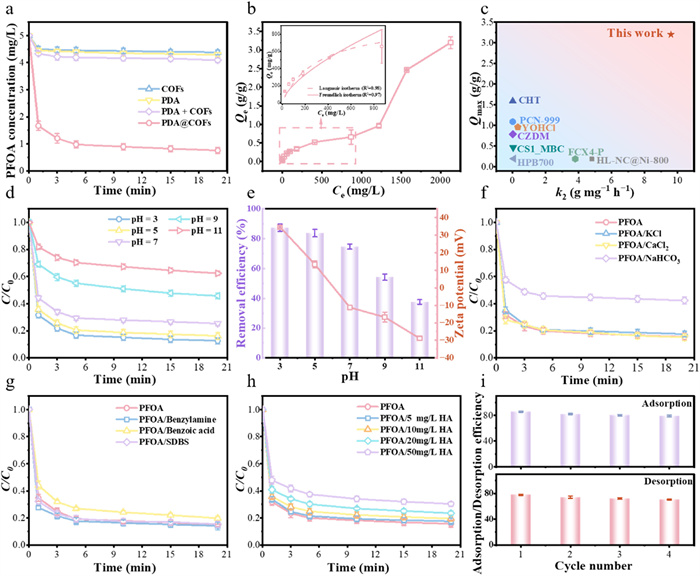
Adsorption capacity was a key indicator for evaluating adsorbent performance, and the adsorption isotherm in Fig. 2b was LS-shaped. PDA@COFs exhibit ultra-high adsorption capacity (3200 mg/g) toward PFOA. Adsorption was not saturated at PFOA concentrations higher than 1000 mg/L, which was due to the cooperative adsorption of electrostatic and hydrophobic interactions. In comparison with other adsorbents (Fig. 2c), including biochar, metal-organic frameworks, and carbon nanosheets [28-34], PDA@COFs exhibit significantly higher adsorption capacity.
As depicted in Fig. 2d, PDA@COFs exhibited stable PFOA adsorption efficiency (>74%) across a pH range of 3–7. For the PDA@COFs adsorbent, the zeta potential value shifted from 34.63 mV to −28.83 mV as pH increased from 3 to 11, indicating the critical role of surface charge in adsorption capacity (Fig. 2e). This correlation emphasized the dominant role of electrostatic attraction in the adsorption mechanism. Under acidic conditions, protonation of adsorption sites on PDA@COFs increased their availability [33]. As shown in Fig. 2f, and Figs. S9 and S10 (Supporting information), the presence of Cl−, SO42−, and PO43− showed negligible interference on the adsorption of PFOA by PDA@COFs, whereas the adsorbed amount of PFOA was significantly reduced when the concentration of HCO3− was 100 times greater than that of PFOA. This decline was attributed to the alkaline nature of NaHCO3, which elevated solution pH and diminished electrostatic attraction.
We selected three representative competing contaminants to evaluate the selective adsorption capability of PFOA by PDA@COFs. Hydrophilic benzylamine and benzoic acid showed no significant interference with PFOA adsorption, whereas hydrophobic sodium dodecylbenzene sulfonate (SDBS) reduced PFOA adsorption by 5% (Fig. 2g and Fig. S11 in Supporting information), demonstrating the strong adsorption affinity of PDA@COFs for PFOA [35]. Additionally, we tested the simultaneous adsorption of different types of PFASs (PFOA, PFOS, and OBS) by PDA@COFs. The results (Fig. S12 in Supporting information) demonstrated that PDA@COFs exhibited excellent adsorption performance for PFASs containing carboxyl and sulfonate functional head. Natural organic matter (NOM), especially HA, was tested for its effect on PFOA adsorption. As the HA concentration rose from 5 mg/L to 50 mg/L, the PFOA adsorption showed a decline from 1.8% to 14.6% in contrast to the blank group (Fig. 2h and Fig. S13 in Supporting information). The reduction in adsorption was attributed to competition for adsorption sites by HA through the interaction of the carboxyl group with PDA@COFs [36]. Moreover, after four adsorption-desorption cycles, PDA@COFs retained 78.9% of its initial PFOA adsorption rate, with only a 6.6% decline (Fig. 2i). Additionally, we determined the number of functional groups remaining after the cyclic process through acid-base back titration experiments (Table S6 in Supporting information). After four cycles, the loss of amine or imine adsorption sites accounted for approximately 7.5% of the total, demonstrating the excellent cycling stability of PDA@COFs.
The adsorption of PFOA onto PDA@COFs were characterized using SEM, EDS, FT-IR, XPS and DFT calculations. The SEM images and corresponding EDS mapping results of PDA@COFs after PFOA adsorption (denoted PFOA@PDA@COFs) revealed that the PFOA molecules were effectively confined within the inner pores of PDA@COFs (Fig. S14 in Supporting information). Furthermore, the water contact angle of PDA@COFs increased from 48° to 79°, indicating that PDA@COFs exhibited enhanced hydrophobicity after PFOA adsorption (Fig. S15 in Supporting information).
FT-IR spectroscopy revealed the binding mechanisms between PFOA and PDA@COFs. As depicted in Fig. 3a, the emergence of new absorption peaks at 1548 cm−1 and 1401 cm−1 in the FT-IR spectrum of PFOA@PDA@COFs was assigned to the asymmetric (υas(COO−)) and symmetric (υs(COO−)) stretching vibrations of the carboxylate group, respectively [33]. The apparent contraction of the C–N stretching band confirmed the electrostatic attraction between the COO− groups in PFOA and the CH3-N+ groups in PDA@COFs. The spectral range from 1142 cm−1 to 1206 cm−1 corresponded to C–Fx [37]. The shift from nearly semi-ionic to covalent C–F in PDA@COFs may be attributed to the hydrophobic interactions. A new peak at C–F3 position of PFOA@PDA@COFs suggested that PFOA has been successfully adsorbed onto PDA@COFs.
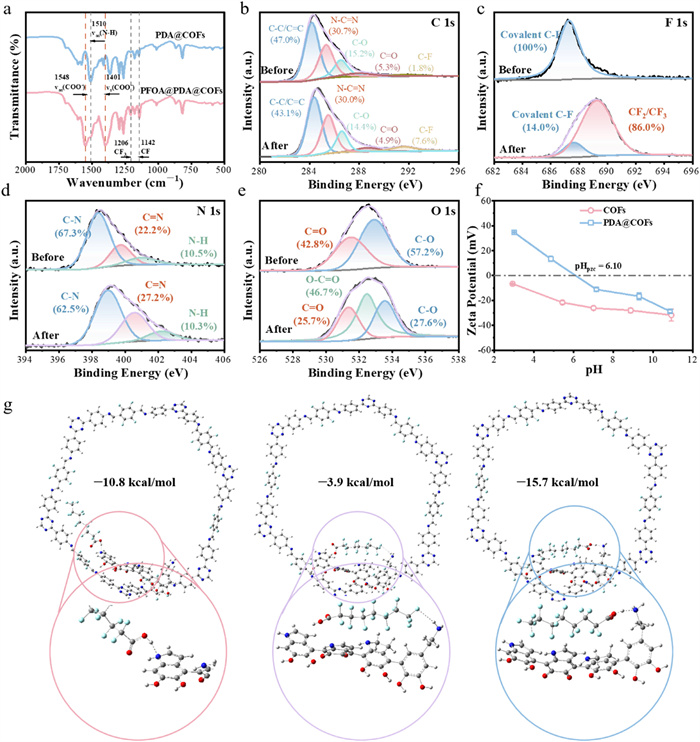
XPS spectra were employed to analyze the elemental composition of PDA@COFs before and after PFOA adsorption, including C, F, N, and O (Fig. S16 in Supporting information). The C 1s spectrum (Fig. 3b) of PDA@COFs showed that the peak at 291.8 eV represented a significant enhancement of the C–F bond, and the peak in the F 1s spectrum (Fig. 3c) exhibited a transition from semi-ionic C–F to covalent C–F bonds [38,39]. After adsorption of PFOA, the peak area of imine–like nitrogen (C=N) in PDA@COFs decreased by 5% (Fig. 3d), indicating that the electrostatic attraction existed between the carboxyl group of PFOA and the surface of PDA@COFs. And the new O-C=O peak representing the carboxyl group further illustrated that PFOA was adsorbed by PDA@COFs successfully (Fig. 3e). Although COFs was negatively charged across pH 3–11 (Fig. 3f), PDA functionalization introduced amine and imine groups that enhanced electrostatic attraction.
The adsorption binding energy (ΔEbinding) was determined using DFT calculation theory. The binding energies between PDA@COFs and PFOA were all negative (Fig. 3g), signifying that the adsorption process was a spontaneous exothermic reaction [40]. DFT calculations revealed that the binding energy of the carboxyl group of PFOA to the amine group on PDA@COFs was the lowest at −15.7 kJ/mol, which was lower than the binding energy between the C–F chain of PFOA and the amine groups, as well as the carboxyl group and pyrrole, which revelated that amine group acts as the primary adsorption site for PFOA.
The adsorption isotherm of PFOA adsorbed by PDA@COFs exhibited a specificity similar to that of the surfactant. This adsorption isotherm was divided into five adsorption stages as shown in Fig. 4a, which was consistent with the LS-shape adsorption isotherm [41,42].
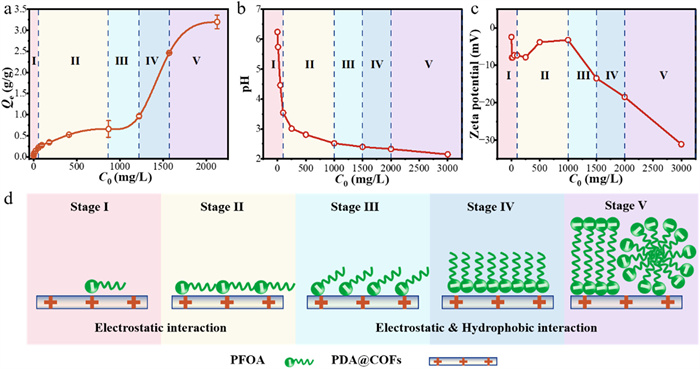
In the initial stage of adsorption (Stage Ⅰ in Fig. 4d), the adsorbate-adsorbate interactions between a few PFOA monomers were negligible because the molecules were far apart, which followed the Henry law (Fig. S17 in Supporting information). The zeta potential at this stage showed a sharp decline (Fig. 4c), attributed to surface neutralization. Significantly, the solution pH dropped rapidly with increasing PFOA concentration in the initial adsorption phase (Fig. 4b). Notably, the zeta potential of the PDA@COFs did not improve, which proved that the first phase was dominated by electrostatic attraction.
As the PFOA concentrations continued to increase, the surface adsorption sites gradually became saturated. Due to the increase in PFOA concentration, the pH of the solution decreases in Fig. 4b, further shifting the zeta potential of the PDA@COFs surface toward positive values. However, due to electrostatic interactions, the zeta potential fluctuates around zero and exhibits upward trend. The isothermal Langmuir model at the Stage Ⅱ exhibited a better fit than the Freundlich (Fig. S18 and Table S3 in Supporting information), demonstrating that the adsorption was monolayer with the adsorption sites on the surface saturated (Stage Ⅱ in Fig. 4d) to a PFOA concentration of around 1000 mg/L.
Stage Ⅲ adsorption isotherm model was more consistent with the Freundlich model (Fig. S19 and Table S4 in Supporting information). Owing to the lengthy hydrophobic carbon chain and compact carboxyl head of the PFOA molecules, the intense electrostatic attraction between the negatively charged carboxylate group and the positively charged surface of PDA@COFs induced the displacement of alkyl chains from adjacent PFOA molecules on the surface (Stage Ⅲ in Fig. 4d). This phenomenon was similar to the adsorption model of nonionic surfactants reported by Paria [42].
The subsequent adsorption stage was dominated by adsorbate-adsorbate interactions. In Stage Ⅳ (Stage Ⅳ in Fig. 4d), the hydrophobic interactions between the alkyl chains of PFOA molecules drive their aggregation, leading to the formation of vertically oriented aggregates within a concentration of 0.001–0.01 critical micellization concentration (CMC) [43], which resulted in a sharp increase in the slope of the adsorption isotherm. Concurrently, the zeta potential decreased significantly again, which aligned with the sharp rise in adsorption capacity observed in the isotherm, further indicating that the formation of aggregates due to the hydrophobic interaction of PFOA contributed to a sudden increase in adsorption capacity.
In the final stage (Stage Ⅴ in Fig. 4d), the isotherm fit was consistent with the LS-Shape adsorption isotherm (Fig. S20 in Supporting information). The adsorption of PFOA onto the surface of PDA@COFs caused a further reduction in zeta potential. Although the concentration of PFOA was lower than the CMC, the significant localized concentration near the adsorbent surface promoted the formation of aggregates of PFOA [44], which was attributed to cooperative adsorption of electrostatic and hydrophobic interactions. It has been shown that aggregates obstruct the diffusion pathway of PFOA to the inner surface of the microporous adsorbent, ultimately hindering adsorption by subsequent adsorbents [10]. However, the COFs-based adsorbent had unobstructed channels, which enhanced the adsorption of PFOA aggregates.
To overcome the challenge of reusing powdered PDA@COFs adsorbent, the adsorbent was severed as an anode in the CDI system (Fig. 5a). Since PFOA molecules existed primarily as anions in aqueous solutions, these negatively charged anions were electrostatically attracted to the PDA@COFs anode under an applied electric field, improving efficient removal via electro-sorption. As shown in Fig. S21 (Supporting information), the FT-IR peaks at 3150–3430 cm−1 before and after preparation have decreased, indicating that some of the PDA@COFs adsorption sites have been encapsulated. However, the clearly identifiable characteristic vibration peaks indicate that the chemical structure of the surface functional groups in PDA@COFs remains intact.
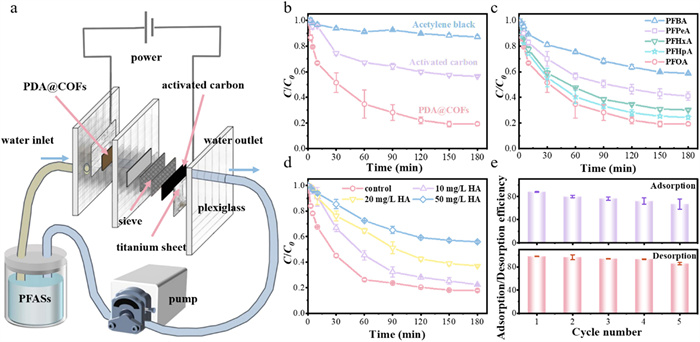
PDA@COFs showed significantly higher adsorption rates for low concentrations of PFOA than activated carbon and acetylene black (Fig. 5b), highlighting their potential application as CDI anodes. In addition, the PDA@COFs anode exhibited excellent adsorption performance for PFASs of different chain lengths (Fig. 5c). The system was able to maintain a high PFOA removal rate when HA concentrations ranged from 0 to 10 mg/L (Fig. 5d). When the concentration of HA was 50 times that of PFOA, competitive adsorption hindered the adsorptive removal of PFOA, which was due to the interaction of HA with ligand exchange sites (e.g., carboxyl groups). Due to the assistance of the electric field, PFOA could still be adsorbed by the PDA@COFs electrode through electrostatic attraction [45,46], albeit with a slightly reduced removal efficiency. Additionally, after five consecutive adsorption and desorption cycles, the PFOA removal decreased slightly to 61.7%, and the PFOA recovery consistently ranged between 85.4% and 98.2% (Fig. 5e), showing the excellent stability and reusability of PDA@COFs in the CDI system. Additionally, to verify the structural stability of the electrodes under long-term electric field action, the PFOA removal attenuation was monitored after 20 consecutive cycles (exceeding 100 h). The PFOA recovery rate decreased from 98.3% to 76.4%, while the PFOA concentration decreased from 98.7% of the initial level to 85.5% over the 20 cycles (Fig. S22 in Supporting information). Furthermore, the chemical state of the PDA layer before and after 20 cycles of CDI was monitored using XPS. The N 1s spectrum (Fig. S23 in Supporting information) revealed no significant reduction in the N-H percentage, indicating the PDA layer possesses excellent chemical stability.
In summary, an efficient and selective porous adsorbent (PDA@COFs) enriched with amine and imine functional groups for PFASs was constructed. The maximum adsorption capacity for PFOA by PDA@COFs was 3200 mg/g, which was far more than that of most adsorbents. Importantly, PDA@COFs exhibited excellent selectivity for PFOA even in complex matrices where NOM and organic contaminants coexist. Theoretical calculations revealed that electrostatic attraction was the dominant driving mechanism for the selective adsorption of PFOA by PDA@COFs. The adsorption isotherm of PFOA conformed to the LS-shaped, with adsorption changing from initial electrostatic attraction to electrostatic and hydrophobic synergistic. Notably, PDA@COFs was a promising CDI anode with high adsorption efficiency and excellent PFOS recovery performance, which had great potential for practical applications. In summary, this study offers novel perspectives on the selective and efficient removal of PFOA and shows great potential for the practical application of PDA@COFs as anodes for capacitive deionization.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Taiwen Chen: Writing – review & editing, Writing – original draft, Methodology, Investigation. Baohua Wang: Methodology, Investigation. Lu Zhang: Writing – original draft, Methodology. Wenbo Bi: Methodology, Data curation. Cong Lyu: Writing – review & editing, Writing – original draft, Visualization, Methodology, Investigation.
The present work was supported by the Science and Technology Development Program of Jilin Province, China (No. 20220101214JC) and the National Natural Science Foundation of China (No. 52070086). The authors would like to thank Associate Professor Ronglin Zhong from the Institute of Theoretical Chemistry-College of Chemistry, Jilin University for the advice on DFT calculation.
Supplementary material associated with this article can be found, in the online version, at doi:
X. Qian, L. Xu, X. Ge, et al., Chin. Chem. Lett. 35 (2024) 109218. doi: 10.1016/j.cclet.2023.109218
R. Lohmann, I.T. Cousins, J.C. DeWitt, et al., Environ. Sci. Technol. 54 (2020) 12820–12828. doi: 10.1021/acs.est.0c03244
K. Singh, N. Kumar, A. Kumar Yadav, et al., Chem. Eng. J. 475 (2023) 145064. doi: 10.1016/j.cej.2023.145064
H. Ryu, B. Li, S. De Guise, et al., J. Hazard. Mater. 408 (2021) 124437. doi: 10.1016/j.jhazmat.2020.124437
H. Li, A.L. Junker, J. Wen, et al., Chem. Eng. J. 452 (2023) 139202. doi: 10.1016/j.cej.2022.139202
H.M. Tan, C.G. Pan, C. Yin, K. Yu, Environ. Res. 233 (2023) 116495. doi: 10.1016/j.envres.2023.116495
C. Ching, M.J. Klemes, B. Trang, et al., Environ. Sci. Technol. 54 (2020) 12693–12702. doi: 10.1021/acs.est.0c04028
S. Kancharla, P. Alexandridis, M. Tsianou, Curr. Opin. Colloid In. 58 (2022) 101571. doi: 10.1016/j.cocis.2022.101571
B. Gao, L. Wu, S. Li, et al., J. Hazard. Mater. 483 (2025) 136698. doi: 10.1016/j.jhazmat.2024.136698
Z. Du, S. Deng, Y. Bei, et al., J. Hazard. Mater. 274 (2014) 443–454. doi: 10.1016/j.jhazmat.2014.04.038
W. Wang, Y. Jia, S. Zhou, S. Deng, J. Hazard. Mater. 460 (2023) 132522. doi: 10.1016/j.jhazmat.2023.132522
S. Kandambeth, K. Dey, R. Banerjee, J. Am. Chem. Soc. 141 (2019) 1807–1822. doi: 10.1021/jacs.8b10334
Y. Yang, H. Niu, L. Xu, et al., Appl. Catal. B 269 (2020) 118799. doi: 10.1016/j.apcatb.2020.118799
M.L. Alfieri, T. Weil, D.Y.W. Ng, V. Ball, Adv. Colloid Interface Sci. 305 (2022) 102689. doi: 10.1016/j.cis.2022.102689
H. Hemmatpour, O. De Luca, D. Crestani, et al., Nat. Commun. 14 (2023) 664–675. doi: 10.1038/s41467-023-36303-8
Z. Wang, Y. Zou, Y. Li, Y. Cheng, Small 16 (2020) 1907042. doi: 10.1002/smll.201907042
Q. Liao, C. Ke, X. Huang, et al., J. Mater. Chem. A 7 (2019) 18959–18970. doi: 10.1039/c9ta06214a
F. Dai, Q. Zhuang, G. Huang, et al., ACS Omega 8 (2023) 17064–17076. doi: 10.1021/acsomega.3c01336
Y. Qian, Y. Yuan, H. Wang, et al., J. Mater. Chem. A 6 (2018) 24676–24685. doi: 10.1039/c8ta09486a
Y. Wan, X. Liu, P. Liu, et al., Sci. Total Environ. 639 (2018) 428–437. doi: 10.1016/j.scitotenv.2018.05.171
J. Li, Q. Zhou, Y. Liu, M. Lei, Sci. Technol. Adv. Mater. 18 (2017) 3–16. doi: 10.1080/14686996.2016.1246941
D. Ishikawa, A. He, Y. Xu, et al., Sci. Rep. 15 (2025) 16991–17002. doi: 10.1038/s41598-025-00774-0
W. Ren, L. Xiong, G. Nie, et al., Environ. Sci. Technol. 54 (2020) 1267–1275. doi: 10.1021/acs.est.9b06208
Y. Zhi, Z. Li, X. Feng, et al., J. Mater. Chem. A 5 (2017) 22933–22938. doi: 10.1039/C7TA07691F
Y. Peng, G. Xu, Z. Hu, et al., ACS Appl. Mater. Interfaces 8 (2016) 18505–18512. doi: 10.1021/acsami.6b06189
J.M. Arana Juve, X. Baami González, L. Bai, et al., Appl. Catal. B: Environ. 349 (2024) 123885. doi: 10.1016/j.apcatb.2024.123885
S. Abbaszadeh, S.R. Wan Alwi, C. Webb, et al., J. Cleaner Prod. 118 (2016) 210–222. doi: 10.1016/j.jclepro.2016.01.054
P.H. Chang, W.T. Jiang, Z. Li, J. Hazard. Mater. 368 (2019) 487–495. doi: 10.1016/j.jhazmat.2019.01.084
L. Chen, K. He, W. Li, et al., ACS ES&T Eng. 4 (2024) 3092–3104. doi: 10.1021/acsestengg.4c00418
R.R. Liang, S. Xu, Z. Han, et al., J. Am. Chem. Soc. 146 (2024) 9811–9818. doi: 10.1021/jacs.3c14487
B. Saawarn, B. Mahanty, S. Hait, Environ. Pollut. 368 (2025) 125734. doi: 10.1016/j.envpol.2025.125734
D. Shetty, I. Jahović, T. Skorjanc, et al., ACS Appl. Mater. Interfaces 12 (2020) 43160–43166. doi: 10.1021/acsami.0c13400
X. Tan, Z. Jiang, W. Ding, et al., Water Res. 230 (2023) 119558. doi: 10.1016/j.watres.2022.119558
Y. Yang, F.S. Cannon, Bioresour. Technol. 344 (2022) 126161. doi: 10.1016/j.biortech.2021.126161
H. Fan, H. Wang, J. Guo, et al., J. Colloid Interface Sci. 414 (2014) 46–49. doi: 10.1016/j.jcis.2013.09.042
Y. Chen, H. Li, Y. Yin, et al., Sci. Total Environ. 905 (2023) 167175. doi: 10.1016/j.scitotenv.2023.167175
H. An, Y. Li, P. Long, et al., J. Power Sources 312 (2016) 146–155. doi: 10.1016/j.jpowsour.2016.02.057
Z. Li, G.D. Del Cul, W. Yan, et al., J. Am. Chem. Soc. 126 (2004) 12782–12783. doi: 10.1021/ja046589+
T. Nakajima, M. Koh, V. Gupta, et al., Electrochim. Acta 45 (2000) 1655–1661. doi: 10.1016/S0013-4686(99)00389-8
H. Zhao, Y. Lyu, J. Hu, et al., Chem. Eng. J. 463 (2023) 142486. doi: 10.1016/j.cej.2023.142486
S. Kalam, S.A. Abu-Khamsin, M.S. Kamal, S. Patil, ACS Omega 6 (2021) 32342–32348. doi: 10.1021/acsomega.1c04661
S. Paria, K.C. Khilar, Adv. Colloid Interface Sci. 110 (2004) 75–95. doi: 10.1016/j.cis.2004.03.001
R.L. Johnson, A.J. Anschutz, J.M. Smolen, et al., Chem. Eng. Data 52 (2007) 1165–1170. doi: 10.1021/je060285g
A. Jrad, G. Das, N. Alkhatib, et al., Nat. Commun. 15 (2024) 10490–10506. doi: 10.1038/s41467-024-53945-4
L. Liu, Y. Liu, N. Che, et al., J. Hazard. Mater. 416 (2021) 125866. doi: 10.1016/j.jhazmat.2021.125866
N. Saeidi, F.D. Kopinke, A. Georgi, Chem. Eng. J. 416 (2021) 129070. doi: 10.1016/j.cej.2021.129070

Figure 1 Synthesis and characterization of the PDA@COFs. (a) Chemical structure and synthesis of the PDA@COFs. (b) Morphology and structure of COFs and PDA@COFs characterized by SEM images. (c) The FT-IR spectra of COFs and PDA@COFs. (d) Crystal structure analysis of COFs and PDA@COFs characterized by XRD spectrum. (e, f) Specific surface area and pore size distribution of COFs and PDA@COFs characterized by N2 adsorption-desorption isotherm. (g) Hydrophobicity of COFs and PDA@COFs characterized by Water contact angles.

Figure 2 Adsorption performance of PFOA. (a) Adsorption kinetics of PFOA by different adsorbents. (b) Adsorption isotherms of PFOA by PDA@COFs. (c) The comparison of capacity and rate constants of PFOA by PDA@COFs against other adsorbents. (d) Effect of pH on the adsorption of PFOA by PDA@COFs. (e) Effect of zeta potential and pH on the adsorption of PFOA. (f) Effect of inorganic ions on the adsorption of PFOA. (g, h) Effect of interfering substance and HA concentration on the adsorption of PFOA. (i) Adsorption and desorption efficiency of PFOA through adsorption/desorption recycles. 50 mL of 5 mg/L PFOA, benzylamine, benzoic acid, and SDBS, 10 mg of PDA@COFs, adsorption for 2 h.

Figure 3 Binding interactions between PFOA and PDA@COFs. (a) FT-IR spectra of the before and after adsorption of PFOA by PDA@COFs. (b) XPS spectra of PDA@COFs C 1s, (c) F 1s, (d) N 1s, and (e) O 1s before and after adsorption of PFOA. (f) The binding energy between PFOA and PDA@COFs.

Figure 4 Mechanism of high adsorption capacity of PFOA by PDA@COFs in aqueous solution. (a) Adsorption isotherm of PFOA for five stages by PDA@COFs. (b) Variation of pH value with initial PFOA concentration in the adsorption solutions. (c) Variation of zeta potential with initial PFOA concentration in the adsorption solutions. (d) Stages Ⅰ–Ⅴ of adsorption isotherms of PFOA by PDA@COFs at increasing PFOA concentrations in aqueous solution. 50 mL of PFOA, 10 mg of PDA@COFs, adsorption for 2 h.

Figure 5 The potential application of PDA@COFs as anode in capacitive deionization (CDI). (a) The schematic diagram of CDI device for selective removal of PFOA. (b) Effect of different electrodes on the adsorption of PFOA, including acetylene black, activated carbon, and PDA@COFs. (c) Effect on the removal of PFASs with different carbon chain lengths. (d) Effects of HA concentration on the adsorption of PFOA. (e) Adsorption and desorption efficiency of PFOA through adsorption/desorption recycles. (10 mL/min of flow rate, 1.2 V of potential).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
 下载:
下载: