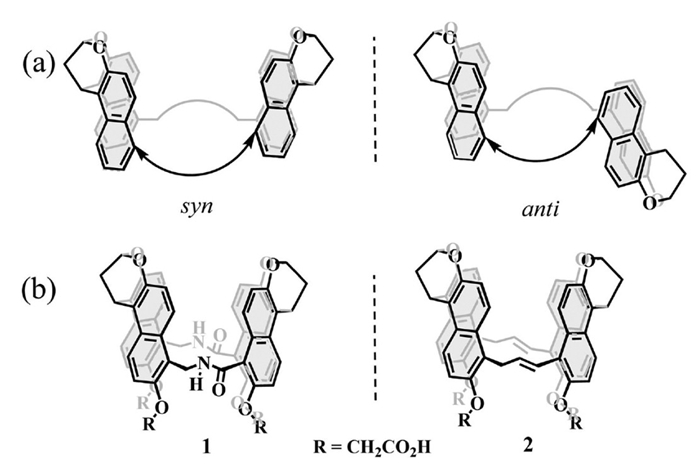
Figure 1.
(a) Syn and anti-naphthalene dimers. (b) Chemical structures of syn amide-linked naphthotube 1 and syn allyl-linked naphthotube 2.
Recent advances in chiral recognition with macrocyclic hosts
Chunhong Liu , Xiaotong Liang , Wanhua Wu , Zhouyu Wang , Cheng Yang

Chirality, a fundamental property inherent in nature, plays a pivotal role in the origin and evolution of life [1]. Many biomolecules exhibit strict chiral preferences: For instance, the sugar units in starch and cellulose adopt exclusively dextrorotatory (D) configurations, whereas amino acids in nature are almost uniformly levorotatory (L). Such asymmetric distribution highlights the decisive role of chirality in biological processes. Homochirality also endows living organisms with the capacity to enantioselectively recognize exogenous chiral substances [2–4]. Chiral small-molecule drugs provide a clear example: Enantiomers often interact differently with biological systems, resulting in distinct absorption, distribution, metabolism, toxicity, and therapeutic outcomes. For instance, levodopa is effective in treating Parkinson's disease, while its enantiomer, dexvodopa, is inactive. Precise enantiomer differentiation is therefore essential not only for fundamental science but also for human health and environmental safety [5–7].
Chiral recognition, a central topic in supramolecular and analytical chemistry, holds broad potential in drug development, environmental monitoring, and asymmetric catalysis [8–10]. Macrocyclic molecules are particularly attractive platforms for chiral recognition due to their well-defined cavities, tunable structures, and strong host–guest properties [11]. However, their practical application is often constrained by limited recognition efficiency, slow response, and narrow scope, which fall short of the selectivity and sensitivity required [12,13]. Developing effective strategies to enhance the chiral recognition performance of macrocyclic receptors is therefore of critical importance [14–17].
This review summarizes recent advances in improving the chiral recognition capabilities of supramolecular macrocycles. Strategies include structural modification to optimize molecular interactions, dynamic regulation of chirality via external stimuli, construction of chirality-specific recognition interfaces, and machine learning-assisted design. Current challenges and limitations are analyzed, and guidelines as well as future perspectives for the development of high-performance chiral recognition materials are proposed. By highlighting recent progress and emerging trends, this article aims to provide insights and inspiration for advancing research in this field.
The chiral recognition of macrocyclic compounds arises from the synergistic interplay between their preorganized cavities and multiple binding sites. Recognition is governed by stereochemical complementarity between host and guest, reinforced by cooperative noncovalent interactions such as hydrogen bonding, electrostatic forces, and π–π stacking. This capability can be further enhanced through the introduction of binding sites within the cavity, functional modification of both the exterior (e.g., incorporation of chiral units) and interior, and precise tuning of the cavity architecture in conjunction with its surrounding chiral microenvironment.
The exceptionally high affinity observed in biological systems is primarily due to the synergistic interaction between strong hydrophobic forces within the binding pocket and cooperative recognition from multiple polar sites. Enzymes, for example, achieve their remarkable recognition capabilities through specific binding sites within their active structures. Accordingly, when designing macrocyclic receptors for molecular recognition, the rational pre-organization of multiple polar interaction sites can effectively harness multi-polar synergies, leading to high-affinity and highly selective recognition.
Conventional macrocyclic hosts, such as cyclodextrins, calixarenes, cucurbiturils, and pillararenes, typically feature either hydrophobic cavities or polar binding sites at their periphery. They are broadly categorized into two types: Those that primarily use a hydrophobic cavity for molecular recognition (e.g., cyclodextrins) [18–21] and those that rely on peripheral polar groups as the main recognition sites (e.g., crown ethers) [22]. Receptors with effective polar binding sites deep within their cavities are relatively rare. Incorporating specific binding motifs into these internal cavities has therefore become a promising strategy to enhance the recognition capabilities of macrocyclic hosts. Macrocycles with inward-facing functional groups possess highly adaptable architectures and distinct binding properties. Their modular synthesis and ease of functionalization make them particularly appealing for advanced applications in supramolecular chemistry, biomedicine, and materials science [23].
In 2004, Glass and co-workers reported a pair of water-soluble, naphthalene-based molecular tubes. Each tube consisted of two bis-naphthalene clefts connected by amide bonds, yielding two distinct isomers (syn and anti, Fig. 1a). The syn isomer exhibited the desired tubular cavity, whereas the anti isomer displayed greater conformational flexibility and adopted a more elongated structure [24]. During macrocyclization, the amide groups were oriented toward the cavity interior, generating a pronounced hydrophobic environment (1, Fig. 1b). By exploiting this feature, the molecular tubes selectively recognized lipid-based guest molecules, exhibiting a strong preference for straight-chain alkanes. In 2012, the group redesigned the macrocyclic framework by replacing the flexible amide linkages with rigid olefin linkers (2, Fig. 1b), thereby enhancing the hydrophobic effect and significantly improving guest recognition in aqueous solution [25].
The synthesis of these macrocyclic compounds relies primarily on ring-closing reactions of bridging segments bearing active functional groups, which introduce binding sites deep within the cavity. Building on this strategy, Jiang and co-workers incorporated diverse functional groups, including imines, ureas, ethers, esters, amides, thioureas, and amines, into the linkages between two naphthalene segments, followed by ring closure. This approach enabled the preparation of a series of structurally related naphthalene-based molecular tubes (naphthalene tubes), which have since found wide application in molecular recognition [26].
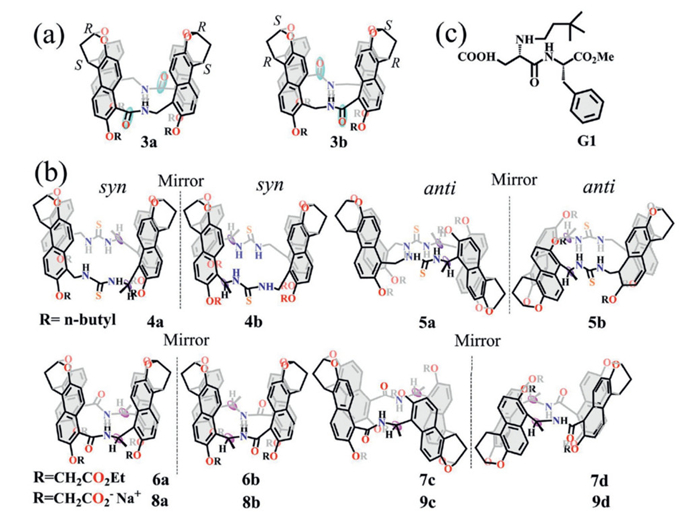
To achieve enantioselective recognition, a chiral environment was introduced into the macrocyclic cavity through the condensation of chiral naphthalene components. Chiral amide naphthotubes (3a/3b) were synthesized from an asymmetric naphthalene amino acid (Fig. 2a), enabling recognition of various chiral epoxides in aqueous solution [27]. However, the enantioselectivity was limited (up to 2.0), mainly because the chiral centers were positioned too far from the hydrogen-bonding site, impeding efficient chiral information transfer. To overcome this limitation, the chiral centers were repositioned in closer proximity to the binding sites, thereby enhancing transfer efficiency through hydrogen-bond interactions. As shown in Fig. 2b, two pairs of chiral thiourea naphthotubes (4a/4b and 5a/5b) have been reported, and amide-based naphthotubes (6–9) have proven to be effective chiral sensors, exhibiting high sensitivity toward a wide range of chiral guests [28,29]. Furthermore, the increased number of inward-oriented hydrogen-bonding sites in compounds 4–9 expanded the scope of detectable chiral guests to include not only epoxides but also pharmaceutical molecules and polar solvents. Notably, amide naphthotube 8 exhibited strong affinity toward guest G1 (Fig. 2c), achieving enantioselectivity of up to 34 [28].
In 2022, Chen and co-workers reported a modular strategy for synthesizing endo-functionalized biphen[n]arenes [30]. Two multi-alkoxyphenyl modules were connected to a central functionalized unit via Suzuki–Miyaura coupling to generate a monomeric precursor, which then underwent Friedel–Crafts alkylation with paraformaldehyde to afford a series of functionalized macrocycles through efficient cyclization and polymerization (Fig. 3a). Single-crystal X-ray diffraction revealed well-defined binding sites within the cavity, enabling selective recognition of neutral azacyclic guests. Owing to its modularity, this approach allowed the properties of the macrocycles to be readily tuned by varying the central module, thereby offering precise control over host–guest affinity and selectivity through rational functionalization.
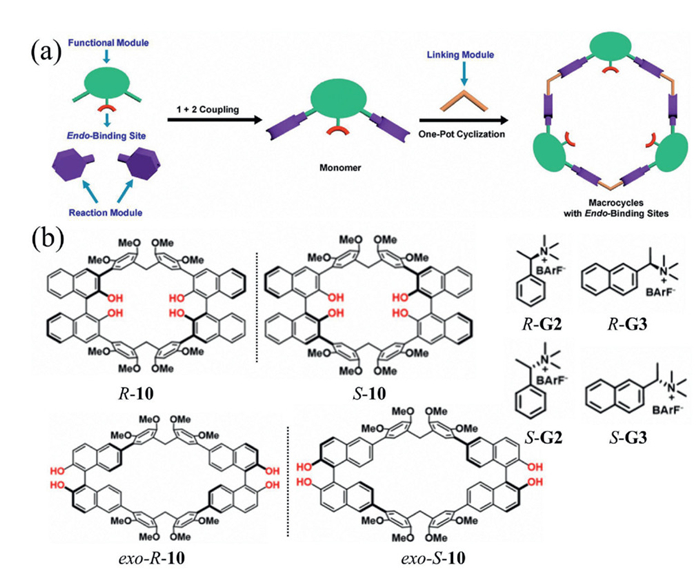
Chiral binaphthol (BINOL) units, renowned for their outstanding chiral induction capabilities, have long served as commercially available chiral scaffolds in a wide range of enantioselective reactions. Incorporating BINOL into modular synthetic strategies has emerged as a promising approach to constructing internally functionalized chiral macrocycles capable of selectively recognizing chiral guest molecules [31,32]. In 2024, Li and co-workers reported the synthesis of a pair of BINOL-based chiral macrocycles, R/S-10 (Fig. 3b), which they applied to the recognition of chiral ammonium salts in aqueous solution [33]. In this macrocyclic framework, inward-oriented hydroxyl groups acted as specific binding sites, enabling stereoselective interactions with guest molecules. Moreover, the direct connection between the recognition site (hydroxyl group, –OH) and the chiral source (binaphthyl backbone) facilitated efficient transfer of chiral information from host to guest, thereby enhancing enantioselectivity. Their experiments showed that R/S-10 exhibited strong binding affinities toward both G2 and G3 guests, with enantioselectivity for G3 reaching 13.2.
Single crystal X-ray diffraction analysis confirmed the formation of a stable host-guest complex mediated by hydrogen bonding. This interaction significantly enhances the binding affinity and represents a key factor in achieving efficient chiral recognition. To further elucidate the crucial role of hydroxyl orientation in the chiral recognition of macrocycles, the research group subsequently synthesized chiral macrocycle hosts exo-(R/S)-10, in which the hydroxyl groups point outward. NMR titration experiments were then performed to investigate the interaction between exo-(R/S)-10 and R/S-G2; however, no notable chemical shift changes were observed. This result indicates the absence of effective recognition sites and underscores the essential role of inward-oriented hydroxyl groups in chiral recognition.
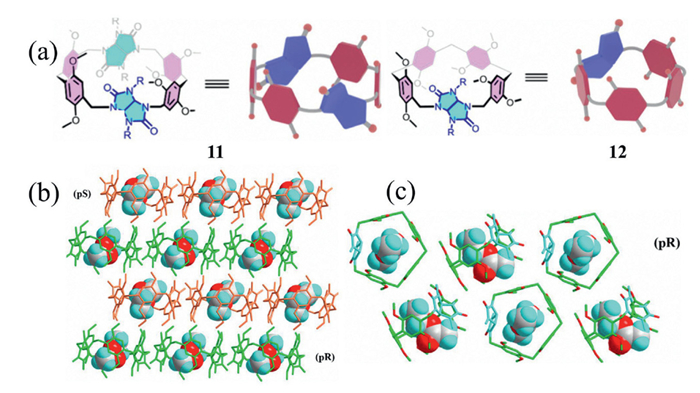
Recently, Wang and co-workers employed a fragment-coupling macrocyclization strategy to incorporate glycoluril units into the sidewalls of pillararenes, successfully synthesizing novel pillarurilarenes 11 and 12 (Fig. 4a) [34]. During cyclization, an upfield shift of the glycoluril methine proton signal was observed in the 1H NMR spectra, providing evidence that these protons reside within the cavities of the resulting pillarurilarenes, indicating their potential as active sites for further functionalization. In addition, the methyl protons in the ethylene glycol urea segment are readily amenable to chemical modification through the introduction of diverse functional groups, allowing precise modulation of the molecular recognition properties of these macrocycles. Single-crystal X-ray diffraction analysis revealed that macrocycle 11 contained two alternately packed enantiomers (Sp and Rp, Fig. 4b), whereas macrocycle 12 exclusively exhibited the Rp enantiomer, likely as a result of chiral self-sorting during crystallization (Fig. 4c).
These distinctive characteristics establish cyclourilarenes as promising platforms for chiral recognition. Subsequent studies demonstrate that the incorporation of additional non-aromatic heterocycles with curved geometries into the macrocyclic sidewalls enables the synthesis of diverse chiral macrocyclic aromatic hydrocarbons. This structural versatility significantly expands their potential applications in chiral recognition [35].
Beyond incorporating endo-binding sites, increasing attention has been directed toward functionalizing the periphery of macrocyclic cavities. Among these, pillar[n]arenes (P[n]s) have emerged as particularly attractive scaffolds owing to their excellent host–guest recognition properties and synthetic versatility, which have stimulated the development of a wide range of P[n] derivatives [36–44]. For instance, a sulfated pillar[5]arene bearing multiple sulfonate groups displayed greatly enhanced affinity toward quaternary ammonium salts, attributed to cooperative hydrophobic and multivalent electrostatic interactions [45].
Chiral racemization of P[n] readily occurs due to the rapid interconversion between chiral conformers (Rp/Sp) via flipping-induced inversion of planar chirality (FIIPC) [46]. A practical strategy to circumvent this challenge involves introducing chirality at the periphery of the macrocyclic cavity through the incorporation of chiral subunits. This approach avoids tedious chiral separation, directly establishes a defined chiral microenvironment, and substantially improves chiral recognition performance. Furthermore, the broad diversity of available chiral building blocks, coupled with the structural versatility of achiral macrocyclic scaffolds, enables modular design strategies for constructing chiral macrocycles with tailored architectures. Consequently, peripheral chiral functionalization has become a powerful method for developing chiral supramolecular macrocycles with enhanced recognition capabilities. This section highlights representative advances in this area, with a focus on chemical modification as a key strategy.
Chemical modification represents an effective means to improve the chiral recognition performance of supramolecular macrocycles. By selectively introducing functional groups into the macrocyclic framework, one can fine-tune stereoelectronic properties and the spatial arrangement of binding sites, thereby promoting selective recognition of chiral guests.
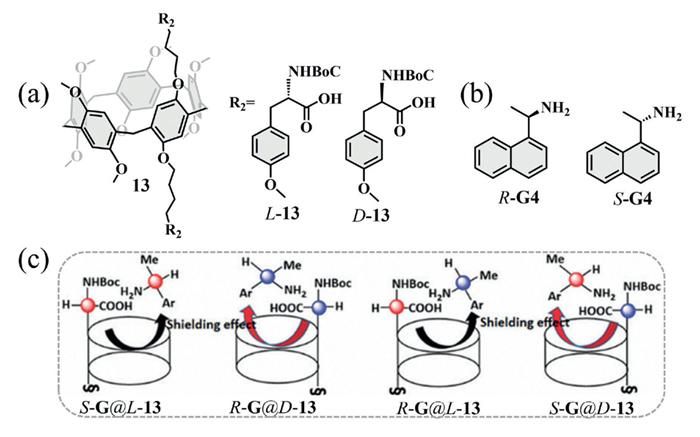
In 2021, Duan and co-workers synthesized two tyrosine-modified pillar[5]arenes L-13 and D-13 (Fig. 5a), which acted as chiral solvating agents for the enantioselective recognition of aromatic amines (Fig. 5b) [47]. 1H NMR experiments showed that L-13 preferentially bound R-G4, while D-13 favored S-G4. Host–guest modeling provided a rational explanation: the bulky Boc group of L-13 reduced steric hindrance with the methyl group of R-G4, thereby enhancing binding. This interaction increased the shielding of aromatic ring protons, producing upfield shifts in the signals of R-diastereomers (Fig. 5c). Hydrogen bonding between the carboxylic acid group of the macrocyclic host and the amine group of the guest further stabilized the complex. Notably, introducing bulky chiral substituents (e.g., Boc-protected tyrosine) at the upper and lower rims of pillar[5]arene created a defined chiral microenvironment within the cavity. This modification enhanced anisotropic shielding, improved NMR resolution of diastereotopic protons, and enabled the detection of multiple distinct proton signals, thereby significantly boosting the chiral discrimination ability of compound 13.
Supramolecular chiral assembly refers to the spontaneous organization of building blocks into ordered chiral structures through non-covalent interactions. Such assemblies can form highly ordered nanoscale architectures via weak intermolecular forces [48], including hydrogen bonding and van der Waals interactions, resulting in a pronounced amplification of chiral signals [49]. More importantly, this strategy allows precise control over molecular arrangement and stacking, thereby optimizing the chiral microenvironment and further enhancing chiral recognition performance.
Cyclodextrins (CDs) have been widely employed as chiral hosts owing to their unique structural features, including inherent chirality, high water solubility, UV transparency, facile chemical modification, and natural abundance, which together confer broad potential in chiral recognition [50,51]. The chiral microenvironment of the CD cavity enables the formation of diastereomeric inclusion complexes with enantiomeric guests, thereby facilitating enantiodiscrimination. Nevertheless, the rotational symmetry of the CD cavity, combined with its reliance on non-directional hydrophobic interactions, constrains its chiral recognition efficiency.
To address this limitation, our group proposed a spin-transformation strategy based on self-assembly, in which the native Cn-symmetric cavity of γ-cyclodextrin (γ-CD) was perturbed by introducing an achiral axial molecule into its cavity. This modification indirectly generated highly asymmetric chiral binding sites. Owing to the relatively large cavity size of γ-CD, incorporation of axial molecules bearing benzene rings not only occupied part of the internal space but also enhanced the binding affinity toward small-molecule guests. As a result, the guest could engage more effectively in host–guest interactions, thereby significantly improving the chiral recognition performance of γ-CD [52].
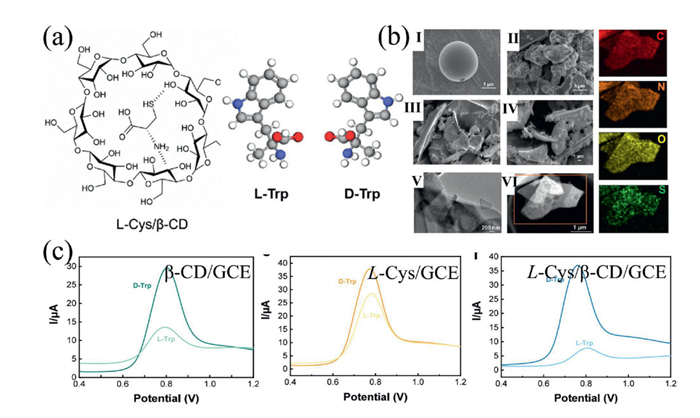
Recently, Wang and co-workers reported a hydrogen-bonding-driven self-assembly strategy to anchor L-cysteine (L-Cys) onto β-CD, yielding a novel chiral cyclic nanostructure with high enantioselectivity toward tryptophan enantiomers (Fig. 6a) [53]. Scanning electron microscopy (SEM) revealed that the L-Cys/β-CD complex exhibited an amorphous morphology with irregular particles (Fig. 6b), arising from intermolecular hydrogen bonding between the hydroxyl groups of β-CD and the thiol (–SH) and amino (–NH2) groups of L-Cys. The resulting composite possessed a large specific surface area, offering abundant chiral recognition sites and thereby enhancing its potential in enantioselective applications. Electrochemical studies showed that the L-Cys/β-CD composite discriminated D-tryptophan (D-Trp) more effectively than L-tryptophan (L-Trp), attributable to stereospecific hydrogen bonding between the hydroxyl group of D-Trp and the β-CD cavity. While the hydrophobic cavity of β-CD alone provided only weak enantiodiscrimination for tryptophan (ID/IL = 1.33), the incorporation of L-Cys markedly enhanced recognition (ID/IL = 2.17; Fig. 6c). This improvement was attributed to the intrinsically poor conductivity of β-CD, which limited its electrochemical performance, whereas the L-Cys/β-CD composite introduced additional chiral sources and strengthened host–guest interactions. Remarkably, this system achieved enantioselectivity values as high as 4.8, highlighting its exceptional chiral discrimination capability.
It is worth noting that both endo- and exo-functionalization present distinct advantages and challenges. Endo-functionalization directly tunes the binding environment within the macrocyclic cavity, enabling precise control over guest affinity and enantioselectivity. For example, incorporating chiral or polar groups into the cavity can significantly enhance recognition capabilities for specific substrates. However, this approach is often limited by synthetic difficulties arising from steric hindrance and restricted cavity accessibility, making selective modifications challenging. In contrast, exo-functionalization primarily influences physicochemical properties such as solubility, aggregation behavior, and compatibility with materials or biological systems, while largely preserving the intrinsic host–guest binding properties. It generally offers greater synthetic versatility and facilitates modular derivatization. Nevertheless, its impact on the core host–guest interaction remains limited.
Looking ahead, both endo- and exo-modifications represent the promising frontier in macrocycle design, paving the way for developing multifunctional systems with precisely tailored properties and holding significant potential for applications in catalysis, drug delivery, and supramolecular therapeutics.
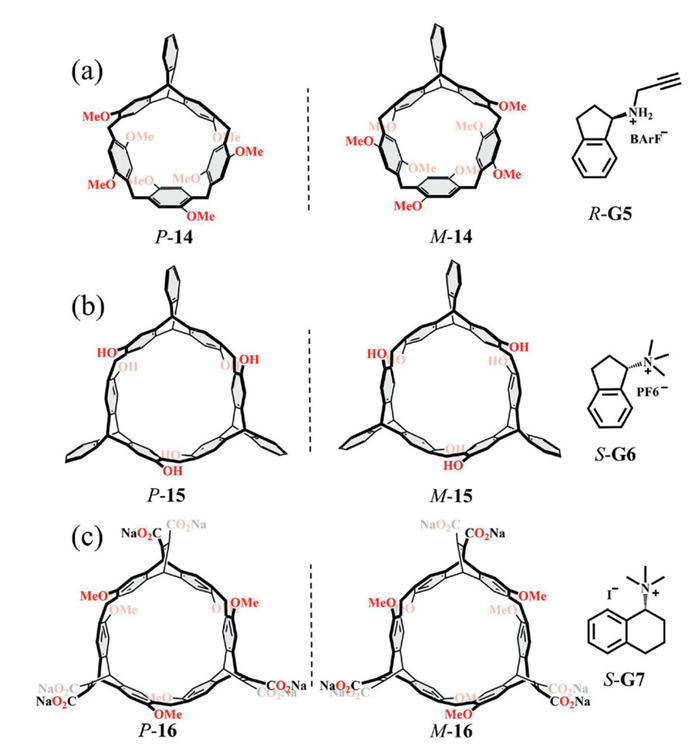
The entropy–enthalpy compensation effect is often invoked as a key factor that diminishes chiral recognition efficiency. Rigid macrocyclic cavities can mitigate entropy loss during host–guest binding by minimizing skeletal conformational changes while still fulfilling the geometric requirements for enantioselectivity. Chen and co-workers have made significant contributions in this area through the design of tripterene-based supramolecular macrocycles. For example, compared with flexible macrocycles derived from enantiopure 2,6-dimethoxy-3,7-dihydroxytripterene 14 (KR/KS = 3.89, Fig. 7a), the more rigid macrocyclic arenes constructed from enantiopure 2,6-dihydroxytripterene 15 exhibited markedly enhanced enantioselectivity (KR/KS up to 6.67, Fig. 7b) in organic media [54,55]. Notably, both systems relied on similar noncovalent interactions, underscoring the pivotal role of structural rigidity in improving chiral discrimination. Building on this strategy, the group further developed water-soluble tripterene-based macrocyclic arenes 16, termed Octopusarenes, which exploited hydrophobic effects as an additional driving force. This design amplified the enantioselective recognition of tetrahydronaphthalene quaternary ammonium salts, achieving KR/KS values as high as 12.89 (Fig. 7c) [56].
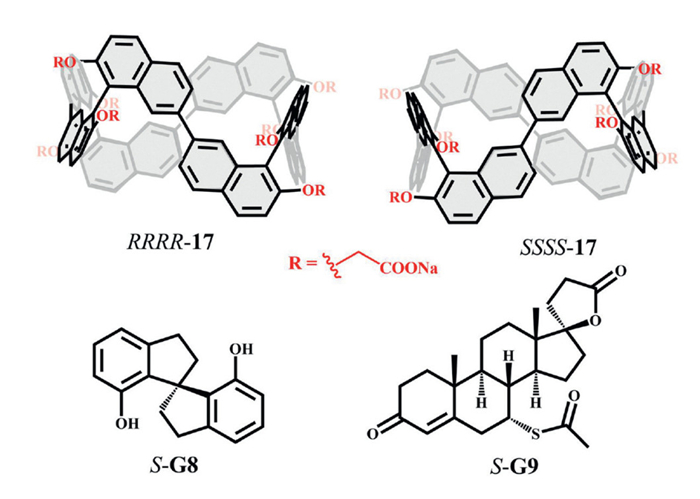
Cai and co-workers employed chiral 1,1′-bi-2,2′-naphthol (BINOL) to construct a pair of water-soluble chiral conjugated hosts, C[4]BINOLs (RRRR-/SSSS-17, Fig. 8). These coral-like macrocycles exhibited greater rigidity than methylene-bridged macrocyclic arenes. The biphenol units generated a conjugated, electron-rich, and deeply hydrophobic cavity, which displayed strong affinity for positively charged hydrophobic guests (association constants up to 1012 L/mol). This rigid deep-cavity design enabled remarkable enantioselectivity, reaching 18.7 for S-G8 and 15.5 for S-G9 [32,57].
Trӧger's base, characterized by its rigid Ⅴ-shaped skeleton and centrally bridged tertiary amine nitrogen atom, is widely employed as a chiral building block in the synthesis of macrocyclic compounds [58,59]. Using achiral 4,4′-diaminophenylmethane as the cyclic monomer, we successfully synthesized a novel Trӧger's base-derived macrocycle (compound 18) via a one-pot approach (Fig. 9a) [60]. Single-crystal analysis revealed that compound 18 consists of multiple Trӧger's base units interconnected by methylene bridges, giving rise to a highly rigid, nearly ortho-hexagonal cavity. This cavity, with a diameter larger than that of pillar[6]arene, provides a robust framework well-suited for efficient chiral recognition (Fig. 9b).
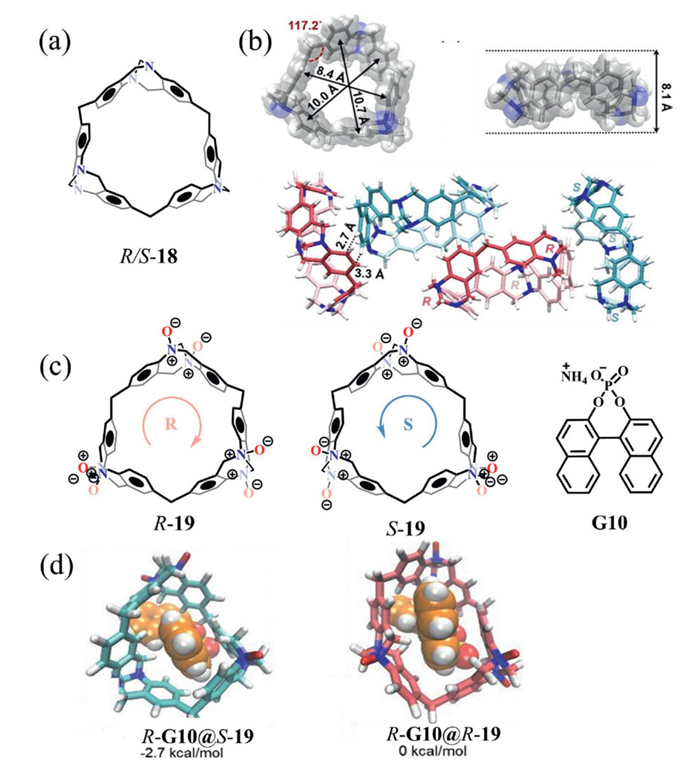
Intermolecular C—H…π interactions between neighboring enantiomers induced an undulating, staggered packing arrangement, resulting in the formation of a helical structure. Additionally, the nitrogen oxide derivatives R/S-19 exhibited excellent water solubility due to the highly polar N═O bond (Fig. 9c), overcoming the pH-related limitations of conventional ion-based functionalization and broadening their applicability in aqueous environments. Notably, compound 19 demonstrated significantly enhanced enantioselective recognition of various chiral guests in water, achieving enantioselectivity as high as 41.0 for G10. Density functional theory (DFT) calculations revealed that the R-G10@S-19 complex was approximately 2.7 kcal/mol lower in energy than R-G10@R-19, indicating stronger interactions between the Sp conformer and the chiral guest (Fig. 9d). Overall, this work establishes a novel strategy for enhancing macrocyclic chiral recognition by constructing rigid cavities.
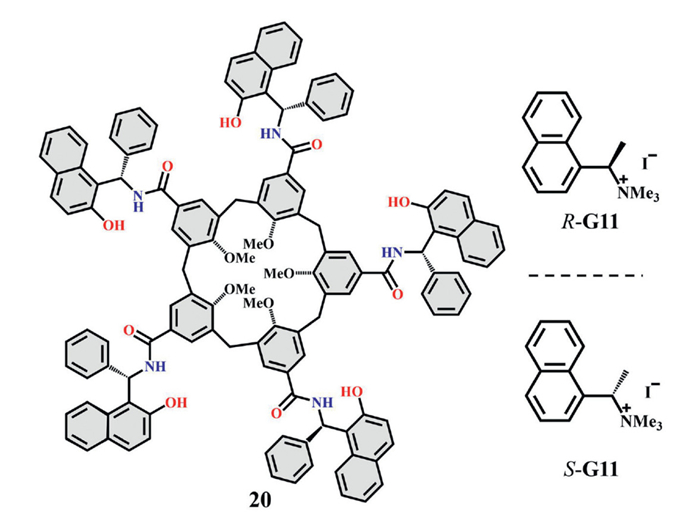
Rigid, pre-organized receptors often sacrifice flexibility and adaptability, departing from the principles underlying a protein's remarkable recognition—namely, induced-fit behavior and conformational heterogeneity [61]. Chiral macrocyclic receptors having a deep cavity, stabilized by hydrogen-bond arrays, reconcile structural closure with conformational flexibility by mimicking the hydrogen-bond networks of protein secondary structures, thereby markedly enhancing chiral recognition performance [62]. For example, the calix[5]arene-based receptor 20 developed by Agustí Lledó and co-workers achieved induced-fit recognition through dynamic conformational adaptation via a self-folding cavity strategy (Fig. 10). By leveraging a synergistic hydrogen-bond network between the calixarene and a chiral Betti base, these receptors exhibited remarkable enantioselectivity toward the guest in chloroform, achieving a KR/KS ratio of 1:18.7. Conformational fluctuations allowed the cavity to adjust its shape, preferentially stabilizing the enantiomer with the higher binding energy [63].
Allosteric regulation involves the interaction of a molecule or structural unit at a specific allosteric site with external stimuli—such as ligands, metal ions, pH, light, or temperature—inducing changes in overall structure or local conformation [64]. These structural alterations, in turn, modulate the activity of another functional site, such as an active or recognition site. Applying allosteric regulation to supramolecular chiral recognition not only elucidates the intrinsic relationship between macrocyclic structures and their recognition performance but also offers a brand-new design strategy, thereby expanding the potential for further enhancing the chiral recognition capabilities of macrocyclic systems.
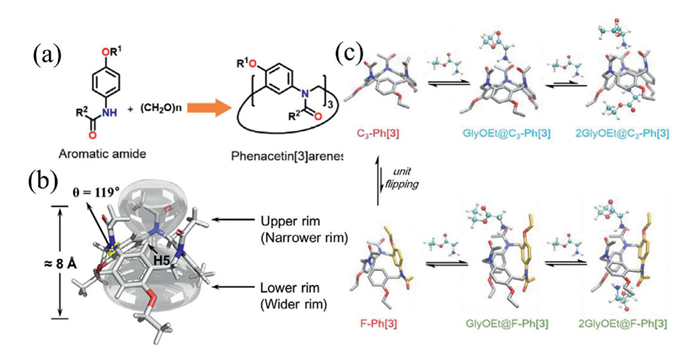
For instance, we have recently synthesized phenacetin[3]arenes (pH[3]) via Mannich reaction, employing phenacetin and paraformaldehyde as the cyclic fragment and methylene source, respectively (Fig. 11a) [65]. This work revealed two competing reaction pathways—Friedel–Crafts and Mannich—and provided mechanistic insights into the factors governing pathway selectivity. The resulting macrocyclic hosts exhibited outstanding chiral recognition and inclusion properties toward various chiral ammonium salts. pH[3] features an asymmetric hourglass-shaped cavity, with amide groups on the upper rim and a hydrophobic alkoxybenzene lower rim (Fig. 11b). This distinctive architecture allows the benzene ring to reversibly flip between two conformations, driving the orientation of the amide groups and establishing a dynamic equilibrium between C3 and F conformations (Fig. 11c). Upon increasing the concentration of organic ammonium guests, the host–guest complexation gradually shifts from a predominant 1:1 ratio to a 1:2 ratio. The binding constant for the 1:1 complex ranges from 104 to 105 L/mol, whereas the total binding constant for the 1:2 complex exceeds 106 M−2. This stepwise complexation, which triggers the C3→F conformational transition, demonstrates significant allosteric amplification and underscores the system's exceptional enantioselectivity.
The rigid structure of macrocycles enables precise complementarity with guest molecules by maintaining a well-defined conformation. This minimizes entropy loss during host–guest complexation and meets the stringent geometric requirements for effective chiral recognition. Their binding behavior is highly predictable, closely resembling the classical "lock-and-key" model. Moreover, high binding specificity is achieved only when the guest closely matches the shape and size of the macrocycle's rigid cavity. Nevertheless, rigid macrocycles face several limitations. Their fixed cavity size restricts substrate adaptability, often precluding the binding of larger biomolecules such as peptides or proteins. Synthetic modification is challenging due to steric hindrance, which impedes multi-step functionalization and limits the number of accessible sites for derivatization. Furthermore, their structural rigidity typically renders them insensitive to external stimuli such as light, temperature, or pH changes.
In contrast, flexible macrocycles exploit conformational adaptability to dynamically adjust their cavity geometry, enabling optimal guest binding, broad substrate scope, and enhanced binding affinities that contribute to complex stability. However, this flexibility carries a cost: it can compromise enantioselectivity, reduce thermal stability due to unfavorable entropic contributions upon binding, and introduce conformational heterogeneity in binding modes. These factors collectively diminish the predictability of host–guest recognition and pose challenges to experimental reproducibility.
Therefore, achieving an optimal balance between rigidity and flexibility in the host framework is crucial for high enantioselectivity in molecular recognition. Overcoming these inherent trade-offs presents a key opportunity for advancing practical chiral recognition technologies.
Precise tuning of the chiral microenvironment, including cavity size, binding sites, and hydrophobicity, is critical for achieving high binding affinity and enantioselective recognition. Yang and co-workers demonstrated that incorporating inward-facing pyridyl nitrogen atoms into an anthracene-based macrocycle significantly altered hydrophobicity and charge distribution, markedly modifying host–guest interactions [66]. This illustrates how subtle structural changes can profoundly affect recognition, akin to a point mutation in proteins.
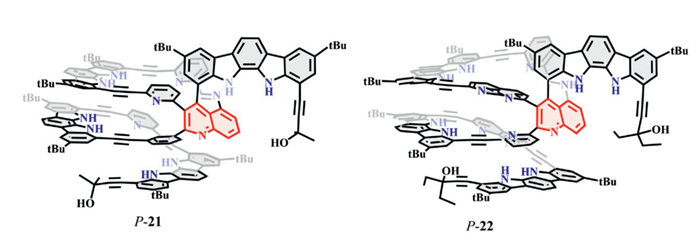
Similarly, Kyu-Sung Jeong and colleagues showed that template-directed synthesis using D- or L-chiral precursors enabled the formation of conformationally locked, protein-like helical macrocycles with exceptional enantioselectivity (KD/KL up to 108.9). Yet, minor chemical modifications, such as oxidation of a 1,2-dihydroquinoline unit, significantly reduced binding affinity and selectivity, despite minimal changes to cavity geometry (P-21 and P-22, Fig. 12). These examples highlight the sensitivity of supramolecular chiral microenvironments, where small modifications can induce major functional differences—a key principle guiding the design of high-performance chiral macrocycles [67,68].
Cui and co-workers synthesized a series of chiral triple-stranded Zn(Ⅱ) helicates (23–26, Fig. 13a) via subcomponent self-assembly. Among them, ΛΛ/ΔΔ−25, featuring an anthracene surface that enhanced hydrophobic effects and facilitated multiple hydrophobic and CH–π interactions, exhibited exceptional chiral discrimination for amino alcohols, reaching enantioselectivity values up to 9.35 for G12. In contrast, helicate 26, containing OMOM (methoxymethyl ether) protecting groups, showed reduced enantioselectivity, highlighting the critical contribution of chiral binding sites within the cavity [69].
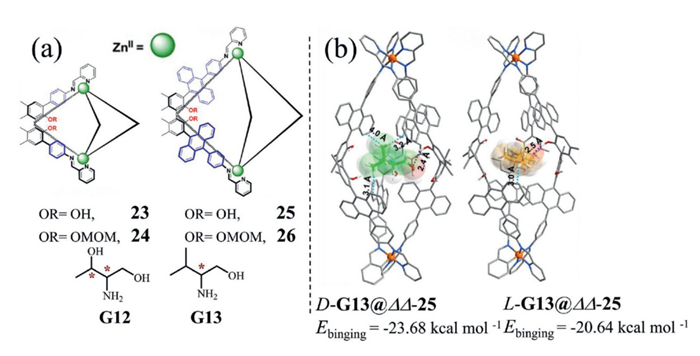
To further elucidate the role of finely tuned chiral microenvironments, energy-minimized DFT calculations were performed. The optimized structures (Fig. 13b) revealed key host–guest interactions, including hydrogen bonds (O–H···N and O–H···O), π–π stacking, and CH···π interactions, which closely aligned with experimental observations. The combination of an optimally sized cavity, abundant chiral binding sites, and dense hydrophobic surfaces enabled 25 to achieve outstanding enantioselectivity toward various chiral amino alcohols, underscoring the dependence of chiral recognition on precisely engineered microenvironments. Moreover, the calculated energy of the ΔΔ−25@D-G13 complex was lower than that of ΔΔ−25@L-G13, indicating stronger binding affinity for D-configured guests relative to their L-counterparts.
In summary, chemical functionalization is a versatile and powerful strategy for enhancing the chiral recognition capabilities of macrocyclic hosts. Achieving an optimal balance between cavity rigidity and flexibility, combined with precise endo- or exo-functionalization, is crucial for high-performance chiral recognition. These strategies, which range from covalent modification to supramolecular assembly and allosteric control, provide powerful tools for designing advanced macrocyclic hosts with applications in sensing, catalysis, and biomedicine. We believe the structural modification to optimize molecular interactions will continue to drive the development of more efficient and functional chiral supramolecular platforms.
External stimuli—such as temperature, solvents, light, or pressure—can modulate intermolecular interactions, reversibly altering the conformation, host–guest recognition behavior, and chiral microenvironment of macrocyclic compounds [70,71]. This enables precise control over the three-dimensional architecture of supramolecular systems and significantly enhances their chiral recognition performance [72–75].
Jiang and co-workers reported that water-soluble chiral naphthotubes R2,S2/S2,R2–27 exhibited enhanced enantioselectivity toward hydrophobic guests under hydrostatic pressure (Fig. 14a). Experimental results showed that the two bis(naphthalene) clefts were linked by flexible methylene-based spacers bearing polar functional groups, enabling dynamic conformational responses to applied pressure. Notably, incremental addition of the achiral guest 1,4-dioxane to the host's aqueous solution induced a pronounced fluorescence enhancement. This turn-on effect resulted from guest-induced disruption of intramolecular naphthalene stacking, with conformational adjustments in the flexible linkers suppressing non-radiative decay pathways (Fig. 14b). Changes in fluorescence intensity thus provided a quantitative measure of host–guest binding affinity and served as an indicator of chiral discrimination capability [76].
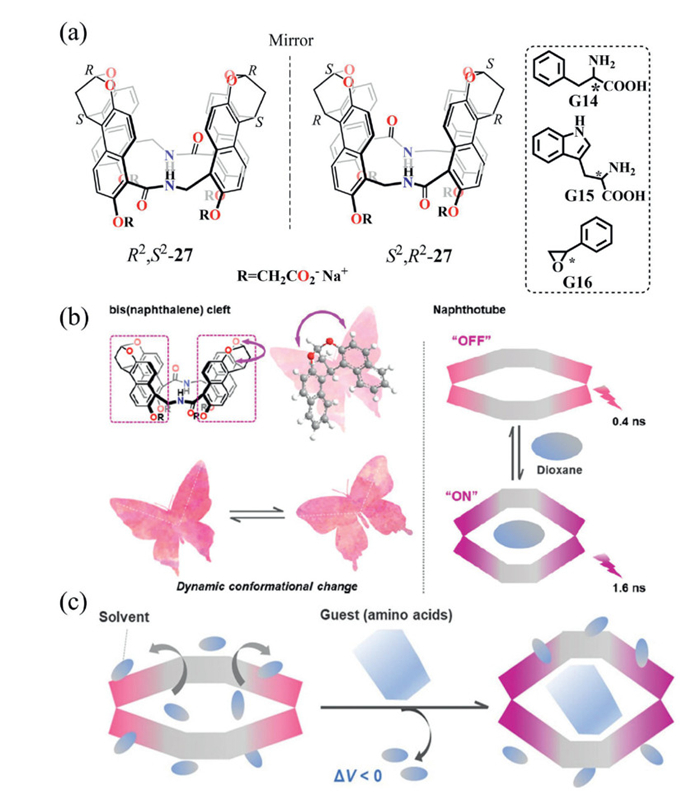
Subsequent chiral discrimination studies comparing hydrophilic amino acid guests (G14/G15, Fig. 14a) with hydrophobic styrene oxide (G16, Fig. 14a) revealed distinct behaviors under hydrostatic pressure. Hydrophilic systems exhibited limited enantioselectivity enhancement in the range of 1.2–1.6 (KL/KD), attributing to desolvation-induced cavity relaxation, where hydrostatic pressure only marginally promoted complexation, as corroborated by the weak fluorescence intensity observed in titration experiments. In contrast, hydrophobic systems demonstrated markedly higher enantioselectivity, with chiral styrene oxide achieving KR/KS = 7.6. This enhancement was ascribed to (ⅰ) solvation-driven contraction of the naphthalene cavity and (ⅱ) pressure-induced-fit binding that optimized steric complementarity with the hydrophobic guest (Fig. 14c).
In conclusion, this stimulus-responsive strategy demonstrates that dynamic control over supramolecular architecture enables tunable and substantially enhanced chiral recognition, establishing a pioneering platform for pressure-amplified enantioselectivity and developing intelligent functional materials.
Chiral interfaces act as reactive platforms for chirality-specific processes. They are typically constructed by immobilizing chiral selectors onto materials such as metal surfaces, inorganic substrates, or polymer nanochannels, endowing the system with distinct chiral recognition capabilities. A key advantage of these interfaces is the confinement effect, which enforces highly ordered arrangements of chiral selectors, enhancing recognition while minimizing entropy increases. This contrasts sharply with solution-phase molecular recognition, where random molecular motion dominates. Moreover, chiral interface materials are cost-effective and operationally straightforward, making them widely applicable in areas such as electrochemical sensing and biosensing [77,78].
Among various chiral selectors, macrocyclic hosts, particularly cyclodextrins, pillararenes, and calixarenes, are widely used in interface construction owing to their well-defined cavities and intrinsic enantioselective recognition capabilities. A common functionalization strategy involves the precise immobilization of these macrocycles onto carrier surfaces via physical adsorption or covalent/non-covalent interactions. This surface modification not only creates an active interface with a tailored chiral microenvironment but also promotes the ordered assembly of macrocycles into a spatially organized recognition layer enriched with multiple binding sites, thereby enhancing chiral discrimination efficiency (Fig. 15).
In 2023, Luo and co-workers reviewed strategies for improving the chiral recognition performance of β-CD using functional materials, including surface immobilization, structural modification, and hybrid material formation. These materials, with their large surface areas, enabled the creation of multiple chiral active sites and high-density immobilization of chiral selectors. Additionally, they amplified output signals, significantly improving the detection sensitivity of β-CD-based enantiomer discrimination [19].
Overall, this review systematizes recent advances in the construction and application of diverse chiral interfaces, focusing on their roles in chiral recognition, separation, and catalysis, and outlines promising directions for future development. Given the persistent challenge of precisely discriminating structurally complex chiral molecules, the design of chiral sensors with high sensitivity, selectivity, and operational reliability is crucial for both current and future research.
In recent years, rapid advancements in artificial intelligence (AI) have opened new avenues for fundamental scientific research. Among its branches, machine learning (ML), a core area of computer science, has demonstrated significant potential across diverse domains, including drug discovery, chemical synthesis optimization, molecular design, novel material development, and spectral analysis [75,79]. This potential is particularly pronounced in chemistry, where ML is increasingly leveraged to drive innovation. One striking example is chiral recognition, where machine learning offers considerable promise. By utilizing existing experimental data and molecular descriptors, ML enables the construction of predictive models to evaluate the feasibility and efficiency of chiral recognition processes.
For instance, Lawtrakul and colleagues combined molecular docking with semiempirical AM1 calculations to investigate the enantioseparation mechanisms of methylated β-CD derivatives toward butylone enantiomers (G17, Fig. 16a) [80]. Their study revealed that butylone adopts two distinct conformations within the cyclodextrin cavity, with methylation at O2, O3, or O6 positions governing conformational preference and complex stability (Fig. 16b). Importantly, specific methylation patterns were found to control enantioselective complexation, providing a predictive framework applicable to > 50 synthetic cathinone derivatives.
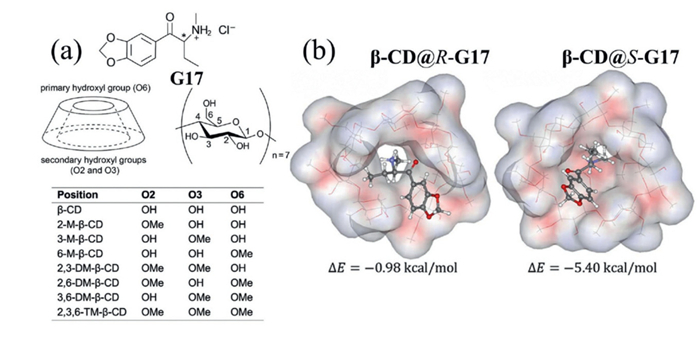
To summarize, the molecular modeling performed in this work provides a clear understanding of the mechanisms governing chiral discrimination of butylone by methylated β-CDs, with results in excellent agreement with experimental observations. These computational insights can be extended to other chiral synthetic cathinones, enabling more accurate prediction of enantioselective behavior and offering a sustainable, resource-efficient alternative to extensive experimental screening. This theoretical approach supports the rational design of chiral selectors and offers the potential to substantially reduce experimental costs in the development of separation technologies.
In supramolecular chemistry, the development of efficient chiral recognition materials remains a highly active area of research. While numerous reviews have examined small molecule-based chiral recognition systems, macrocyclic compounds offer exceptional potential for enantioselective applications owing to their deep cavities, intrinsic chirality, and distinctive optoelectronic properties (Table 1). These features allow macrocyclic hosts to achieve selectivity and sensitivity that often surpass those of small-molecule systems. As a result, increasing efforts are directed toward enhancing the chiral discrimination performance of macrocycles, advancing their applications in chemical sensing, pharmaceutical separation, and biomedical analysis.
 DownLoad:
CSV
DownLoad:
CSV
| Macrocyclic host | Targeted guest | Method | Host-guest binding constants Ka (L/mol) | Enantioselectivity KR/KS |
| 8 | G1 | Fluorometric titration | (4.44 ± 0.14) × 103 (1.29 ± 0.09) × 102 |
34.4/1a |
| 10 | G3 | NMR titration | (4.10 ± 0.30) × 104 (3.10 ± 0.30) × 103 |
13.2/1 for S-10 |
| 14 | R-G5 | NMR titration | (7.63 ± 0.75) × 103 (1.96 ± 0.18) × 103 |
3.89/1 for P-14 |
| 15 | S-G6 | NMR titration | (0.25 ± 0.01) × 103 (1.66 ± 0.21) × 103 |
1/6.67 for M-15 |
| 16 | S-G7 | ITC titration | (1.52 ± 0.03) × 104 (1.95 ± 0.11) × 105 |
1/12.89 for M-16 |
| 17 | S-G8/S-G9 | Fluorometric titration | (5.30 ± 1.10) × 103 (9.90 ± 2.50) × 104 (2.00 ± 0.40) × 109 (3.10 ± 0.50) × 1010 |
1/18.7 for (S-G8)a 1/15.5 for (S-G9)a |
| 19 | G10 | ITC titration | (1.21 ± 0.04) × 104 (2.95 ± 0.33) × 102 |
41/1 for (S-G10)a |
| 20 | G11 | NMR titration | (0.31 ± 0.03) × 102 (5.87 ± 0.73) × 102 |
1/18.7 for (R-G11)a |
| 25 | G12 | Fluorometric titration | (4.33 ± 0.16) × 102 (4.04 ± 0.17) × 103 |
1/9.35 for (L-G12)b |
| 27 | G16 | Fluorometric titration | (1.79 ± 0.32) × 103 (1.36 ± 0.11) × 104 |
1/7.6 |
| a For the same chiral guest, the enantioselectivity is calculated through dividing the association constant of the R-configured host by that of the S-configured host. b ΔΔ-configured host by that of the ΛΛ-configured host. | ||||
Several strategies have been employed to enhance enantioselectivity in macrocyclic systems: (ⅰ) Structural modification through the introduction of intramolecular binding sites and chiral units; (ⅱ) Synergistic multi-force design leveraging hydrogen bonding, electrostatic, and hydrophobic interactions; (ⅲ) Dynamic conformational control via allosteric or stimulus-responsive mechanisms; (ⅳ) Construction of biomimetic intelligent materials; (ⅴ) Machine learning (ML)-assisted approaches. These strategies effectively improve chiral recognition capabilities and enantioselectivity.
Despite these advances, the field faces several challenges: (1) Complexity of recognition mechanisms, as precise control remains difficult due to the interplay of multiple non-covalent interactions; (2) Limited environmental adaptability, with recognition stability and selectivity often compromised in aqueous or complex physiological environments; (3) Scarcity of versatile platforms, as universal macrocyclic systems capable of recognizing diverse chiral molecules remain rare.
To address these challenges, future research should focus on developing multifunctional synergistic recognition systems that integrate complementary mechanisms, advancing intelligent design strategies such as ML-assisted structural optimization, establishing high-throughput screening methods for rapid performance evaluation, and promoting practical applications in biomedicine (e.g., chiral drug separation) and environmental monitoring (e.g., chiral pollutant analysis). This progress is expanding the utility of macrocycle-based systems in high-precision applications, including enantioselective chemical sensing, chiral separation techniques (e.g., chromatographic and membrane-based processes), and real-time monitoring in pharmaceutical development and biomedical analysis. A critical challenge lies in achieving reliable performance in complex biological environments; therefore, future efforts should prioritize the design of water-soluble chiral macrocyclic systems capable of stable operation in biological media such as serum or plasma, where high protein concentrations, variable pH, and competitive binding can severely compromise recognition accuracy. Additionally, significant potential exists in the development of macrocycle-based chiral separation membranes for scalable and energy-efficient enantiopurification. Collectively, these advancements position macrocycle-based chiral recognition platforms to play an increasingly central role in next-generation analytical tools and therapeutic strategies.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Chunhong Liu: Writing – original draft, Conceptualization. Xiaotong Liang: Investigation. Wanhua Wu: Supervision, Investigation. Zhouyu Wang: Supervision, Investigation. Cheng Yang: Writing – review & editing, Supervision, Investigation.
This work was supported by the National Natural Science Foundation of China (Nos. 22471182, 22271201, 92056116, 22422108, 22171194, 22201194), the Fundamental Research Funds for the Central Universities (No. 20826041D4117) and Science & Technology Department of Sichuan Province (No. 2025ZNSFSC0125), Science and Technology Department of Yibin Program (2023SF009). The authors would like to thank the Comprehensive Training Platform of Specialized Laboratory, College of Chemistry.
M. Zhang, M. Kim, W. Choi, et al., Prog. Polym. Sci. 151 (2024) 101800. doi: 10.1016/j.progpolymsci.2024.101800
X. Niu, Y. Liu, R. Zhao, et al., Adv. Colloid Interface Sci. 335 (2025) 103342. doi: 10.1016/j.cis.2024.103342
G. Wang, A. Qu, M. Sun, et al., Acc. Mater. Res. 5 (2024) 1221–1236. doi: 10.1021/accountsmr.4c00158
H. Jędrzejewska, A. Szumna, Chem. Rev. 117 (2017) 4863–4899. doi: 10.1021/acs.chemrev.6b00745
J. Sun, G. Hu, L. Jiang, et al., Small 21 (2025) 2410895. doi: 10.1002/smll.202410895
Y.X. Zhang, F.Q. Zhang, A.P. Peng, et al., Chin. Chem. Lett. 37 (2026) 111500. doi: 10.1016/j.cclet.2025.111500
F. Ma, H. Dou, D. Luo, et al., Chin. Chem. Lett. 37 (2026) 111632. doi: 10.1016/j.cclet.2025.111632
R. Cui, Z. Wang, L. Li, et al., Anal. Chim. Acta 1318 (2024) 342960. doi: 10.1016/j.aca.2024.342960
V. Dašková, D. Padín, B.L. Feringa, J. Am. Chem. Soc. 144 (2022) 23603–23613. doi: 10.1021/jacs.2c10911
Y. Che, K. Li, C. Xie, et al., Adv. Funct. Mater. 35 (2025) 2506753. doi: 10.1002/adfm.202506753
X.N. Han, Y. Han, C.F. Chen, Chem. Soc. Rev. 52 (2023) 3265–3298. doi: 10.1039/d3cs00002h
C.B. Du, Y.J. Long, X.N. Han, et al., Chem. Commun. 60 (2024) 13492–13506. doi: 10.1039/d4cc05084c
X. Mao, S. Zhang, Q. Shi, et al., Chin. Chem. Lett. 36 (2025) 110950. doi: 10.1016/j.cclet.2025.110950
X. Liang, W. Liang, W. Wu, et al., Chem. Commun. 61 (2025) 7573–7584. doi: 10.1039/d5cc00828j
C. Liu, L. Wang, Z. Wang, et al., Supramol. Mater. 4 (2025) 100107.
X. Liang, W. Liang, P. Jin, et al., Chemosensors 9 (2021) 279. doi: 10.3390/chemosensors9100279
X. Liang, Y. Shen, D. Zhou, et al., Chem. Commun. 58 (2022) 13584–13587. doi: 10.1039/d2cc05690a
J. Ji, X. Wei, W. Wu, et al., Acc. Chem. Res. 56 (2023) 1896–1907. doi: 10.1021/acs.accounts.3c00234
J. Guo, J. Hou, J. Hu, et al., Chem. Commun. 59 (2023) 9157–9166. doi: 10.1039/d3cc01962d
M.V. Rekharsky, Y. Inoue, J. Am. Chem. Soc. 124 (2002) 813–826. doi: 10.1021/ja010889z
Q. Huang, L. Jiang, W. Liang, et al., J. Org. Chem. 81 (2016) 3430–3434. doi: 10.1021/acs.joc.6b00130
Y. Mao, J. Ma, J. Ji, et al., Chin. Chem. Lett. 35 (2024) 109927. doi: 10.1016/j.cclet.2024.109927
X. Sun, S. Li, R. Wang, et al., Chin. Chem. Lett. 36 (2025) 110806. doi: 10.1016/j.cclet.2024.110806
B.J. Shorthill, C.T. Avetta, T.E. Glass, J. Am. Chem. Soc. 126 (2004) 12732–12733. doi: 10.1021/ja047639d
C.T. Avetta, B.J. Shorthill, C. Ren, et al., J. Org. Chem. 77 (2012) 851–857. doi: 10.1021/jo201791a
L.P. Yang, X. Wang, H. Yao, et al., Acc. Chem. Res. 53 (2020) 198–208. doi: 10.1021/acs.accounts.9b00415
H. Chai, Z. Chen, S.-H. Wang, et al., CCS Chem. 2 (2020) 440–452. doi: 10.31635/ccschem.020.202000160
X. Yang, W. Jiang, J. Am. Chem. Soc. 146 (2024) 3900–3909. doi: 10.1021/jacs.3c11492
S.M. Wang, Y.F. Wang, L. Huang, et al., Nat. Commun. 14 (2023) 5645. doi: 10.1038/s41467-023-41390-8
K. Xu, B. Li, S. Yao, et al., Angew. Chem. Int. Ed. 61 (2022) e202203016. doi: 10.1002/anie.202203016
L. Pu, Chem. Rev. 124 (2024) 6643–6689. doi: 10.1021/acs.chemrev.4c00132
R. Fu, Q.Y. Zhao, H. Han, et al., Angew. Chem. Int. Ed. 62 (2023) e202315990. doi: 10.1002/anie.202315990
G. Sun, X. Zhang, Z. Zheng, et al., J. Am. Chem. Soc. 146 (2024) 26233–26242. doi: 10.1021/jacs.4c07924
C. Ruan, Z. Li, W. Lin, et al., Org. Lett. 26 (2024) 4122–4126. doi: 10.1021/acs.orglett.4c01243
K. Diao, W. Xie, M. Wen, et al., Org. Lett. 27 (2025) 9738–9743. doi: 10.1021/acs.orglett.5c03042
T. Ogoshi, S. Kanai, S. Fujinami, et al., J. Am. Chem. Soc. 130 (2008) 5022–5023. doi: 10.1021/ja711260m
J. Ji, Y. Li, C. Xiao, et al., Chem. Commun. 56 (2020) 161–164. doi: 10.1039/c9cc08541f
F. Gao, X. Yu, L. Liu, et al., Chin. Chem. Lett. 34 (2023) 107558. doi: 10.1016/j.cclet.2022.05.072
Y. Lv, C. Xiao, J. Ma, et al., Chin. Chem. Lett. 35 (2024) 108757. doi: 10.1016/j.cclet.2023.108757
J. Ji, X. Wei, W. Wu, et al., J. Am. Chem. Soc. 144 (2022) 1455–1463. doi: 10.1021/jacs.1c13210
C. Liu, J. Yao, C. Xiao, et al., Org. Lett. 23 (2021) 3885–3890. doi: 10.1021/acs.orglett.1c01016
R. Huang, X. Wei, P. Wang, et al., Org. Lett. 26 (2024) 1405–1409. doi: 10.1021/acs.orglett.3c04367
T. Zhao, J. Yi, C. Liu, et al., Angew. Chem. Int. Ed. 62 (2023) e202302232. doi: 10.1002/anie.202302232
C. He, R. Huang, L. Wei, et al., Chin. Chem. Lett. 36 (2025) 110103. doi: 10.1016/j.cclet.2024.110103
Y. Ma, X. Ji, F. Xiang, et al., Chem. Commun. 47 (2011) 12340–12342. doi: 10.1039/c1cc15660h
T. Zhao, W. Wu, C. Yang, Chem. Commun. 59 (2023) 11469. doi: 10.1039/d3cc03829g
L. Liu, C. Ma, Q. He, et al., Org. Chem. Front. 8 (2021) 4144–4152. doi: 10.1039/d1qo00525a
L. Wei, F. Gao, C. He, et al., Sci. China Chem. 66 (2023) 3546–3554. doi: 10.1007/s11426-023-1786-5
Y. Shen, W. Wu, Z. Yu, et al., Sci. China Chem. 67 (2024) 2842–2863. doi: 10.1007/s11426-024-2008-y
X. Wei, J. Ji, Y. Nie, et al., Nat. Protoc. 17 (2022) 2494–2516. doi: 10.1038/s41596-022-00722-6
Z. Yan, Q. Huang, W. Liang, et al., Org. Lett. 19 (2017) 898–901. doi: 10.1021/acs.orglett.7b00057
L. Dai, W. Wu, W. Liang, et al., Chem. Commun. 54 (2018) 2643–2646. doi: 10.1039/c8cc00840j
H. Zhao, J. Wang, H. Li, et al., ACS Appl. Mater. Interfaces 17 (2025) 15067–15079. doi: 10.1021/acsami.4c21850
J. Li, Y. Han, C.F. Chen, Molecules 26 (2021) 536. doi: 10.3390/molecules26030536
G.W. Zhang, P.F. Li, Z. Meng, et al., Angew. Chem. Int. Ed. 55 (2016) 5304–5308. doi: 10.1002/anie.201600911
X.N. Han, P.F. Li, Y. Han, et al., Angew. Chem. Int. Ed. 61 (2022) e202202527. doi: 10.1002/anie.202202527
R. Fu, D.Y. Li, J.H. Tian, et al., Angew. Chem. Int. Ed. 63 (2024) e202406233. doi: 10.1002/anie.202406233
J. Tröger, J. Prakt. Chem. 36 (1887) 225–245. doi: 10.1002/prac.18870360123
C. Shi, G. Xu, H. Qiu, et al., Chem. Commun. 61 (2025) 2450–2467. doi: 10.1039/d4cc05134c
X. Liang, T. Zhao, Y. Shen, et al., Angew. Chem. Int. Ed. 64 (2024) e202416975.
Y. Qian, S. Huang, Y. Su, et al., Chin. Chem. Lett. 37 (2026) 111354. doi: 10.1016/j.cclet.2025.111354
T. Kida, T. Iwamoto, H. Asahara, et al., J. Am. Chem. Soc. 135 (2013) 3371–3374. doi: 10.1021/ja312367k
R. Álvarez-Yebra, R. López-Coll, P. Galán-Masferrer, et al., Org. Lett. 25 (2023) 3190–3194. doi: 10.1021/acs.orglett.3c00463
Y. Yang, Y. Du, A.W. Heard, et al., Nat. Synth. 4 (2025) 537–551. doi: 10.1038/s44160-025-00799-3
Y. Shen, X. Liang, T. Ma, et al., Angew. Chem. Int. Ed. 64 (2025) e202504211. doi: 10.1002/anie.202504211
S.M. Wang, H. Nian, Y.F. Wang, et al., Sci. China Chem. 68 (2025) 369–376. doi: 10.1007/s11426-024-2120-0
K.M. Kim, G. Song, S. Lee, et al., Angew. Chem. Int. Ed. 59 (2020) 22475–22479. doi: 10.1002/anie.202011230
G. Song, K.M. Kim, S. Lee, et al., Chem. Asian J. 16 (2021) 2958–2966. doi: 10.1002/asia.202100768
J. Jiao, J. Dong, Y. Li, et al., Angew. Chem. Int. Ed. 60 (2021) 16568–16575. doi: 10.1002/anie.202104111
H. Yang, S. Yang, X.F. Wu, et al., Green Synth. Catal. 6 (2025) 81–85.
D. Wang, J. Yao, W. Zhang, et al., Green Synth. Catal. 5 (2024) 324–328.
J. Yao, W. Wu, C. Xiao, et al., Nat. Commun. 12 (2021) 2600. doi: 10.1038/s41467-021-22880-z
J. Yao, W. Wu, W. Liang, et al., Angew. Chem. Int. Ed. 56 (2017) 6869–6873. doi: 10.1002/anie.201702542
C. Xiao, W. Wu, W. Liang, et al., Angew. Chem. Int. Ed. 59 (2020) 8094–8098. doi: 10.1002/anie.201916285
C. Liu, X. Chen, Y. Shen, et al., Sci. China Chem. 68 (2025) 610–621. doi: 10.1007/s11426-024-2190-5
J. Motoori, T. Kinoshita, H. Chai, et al., ACS Nano Sci. Au 4 (2024) 435–442. doi: 10.1021/acsnanoscienceau.4c00052
W. Xu, M. Cheng, S. Zhang, et al., Chem. Commun. 57 (2021) 7480–7492. doi: 10.1039/d1cc01501j
W. Liang, Y. Rong, L. Fan, et al., J. Mater. Chem. C 6 (2018) 12822–12829. doi: 10.1039/c8tc04448a
L. Fu, R. Wang, Q. Zhu, et al., J. Chem. Theory Comput. 19 (2023) 4364–4376. doi: 10.1021/acs.jctc.2c01265
L. Lawtrakul, P. Toochinda, ACS Omega 10 (2025) 2003–2011. doi: 10.1021/acsomega.4c07879

Figure 1 (a) Syn and anti-naphthalene dimers. (b) Chemical structures of syn amide-linked naphthotube 1 and syn allyl-linked naphthotube 2.

Figure 2 Chemical structures of (a) chiral amide naphthotubes 3, (b) chiral thiourea naphthotubes 4–5, chiral amide naphthotubes 6–9 and (c) chiral guest G1.

Figure 3 (a) Schematic illustration of modular introduction of endo-binding sites in macrocycle cavity. Copied with permission [30]. Copyright 2022, the Wiley. (b) Chemical structures of BINOL-based chiral macrocycles 10 with their chiral guests G2/G3. Copied with permission [33]. Copyright 2024, American Chemical Society.

Figure 4 (a) Chemical structures of pillarurilarenes 11/12. The two-dimensional (2D) packing mode of (b) ethyl acetate@11, (c) ethyl acetate@12. Copied with permission [34]. Copyright 2024, American Chemical Society.

Figure 5 Chemical structures of (a) chiral tyrosine-modified pillar[5]arenes 13 and (b) chiral guests G4. (c) Chiral recognition model of 13 with an aromatic amine. Copied with permission [47]. Copyright 2021, the Royal Society of Chemistry.

Figure 6 (a) Chemical structures of L-Cys/β-CD composite and L/D-Trp. (b) SEM images of Ⅰ: β-CD; Ⅱ: L-Cys; Ⅲ/Ⅳ: L-Cys/β-CD. Ⅴ: TEM and Ⅵ: low-magnification TEM image and EDS elemental mapping of C, N, O, and S in L-Cys/β-CD. (c) DPVs of different modified electrodes. Copied with permission [53]. Copyright 2025, the American Chemical Society.

Figure 7 Chemical structures of (a) chiral helic[1]triptycene[3]arenes 14 and their chiral guest R-G5. (b) 2,6-Helic[6]arenes 15 and their chiral guest S-G6. (c) water-soluble octopus[3]arenes 16 and their chiral guest S-G7.

Figure 9 Chemical structures of Tröger's base (TB)-containing macrocyclic arenes (a) 18 and (c) 19 and chiral guest G10. (b) Crystal structure of 18. (d) IGMH maps of the energy-minimized conformation of R-G10@(R/S)-19. Copied with permission [60]. Copyright 2024, Wiley.

Figure 10 Chemical structures of calix[5]arene-based receptor 20 with chiral guests G11.

Figure 11 (a) Chemical structures and (b) crystal structures of phenacetin[3]arenes. (c) Schematic representation of the equilibria between C3 and F conformations of phenacetin[3]arenes and chiral guests. Copied with permission [65]. Copyright 2025, Wiley.

Figure 13 (a) Chemical structures of triple-stranded helicates 23–26 and their chiral guests G12/G13; (b) Energy-minimized DFT calculations of ΔΔ−25@G13 host-guest complexes. Copied with permission [69]. Copyright 2021, Wiley.

Figure 14 (a) Chemical structures of chiral naphthotubes R2,S2/S2,R2–27 and their chiral guests G14-G16. (b) Schematic illustration of dynamic flapping (left) and turn-on mechanism of 27 (right). (c) Dynamic stretch of 27 stimulated by guest inclusion upon hydrostatic pressurization. Copied with permission [76]. Copyright 2024, American Chemical Society.

Figure 15 Schematic illustration of host–guest enhanced chiral discrimination on chiral interfaces. Copied with permission [77]. Copyright 2021, Royal Society of Chemistry.

Figure 16 (a) Chemical structures of methylated β-CD derivatives and chiral guest G17. (b) Molecular docking structures β-CD@(R/S)-G17. Copied with permission [80]. Copyright 2025, American Chemical Society.
Table 1. Summary of the enantioselectivity of macrocyclic hosts for chiral guests.
| Macrocyclic host | Targeted guest | Method | Host-guest binding constants Ka (L/mol) | Enantioselectivity KR/KS |
| 8 | G1 | Fluorometric titration | (4.44 ± 0.14) × 103 (1.29 ± 0.09) × 102 |
34.4/1a |
| 10 | G3 | NMR titration | (4.10 ± 0.30) × 104 (3.10 ± 0.30) × 103 |
13.2/1 for S-10 |
| 14 | R-G5 | NMR titration | (7.63 ± 0.75) × 103 (1.96 ± 0.18) × 103 |
3.89/1 for P-14 |
| 15 | S-G6 | NMR titration | (0.25 ± 0.01) × 103 (1.66 ± 0.21) × 103 |
1/6.67 for M-15 |
| 16 | S-G7 | ITC titration | (1.52 ± 0.03) × 104 (1.95 ± 0.11) × 105 |
1/12.89 for M-16 |
| 17 | S-G8/S-G9 | Fluorometric titration | (5.30 ± 1.10) × 103 (9.90 ± 2.50) × 104 (2.00 ± 0.40) × 109 (3.10 ± 0.50) × 1010 |
1/18.7 for (S-G8)a 1/15.5 for (S-G9)a |
| 19 | G10 | ITC titration | (1.21 ± 0.04) × 104 (2.95 ± 0.33) × 102 |
41/1 for (S-G10)a |
| 20 | G11 | NMR titration | (0.31 ± 0.03) × 102 (5.87 ± 0.73) × 102 |
1/18.7 for (R-G11)a |
| 25 | G12 | Fluorometric titration | (4.33 ± 0.16) × 102 (4.04 ± 0.17) × 103 |
1/9.35 for (L-G12)b |
| 27 | G16 | Fluorometric titration | (1.79 ± 0.32) × 103 (1.36 ± 0.11) × 104 |
1/7.6 |
| a For the same chiral guest, the enantioselectivity is calculated through dividing the association constant of the R-configured host by that of the S-configured host. b ΔΔ-configured host by that of the ΛΛ-configured host. | ||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: