Citation:
Yuan Tang, Chunyang Wang, Yuchen Guo, Liang Mao, Xiaoyan Cai, Yanfang Li, Boxin Liu, Xin Tan, Jinhua Ye, Tao Yu. Modulating coordination environment of Bi active center atom for efficient photocatalytic water oxidation[J]. Chinese Chemical Letters,
2026, 37(7): 112275.
doi:
10.1016/j.cclet.2025.112275
Modulating coordination environment of Bi active center atom for efficient photocatalytic water oxidation
English
Modulating coordination environment of Bi active center atom for efficient photocatalytic water oxidation
School of Chemical Engineering and Technology, Tianjin University, Tianjin 300350, China
b.
School of Environmental Science and Engineering, Tianjin University, Tianjin 300350, China
c.
School of Materials Science and Physics, China University of Mining and Technology, Xuzhou 221116, China
d.
Research Center for Solar Driven Carbon Neutrality, Hebei University, Baoding 071002, China
e.
China Wuzhou Engineering Group Corporation Limited, Beijing 100053, China
f.
Advanced Catalytic Materials Research Center, School of Material Science and Engineering, Tianjin University, Tianjin 300072, China
* Corresponding author. E-mail address: yutao@tju.edu.cn (T. Yu). 1 These authors contributed equally to this work.
Received Date:
27 June 2025 Accepted Date:
11 December 2025 Revised Date:
22 November 2025 Available Online:
15 July 2026
Abstract:
Photocatalytic water oxidation is considered as the bottleneck step for overall artificial photosynthesis, due to the slow photocarrier transfer kinetics and climbing thermodynamics. Developing the electronic structure of catalytic with more dynamic-thermodynamic advantages is crucial to deal with the above dilemma and realize the photocatalytic water oxidation. This work successfully explores an S-induced modification strategy to induce electronic delocalization and non-equilibrium states for unique Bi active sites. The introduction of V4+ species leads to asymmetric coordination of Bi atoms in the S-Bi-V4+ structure to provide more nonlocalized electrons, which initiates a concerted electron transfer in the system. The fine regulation on the local electron density and energy level structure of the Bi active site optimizes the reaction energy barriers to construct thermodynamically more favourable active centers for water oxidation. The optimized 2% S-BVO catalyst achieves outstanding water oxidation rate of 70.25 μmol/h (40 mg catalyst) triggering up to 11.06% apparent quantum efficiency at 420 nm. This work elucidates the influence of synergistic electron interaction of asymmetric coordination structure on both photocarrier transfer kinetics and reaction thermodynamic.
Nowadays, with the deepening contradiction between energy crisis and environmental pollution, the transformation of energy structure to low carbonization is becoming more and more serious against the backdrop of globalization [1-4]. The solar-driven photocatalytic CO2 reduction, water splitting, and biomass valorization for the production of clean and safe energy-containing chemicals are expected to step onto the stage as early as possible [5-8]. However, the realization of the above applications is largely limited by the proton-coupled electron transfer process as water oxidation reaction [9-11], due to the slow photocarrier transfer kinetics and climbing thermodynamics. Specifically, the water oxidation requires four holes with higher effective mass to participate in the reaction, and the adsorption energy of oxygen-containing intermediates should be optimized according to Sabatier principle, which is considered as a bottleneck step for achieving the high-efficiency overall artificial photosynthesis [12,13]. Thus, designing an ideal catalyst to enhance the water oxidation ability becomes quite a difficult but indispensable task to accomplish [14-16].
Through the modification of low electronegativity elements can enhance the electron cloud density around the active metal center of the catalyst, and the formation of orbital hybridization is conducive to the promotion of the charge delocalization [17,18]. Compared with other non-metallic elements such as nitrogen, fluorine and oxygen, elemental sulfur has a relatively low electronegativity and unique orbital properties, which are conducive to the formation of multi-orbital hybridization to significantly enhance the off-domain nature of the electrons, facilitating the redistribution of asymmetric charges and optimizing the local coordination environment of the active centers. Such as the 3p orbitals of S that can form multidimensional hybridization with metal d/f orbitals and O 2p orbitals to extend the range of electron delocalization, weaken the local carrier confinement, optimize the charge separation and migration efficiency, and induce asymmetric coordination to the active centers [19,20]. Such doping modification strategies can break the pristine metal-oxygen coordination symmetry and modulate the valence and orbital occupancy of the metal center, which reduces the free energy change of the water oxidation decisive step and thermodynamically drives the reaction.
As a classic model photocatalyst, BiVO4 has attracted widespread attention among various bismuth-based semiconductors photocatalysts because of suitable band gap, providential oxidation potential, easy preparation and nontoxicity [21,22]. However, BiVO4-based photocatalysts are limited in charge delocalization and photogenerated carrier separation efficiency due to insufficient electron cloud density at the metal center and excessive metal-oxygen coordination symmetry, which leads to lower photocatalytic activity. The photocatalytic properties of BiVO4 are strongly related to its crystal structures and the local polarization pronounced by crystalline polyhedron distortion would favorably affect the electron–hole separation [23,24] Through modulating the electronic environment of the active site, breaking the symmetric coordination structure and optimizing the orbital occupancy state are conducive to reducing the free energy change of the water oxidation decisive step and enhancing the reaction efficiency. Vδ+ (δ = 3 or 4) species generated during the process of synthesis, morphology modulation using surfactants, and doping engineering over BiVO4 have been reported early on, which induced the changes in crystal structure and electron distribution, but only loosely interpreted as the by-products of lattice defects construction [25,26]. Liu et al. reported the formation of unsaturated coordinate oxygen bonds at the interface, which optimized the O—O coupling energy barrier at the V active site, enhancing the efficiency of O—H bond cleavage and promoting water oxidation [27]. The development of controllable Vδ+ species construction combined with atomic-level modulation strategy can bridge the theoretical gap in the intrinsic structure of BiVO4 photocatalysts, which is of great significance for realizing highly efficient solar-driven photocatalytic water oxidation.
In this work, to reveal the importance of modulating electronic structure for water oxidation, a novel S-induced doping strategy is demonstrated to realize the introduction of S dopant coupling with V4+ atom. The introduction of S atom with low electronegativity endows the Bi-S bond with more covalent bonding property and regulates the electron with delocalization state. Furthermore, V4+ with excess electrons filled into 3d orbital forms multidimensional hybridization with O 2p, leading to the intrinsic electron migration. Under the influence of asymmetric coordination, synergistic electron interaction based on S-Bi-V4+ structure is realized and enables fine tuning of the chemical environment around Bi atom. The efficient photocarrier transfer and favorable thermodynamics of oxygen-containing intermediates at the active site synergistically achieve the outstanding water oxidation activity. 2%S-BVO possesses a 2.31-fold water oxidation activity relative to the pristine BiVO4 accompanied by an apparent quantum efficiency (AQE) of 11.06% (λ = 420 nm).
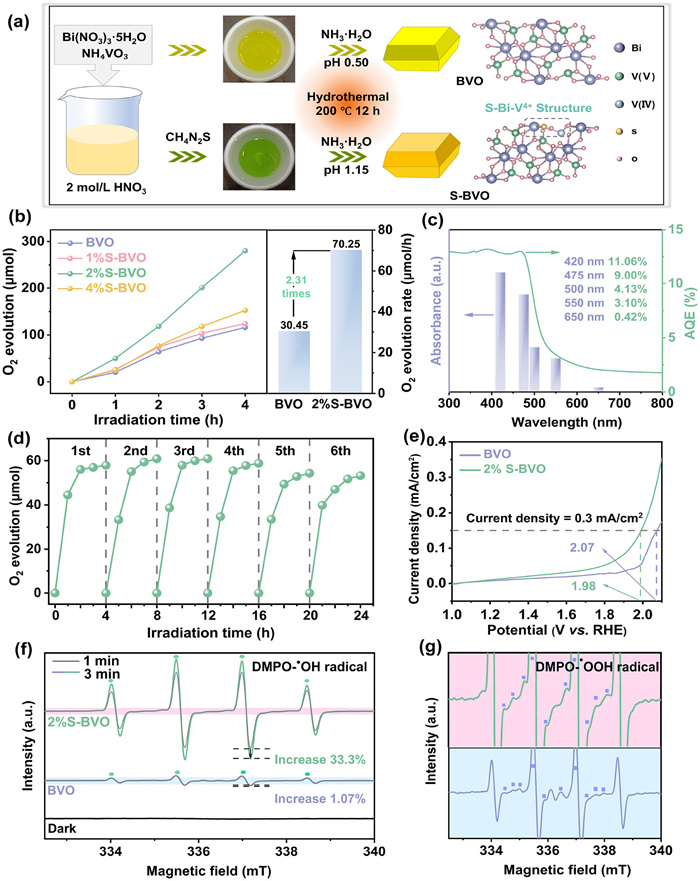
The investigation for enhanced water oxidation activity starts from the preparation of photocatalyst. The hydrothermal method to synthesis BiVO4 (BVO) and S-induced doping BiVO4 (S-BVO) are shown schematically in Fig. 1a. In the S-induced doping process, thiourea acts as both dopant and reducing agent, which contributes BVO precursor solution turns to green color owing the generation of V4+ species. To objectively evaluate the photocatalytic water oxidation activity, the relationship of photocatalyst amounts and water oxidation rates were firstly investigated to determine the stationary point. The water oxidation rate no longer follows linear progression when the amount of photocatalyst exceeds 40 mg (Fig. S1 in Supporting information), due to the incident light cannot effectively penetrate the turbid liquid layer to excite the superfluous photocatalysts [28]. The stationary point of 2%S-BVO for this reaction system is confirmed to be 40 mg. Under visible light irradiation (λ > 420 nm, 300 W Xe), the pristine BVO exhibits a low water oxidation rate of 30.45 μmol/h due to the sluggish carrier migration within BVO (Fig. 1b). By comparison, S-BVO samples show an approximately linear increasing trend with the S-induced doping amount, and 2%S-BVO possesses the highest photocatalytic water oxidation rate of 70.25 μmol/h, which is 2.31-fold higher than that of pristine BVO. The improved activity originates from the synergistic electron interaction of S-Bi-V4+ to accelerate photocarrier transfer kinetics and reaction thermodynamic. Nevertheless, the excess S-induced doping results a decrease of photocatalytic activity, since the extra S dopant and V4+ species introduce excess defects acting as recombination centers [29], which indicates the moderation of S-induced doping is indispensable for achieving the desired photocatalytic water oxidation activity.
Figure 1
Figure 1.
Photocatalytic water oxidation property. (a) Schematic diagram for the synthesis of pristine BVO and S-BVO by S-induced doping process. (b) Photocatalytic water oxidation activity of pristine BVO and S-BVO at stationary point (40 mg). (c) AQE and DRS spectrum of 2%S-BVO. (d) Cycling experiments of 2%S-BVO. (e) Linear scanning voltammogram (LSV) curves of BVO and 2%S-BVO. (f) In-situ ESR spectra of pristine BVO and 2%S-BVO (DMPO-•OH). (g) Enlarged image for in-situ ESR spectra (DMPO-•OOH).
As another key factor in the regulation of S-induced doping process, pH of precursor solution was optimized accordingly (Fig. S2 in Supporting information). The water oxidation rate of 2%S-BVO shows a volcano-shaped distribution with the increase of pH, indicating the optimal equilibrium of V4+ construction and S introduction is reached at pH 1.15, which ultimately improves the water oxidation activity (Fig. S3 in Supporting information). To evaluate the photocatalytic activity from the viewpoint of light response property, the apparent quantum efficiency (AQE) under monochromatic light irradiation of different wavelengths were carried out systematically. AQE of 2%S-BVO agrees well with the optical absorption spectrum, and the maximum value reaches 11.06% at 420 nm, demonstrating the light-driven photocatalytic water oxidation process coupled with the excellent utilization capability of photons (Fig. 1c). For the photocatalytic water oxidation system using AgNO3 as sacrificial agent, the deposition of Ag nanoparticles on the surface of photocatalyst will unavoidably hinder the light absorption, which impacts the objectivity of stability evaluations [30-32]. Therefore, the amount of AgNO3 in the solution was reduced to 70 mg and supplemented equal amount after each cycle to exclude the light shielding effect (Fig. 1d). The water oxidation rate of 2%S-BVO remains at about 87% after 24 h illumination (six cycles), indicating the satisfactory stability of as-synthesis sample. In addition, the photocatalytic oxygen evolution cycle experiment of 2% S-BVO is investigated with different sacrificial agents, which further confirms the excellent catalytic stability of 2% S-BVO (Fig. S4 in Supporting information). In addition, the composition and chemical structure of the catalyst exhibit no significant changes before and after the reaction, further demonstrating the outstanding stability of 2%S-BVO (Fig. S5 in Supporting information). Linear scanning voltammogram (LSV) curves indicate the overpotential for water oxidation of 2%S-BVO decreases significantly, indicating the water oxidation kinetic is improved by synergistic electron interactions of S-Bi-V4+ structure, reconfirming the excellent photocatalytic performance of 2%S-BVO (Fig. 1e and Fig. S6 in Supporting information).
To capture the pivotal intermediates in water oxidation and investigate the impact of synergistic electron interactions on the generation of oxygen-containing intermediates under visible light irradiation, in-situ electron spin resonance (ESR) was performed. By taking DMPO as the spin trapping agent, no significant radical signal is detected in dark. After 1 min irradiation, both BVO and 2%S-BVO display four characteristic peaks attributing to DMPO-•OH adduct (Fig. 1f) [33,34]. Here, •OH radicals originate from the oxidation of OH−/H2O by photogenerated holes, indicating the band structure of both BVO and 2%S-BVO can activate and drive the photocatalytic water oxidation under visible light irradiation. 2% S-BVO possesses obvious intermediate signals and increases by 33.3% after 3 min, whereas BVO only exhibits a marginal improvement of 1.07%. More intense signals of 2%S-BVO indicate more powerful water oxidation activity of 2%S-BVO, in accord with the above water oxidation tests. Signals for DMPO-•OOH in the enlarged images are observed simultaneously (Fig. 1g), and the peak intensity increases obviously from BVO to 2%S-BVO, also demonstrating 2%S-BVO possesses a more superior photocatalytic water oxidation activity [35].
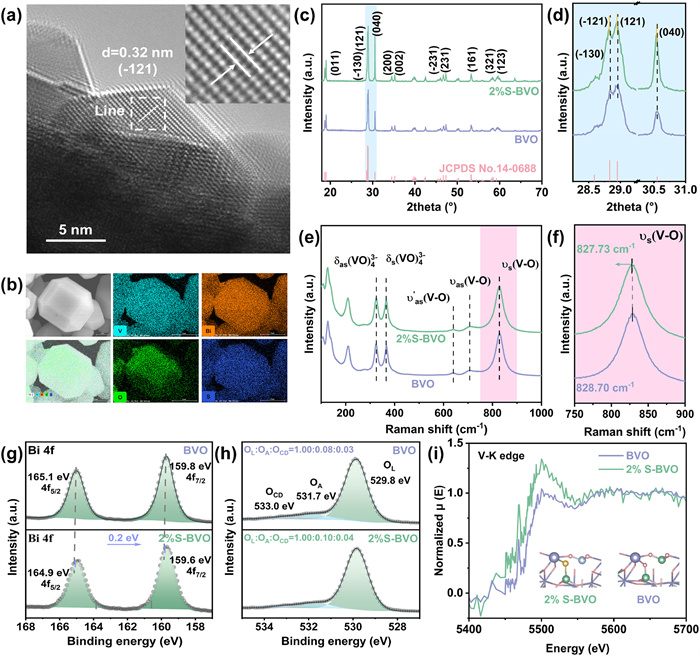
The morphology of as-synthesized samples was characterized by scanning electron microscopy (SEM). The pristine BVO presents a typical decahedral structure with well-defined crystal faces (Fig. S7a in Supporting information). After S-induced doping reaction, 1%S-BVO and 2%S-BVO retain the mentioned morphology (Figs. S7b and c in Supporting information), which enables the spatial separation of photogenerated electrons and holes. However, the morphology changes drastically into irregular clusters with the S-induced doping amount raised to 4% (Fig. S7d in Supporting information), which inevitably leads to the decrease of photocarrier transfer efficiency, indicating the moderate S-induced doping amount is of great significance to the enhancement of photocarrier transfer kinetic. The crystal structures of samples were further analyzed by transmission electron microscopy (TEM). The lattice spacing of 0.31 nm at the edge of BVO is consistent with (1¯21) plane of monoclinic scheelite BiVO4 (Fig. S8 in Supporting information) [36]. Notably, the lattice spacing of (1¯21) plane is enlarged from 0.31 nm to 0.32 nm, which is ascribed to the S-induced doping effect (Fig. 2a and Fig. S9 in Supporting information). Dense and well-distributed Bi, V, and O elements are observed in 2%S-BVO via the elemental mappings, while the S elements are relatively sparse but still homogeneously distributed (Fig. 2b and Table S1 in Supporting information), indicating the success of S-induced doping.
Figure 2
Figure 2.
Morphology and structural characterization. (a) HRTEM image of 2%S-BVO. (b) Elements mappings of 2%S-BVO. (c) XRD patterns of pristine BVO and 2%S-BVO. (d) Enlarged image for XRD patterns. (e) Raman spectra of pristine BVO and 2%S-BVO. (f) Enlarged image for Raman spectra. (g) XPS spectra of Bi 4f. (h) XPS spectra of O 2p (i) Normalized V K-edge XANES μ(E) spectra of pristine BVO and 2%S-BVO.
To comprehensively identify the crystal phase after S-induced doping process, X-ray diffraction (XRD) was conducted. XRD diffraction peaks of pristine BVO and 2%S-BVO located at 19.0°, 25.6°, 28.9°, 30.5°, 34.5°, 45.6°, 53.3°, 58.5° and 59.3° can be respectively assigned to (011), (−130), (121), (040), (200), (231), (161), (321) and (123) planes of monoclinic scheelite BiVO4 (JCPDS #14–0688) (Fig. 2c and Fig. S10 in Supporting information). The relative intensity of XRD peak of (040) plane increases with S dopant, indicating the S-induced doping effect induces changes in the crystal unit growth inclination of the BVO structure. Slight shifts to lower degree of (−121), (121) and (040) diffraction peaks are observed (Fig. 2d), demonstrating S atoms incorporate into the lattice of BVO in view of the larger radius of S (1.02 Å) compared to O (0.70 Å), according to the Bragg equation. Above XRD analysis matches well with the lattice expansion measured by TEM. In Raman spectra, the intense bands centered at 326 and 365 cm−1 are assigned to the asymmetric bending vibration mode and symmetric bending vibration mode of VO4 tetrahedral unit, and the intense bands centered at 707 and 640 cm−1 are attributed to the asymmetric stretching modes of the V-O bonds (Fig. 2e and Fig. S11 in Supporting information) [37]. Peculiarly, the intense band located at 820–830 cm−1 can pinpoint crucial variations of V-O bond corresponding to the symmetric stretching mode. In the enlarged image (Fig. 2f), the S-induced doping decreases the shift position of V-O vibrational mode from 828.70 cm−1 to 827.19 cm−1 (Table S1 in Supporting information) [38]. Based on the above functional relationship, the Raman stretching frequencies and the respective metal−oxygen bond lengths have an inverse relationship, as summarized in Table S2 (Supporting information). The S-induced doping effect lengthens the V-O bond from 1.6942 Å (BVO) to 1.6952 Å (4% S-BVO), which can be attributed to the coordination behavior of the exogenous S dopant with matrix atoms and the electronic structure changes caused by the construction of V4+ species. No impurity peak belonging to S-containing compound is observed in XRD and Raman, implying the successful of S-induced doping for synthesizing S-BVO.
To characterize the light-absorption property and band structures, UV–vis absorption spectroscopy and electrochemical tests were implemented. All samples show obvious absorption edges around 500 nm, and Tauc plots exhibit the decreasing trend of band gap values for S-BVO with the increase of S-induced doping amount, which indicating the S-induced doping effect is responsible for enhancing the visible light absorption property (Fig. S12 in Supporting information). The flat band potentials (Efb) of BVO and 2%S-BVO obtained by Mott-Schottky plots are confirmed to be −0.03 and 0.06 V vs. Ag/AgCl and calculated as 0.167 and 0.257 V vs. NHE (Fig. S13a in Supporting information), which is nearly equal to its Fermi level (EF). For n-type semiconductors, the valence band potential (EVB) is positive about 0.2 V than EF. Thus, according to the above analyses, 2% S-BVO possesses a more positive valence band potential (2.457 V vs. NHE) than that of pristine BVO (2.377 V vs. NHE), suggesting a more advantageous water oxidization potential (Fig. S13b in Supporting information).
To probe the coordination relationship and the surface chemical state, X-ray photoelectron spectroscopy (XPS) was employed. The obvious characteristic peaks ascribed to Bi, V, and O elements are detected in both BVO and 2%S-BVO (Fig. S14a in Supporting information). For 2%S-BVO, the deconvoluted peaks of Bi 4f located at 165.0 eV and 159.7 eV are consistent with Bi3+ state (Fig. 2g) [39]. Impressively, the binding energy of Bi 4f in 2%S-BVO exhibits a negative shift of 0.2 eV comparing to pristine BVO, which indicates that the electron accumulation around Bi atom in Bi-S coordination is higher than that in Bi-O coordination, due to the electronegativity of O is greater than S. The peaks located at 162.5 eV and 161.2 eV of 2% S-BVO and 4% S-BVO are assigned to S 2p1/2 and 2p3/2 belonging to the S2− state, which suggests that the process of thiourea-modified BVO has partially doped the S atoms into the lattice to form the Bi-S and V-S coordination (Figs. S14b and S15b in Supporting information) [40]. In contrast to the V 2p XPS spectra of BVO, the characteristic peaks of V4+ corresponding to the V 2p3/2 and V 2p1/2 orbitals are detected in 2% S-BVO and 4% S-BVO, and the content of V4+ in 4% S-BVO is significantly increased based on the calculation of the peak area (Fig. S14c and S15c in Supporting information). It suggests that the reduction of the V species due to S modification provides a prerequisite for the asymmetric coordination modulation of the formation of Bi atoms in the S-BVO structure [26,41]. Three peaks of O 1s spectra centered at 529.8, 531.7 and 533.0 eV can be divided from lattice oxygen (OL: O2−), adsorbed hydroxyl or water (OA: OH−, H2O), and chemisorbed or dissociated oxygen species (OCD: O2−, O22−, O−), respectively (Fig. 2h) [42]. 2%S-BVO possesses moderately higher amount of OA and OCD comparing to pristine BVO, suggesting the surface of 2%S-BVO is comparatively more active in interacting with adsorbed hydroxyl or water than that of pristine BVO, which is beneficial to activate the reaction of water oxidation and generate oxygen species.
To further reveal the chemical state of V species, X-ray absorption spectroscopy (XAS) was collected. K-edge X-ray absorption near edge structure (XANES) spectra of V atom exhibit that 2%S-BVO possesses a positive shift of absorption edge energy comparing to pristine BVO (Fig. 2i), further demonstrating the existence of V4+ induced by S-induced doping reaction, which is indispensable to the regulation of electronic structure.
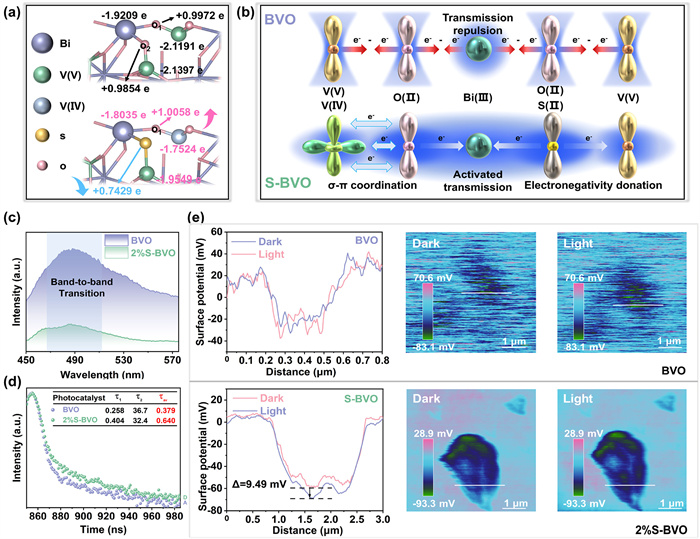
The electronic structure directly contributes to the ability of separation and transfer kinetics of photocarrier. To verify the contribution of S-induced doping effect to the regulation of electronic structure, density functional theory (DFT) calculations were implemented. The tendency of S doping and V4+ formation in different sites of BVO is discussed, and the subsequent calculations are based on the stable structure with the lowest formation energy (Fig. S16 in Supporting information). Bader charge was hired to further analyze the electron cloud density changes in spatial and quantitative after S-induced doping reaction (Fig. 3a). Comparing with V5+ atom (−2.1191 e) in pristine BVO, V4+ atom (−1.7524 e) of S-BVO possesses more electrons, indicating its lower valence. Compared to more distant V atoms, the V directly coordinated to S gains 0.1848 e after doping less than the 0.3667 e of more distant atoms, which reveals an insignificant reduction in its valence. Therefore, more distant V atoms with greater valence reduction contribute more actively to the synergistic electron interaction. Hence, the charge of neighboring O1 atom increases from +0.9972 e in BVO to +1.0058 e in S-BVO, triggering electron migration toward the coordinating Bi atom. In parallel, S atom (+0.7429 e) is more difficult to bind electrons comparing to the pristine O2 atom (+0.9854 e) due to the lower ionization energy and stronger electron affinity [43], resulting in the electron migration behavior towards the ligand transition metal atoms. Due to the asymmetric coordination of Bi atom in S-Bi-V4+ structure, the electronic structure of Bi atom is finely tuned, and the redistribution of electron density at Bi and V atoms are consistent with XPS analyses.
Figure 3
Figure 3.
Photocarrier Transfer Kinetics. (a) Calculated Bader charge of surface atoms on BVO and S-BVO. (b) Schematic representation of the synergistic electron interaction among S-Bi-V4+ in S-BVO. (c) Steady-state photoluminescence spectra of pristine BVO and 2%S-BVO. (d) Time-resolved transient PL decay spectra of pristine BVO and 2%S-BVO at the excitation wavelength of 475 nm. (e) KPFM analysis of pristine BVO and 2%S-BVO. Shown from left to right are the surface photocurrent voltage variation at the sample marking position, the surface potential distribution under dark conditions, and the surface potential distribution under illumination. Δ value was obtained by subtracting the potential of the samples in dark condition from the potential under visible light irradiation.
Based on the spatial and quantitative electron distribution analyses obtained by Bader charge, and combined with empirical evidence of characterization on chemical coordination, we propose the synergistic electron interaction of S-Bi-V4+ structure (Fig. 3b). In the V-O-Bi-O-V structural unit of pristine BVO, the single hybridization between the p and s orbits results in severe electronic localization [44], which makes it difficult for electrons to transfer across the highly symmetric periodic potential field. After S-induced doping process, the excess electrons around V4+ fill in 3d orbits and form multidimensional hybridization with O 2p The above asymmetric coordination possesses a stronger interaction, leading to an electron migration behavior towards O atom as well as the neighboring Bi atom. Meanwhile, S atom with lower electronegative substitutes for O atom, motivating electron delocalize from ligand to transition metal atom. Thus, redistribution of electron density promoted by the synergistic electron interaction of S-Bi-V4+ structure breaks the local symmetry of potential field and modulates the local chemical environment of Bi atom. It constructs an efficient charge transfer channel while mediating the generation of active sites with thermodynamic advantages, which is responsible for improving the separation and transfer kinetics of photocarrier and boosting the water oxidation activity.
To further investigate the origination of the improved separation and transfer kinetics of photocarrier, photophysical and photochemical methods were used. By fitting and comparing the electrochemical impedance spectra (EIS), 2%S-BVO expresses the smallest semicircle and fitted value of Rt, demonstrating the fastest electron transfer capacity (Fig. S17 in Supporting information). The superior conductivity property of 2%S-BVO originates from the redistribution of periodic potential field induced by the introduction of S dopant and the construction of V4+ construction, which reduces the photocarrier transfer resistance. The increased response of transient photocurrent (Fig. S18 in Supporting information) demonstrates the enhanced efficiency of the separation and transfer of photocarrier. To further reveal the influences of asymmetric coordination of S-Bi-V4+ structure on photocarrier separation kinetics, Steady-state photoluminescence (PL) emission spectroscopy was performed. The pristine BVO displays a prominent emission peak at around 500 nm corresponding to band-to-band transition, and a much lower intensity is detected on 2%S-BVO due to the effectively suppressed electro-hole recombination (Fig. 3c and Fig. S19 in Supporting information). Time-resolved transient PL decay spectroscopy (TRPL) of as-prepared samples were measured at the excitation wavelength of 475 nm (Fig. 3d and Fig. S20 in Supporting information). All samples obtained through S-induced doping reaction have larger fitting attenuation values than pristine BVO, and due to the optimal balance between V4+ structure and S dopant, the longest photogenerated carrier lifetime (0.528 ns) was detected on 2%S-BVO. The lifetime of 4%S-BVO (0.223 ns) is even lower than that of 2%S-BVO, indicating that excessive S doping amount may lead to the formation of electron trap centers, and then shorten its lifetime. This indicates that the appropriate amount of S-induced doping is a prerequisite for maximizing the suppression of photocarrier recombination.
Kelvin probe force microscopy (KPFM) was hired to visualize the separation and migration behavior of photocarriers reflected in surface potential distribution. Under visible light irradiation (λ > 420 nm), pristine BVO exhibits no obvious change of surface potential (Fig. 3e), due to the recombination of photocarrier within the bulk phase, which the photogenerated electrons cannot enrich toward the surface. Contrastively, the surface potential of 2%S-BVO exhibits more significant change of 9.49 mV, indicating more photogenerated electrons converge toward the surface of 2%S-BVO, which is attributed to the efficient photocarriers migration of S-Bi-V4+ structure. In brief, the S-induced doping effect couples S dopant with V4+ construction, which forms the asymmetric coordination of S-Bi-V4+ structure and provides more delocalized electrons around asymmetric structure by synergistic electron interaction, thereby suppressing the recombination of photocarriers, promoting its migration, and thus contributing to the excellent performance of photocatalytic water oxidation of 2%S-BVO.
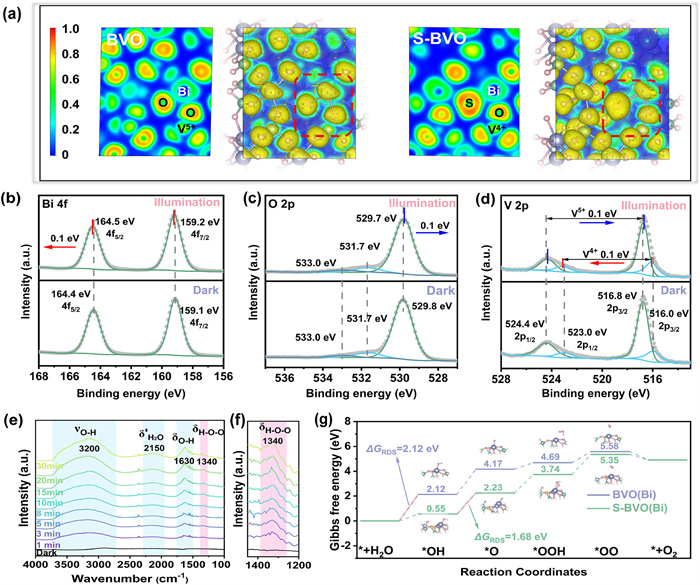
The electronic structure not only affects the electronic localization state for photocarrier transfer kinetics, but also relates the surface reaction thermodynamics. To intrinsically verify the photocatalytic mechanism from the angle of atomic-level, DFT calculations were performed accordingly. To determine the electron cloud distribution due to the formation of S-Bi-V4+ structure, electron localization function (ELF) was employed (Fig. 4a). Comparing to pristine BVO, the construction of V4+ species does not change the nature of V-O covalent bond of S-BVO. It should be noted that the presence of highly localized electrons (ELF ≈ 0.9) packed around S dopant and the expansion of electron region (ELF ≈ 0.5) revolved around Bi-S bond, indicating the uniform distribution of delocalized electrons along the Bi-S bond relative to the original Bi-O bond. The above well-designed structure of S-Bi-V4+ constructs an efficient transfer channel for accelerating the photocarriers migration. The energy band configurations of samples were logically verified by calculated density of states (DOS) (Fig. S21a in Supporting information). In pristine BVO, the conduction band is comprised of V 3d, and the valence band is comprised of the hybridization orbitals of Bi 6s−O 2p According to the PDOS of S-BVO (Fig. S21b in Supporting information), excess electrons filling induced by V4+ causes a new hybrid state located near the conduction band. Meanwhile, the existence of another hybrid energy levels mainly come from the localized S 2p, which is hybridized with O 2p and Bi 6 s around the valance band [45]. The above newly constructed hybrid states within the bandgap of S-BVO promote the photocarriers separation at the atomic level and reduce its transition energy, which contribute to enhance the photocatalytic activity of S-BVO.
Figure 4
Figure 4.
Photocatalytic mechanism and verification. (a) Electron localization function of pristine BVO and S-BVO. In-situ XPS spectra of 2%S-BVO: (b) Bi 4f, (c) O 2p, (d) V 2p. (e) In-situ ATR-IR spectra of 2%S-BVO in contact with AgNO3 solution. (f) Enlarged image for δH–O–O. (g) Reaction free energy diagrams for the water oxidation reaction on pristine BVO and S-BVO.
To convincingly analyse the above migration mechanisms of photocarriers, in-situ XPS was employed. For pristine BVO, the binding energy of Bi 4f, O 2p, and V 2p exhibit no significant change under visible light irradiation (λ > 400 nm) (Fig. S22 in Supporting information), which is attributed to the separation and migration of photocarriers within the pristine BVO are inert as described above due to the lack of electronic state modulation. The peak of Bi 4f shows a positive shift of 0.1 eV under irradiation with 2% S-BVO, suggesting that the Bi site is in an electron-consuming state during illumination, and the modulation of the local coordination environment of Bi is conducive to driving water oxidation at the Bi active site (Fig. 4b). In addition, O 2p exhibits the expected negative shift indicating that the O site is electron-rich during illumination (Fig. 4c). V species show the valence-neutralization tendency under irradiation, which specifically demonstrates that the migration of photocarriers via the modified structure of S-Bi-V4+ is efficient and benign (Fig. 4d).
In order to clarify the intermediates of the reaction and achieve in-depth analysis of the mechanism, attenuated total reflection infrared spectroscopy (ATR-IR) was conducted (Fig. 4e). For 2%S-BVO, the broad band around 3200 cm−1 is correspond to the OH stretching vibration (νO–H), and the sharp band at 1630 cm−1 can be ascribed to the OH bending modes (δO–H) [46,47]. A positive trending peak in this region suggests an increase in the concentration of OH species either at or close to the surface with the increased illumination time. A slight swelling from 2000 cm−1 to 2300 cm−1 can be ascribed to the second overtone of the vibrational mode of water (δ’H2O) [48]. A weak band at 1340 cm−1 is observed (Fig. 4f), which can be interpreted as H–O–O bending mode (δH–O–O) for *OOH intermediate in water oxidation reaction [49]. Contrastively, pristine BVO only exhibits a broad band around 3200 cm−1, whose intensity displays no obvious increase as the irradiation (Fig. S23 in Supporting information), confirming 2%S-BVO possesses superior photocatalytic water oxidation activity relative to pristine BVO.
Gibbs free energy over the water oxidation pathway were calculated based on the optimized models of BVO and S-BVO. The negative values of water adsorption energy on both BVO and S-BVO indicate the advantage for activating the reaction of water oxidation (Fig. S24 in Supporting information) [50,51]. Water oxidation involves a multielectron transfer process, thus the step with the largest reaction free energy difference is typically considered as the rate-determining step (RDS) [52-54]. Combining with the analyses of in-situ XPS, Bi atom was selected as the active site for calculating the subsequent Gibbs free energy. For the pristine BVO (110) model, the formation of *OH with the largest reaction free energy difference (2.12 eV) is the RDS of water oxidation (Fig. 4g). Impressively, the free energy difference of this step for S-BVO is significantly reduced to 0.55 eV, which is originated the thermodynamic property of active site is regulated by the synergistic electron interaction of S-Bi-V4+. According to the widely accepted volcano theory proposed by Nørskov, the binding energy of each oxygen-containing intermediates is linearly correlated and cannot be independently modulated [55,56]. As a result, the RDS of S-BVO changes to the dissociation from *OH to *O, with a lower energy barrier of 1.68 eV, which is still lower than that of pristine BVO (2.05 eV). In summary, the balanced reaction energy barrier of each step in S-BVO is highly favourable for the stable and high-efficiency water oxidation.
Systematically summarizing the verified theoretical calculations and the characterization of the photocarriers transfer behaviors, the following mechanism of photocatalytic water oxidation on 2%S-BVO is proposed. The asymmetric coordination of S-Bi-V4+ structure was constructed via S-induced doping strategy to enhance the photochemical and photophysical properties of BVO, which provides the structural foundation for the desired photocatalytic water oxidation. Under visible light irradiation (λ > 420 nm), the photogenerated electrons migrate to the conduction band for activating the reduction reaction with the sacrificial agent, and the holes remain in the valance band for oxidizing H2O to O2 at thermodynamically favourable Bi site. Under the synergistic electron interaction, the asymmetric coordination structure of S-Bi-V4+ possesses more delocalized electrons for accelerating the photocarriers migration kinetics and the electronic environment of Bi is modulated for generating a thermodynamically favourable active site, which synergistically lead 2%S-BVO achieves an excellent performance of photocatalytic water oxidation.
In Conclusion, this work proposes a novel S-induced modification strategy to illustrate the decisive role of asymmetric coordination structures with the synergistic electronic interactions for photocatalytic water oxidation. Controllable introduction of V4+ species in BiVO4 can construct a distinct S-Bi-V4+ asymmetric coordination structure, which effectively facilitates the delocalization of electrons in the coordination environment of Bi atoms. This dynamic reconfiguration of the electronic structure significantly reduces the reaction energy barrier for water oxidation and constructs a more thermodynamically favourable catalytic active site. As a result, the optimized 2% S-BVO produces an excellent photocatalytic water oxidation performance of 70.25 μmol/h (40 mg) with an apparent quantum efficiency of 11.06% at 420 nm. The findings of this work provide valuable and meaningful perspective for finely regulating electronic structure on both photocarrier transfer kinetics and reaction thermodynamic, which is of significance for artificial photosynthesis.
Declaration of competing interest
The authors declare that they have no known competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 22372116 and 22309204), the National Key R&D Program of China (No. 2021YFA1500704).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.112275.
M. Kuang, J. Zhang, D. Liu, et al., Adv. Energy Mater. 10 (2020) 2002215. doi: 10.1002/aenm.202002215
[56]
Z.L. Zhao, Q. Wang, X. Huang, et al., Energy Environ. Sci. 13 (2020) 5143–5151. doi: 10.1039/d0ee01960g
Figure 1
Photocatalytic water oxidation property. (a) Schematic diagram for the synthesis of pristine BVO and S-BVO by S-induced doping process. (b) Photocatalytic water oxidation activity of pristine BVO and S-BVO at stationary point (40 mg). (c) AQE and DRS spectrum of 2%S-BVO. (d) Cycling experiments of 2%S-BVO. (e) Linear scanning voltammogram (LSV) curves of BVO and 2%S-BVO. (f) In-situ ESR spectra of pristine BVO and 2%S-BVO (DMPO-•OH). (g) Enlarged image for in-situ ESR spectra (DMPO-•OOH).
Figure 2
Morphology and structural characterization. (a) HRTEM image of 2%S-BVO. (b) Elements mappings of 2%S-BVO. (c) XRD patterns of pristine BVO and 2%S-BVO. (d) Enlarged image for XRD patterns. (e) Raman spectra of pristine BVO and 2%S-BVO. (f) Enlarged image for Raman spectra. (g) XPS spectra of Bi 4f. (h) XPS spectra of O 2p (i) Normalized V K-edge XANES μ(E) spectra of pristine BVO and 2%S-BVO.
Figure 3
Photocarrier Transfer Kinetics. (a) Calculated Bader charge of surface atoms on BVO and S-BVO. (b) Schematic representation of the synergistic electron interaction among S-Bi-V4+ in S-BVO. (c) Steady-state photoluminescence spectra of pristine BVO and 2%S-BVO. (d) Time-resolved transient PL decay spectra of pristine BVO and 2%S-BVO at the excitation wavelength of 475 nm. (e) KPFM analysis of pristine BVO and 2%S-BVO. Shown from left to right are the surface photocurrent voltage variation at the sample marking position, the surface potential distribution under dark conditions, and the surface potential distribution under illumination. Δ value was obtained by subtracting the potential of the samples in dark condition from the potential under visible light irradiation.
Figure 4
Photocatalytic mechanism and verification. (a) Electron localization function of pristine BVO and S-BVO. In-situ XPS spectra of 2%S-BVO: (b) Bi 4f, (c) O 2p, (d) V 2p. (e) In-situ ATR-IR spectra of 2%S-BVO in contact with AgNO3 solution. (f) Enlarged image for δH–O–O. (g) Reaction free energy diagrams for the water oxidation reaction on pristine BVO and S-BVO.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
