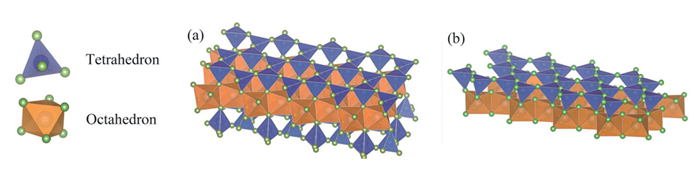
Figure 1.
Crystal structure of Fe-bearing clay minerals: (a) Montmorillonite, (b) kaolinite.
Fe-bearing clay minerals as catalysts for oxidant activation in organic pollutant degradation: ROS generation driven by Fe(Ⅱ)/Fe(Ⅲ) redox cycling
Mingli Jiang , Weihua Xu , Xin Li , Xiaofei Tan , Wei Zhang

Fe-bearing clay minerals were ubiquitous in sediment, soil, and water environments, where they played a vital role in iron cycling within biogeochemical processes [1,2]. The emergence of •OH in underground environments, generated through the activation of O2 by Fe-bearing clay minerals, had recently attracted increasing attention [3]. Due to natural processes such as surface water-groundwater interactions, groundwater level fluctuations, rainfall, alternating dry and wet conditions, and human activities, O2 could infiltrate previously reductive environments [4]. In the presence of O2, reducible Fe(Ⅱ) could activate O2 to produce hydrogen peroxide (H2O2), which subsequently generated •OH [5]. Hydroxyl radicals might efficiently oxidize a range of challenging contaminants, including Cr(Ⅵ), phenol, and antibiotics [6–8]. Consequently, elucidating the mechanisms of hydroxyl radical generation in subsurface anoxic environments improved our comprehension of soil environmental chemistry and provided practical techniques for in situ remediation of contaminated soils [9]. Moreover, this review offered novel avenues for advancing catalyst materials in advanced oxidation processes (AOPs).
Fe-bearing clay minerals exhibited primary traits similar to those of layered clay minerals, characterized by distinct silicate tetrahedra and aluminum octahedra, arranged in precise ratios, such as 2:1 in montmorillonite and 1:1 in kaolinite (Fig. 1) [10]. Compared with the 1:1 layered structure, the 2:1 configuration not only offered a larger interlayer spacing and more abundant surface hydroxyl groups but also facilitated the incorporation of redox-active Fe species into the octahedral sheets, which was critical for catalytic activity. Clay minerals had long been recognized as natural adsorbents owing to the extensive interlayer surface area and capacity for water absorption and swelling [11]. In Fe-bearing clays, Fe(Ⅱ) and Fe(Ⅲ) typically substituted for Al in the octahedral layers via heterovalent exchange, generating redox-active centers that were essential for Fenton-like reactions [12]. In contrast, minerals such as kaolinite and talc either lacked iron entirely or contained it in isolated, catalytically inert forms, rendering them ineffective under advanced oxidation conditions. The excellent swelling behavior and high cation exchange capacity of Fe-montmorillonite significantly enhanced its accessibility to oxidants and organic pollutants. In contrast, minerals such as illite and kaolinite exhibit poor swelling due to strong interlayer bonding (e.g., K+ fixation or hydrogen bonding), which limited reactant diffusion and active site exposure.
Montmorillonite was a characteristic Fe-bearing clay mineral that strongly exhibited the features of layered clay minerals. The amalgamation in montmorillonite of substantial layer expandability, interlayer cation exchange, and redox-active iron rendered it exceptionally useful in environmental applications. These structural characteristics not only determined the adsorption properties of Fe-bearing clay minerals but also imparted unique catalytic abilities, particularly in O2 activation leading to the generation of ROS.
Fe-bearing clay minerals represented a distinct category within layered clay minerals, distinguished by the replacement of aluminum by iron in the octahedral layers, which profoundly affected the composition, structure, and properties of clay minerals. Iron existed in several forms, including structural Fe, edge-bound Fe, and exchangeable Fe [2,13,14]. The iron replacement of aluminum modified the layer charge and cation exchange capacity, introducing redox activity and enabling Fe-bearing clay minerals to participate in biogeochemical processes. Specifically, structural Fe(Ⅲ) in the dioctahedral coordination could be reduced to Fe(Ⅱ) by microbial or chemical reducers [15,16]. The Fe(Ⅱ)/Fe(Ⅲ) redox cycle played a central role in subsurface environments by acting as a redox buffer, influencing biogeochemical cycles, and affecting the transformation and transport of pollutants [17]. The redox activity allowed Fe-bearing clay minerals to act as adequate natural catalysts, influencing the transformation and transport of contaminants in subsurface environments.
Persistent organic pollutants (POPs) were a class of toxic organic compounds characterized by resistance to degradation, high bioaccumulation potential, and long-range environmental transport [18]. Exposure to POPs had been linked to a range of adverse health effects, including endocrine disruption, reproductive issues, and carcinogenesis [19,20]. Given the robust oxidation capability and elevated mineralization efficiency of ROS, advanced oxidation processes were extensively used in both research and practical applications for the degradation of organic pollutants [21,22]. The large-scale application of AOPs was restricted by high energy requirements and the limited cost effectiveness of catalytic materials [23]. Electrode materials used in electrochemical AOP systems were sensitive to operational conditions and were easily damaged, requiring frequent replacement [24]. Ozone-based oxidation, although effective, involved complex infrastructure and high energy input. Additionally, the compositional complexity of real wastewater imposed further constraints on process scalability and operational stability [25].
Fe-bearing clay minerals had emerged as promising candidates for catalytic applications due to low extraction and processing costs, as well as strong environmental compatibility. These minerals were naturally abundant and could be obtained with minimal chemical treatment, which significantly reduced the overall cost. Their layered silicate structures provided strong confinement for iron species, which stabilized redox-active centers and minimized iron leaching during reactions. In the context of AOPs, Fe-bearing clay minerals had demonstrated the ability to activate O2 to produce ROS, offering a pathway to avoid secondary pollution caused by excessive use of chemical oxidants [26,27]. Moreover, their successful implementation in situ pollutant remediation highlighted strong potential for large-scale environmental applications [28].
Despite the substantial documentation of the redox characteristics of Fe-bearing clay minerals in prior research, there had been a deficiency of thorough investigations connecting their redox activity to pollutant remediation in environmental engineering contexts [29,30]. Current research predominantly examined the interaction between Fe-bearing clay minerals and pollutants [3]. However, there had been inadequate emphasis on systematically summarizing and theorizing the role of ferrous iron in activating O2 to produce •OH for pollutant degradation. This review comprehensively investigated the function of structural Fe(Ⅱ)/Fe(Ⅲ) cycling in activating O2 by elucidating the electron transfer mechanisms between O2 and clay minerals, laying a robust theoretical foundation to guide future research on Fe-bearing clay minerals.
Structural Fe(Ⅱ) was crucial for activating oxidants in Fe-bearing clay minerals. In contrast, ion-exchanged and surface-adsorbed Fe(Ⅱ) constituted only a minor fraction of the total reduced iron [31]. Under natural reducing conditions, structural Fe(Ⅱ) in Fe-bearing clay minerals such as Montmorillonite transferred electrons for O2 activation and demonstrated the ability to produce ROS [32]. However, this process typically occurred only when a previously anoxic environment was disturbed and O2 became available. Montmorillonite possessed up to 18.0 wt% Fe, whereas SWy-2 montmorillonite contained approximately 2.3 wt% Fe [7]. Even in Fe-rich clay minerals under natural subsurface conditions, the maximum documented reduction extent was approximately 1.0 mmol Fe(Ⅱ) g-1 [29]. Controlled laboratory investigations were essential to systematically replicate and optimize the reduction of Fe-bearing clay minerals, with the goal of maximizing structural Fe(Ⅱ) content for effective application in AOPs. Accordingly, it was critical to minimize direct competition between reductants and oxidants by temporally separating the pre-reduction and oxidation stages, thereby preventing premature consumption of Fe(Ⅱ). Prior research had suggested that the reduction of Fe(Ⅲ) occurred primarily through biological or chemical mechanisms [33,34]. Given the crucial role of structural Fe(Ⅱ) in oxidant activation, it was essential to investigate how various reduction conditions impacted the efficiency of Fe(Ⅱ) reduction in these minerals.
In recent years, the role of microorganisms in reducing ferric iron (Fe3+) had gained significant attention. Research had demonstrated that various microbes could metabolically reduce structural Fe(Ⅲ) to ferrous iron (Fe(Ⅱ)) (Table S1 in Supporting information). They derived energy by oxidizing organic compounds or hydrogen, with Fe(Ⅲ) as the electron acceptor, which was reduced to Fe(Ⅱ) in the process.
Microbial reduction of Fe-bearing clay minerals was classified into solid-state reduction and the dissolution-precipitation pathway [35]. Microorganisms transferred electrons to modify the bonding or symmetry within the octahedral and tetrahedral layers of Fe-bearing clay minerals without causing substantial disruption to the clay structure (Fig. 2a). Structural Fe(Ⅲ) served as an electron acceptor, interacting with organic electron donors to complete the structural Fe(Ⅲ) reduction process. A prominent characteristic of the solid-state reduction mechanism was the reversibility of the cation exchange capacity of clay, which was observed under both laboratory conditions and natural environments [36,37]. In Fe(Ⅲ)-bearing smectites, reduction altered -OH and Si-O vibrational frequencies, which predominantly was reinstated upon reoxidation [38].
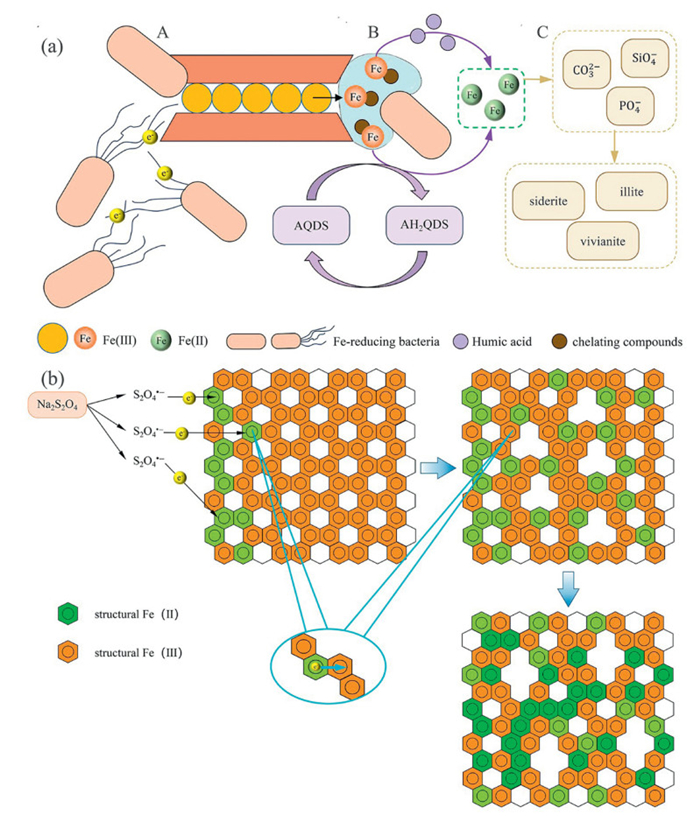
The dissolution-precipitation mechanism functioned independently of the solid-state reduction mechanism [35,39,40]. Dissolution and precipitation were the two separate phases of the dissolution-precipitation process. During the dissolution stage, microbial metabolic byproducts, such as organic acids, siderophores, and extracellular polymeric substances, disrupted the crystalline structure of the clay mineral and released Fe(Ⅲ) into the solution (Fig. 2a). In the subsequent precipitation stage, Fe(Ⅲ) was reduced to Fe(Ⅱ) by microorganisms and interacted with solution constituents like phosphate and carbonate to form secondary biogenic minerals, such as siderite, vivianite, and amorphous silica (Fig. 2a). Unlike solid-state reduction, the dissolution-precipitation mechanism was characterized by significant structural damage to the clay mineral, which rendered the reduction of structural Fe(Ⅲ) irreversible.
Restrictions had constrained the two mechanisms mentioned above. The efficacy of microbial reduction was influenced by parameters such as microbial activity, the presence of metabolic byproducts, and microbial population density. The structural availability of Fe(Ⅲ) had significantly influenced microbial reduction under the dissolution-precipitation mechanism, with reduction primarily occurring at the exposed edges of clay particles or in highly soluble minerals [30]. Moreover, reduction byproducts, including Fe(Ⅱ) or secondary minerals, might have adsorbed onto clay surfaces or microbial cells, obstructing electron transport and impeding further reduction. Furthermore, microbial reduction necessitated stringent experimental settings, including the kind of growth medium and the structural Fe(Ⅲ) reduction process duration of up to two weeks, which might have influenced the reduction outcomes [41,42].
Chemical reduction presented considerable advantages over biological reduction, such as shorter reaction times, simpler operational procedures, and lower experimental costs [43]. Biological reduction typically required several days to weeks and often achieved < 50% reduction, whereas chemical reducing agents completed the process within several hours, with reduction efficiencies exceeding 80% [44–46]. Because chemical reductants were much smaller than microorganisms, they penetrated more deeply into clay layers, thereby enabling more extensive Fe(Ⅲ) reduction [16]. Consequently, chemical reduction methods were widely adopted to reduce Fe-bearing clay minerals. Prior research showed that reductants, including dithionite in citrate-bicarbonate buffer, hydrazine, sulfide, hydrosulfide, benzidine, hydrogen gas (300 ℃), phenylenediamine, thiourea, tetraphenyl boron, and ascorbic acid, could reduce structural Fe(Ⅲ) [47–51]. Sodium dithionite was a common chemical reductant known for low toxicity, minimal dose requirements, high reduction efficiency, and the ability to penetrate the internal structure of clay minerals to effectively convert Fe(Ⅲ) to Fe(Ⅱ) [52]. In an aqueous solution, sodium dithionite decomposed into sulfoxylate free radicals (S2O4•–, E0(S2O42–/HSO3–) = −0.66 V), which possessed vigorous electron activity and reduction potential [53]. Studies indicated that the rate and extent of reduction significantly increased with elevated temperatures and higher reductant concentrations [16]. Moreover, the extent of reduction could be precisely controlled by adjusting reductant dosage and reaction conditions, which was beneficial for investigating the structure-activity relationships of Fe-bearing clay catalysts in AOPs.
Iron in Fe-bearing clay minerals was present in tetrahedral and octahedral coordination, with the reduction of Fe(Ⅲ) predominantly occurring in the octahedral layer [16]. The process of Fe(Ⅲ) reduction to Fe(Ⅱ) occurred on the mineral surface, penetrating to depths of several nanometers, as well as within the internal structure (Fig. 2b). Electrons provided by sulfoxylate free radicals preferentially reduced dispersed Fe(Ⅲ) sites within the octahedral layer [52,54]. This Fe(Ⅲ) reduction process followed a nearest-neighbor exclusion mechanism, which minimized the formation of Fe(Ⅱ)-O-Fe(Ⅱ) pairs during the reduction process. Instead, Fe(Ⅱ)-O-Fe(Ⅲ) pairs predominated in the partially reduced state, gradually transitioning to Fe(Ⅱ)-O-Fe(Ⅱ) pairs as Fe(Ⅲ) content decreased [52]. The Fe(Ⅱ) distribution was uniform across the bulk clay mineral but exhibited a pseudo-random arrangement within localized structures. The pseudo-random arrangement improved charge compensation within the mineral layers, thereby enhancing structural stability [55].
Sodium dithionite effectively reduced Fe-bearing clay minerals, regardless of the iron content in the samples [54,56,57]. After reduction, Fe(Ⅱ) tended to be uniformly distributed throughout the bulk mineral, while exhibiting a pseudo-random arrangement within local structural domains [55]. This arrangement facilitated interlayer charge compensation, thereby stabilizing the mineral lattice. Structural Fe(Ⅲ) reduction in lower Fe content minerals like SWy-2 proceeded primarily at dispersed active sites [56]. The observed minor structural distortions were reversible through re-oxidation, indicating structural reversibility [52]. Conversely, for higher Fe-content minerals like SWa-1 [56], the reduction reaction could occur concurrently at relatively dispersed sites and within dioctahedral and trioctahedral clusters.
The distribution of dioctahedral vacancies was influenced by interlayer charge balance and local electronic density, resulting in a pseudo-random spatial pattern governed by symmetry constraints [54]. During the structural Fe(Ⅲ) reduction process, certain areas of the dioctahedral structure were partially degraded as iron ions migrated and aggregated, forming trioctahedral clusters [53]. The formation of trioctahedral clusters involved localized dehydroxylation and irreversible structural reorganization [48,56]. As a result, dioctahedral and trioctahedral domains dynamically coexisted in the reduced state. While partial reoxidation might have converted some trioctahedral structures back to the dioctahedral form, complete structural recovery to the original state was generally unattainable.
The use of sodium dithionite as a reductant in structural Fe(Ⅲ) reduction presented several inherent risks. Experimental results indicated that optimal reductive performance is typically observed under mildly alkaline conditions (pH ≈ 9), whereas lower pH accelerated dithionite decomposition, thereby diminishing its electron-donating capacity (EDC), and higher pH values reduced the accessibility and reactivity of structural Fe(Ⅲ), thereby limiting reduction efficiency. Additionally, the decomposition of dithionite yielded various sulfurous byproducts (e.g., sulfite, thiosulfate), which might have accumulated in the system and posed risks of secondary pollution or interfered with downstream oxidative processes. Moreover, excessive dosage or repeated treatments might have destabilized the crystalline framework of clay minerals, ultimately compromised structural integrity and impaired the long-term redox reusability of Fe-bearing minerals.
Fe-bearing clay minerals demonstrated remarkable catalytic properties in the degradation of organic pollutants, making them highly effective for environmental remediation (Table S2 in Supporting information). Fe-bearing clay minerals enhanced the degradation efficiency of persistent contaminants and showed versatility across a wide range of pollutant types. For instance, 2.5 g/L montmorillonite activated peroxymonosulfate (PMS), achieving 94.1% degradation of 5 mmol/L atrazine within 60 min [58]. Soil components containing 12% montmorillonite (total iron content of 91.29 g/kg) activated PS to achieve 85.5% Sulfamethoxazole (SMX) degradation within 8 h, with an initial mineralization efficiency of 29.4% [59,60]. Using 1 g/L of a type of reduced nontronite (rNAu-2) to activate PMS for the degradation of diethyl phthalate (DEP), the pseudo-first-order rate constant (kobs) of DEP degradation was 0.0017 ± 0.001 s-1 [61]. Under anoxic and dark conditions, Fe(Ⅲ) in Fe-rich montmorillonite facilitated electron transfer reactions that promoted the degradation of tetracycline antibiotics. Notably, clays with higher Fe content demonstrated greater efficiency in pollutant degradation [53]. These promising results underlined the versatility and effectiveness of Fe-bearing clay minerals across various contaminants, demonstrating their potential for large-scale environmental applications.
Compared with natural conditions, Fe-bearing clay minerals significantly enhanced the degradation efficiency of nitroaromatic compounds, achieving improvements of several orders of magnitude. Fe-bearing clay minerals activated at 70 ℃ could increase the degradation efficiency of phenol from 25.8% at room temperature to 74.4% [62]. For trichloroethylene (TCE), the dechlorination rate constant under anoxic conditions was 3.1 × 10–5d-1 [63], whereas under aerobic conditions, Fe-bearing clay minerals accelerated O2 activation to achieve 89% degradation of 1 mg/L TCE within 3 h, corresponding to a kobs of 7.4 × 10–3d-1 [64]. The degradation efficiency of DEP achieved 66.7% efficiency within 30 s and 69.6% after 300 s, driven by the Fe-bearing clay minerals active O2 mechanism [61]. For benzoic acid, phenol, and nitrobenzene, the action of Fe-bearing clay minerals achieved degradation efficiencies exceeding 80% by activating O2 or other oxidizing agents [6,28,65].
Fe-bearing clay minerals, such as rNAu-2, exhibited significant catalytic properties in AOPs due to their structural Fe(Ⅱ), which was the electron donor [64]. In Fe-bearing clay mineral-based advanced oxidation processes, oxidants such as H2O2, PMS, and O2 reacted with Fe(Ⅱ) to produce highly reactive •OH [8,60,66,67]. The subsequent generation and reactions of ROS proceeded through a sequential process [68–70].
The oxidation of Fe(Ⅱ) in rNAu-2 proceeded via two interconnected pathways: direct reaction with O2 and the redox cycle of Fe(Ⅱ)/Fe(Ⅲ) [69]. Prior research highlighted the role of O2 in generating •OH through a single-electron transfer mechanism [69]. The direct reaction with O2 predominantly occurred on the exposed Fe(Ⅱ) sites at the basal planes and fractured edges of clay minerals [62,71]. During the reoxidation reaction, structural Fe(Ⅱ) reacted with O2 to generate structural Fe(Ⅲ) and superoxide anions (O2•–) (Eq. 1). After the oxidation of these exposed Fe(Ⅱ) to Fe(Ⅲ), exposed Fe(Ⅲ) acted as Lewis acid centers, promoted the dissociation of water molecules and the reduction of O2, which led to the formation of H2O2 (Eq. 5) [72,73]. Superoxide radicals not only neutralized certain ROS but also produced H2O2, thereby sustaining the oxidative capacity of the system (Eq. 2). Furthermore, structural Fe(Ⅱ) could react with H2O2 to generate structural Fe(Ⅲ) and •OH, which further drove the oxidative reactions (Eq. 3) [74]. Hydroxyl radicals as a non-selective ROS could be consumed by reacting with Fe(Ⅱ), producing Fe(Ⅲ) and hydroxide ions (OH-) (Eq. 4) [69], which lowered the effective concentration of •OH in the reaction system. However, the quenching effect of structural Fe(Ⅱ) on •OH was relatively slow and did not significantly inhibit the degradation of organic pollutants, as •OH reacted more readily with the pollutants than with the structural Fe(Ⅱ) [69].
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
In Fe-bearing clay minerals catalyzed oxidation systems, •OH directly attacked pollutants, initiating the conversion or mineralization of organic pollutants. The radical reacted with aromatic rings or unsaturated bonds and formed hydroxylated products [75,76]. In addition to direct oxidative mechanisms, radical-assisted surface electron transfer pathways contributed to pollutant degradation. Fe(Ⅲ) generated during oxidation could be reduced back to Fe(Ⅱ) via interactions with other reducing agents, maintaining the redox cycle [77,78]. For instance, tetracycline (CTC) molecules adsorbed on clay surfaces could transfer electrons to structural Fe(Ⅲ), reducing Fe(Ⅲ) to Fe(Ⅱ) [79]. The electrons donated by CTCs might also participate in O2 reduction, forming H2O2. Similarly, PMS molecules could adsorb onto hydroxyl groups on the clay surface, forming surface complexes. These complexes activated PMS through electron transfer, generating intermediates such as SO5•–, which subsequently produced SO4•– and •OH [66]. This dual contribution of surface interactions and radical production enhanced the degradation efficiency of pollutants [61].
Mössbauer spectroscopy, a powerful tool for analyzing the oxidation states and coordination environments of iron, provided crucial insights into the role of structural iron in Fe-bearing clay minerals [71,80,81]. Mössbauer spectroscopy revealed that rNAu-2 contained four octahedral iron (OctFe) sites: OctFe(Ⅲ)-1, OctFe(Ⅲ)-2, OctFe(Ⅱ)-1, and OctFe(Ⅱ)-2 [82]. OctFe(Ⅱ)-1 and OctFe(Ⅱ)-2 were iron species in the reduced state, and OctFe(Ⅱ)-2 was the primary reactive species during the structural Fe(Ⅱ) oxidation process due to significant structural distortion [54,83]. Structural Fe in Fe-bearing clay minerals played a crucial role in generating reactive species [51]. Among the oxidized iron species, OctFe(Ⅲ)-2 exhibited uneven electron distribution, which greatly enhanced the reactivity during the Fe(Ⅱ)/Fe(Ⅲ) cycle [84]. In contrast, the reactivity of OctFe(Ⅲ)-1 depended on the degree of distortion in the coordination environment.
Surface-adsorbed Fe(Ⅱ) primarily formed through ion exchange between mineral surface functional groups (e.g., ≡SiO–, ≡AlO–, and ≡Fe(Ⅲ)O–) and [Fe(H2O)6]2+ or similar complexes [85]. The reactivity of Fe(Ⅱ) at these adsorption sites depended on the electron-donating ability of the coordinating ligands. Ligands with higher electron density could enhance the reactivity of Fe(Ⅱ). In the presence of highly reactive ligands, surface-adsorbed Fe often favored the production of oxidants other than •OH [6]. Those oxidants were less effective in driving pollutant oxidation, limiting the role of surface-adsorbed Fe(Ⅱ) in such processes. This phenomenon indicated that structural Fe(Ⅱ) and structural Fe(Ⅲ) were the predominant oxidation states driving the catalytic processes [86].
During the structural Fe(Ⅱ) oxidation process, the dynamic changes in octahedral Fe(Ⅱ) coordination highlighted the critical role of OctFe(Ⅱ)-2 in driving catalytic activity. Following a complete redox cycle that coupled chemical reduction with O2 activation for organic pollutant degradation, most Fe(Ⅱ) in reduced clay minerals was oxidized. The oxidized Fe(Ⅲ), with a smaller ionic radius and higher charge density, interacted strongly with surrounding oxygen or hydroxyl ligands. Additionally, the higher oxidative potential of Fe(Ⅲ) compared with Fe(Ⅱ) led to the release of structural hydroxyl groups, producing water or other byproducts. Structural distortion and hydroxyl release mechanisms weakened the original structure, augmented local disorder, and facilitated the migration of iron atoms from dioctahedral to trioctahedral locations. The migration formed localized trioctahedral structures, resulting in irreversible changes to the framework of the mineral.
Studies revealed that within the first 15 s of oxidation, the proportion of OctFe(Ⅱ)-2 dropped sharply from 16.1% to 2.0%, accompanied by an increase in OctFe(Ⅲ)-1 and OctFe(Ⅲ)-2 [61]. This indicated that OctFe(Ⅱ)-2 was rapidly consumed and partially converted into other iron species. As the reaction progressed, OctFe(Ⅱ)-2 was almost completely depleted by 300 s, while OctFe(Ⅱ)-1 became the primary driving source for further oxidation. The proportion of OctFe(Ⅱ)-1 steadily decreased from 9.6% to 2.4%. At the same time, OctFe(Ⅲ)-2 increased significantly, from 9.6% to 46.0% [61]. The notable formation of OctFe(Ⅲ)-2, identified as a distorted cis-octahedral Fe(Ⅲ) species, underscored the structural rearrangements occurring during oxidation and highlighted the stability of dioctahedral coordination structures [79,87].
The FTIR method is especially effective for identifying iron coordination structures and detecting minor but diagnostically significant vibrational shifts that indicated transformations in hydroxyl groups [80,81]. In the FTIR spectra, OctFe(Ⅱ)-2 and OctFe(Ⅱ)-1 corresponded to Fe(Ⅱ)Fe(Ⅱ)Fe(Ⅱ)-OH and Fe(Ⅱ)Fe(Ⅱ)-OH entities, respectively [86]. The pre-reduced samples typically exhibited characteristic vibrational peaks for Fe(Ⅱ)Fe(Ⅱ)Fe(Ⅱ)-OH and Fe(Ⅲ)Fe(Ⅲ)-OH. During oxidation, the vibrational peaks underwent significant changes. In particular, the peaks corresponding to Fe(Ⅱ) in the octahedral structure gradually disappeared, replaced by new peaks corresponding to Fe(Ⅱ)Fe(Ⅱ)-OH or Fe(Ⅱ)-Fe(Ⅲ)-OH. For example, a previous study reported that the bending vibration peaks at 627 cm-1 and 637 cm-1 and the stretching vibration at 3638 cm-1 diminished markedly during the structural Fe(Ⅱ) oxidation process. Simultaneously, a new peak appeared at 843 cm-1, attributed to the formation of Fe(Ⅱ)Fe(Ⅱ)-OH structures [8].
The oxidation of structural Fe(Ⅱ) in minerals occurred in different coordination environments. Structural Fe(Ⅱ) was more readily oxidized to produce •OH in dioctahedral coordination. In trioctahedral coordination, structural Fe(Ⅱ) oxidation was constrained by structural disruption, which made structural Fe(Ⅱ) more resistant to oxidation. The oxidation resistance was evidenced by the fact that, after a complete redox cycle, even under the same potential (E = −0.20 V), the Fe2+/Fe3+ ratio in Fe-bearing clay minerals (Fe2+/Fe3+ ≈ 0.40) was higher than that of the original mineral (Fe2+/Fe3+ ≈ 0.02) [18]. Notably, structural Fe(Ⅱ) was rapidly oxidized during the initial oxidation stage, while in the later stages, the oxidation rate of structural Fe(Ⅱ) slowed significantly. The change in reaction rate over time might have indicated a regeneration process for surface-structured Fe(Ⅱ). The regeneration was closely related to the dynamic interactions between structural and surface iron species, which would be discussed in detail in subsequent analyses.
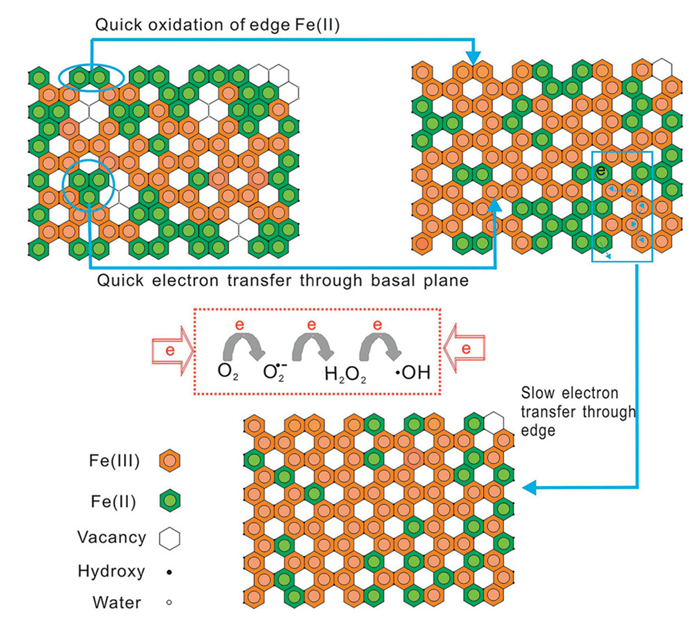
Research results demonstrated a strong correlation between the electron transfer rate and the redox potential (E0) of Fe-bearing minerals (Fig. S1 in Supporting information). For different Fe-bearing clay minerals, E0 ranged from −0.17 V to 0.02 V, indicating that different distributions of active sites contributed differently to the efficiency of electron migration (Text S1 in Supporting information) [88]. During the initial oxidation phase (rapid phase), the oxidation efficiency of edge structural Fe(Ⅱ) typically reached the maximum value within the first 15 min [69]. The rapid reaction was attributed to the high reactivity of edge structural Fe(Ⅱ) and the direct exposure to oxidants, rendering structural Fe(Ⅱ) the dominant electron donor.
Electrons were quickly transferred to oxidants, leading to a stepwise single electron transfer (SET) process that generated ROS such as O2•–, H2O2, and ultimately highly reactive species like •OH and SO4•–. Following the rapid oxidation phase, surface structural Fe(Ⅲ) participated in a new pathway for electron transfer by channeling electrons from the mineral interior to the edge. The edge structural Fe(Ⅱ) was continuously regenerated via electron shuttling from the mineral interior to the edge, thereby sustaining continued oxidation reactions. The ongoing edge structural Fe(Ⅱ) oxidation process, supported by electron transfer from the interior to the edge, was referred to as the slow phase.
During the slow phase of oxidation, electrons migrated from the mineral interior toward edge sites through Fe(Ⅱ)-O-Fe(Ⅲ) connections. In the slow stage, electrons were transferred by a "hopping" mechanism, where edge structural Fe(Ⅲ) accepted electrons from the mineral interior, leading to the regeneration of edge structural Fe(Ⅱ) (Fig. 3) [69]. The rate of electron "hopping" was significantly influenced by the diffusion coefficient [62]. Fe-bearing clay minerals with high diffusion coefficients (e.g., SWy-2) quickly replenished Fe(Ⅱ) at edge sites, ensuring the continuity of the electron transfer process. The redox profiles of Fe-bearing clay minerals were asymmetrical, indicating that the oxidation and reduction processes did not follow the same path. This asymmetry displayed a lag effect caused by insufficient electron migration time, referred to as the "hysteresis effect" of Fe-bearing clay minerals [88]. Research on SWa-1 showed that extending the reaction time to 4.75 days allowed redox reactions to reach equilibrium, while shorter experiments (e.g., 30 min) often failed to capture the diffusion process fully [43].
Structural Fe(Ⅱ) in Fe-bearing clay minerals served as the primary electron donor in the oxidation and degradation of organic pollutants, highlighting the significance of studying electron transfer dynamics. rNAu-2 served as a model for Fe(Ⅱ)-bearing clay minerals [12,16,89]. Experimental data showed that during the oxidation of rNAu-2, the total oxidation of structural Fe(Ⅱ) accounted for 15%, while surface structural Fe(Ⅱ) oxidation reached approximately 50% [90]. This research result indicated that surface structural Fe underwent continuous redox cycling, supported by electron supply from interior structural Fe(Ⅱ). The limited contribution of initial structural Fe(Ⅱ) to radical generation was attributed to the small surface area of edge sites, constituting only 2%−5% of the total surface area [78]. Conversely, the interior structural Fe(Ⅱ), which comprised the predominant portion of structural Fe(Ⅱ) in rNAu-2, was crucial for the regeneration of edge structural Fe. Interior structural Fe(Ⅱ) served as reactive sites for degrading organic pollutants when pollutants such as nitrobenzene could interact with mineral basal planes [91].
As the slow phase progressed, the regeneration of edge structural Fe(Ⅱ) approached saturation, thereby limiting the opportunity for oxidants to react effectively with Fe(Ⅱ). Additionally, secondary products, such as iron oxides or green rust, coated the active sites, further reducing reaction efficiency [92]. As the reaction continued, the redox potential gradient within the mineral gradually diminished, leading to a reduced driving force for electron migration from the interior structural Fe(Ⅱ) to the edge structural Fe(Ⅲ). The reaction rate stabilized or became extremely slow when the system approached equilibrium.
Oxidation experiments on NAu-2 revealed changes in Fe(Ⅱ) content over time, reflecting the regeneration process. The reaction within the first 15 min was referred to as the rapid phase. During the rapid phase, Fe(Ⅱ) content decreased from 54.5% to 32.7% [69]. Between 15 and 90 min, the reaction entered the slow phase. Fe(Ⅱ) content further decreased to 25.7%, while the oxidation rate significantly slowed. The reaction from 90 min to 180 min was referred to as the later phase, during which the Fe(Ⅱ) content slightly decreased to 21.6% [69].
Different types of Fe-bearing clay minerals (NAu-1, NAu-2, SAz-2) exhibited significant differences in iron content. Fe-rich clay minerals typically contained more structural Fe(Ⅱ) after experimental reduction, which allowed for the provision of more electrons during the oxidative degradation process to form radicals. The number of electrons involved in clay minerals indicated that the iron content directly influenced pollutant degradation efficiency. Studies showed that NAu-2 contained 41.7% total iron, of which 96% was octahedral Fe (OctFe) and 4% was tetrahedral Fe (TetFe) [28]. NAu-1 contained 21.1% total iron, entirely in the form of OctFe, while SAz-2 contained only 0.12% total iron, all of which was OctFe. Although the proportion of Fe(Ⅱ) to total iron was higher in reduced low-iron clay mineral rSWy-3 (62.6%), the high-iron clay mineral rNAu-2 exhibited greater reactivity [28]. In the rNAu-2 system, the value of •OH formation efficiency (E•OH) reached over 6 μmol/L, which was markedly greater compared to the values recorded for rNAu-1 (< 6 μmol/L) and rSAz-2 (< 4 μmol/L) systems [79].
For the same type of Fe-bearing clay mineral, under similar reduction levels, the dosage of the clay mineral became a critical factor influencing the efficiency of pollutant degradation. When the dosage of rNAu-2 increased from 0.5 g/L to 2 g/L, the Trichloroethylene (TCE) oxidation efficiency increased significantly from 35% to over 65%. However, increasing the dosage to 4 g/L did not significantly improve oxidation efficiency [64]. As the dosage increased, more Fe(Ⅱ) became available to participate in oxidation reactions and generate •OH. However, Fe(Ⅱ) also acted as a scavenger of •OH, leading to reduced availability of •OH for TCE degradation. When the dosage increased from 1 g/L to 2 g/L, the enhanced •OH production dominated, leading to significantly improved degradation efficiency [66]. However, when the dosage reached 4 g/L, the scavenging effect of excess Fe(Ⅱ) offset the additional •OH generation, resulting in a plateau in degradation efficiency [64]. Thus, optimal dosages balanced generating sufficient •OH and minimizing the consumption of Fe(Ⅱ).
The electron transfer properties of iron minerals were significantly influenced by the coordination environments of Fe(Ⅱ) and Fe(Ⅲ) within mineral structures [93]. For example, in biotite and chlorite, Fe(Ⅱ) predominantly adopted an octahedral coordination, which exhibited relatively low reactivity in oxidation reactions. In contrast, Fe(Ⅱ) in montmorillonite formed a tetrahedral coordination structure ([Fe(Ⅱ)(OSi)4(OH)2]), which, due to its more loosely bound electron cloud, facilitated oxidation reactions more readily and generated higher amounts of •OH radicals. The data demonstrated that minerals with higher Fe(Ⅱ) content exhibited superior degradation rates. The generation rates of •OH radicals in the PS systems of IRS1 (IRS, Fe-rich mineral) and IRS2 minerals were 0.15 h-1 and 0.24 h-1, respectively, compared to only 0.09 h-1 in the IPS (Fe-poor mineral) system, highlighting the strong dependence of degradation rates on the concentration of divalent iron [59].
In Na-SAz-2 (Fe-poor mineral), Fe(Ⅲ) was primarily located deep within the mineral structure, limiting its involvement in reduction reactions. In contrast, Fe(Ⅱ)-rich minerals, such as Na-NAu-2, had Fe(Ⅱ) present in more accessible surface structures, where the structural Fe(Ⅱ) could more effectively transfer electrons to the mineral surface. Experimental data showed that after 12 h of reaction, Na-NAu-2 achieved a CTC degradation rate of 56.8%, whereas the Fe-poor Na-SAz-2 reached only 13.1% [79]. These results underscored the significant catalytic efficiency of Fe(Ⅱ)-rich minerals. The regeneration of Fe(Ⅱ) on the mineral surface was essential for persulfate activation, leading to the formation of ROS that promoted pollutant degradation.
The non-radical degradation pathway primarily generated oxidants, such as H2O2, through surface electron transfer reactions, which oxidized the pollutants. Experimental results showed that in Fe(Ⅱ)-rich minerals, PS activation led to the formation of Fe(Ⅱ)-PS complexes on the mineral surface. The formation of these complexes not only enhanced the electron transfer capability of the mineral but also significantly increased the potential of the mineral surface. The open-circuit potential of ilmenite reached 0.082 V. Hematite showed a value of 0.063 V, and goethite had 0.059 V [59]. Ilmenite demonstrated stronger surface electron transfer capacity than the other two minerals. The change of open-circuit potential further confirmed that Fe(Ⅱ)-rich minerals could initiate non-radical degradation pathways through surface electron transfer. In the IPS/PS (Fe-poor mineral) system, the non-radical reaction contributed minimally, whereas in the IRS1/PS and IRS2/PS systems, surface electron transfer reactions dominated the degradation pathway, contributing approximately 52.9% in the IRS2/PS system [59]. Moreover, the non-radical reaction in the IRS2/PS system significantly outperformed the radical reactions, demonstrating the dominant role of Fe(Ⅱ) minerals in SMX degradation. Analysis of SMX degradation products, such as sulfanilic acid and 3-amino-5-methylisoxazole, revealed that surface electron transfer reactions in Fe(Ⅱ) minerals were more efficient in decomposing SMX compared to other oxidation pathways. Particularly in the IRS2/PS system, the surface electron transfer pathway contributed predominantly to the reaction, while in the Fe-poor IPS/PS system, the free radical pathways (•OH and SO4•–) dominated.
Reduction extent referred to the proportion of Fe(Ⅲ) reduced to Fe(Ⅱ) in a mineral. In minerals with a high reduction extent, structural Fe(Ⅱ) did not exist in a naturally stable state, but rather in an elevated reactive state that could activate oxidants and generate •OH. This state allowed for electron transfer from the interior of the mineral to its surface, ensuring a continuous generation of Fe(Ⅱ) and thereby a sustained production of •OH [2]. In contrast, in minerals with a lower reduction extent, even clay minerals with a high Fe(Ⅱ) content, Fe(Ⅱ) was predominantly located within the inaccessible crystal lattice. This limited the efficiency of oxidation reactions and restricted the production of •OH. NAu-2 with a 50% reduction extent generated more •OH during the oxidation process, achieving a TCE degradation rate approaching 90% after 3 h of oxidation. Under the same conditions, the TCE degradation rate in the experimental group with 25% reduction in NAu-2 was only 54.5% [64]. In reduced NAu-2 particles, increasing the reduction level from 16.5% to 54.3% led to an increase in accumulated •OH concentration from 1.28 ± 0.07 μmol/L to 9.17 ± 0.04 μmol/L over 3 h [79].
While higher dosages provided more Fe(Ⅱ), the competitive scavenging of •OH by Fe(Ⅱ) diminished the efficiency of organic pollutant oxidation. A higher reduction level, however, optimized the balance between •OH generation and consumption, enhancing pollutant degradation efficiency even at moderate Fe(Ⅱ) concentrations. At low reduction levels (< 30%), the limited amount of structural iron and insufficient reactive sites resulted in a less pronounced effect on pollutant degradation [86,94].
Under mildly acidic to slightly basic conditions, the generation rate of active radicals (SO4•– and •OH) increased significantly, with minimal scavenging effects. Under extreme pH conditions, radical generation and retention were notably suppressed. The stability of the mineral structure might be altered under certain conditions. The capture of SO4•– by H+ to form H2SO5– significantly inhibited the generation of SO4•–, while the acidic environment limited the oxidation rate of Fe2+, further reducing the formation of •OH [66,92,95]. Protonation on the Fe-bearing clay minerals surface could block active reaction sites, weakening effective interactions with oxidants [96]. Under high pH (> 11), PMS decomposition tended to produce SO52-, which significantly suppressed the generation of SO4•– and •OH, directly reducing the efficiency of radical-driven oxidation and pollutant degradation [95]. Additionally, high OH- concentrations might alter the form or configuration of hydroxyl groups on the surface of minerals, further impairing radical generation. In the slightly acidic to slightly basic range (pH 5–8), the surface of minerals maintained a moderate charge balance, avoiding site blocking caused by excessive protonation or high OH– concentrations [65,95,97]. The moderate charge balance on the surface of minerals created an optimal environment for PMS activation, enhancing the generation of radicals, particularly SO4•– and •OH.
The initial pH significantly affected the degradation of DEP in the PMS/r-NAu-2 system [61,79]. Within a pH range from 4.3 to 6.5, DEP degradation efficiency remained between 63.7% and 70.4%, with slight variation in the normalized kobs (0.051 ± 0.006 to 0.057 ± 0.005 s-1) [61]. When the pH increased to 8.3 and 10.3, kobs decreased to 0.022 ± 0.001 s-1 and 0.019 ± 0.001 s-1, respectively, and degradation efficiency dropped significantly [79]. Studies on atrazine degradation indicated that the degradation efficiency decreased significantly at pH values below 3 or above 11 while increasing within the pH range from 3 to 11 [61].
Under slightly acidic to mildly basic conditions (pH 5–8), Fe-bearing clay minerals exhibited enhanced catalytic activity for PMS activation. This favorable range ensured balanced surface charge, minimized surface site deactivation caused by protonation or hydroxide adsorption, and favored efficient and sustained generation of ROS, including SO4•– and •OH. Beyond this range, especially under strongly acidic (pH < 3) or highly basic (pH > 11) conditions, radical formation was significantly suppressed due to scavenging reactions and unfavorable surface interactions. This favorable pH range also facilitated redox cycling of Fe(Ⅱ)/Fe(Ⅲ), which was essential for maintaining catalytic activity and radical propagation.
The combination of synergistic additives with Fe-bearing clay minerals garnered significant attention. Synergistic additives, such as humic acids (HA), nitrilotriacetic acid (NTA), and EDTA, were shown to significantly enhance the oxidation performance of Fe-bearing clay minerals [79,98–101]. These additions affected the surface characteristics and electron transfer routes of the minerals by methods including chemical complexation, facilitating electron transfer, and interfacial electron transfer.
NTA and EDTA were organic ligands that formed stable complexes with Fe(Ⅱ) by coordinating via carboxyl (–COOH) and amino (–NH2) groups, leading to the production of soluble complexes (NTA-Fe2+ and EDTA-Fe2+) [99,101]. These complexes significantly enhanced the solubility of structural Fe(Ⅱ) on the mineral surface. Likewise, the –COOH and –OH groups in Pahokee peat humic acid (PPHA) coordinated with Fe(Ⅱ) to create soluble Fe(Ⅱ)-HA complexes (Fig. 4a) [98].

In contrast to structural Fe(Ⅱ), soluble Fe(Ⅱ)-ligand complexes demonstrated enhanced oxidative reactivity, producing a greater quantity of ROS, including •OH. Experimental findings demonstrated that the oxidation rate of soluble Fe(Ⅱ)-EDTA complexes escalated by a factor of 3.5 under 2 mmol/L EDTA conditions, with a resultant •OH concentration of 204 μmol/L within 120 min, markedly exceeding the 17 μmol/L recorded in the absence of ligands (Fig. 4b) [101]. NTA demonstrated comparable performance, yielding an accumulated •OH concentration of 198 μmol/L at 2 mmol/L [100]. The production of p-HBA was used as an indicator for the generation of •OH. The k value represented the amount of p-HBA produced per unit of Fe(Ⅱ) oxidation, with units of μmol/L, indicating the amount of p-HBA generated for each Fe(Ⅱ) oxidation unit. In another experiment, the k values were 6.64 μmol/L without ligands and 55.08 μmol/L with NTA, indicating that the NTA ligand markedly enhanced the oxidation of Fe(Ⅱ) and the generation of •OH [101].
In oxidative conditions, soluble NTA-Fe2+, EDTA-Fe2+, and HA-Fe2+ complexes were swiftly oxidized to Fe(Ⅲ)-ligand complexes, which then produced potent oxidants, including H2O2 and •OH, via homogeneous reactions. Moreover, investigations indicated that the functional groups in ligands, including carboxyl and phenolic hydroxyl groups, could promote internal electron transfer, converting Fe(Ⅲ)-ligand complexes to Fe(Ⅱ)-ligand complexes. The redox cycling of iron markedly increased the generation of oxidants while prolonging the reaction time. In pollutant degradation studies, NTA and EDTA exhibited significant synergistic effects on TCE breakdown. The introduction of 2 mmol/L EDTA increased the TCE degradation efficiency to 60%, which was 13% without ligands. Meanwhile, 2 mmol/L NTA achieved a similar degradation efficiency of 57% [100].
PPHA and Elliott Soil humic acid (ESHA) demonstrated considerable electron-accepting capacity (EAC) and EDC owing to abundant quinone, hydroxyl, and aromatic structures [102]. PPHA and ESHA facilitated electron transfer between surface Fe(Ⅱ) and O2. This transformation converted the SET process into a more efficient double electron transfer process (Fig. 4b) [101], with H2O2 serving as the key intermediate [103].
Quinones and hydroxyl groups in humic acids specifically took electrons from surface Fe(Ⅱ), forming reduced humic acids (H2Q or SQ). The reduced humic acids subsequently interacted with aqueous oxygen, promoting rapid electron transfer and producing •OH. These reactions occurred primarily through electron transfer pathways involving both heterogeneous and homogeneous processes. PPHA and ESHA facilitated the direct reduction of O2 to H2O2 by circumventing the intermediate steps of SET, thereby augmenting the production rate of both H2O2 and •OH. Experimental findings showed that ESHA possessed a superior EAC compared to PPHA, hence markedly improving the oxidation of Fe(Ⅱ) and the efficacy of oxidant production. In a system comprising 20 mg C/L ESHA and rNAu-2, the EAC of ESHA diminished from 158.4 μmol e-/L to 89.6 μmol e-/L within 5 min and further reduced to 78.2 μmol e-/L after 60 min, indicating a rapid reduction of Fe(Ⅱ) inside the mineral matrix [98]. This fast decrease in EAC suggested that ESHA enhanced the electron transfer from Fe(Ⅱ) to O2 rapidly, demonstrating its high reactivity in promoting the oxidation process. The elevated EAC of ESHA relative to PPHA enabled it to play a more prominent role in the early phases of the structural Fe(Ⅱ) oxidation process. Furthermore, in systems with 20–40 mg C/L ESHA, the efficiency of Fe(Ⅱ) oxidation, quantified as the yield of •OH, rose from 2.7% (2.7 μmol/L •OH per 100 μmol/L Fe(Ⅱ)) oxidized to 3.4%−3.8%, demonstrating that ESHA markedly improved •OH production efficiency [98]. This efficiency increase indicated that higher concentrations of ESHA led to more efficient electron transfer and thus more effective •OH production at higher Fe(Ⅱ) oxidation rates. At a low concentration (5 mg C/L), the ESHA-mediated route contributed roughly 15.2% to H2O2 production. As the ESHA concentration increased to 40 mg C/L, this contribution significantly increased to 72.5%, demonstrated the positive association between humic acid concentration and oxidant production efficiency [98].
Despite the significant improvements in oxidative performance achieved by synergistic additives, potential limitations warranted careful consideration. For organic ligands, the optimal type and dosage varied considerably depending on the mineral substrate and reaction conditions, making generalized application challenging. Excessive ligand concentrations might compete with target contaminants for •OH, thereby reducing degradation efficiency. Furthermore, ligand-induced mobilization of structural iron from clay mineral matrices could compromise the reusability and long-term stability of the catalytic materials. The ecological risks associated with unreacted ligands and transformation byproducts also remained poorly understood, raising concerns about potential secondary pollution.
The basic pathway of interfacial electron transfer involved the movement of electrons from a solid surface electron-donating species to an electron-accepting species in the solution. The synergistic effect between Fe-bearing clays and nanoscale zero-valent aluminum (nZVAl) was not a simple additive combination. Instead, it was driven by interfacial electron transfer and chemical interactions within the reaction environment. The ball milling process during material preparation enhanced the synergy by reducing the electron transfer distance between nZVAl and Fe-bearing clays, which helped nZVAl donate electrons to the structural Fe(Ⅲ) in the clays [91]. As a result, the electron transfer distance was shortened, and the Fe(Ⅱ)/Fe(Ⅲ) cycling efficiency at the edge sites was also enhanced, thereby improving the catalytic activity of the clay minerals (Fig. 4c) [91]. Additionally, the high surface area and buffering properties of Fe-bearing clays played a crucial role in stabilizing the reaction environment. The clays adsorbed H+ and Al3+ ions from the solution, maintaining a stable pH condition (Fig. 4d) [97]. The buffering effect minimized the oxidative passivation of nZVAl, preserving the long-term reactivity. The combined effects underscored the importance of clay minerals in amplifying the degradation efficiency of nZVAl systems.
In the system where nZVAl was combined with Fe-bearing clay minerals to activate O2 for degrading 4-chlorophenol (4-CP), the reaction demonstrated remarkable synergy between Fe-bearing clay minerals and nZVAl. At an initial pH of 6.08 [104], the observed 4-CP degradation kobs was determined to be 0.0852 min-1. In comparison, nZVAl alone showed negligible activity in degrading 4-CP, highlighting the significant catalytic role of Fe-bearing clay minerals [97]. In another research, the reaction achieved even higher rate under more acidic conditions (pH 3.0), with kobs, ZVAl−NaSWy−3 = 1.095 h-1 and kobs, ZVAl−NaNAu−2 = 0.741 h-1. Furthermore, the total organic carbon removal efficiency confirmed the efficiency of the nZVAl and Fe-bearing clay minerals system, as ZVAl-NaSWy-3 achieved a mineralization efficiency of 48.9% and ZVAl-NaNAu-2 reached 40.0% [105].
For nanoscale zero-valent metals, particularly aluminum-based additives, the reactivity was highly pH-dependent. In mildly acidic to neutral environments, surface passivation significantly inhibited electron transfer. Under highly acidic conditions (pH < 1), rapid corrosion and structural degradation of the metal could occur, impairing the stability of the composite system. These challenges underscored the need for a balanced design strategy that enhanced catalytic efficiency while mitigating environmental risks, thereby promoting the safe and sustainable application of additive-assisted advanced oxidation processes in environmental remediation.
Fe-bearing clay minerals had substantial potential as catalysts in AOPs for treating environmental pollutants. The effectiveness of Fe-bearing clay minerals was attributed to their capacity to activate oxidants, resulting in the generation of ROS. Fe-bearing clay minerals played a dominant role in radical-based reactions, enabling the efficient degradation of organic contaminants. The redox cycle of Fe(Ⅱ)/Fe(Ⅲ) within Fe-bearing clay minerals was critical in sustaining catalytic activity.
Structural and surface-associated iron was essential for electron transfer, which drove the production of ROS. This review focused on the vital mechanisms of electron transfer processes in Fe-bearing clay minerals, illustrated the role of Fe-bearing clay minerals in initiating and maintaining the degradation of various contaminants. This review highlighted that incorporating synergistic substances, including humic acids and nZVAl, improved electron transfer efficiency, accelerated the production of ROS and significantly enhanced pollutant degradation efficiency. This review connected the structural properties of Fe-bearing clay minerals to catalytic functions, improving the understanding of their role in organic pollutant oxidation processes.
Considering the insights gained from the catalytic mechanisms and the application of Fe-bearing clay minerals in pollutant degradation, future research can address many crucial areas to improve our understanding and use of Fe-bearing clay minerals. The following directions could lead to significant field breakthroughs.
The activation of oxidants like PMS, PI, and O₃ by Fe-bearing clay minerals is still being studied. Activation mechanisms, electron transfer procedures, and optimization of reaction conditions for various oxidants could support catalyst design in the future.
Structural characteristics and external environmental conditions may limit the catalytic efficiency and stability of Fe-bearing clay minerals. Synergistic pairings with other functional materials can boost catalytic performance. Biochar, graphene, and MOFs have excellent research potential.
Future research could optimize oxidation reaction initiation, reaction kinetics, and adaptability to complex pollution environments like the combined pollution of organic pollutants and heavy metals for in situ remediation of polluted rivers, groundwater, and soils.
Fe-bearing clay minerals are ecologically favorable catalysts, but their complete life cycle, including preparation, use, and disposal, must be evaluated to ensure technical sustainability. Quantifying resource use (mineral extraction, energy use) and environmental emissions during material preparation is key. Researchers can assess natural and synthetic clay mineral production techniques based on carbon footprint and energy efficiency to design catalysts with fewer environmental costs.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Mingli Jiang: Writing – review & editing, Writing – original draft, Visualization, Data curation, Conceptualization. Weihua Xu: Writing – review & editing, Supervision, Resources, Project administration, Funding acquisition. Xin Li: Methodology, Investigation, Formal analysis, Data curation. Xiaofei Tan: Supervision, Software, Resources, Methodology. Wei Zhang: Validation, Supervision, Methodology.
This work was financially supported by Hunan Provincial Natural Science Foundation of China (No. 2025JJ50305).
Supplementary material associated with this article can be found, in the online version, at doi:
B.O. Otunola, O.O. Ololade, Environ. Technol. Innov. 18 (2020) 100692. doi: 10.1016/j.eti.2020.100692
J. Huang, A. Jones, T.D. Waite, et al., Chem. Rev. 121 (2021) 8161–8233. doi: 10.1021/acs.chemrev.0c01286
Q. Fan, L. Wang, Y. Fu, et al., Sci. Total Environ. 855 (2023) 159003. doi: 10.1016/j.scitotenv.2022.159003
K.H. Knorr, G. Lischeid, C. Blodau, Geoderma 153 (2009) 379–392. doi: 10.1016/j.geoderma.2009.08.023
H. Zhou, J. Peng, X. Duan, et al., Environ. Sci. Technol. 57 (2023) 3334–3344. doi: 10.1021/acs.est.2c07447
W. Xie, S. Yuan, M. Tong, et al., Environ. Sci. Technol. 54 (2020) 2975–2984. doi: 10.1021/acs.est.9b04870
M.E. Bishop, P. Glasser, H. Dong, B. Arey, L. Kovarik, Geochim. Cosmochim. Acta 133 (2014) 186–203. doi: 10.1016/j.gca.2014.02.040
N. Chen, M. Huang, C. Liu, et al., Water Res. 165 (2019) 114997. doi: 10.1016/j.watres.2019.114997
M. Usman, J.M. Byrne, A. Chaudhary, et al., Chem. Rev. 118 (2018) 3251–3304. doi: 10.1021/acs.chemrev.7b00224
R.A. Schoonheydt, Appl. Clay Sci. 131 (2016) 107–112. doi: 10.1016/j.clay.2015.12.005
D.B. França, L.S. Oliveira, F.G.N. Filho, et al., J. Environ. Chem. Eng. 10 (2022) 107341. doi: 10.1016/j.jece.2022.107341
M.E. Bishop, H. Dong, R.K. Kukkadapu, C. Liu, R.E. Edelmann, Geochim. Cosmochim. Acta 75 (2011) 5229–5246. doi: 10.1016/j.gca.2011.06.034
T. Peretyazhko, J.M. Zachara, S.M. Heald, et al., Geochim. Cosmochim. Acta 72 (2008) 1521–1539. doi: 10.1016/j.gca.2008.01.004
H. Liu, T. Liu, S. Chen, et al., Sci. Total Environ. 951 (2024) 175722. doi: 10.1016/j.scitotenv.2024.175722
P. Komadel, P.R. Lear, J.W. Stucki, Clays Clay Miner 38 (2024) 203–208.
P. Komadel, J. Madejová, J.W. Stucki, Appl. Clay Sci. 34 (2006) 88–94. doi: 10.1016/j.clay.2005.10.016
K.B. Gregory, P. Larese-Casanova, G.F. Parkin, M.M. Scherer, Environ. Sci. Technol. 38 (2004) 1408–1414. doi: 10.1021/es034588w
C.A. Gorski, L. Klupfel, A. Voegelin, M. Sander, T.B. Hofstetter, Environ. Sci. Technol. 46 (2012) 9369–9377. doi: 10.1021/es302014u
E.J. Calabrese, E.J. Stanek, P. Pekow, R.M. Barnes, Ecotox. Environ. Safe. 36 (1997) 258–268. doi: 10.1006/eesa.1996.1511
B. Petrie, R. Barden, B. Kasprzyk-Hordern, Water Res. 72 (2015) 3–27. doi: 10.1016/j.watres.2014.08.053
C. Dong, W. Fang, Q. Yi, J. Zhang, Chemosphere 308 (2022) 136205. doi: 10.1016/j.chemosphere.2022.136205
S. Li, Y. Yang, H. Zheng, et al., Chemosphere 297 (2022) 134214. doi: 10.1016/j.chemosphere.2022.134214
Q. Zhang, D. Zheng, B. Bai, Z. Ma, S. Zong, Chem. Eng. J. 500 (2024) 157134. doi: 10.1016/j.cej.2024.157134
B.O. Orimolade, A.O. Oladipo, A.O. Idris, et al., Sci. Total Environ. 881 (2023) 163522. doi: 10.1016/j.scitotenv.2023.163522
Z. Mahmood, S. Garg, Y. Yuan, et al., Water Res. 254 (2024) 121410. doi: 10.1016/j.watres.2024.121410
M. Manaka, M. Takeda, Appl. Geochem. 131 (2021) 105034. doi: 10.1016/j.apgeochem.2021.105034
C.R. Blanco Zúñiga, N. Rojas-Arias, L.Y. Peña Pardo, M.E. Mendoza Oliveros, S.A. Martínez Ovalle, Ingeniería 26 (2020) 5–14. doi: 10.14483/23448393.15846
W. Zhang, X. Li, J. Shen, et al., J. Hazard. Mater. 444 (2023) 130401. doi: 10.1016/j.jhazmat.2022.130401
L. Pentráková, K. Su, M. Pentrák, J.W. Stucki, Clay Min. 48 (2018) 543–560.
H. Dong, D.P. Jaisi, J. Kim, G. Zhang, Am. Mineral 94 (2009) 1505–1519. doi: 10.2138/am.2009.3246
T.B. Hofstetter, R.P. Schwarzenbach, S.B. Haderlein, Environ. Sci. Technol. 37 (2003) 519–528. doi: 10.1021/es025955r
R. Singh, H. Dong, Q. Zeng, L. Zhang, K. Rengasamy, Geochim. Cosmochim. Acta 210 (2017) 25–41. doi: 10.1016/j.gca.2017.04.030
G. Liu, S. Qiu, B. Liu, et al., J. Zhou, Sci. Rep. 7 (2017) 45354. doi: 10.1038/srep45354
A.M. Jones, C.A. Murphy, T.D. Waite, R.N. Collins, Environ. Sci. Technol. 51 (2017) 12573–12582. doi: 10.1021/acs.est.7b01793
S.N. Yong, S. Lim, C.L. Ho, S. Chieng, S.H. Kuan, Appl. Clay Sci. 228 (2022) 106653. doi: 10.1016/j.clay.2022.106653
W.P. Gates, J.W. Stucki, R.J. Kirkpatrick, Phys. Chem. Miner. 23 (1996) 535–541.
F. Favre, D. Tessier, M. Abdelmoula, et al., Eur. J. Soil Sci. 53 (2002) 175–183. doi: 10.1046/j.1365-2389.2002.00423.x
K. Lee, J.E. Kostka, J.W. Stucki, Clays Clay Miner 54 (2024) 195–208.
M. Grybos, P. Billard, S. Desobry-Banon, et al., J. Colloid Interface Sci. 362 (2011) 317–324. doi: 10.1016/j.jcis.2011.07.031
H. Dong, J.E. Kostka, J. Kim, Clays Clay Miner 51 (2024) 502–512. doi: 10.3390/jmse12030502
H. Dong, Elements 8 (2012) 113–118. doi: 10.2113/gselements.8.2.113
J.E. Kostka, E. Haefele, R. Viehweger, J.W. Stucki, Environ. Sci. Technol. 33 (1999) 3127–3133. doi: 10.1021/es990021x
X.N. Wang, G.X. Sun, X.M. Li, T.A. Clarke, Y.G. Zhu, J. Soils Sediments 18 (2017) 159–168.
J.E. Amonette, J. Charles Templeton, Clays Clay Miner 46 (2024) 51–62.
C. Moon, B.-C. Kim, K. Nam, J. Environ. Chem. Eng. 13 (2025) 117250. doi: 10.1016/j.jece.2025.117250
Y. Liu, H. Chen, Y. Sheng, et al., Geochim. Cosmochim. Acta. 395 (2025) 44–63. doi: 10.1117/12.3065193
J. Manjanna, Appl. Clay Sci. 42 (2008) 32–38. doi: 10.1016/j.clay.2008.02.005
I. Rozenson, L. Heller-Kallai, Clays Clay Miner 24 (2024) 271–282.
I. Rozenson, L. Heller-Kallai, Clays Clay Miner 24 (2024) 283–288.
J.W. Stucki, G.W. Bailey, H. Gan, Appl. Clay Sci. 10 (1996) 417–430. doi: 10.1016/0169-1317(96)00002-6
F. Luan, Y. Liu, A.M. Griffin, C.A. Gorski, W.D. Burgos, Environ. Sci. Technol. 49 (2015) 1418–1426. doi: 10.1021/es504149y
F.R. Ribeiro, J.D. Fabris, J.E. Kostka, P. Komadel, J.W. Stucki, Pure Appl. Chem. 81 (2009) 1499–1509. doi: 10.1351/pac-con-08-11-16
J.W. Stucki, K. Lee, L. Zhang, R.A. Larson, Pure Appl. Chem. 74 (2002) 2145–2158. doi: 10.1351/pac200274112145
A. Manceau, V.A. Drits, B. Lanson, et al., Am. Mineral 85 (2000) 153–172. doi: 10.2138/am-2000-0115
A. Manceau, B. Lanson, V.A. Drits, et al., Am. Mineral 85 (2000) 133–152. doi: 10.2138/am-2000-0114
A. Neumann, S. Petit, T.B. Hofstetter, Geochim. Cosmochim. Acta 75 (2011) 2336–2355. doi: 10.1016/j.gca.2011.02.009
J.E. Kostka, J. Wu, K.H. Nealson, J.W. Stucki, Geochim. Cosmochim. Acta 63 (1999) 3705–3713. doi: 10.1016/S0016-7037(99)00199-4
P. Wang, X. Liu, W. Qiu, et al., Water Res. 182 (2020) 116030. doi: 10.1016/j.watres.2020.116030
J. Liang, X. Duan, X. Xu, et al., Environ. Sci. Technol. 58 (2023) 915–924. doi: 10.1007/978-981-19-7826-5_88
H.-J. Cui, Y. Ning, C. Wu, et al., J. Environ. Sci. 124 (2023) 688–698. doi: 10.1016/j.jes.2022.02.012
N. Chen, G. Fang, C. Zhu, et al., J. Hazard. Mater. 389 (2020) 121819. doi: 10.1016/j.jhazmat.2019.121819
T.B. Hofstetter, A. Neumann, R.P. Schwarzenbach, Environ. Sci. Technol. 40 (2006) 235–242. doi: 10.1021/es0515147
J. Entwistle, D.E. Latta, M.M. Scherer, A. Neumann, Environ. Sci. Technol. 53 (2019) 14308–14318. doi: 10.1021/acs.est.9b04665
X. Liu, S. Yuan, M. Tong, D. Liu, Water Res. 113 (2017) 72–79. doi: 10.1016/j.watres.2017.02.012
N. Chen, M. Geng, D. Huang, et al., J. Hazard. Mater. 434 (2022) 128861. doi: 10.1016/j.jhazmat.2022.128861
C. Li, Y. Huang, X. Dong, et al., Appl. Catal. B: Environ. 247 (2019) 10–23. doi: 10.1016/j.apcatb.2019.01.079
L. Huang, Z. Liu, H. Dong, et al., Appl. Clay Sci. 188 (2020) 105504. doi: 10.1016/j.clay.2020.105504
G.D. Fang, D.M. Zhou, D.D. Dionysiou, J. Hazard. Mater. 250-251 (2013) 68–75. doi: 10.1016/j.jhazmat.2013.01.054
S. Yuan, X. Liu, W. Liao, et al., Geochim. Cosmochim. Acta 223 (2018) 422–436. doi: 10.3390/icem18-05277
A.N. Pham, T.D. Waite, Geochim. Cosmochim. Acta 72 (2008) 3616–3630. doi: 10.1016/j.gca.2008.05.032
A. Neumann, T.L. Olson, M.M. Scherer, Environ. Sci. Technol. 47 (2013) 6969–6977. doi: 10.1021/es304744v
Y. Qian, A.C. Scheinost, S. Grangeon, et al., ACS Earth Space Chem. 7 (2023) 1868–1881. doi: 10.1021/acsearthspacechem.3c00136
C. Chen, Y. Dong, A. Thompson, Environ. Sci. Technol. 57 (2023) 10696–10707. doi: 10.1021/acs.est.3c01876
A. Zhu, Y. Guo, G. Liu, et al., Chin. Chem. Lett. 30 (2019) 2241–2244. doi: 10.1016/j.cclet.2019.09.003
H. Liu, T.A. Bruton, W. Li, et al., Environ. Sci. Technol. 50 (2016) 890–898. doi: 10.1021/acs.est.5b04815
C.E. Schaefer, P. Ho, E. Berns, C. Werth, Environ. Sci. Technol. 52 (2018) 13747–13755. doi: 10.1021/acs.est.8b04108
W. Liao, S. Yuan, X. Liu, M. Tong, Geochim. Cosmochim. Acta 257 (2019) 96–109. doi: 10.1016/j.gca.2019.04.027
D.E. Latta, A. Neumann, W.A.P.J. Premaratne, M.M. Scherer, ACS Earth Space Chem. 1 (2017) 197–208. doi: 10.1021/acsearthspacechem.7b00013
N. Chen, G. Fang, G. Liu, et al., Chem. Eng. J. 355 (2019) 333.
S. Petit, F. Baron, O. Grauby, A. Decarreau, Vib. Spectrosc. 87 (2016) 137–142. doi: 10.1016/j.vibspec.2016.09.022
C.I. Fialips, D. Huo, L. Yan, J. Wu, J.W. Stucki, Am. Mineral 87 (2002) 630–641. doi: 10.2138/am-2002-5-605
M.D. Dyar, Am. Mineral 72 (1987) 102–112.
L. Heller-Kallai, I. Rozenson, Phys. Chem. Miner. 7 (1981) 223–238. doi: 10.1007/BF00311893
G.L. Taylor, A.P. Ruotsala, R.O. Keeling, Clays Clay Miner 16 (2024) 381–391. doi: 10.1080/14708477.2024.2355736
A.Y. de Llano, G. Bidoglio, A. Avogadro, P.N. Gibson, P. Rivas Romero, J. Contam. Hydrol. 21 (1996) 129–139. doi: 10.1016/0169-7722(95)00038-0
K.A. Rothwell, M.P. Pentrak, L.A. Pentrak, J.W. Stucki, A. Neumann, Environ. Sci. Technol. 57 (2023) 10231–10241. doi: 10.1021/acs.est.3c01655
W. Zhang, X. Li, Y. Zhao, et al., Sci. Total Environ. 902 (2023) 247–254.
V. Pothanamkandathil, A. Neumann, A. Thompson, C.A. Gorski, Environ. Sci. Technol. 58 (2024) 19702–19713. doi: 10.1021/acs.est.4c07835
V. Alexandrov, A. Neumann, M.M. Scherer, K.M. Rosso, J. Phys. Chem. C 117 (2013) 2032–2040. doi: 10.1021/jp3110776
B. Shi, K. Liu, L. Wu, et al., Environ. Sci. Technol. 50 (2016) 8661–8669. doi: 10.1021/acs.est.6b02019
R. Yang, Q. Chang, N. Li, H. Yang, Chem. Eng. J. 433 (2022) 133682. doi: 10.1016/j.cej.2021.133682
B. Morgan, O. Lahav, Chemosphere 68 (2007) 2080–2084. doi: 10.1016/j.chemosphere.2007.02.015
S. Cui, R. Wang, Q. Chen, L. Pugliese, S. Wu, Environ. Sci. Ecotechnol. 22 (2024) 100446. doi: 10.1016/j.ese.2024.100446
D.P. Jaisi, R.K. Kukkadapu, D.D. Eberl, H. Dong, Geochim. Cosmochim. Acta 69 (2005) 5429–5440. doi: 10.1016/j.gca.2005.07.008
T. Xie, S. Lu, J. Zeng, et al., Sci. Total Environ. 726 (2020) 138650. doi: 10.1016/j.scitotenv.2020.138650
H. Dong, Y. Li, S. Wang, et al., Environ. Sci. Technol. Lett. 7 (2020) 219–224. doi: 10.1021/acs.estlett.0c00025
R. Yang, J. Cai, H. Yang, Sci. Total Environ. 773 (2021) 145661. doi: 10.1016/j.scitotenv.2021.145661
C. Yu, Y. Zhang, Y. Lu, et al., Environ. Sci. Technol. 55 (2021) 13366–13375.
Q. Zeng, X. Wang, X. Liu, et al., Environ. Sci. Technol. 54 (2020) 15013–15023. doi: 10.1021/acs.est.0c04463
W. Xie, P. Zhang, W. Liao, M. Tong, S. Yuan, Environ. Sci. Technol. 55 (2021) 7044–7051. doi: 10.1021/acs.est.1c00136
Q. Zeng, H. Dong, X. Wang, Geochim. Cosmochim. Acta 251 (2019) 136–156. doi: 10.1016/j.gca.2019.02.032
L. Klüpfel, A. Piepenbrock, A. Kappler, M. Sander, Nat. Geosci. 7 (2014) 195–200. doi: 10.1038/ngeo2084
M. Fujii, A. Imaoka, C. Yoshimura, T.D. Waite, Environ. Sci. Technol. 48 (2014) 4414–4424. doi: 10.1021/es405496b
N.S. Shah, J.A. Khan, M. Sayed, et al., J. Clean Prod. 246 (2020) 119032. doi: 10.1016/j.jclepro.2019.119032
A.D. Bokare, W. Choi, Environ. Sci. Technol. 43 (2009) 7130–7135. doi: 10.1021/es9013823

Figure 1 Crystal structure of Fe-bearing clay minerals: (a) Montmorillonite, (b) kaolinite.

Figure 2 (a) Microbial reduction process of structural Fe(Ⅲ): (A) Microorganisms transferred electrons through direct contact and nanowires. (B) Metabolites produced by microorganisms dissolved structural Fe(Ⅲ) and reduced it to ferrous iron (Fe2+). (C) The precipitation of biogenic secondary minerals. (b) Electron transfer and structural oxygen vacancy formation during chemical reduction of structural Fe(Ⅲ) by sodium dithionite.

Figure 3 Electron transfer pathways from structural Fe(Ⅱ) in reduced NAu-2 to O2, leading to •OH radical generation. Reproduced with permission [69]. Copyright 2018, Elsevier.

Figure 4 (a) Coexisting HA led to two-electron transfer process with H2O2 as the main intermediate. Reproduced with permission [98]. Copyright 2021, American Chemical Society. (b) Electron transfer mechanisms of reduced NAu-2 during oxygenation depending on the type of ligands. Reproduced with permission [101]. Copyright 2019, Elsevier. (c) Suggested mechanisms for persulfate (PS) activation using mZVAl-modified NaNAu-2 to generate reactive species. Reproduced with permission [91]. Copyright 2021, Elsevier. (d) Mechanism of ROS generation and the degradation of 4-CP using ZVAl-Clay composites. Reproduced with permission [97]. Copyright 2022, Elsevier.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: