Citation:
Mei Wang, Kai Song, Yuning Song, Haiyong Jia, Peng Zhan. Advances in nucleotide metabolism-targeted drug research[J]. Chinese Chemical Letters,
2026, 37(7): 112238.
doi:
10.1016/j.cclet.2025.112238
Advances in nucleotide metabolism-targeted drug research
English
Advances in nucleotide metabolism-targeted drug research
Targeting nucleotide metabolic enzymes has seen significant advancements in recent years, demonstrating transformative potential across multiple disease areas. In oncology, approaches such as the dihydroorotate dehydrogenase (DHODH) inhibitor 416 for all-trans retinoic acid (ATRA)-resistant acute promyelocytic leukemia (APL) and the Brequinar-DT2216 combination for pancreatic ductal adenocarcinoma (PDAC) illustrate a growing emphasis on synergistic "metabolism-apoptosis" strategies. Nucleotide metabolism-targeted therapies have advanced across diseases: compound 54 improves activity/selectivity in rheumatoid arthritis (RA); JNJ-6640 validates amidophosphoribosyltransferase (PurF) for drug-resistant tuberculosis (TB); and antiviral agents like ASLAN003 plus metabolic combinations address resistance/efficacy gaps. Key challenges include metabolic compensation, tissue-specific selectivity, and optimal combinations. Future directions involve dual-action molecules (de novo/salvage pathway inhibition), targeted delivery via antibody-drug conjugates (ADCs) or proteolysis-targeting chimeras (PROTACs), and synergistic combinations [1]. Enabled by cryo-electron microscopy (cryo-EM), artificial intelligence (AI)-driven design, and single-cell metabolomics, these therapies are pivotal for cancers, autoimmune disorders, TB, and viral infections, with clinical candidates expected to yield breakthroughs in five years.
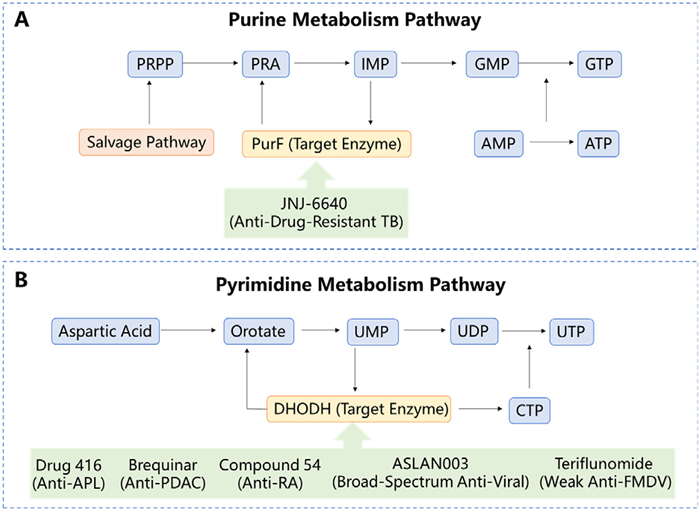
In recent years, nucleotide metabolism-targeted drugs have become a research focus due to the close association of nucleotide metabolism disorders with tumors, infectious diseases, and other pathologies; to clarify their action mechanism, we present a schematic of the core purine and pyrimidine metabolic pathways (Fig. 1), where the purine pathway (Fig. 1A) starts from phosphoribosyl pyrophosphate pentasodium (PRPP) and involves metabolites like 5-phosphoribosyl-1-amine (PRA), inosine monophosphate (IMP), guanosine monophosphate (GMP), and adenosine monophosphate (AMP), with drug JNJ-6640 targeting PurF to treat drug-resistant TB and the salvage pathway serving as a metabolic supplement, while the pyrimidine pathway (Fig. 1B) is initiated by aspartic acid and involves metabolites such as orotate, uridine monophosphate (UMP), uridine triphosphate (UTP), and cytidine triphosphate (CTP), with drugs like Drug 416, Brequinar, and ASLAN003 targeting DHODH for conditions like APL, PDAC, and viral infections, these drugs exert therapeutic effects by inhibiting key enzymes like PurF and DHODH to block nucleotide synthesis, providing a framework for subsequent analysis of drug chemical structures and pharmacological activities.
Figure 1
Figure 1.
Schematic diagram of the mechanism of action of nucleotide metabolism-targeted drugs. (A) Purine metabolism pathway: the core metabolic flow starting from PRPP, with key metabolites including PRA, IMP, GMP, AMP, GTP, and ATP. Key target enzymes are PurF and IMPDH; the corresponding targeted drug is JNJ-6640 (anti-drug-resistant TB). The "salvage pathway" is labeled to reflect alternative metabolic supplementation for purines. (B) Pyrimidine metabolism pathway: the core metabolic flow initiated by aspartic acid, with key metabolites including orotate, UMP, UDP, UTP, and CTP. The key target enzyme is DHODH; corresponding targeted drugs include ASLAN003 (broad-spectrum anti-viral), teriflunomide (anti-FMDV), Drug 416 (anti-APL), Brequinar (anti-PDAC), and compound 54 (anti-RA).
Tumor proliferation relies on nucleotide synthesis, making key metabolic enzymes attractive anti-cancer targets. However, tumor metabolic plasticity limits monotherapy efficacy, with nucleotide salvage pathway compensation as a core challenge: tumor cells (e.g., pancreatic ductal adenocarcinoma (PDAC)) upregulate transporters (e.g., SLC29A1) to utilize 30-50 µmol/L tumor microenvironment uridine, bypassing de novo pathway inhibition. Long-term treatment of PDAC with dihydroorotate dehydrogenase (DHODH) inhibitor Brequinar induces a 2.1-fold SLC29A1 increase, enhancing uridine uptake and reducing drug efficacy.
In APL, the use of ATRA has raised the 5-year survival rate to ~90%. However, challenges remain due to primary resistance (about 5%) and relapse-related resistance (~30%). In 2023, Yang et al. reported a major breakthrough using compound 416, a highly selective DHODH inhibitor (Fig. 2) [2]. With a half maximal inhibitory concentration (IC50) of 7.5 nmol/L against DHODH, it is 40 times more potent than teriflunomide (IC50 = 307.1 nmol/L) and effectively overcomes ATRA resistance in NB4-R1 cells. At 0.057 µmol/L, compound 416 causes significant S-phase cell cycle arrest, increasing S-phase cells from 20% to 55%, and activates the caspase-3/8/9 apoptotic pathway, leading to an apoptosis rate of 86.59%. It works by inhibiting DHODH, which lowers intracellular uridine levels and bypasses resistance caused by promyelocytic leukemia/retinoic acid receptor alpha (PML/RARα) mutations. In ATRA-resistant NB4-R1 xenograft models, 416 at 10 mg/kg improved survival to 62.5% and reduced liver and spleen infiltration. It shows strong selectivity for cancer cells (75 times higher than for normal Wi-38 cells) and is orally stable, supporting its clinical potential. However, further studies are needed to assess long-term toxicity and possible synergies with ATRA.
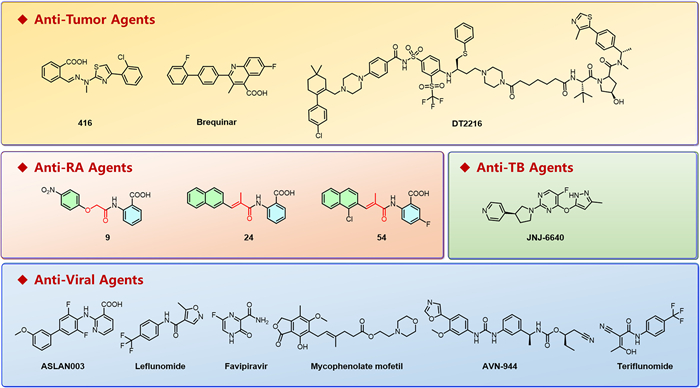
Figure 2
Figure 2.
Chemical structures of representative compounds or drugs mentioned in this editorial, include: 416 (a selective DHODH inhibitor for APL resistant to ATRA); Brequinar (a DHODH inhibitor) and DT2216 (a BCL-XL PROTAC degrader) for combined treatment of PDAC; compounds 9, 24 and 54 (potent hDHODH inhibitors for RA); JNJ-6640 (a PurF inhibitor targeting drug-resistant Mycobacterium tuberculosis for TB treatment); ASLAN003 (a broad-spectrum antiviral DHODH inhibitor); AVN-944 (an IMPDH inhibitor) and teriflunomide (a DHODH inhibitor) for FMDV inhibition; MMF (an IMPDH inhibitor) used in combination with favipiravir against influenza. These compounds target key enzymes in nucleotide metabolism, providing novel therapeutic strategies for cancers, autoimmune diseases, TB, and viral infections.
PDAC, driven by Kirsten ratsarcoma viral oncogene homolog (KRAS) mutations, relies on de novo pyrimidine synthesis and shows limited response to DHODH inhibitor Brequinar monotherapy (tumor inhibition < 30%) due to resistance. In 2025, Zhang et al. found that long-term Brequinar treatment led to adaptive increases in SLC29A1 (2.1-fold) and BCL-XL (45%) through multi-omics analysis (Fig. 2) [3]. To counter this, they combined Brequinar with DT2216, a BCL-XL PROTAC degrader (degradation half-life 2.1 h), achieving strong synergy (human serum albumin (HSA) index > 10) in PDAC cells, raising Annexin V+ cells from 22% to 68% (Fig. 2). In patient-derived organoids, the combination reduced growth by 79%, and in C57BL/6 models, it inhibited tumors by 62% without causing severe thrombocytopenia (platelets ≥ 150 × 109/L). This dual approach targeting pyrimidine synthesis and apoptosis offers promise for gemcitabine-resistant/KRAS G12D+ PDAC [2]. However, high uridine levels in the tumor microenvironment (30–50 µmol/L) may reduce efficacy, and long-term myelosuppression risks require monitoring.
RA, characterized by chronic synovitis and joint destruction, lacks effective treatments due to the long half-life (> 2 weeks) and hepatotoxicity of DHODH inhibitors (e.g., leflunomide). Activated lymphocytes in RA undergo metabolic compensation via pyrimidine salvage pathway bypass, upregulating enzymes (e.g., thymidine kinase 1) to utilize synovial preformed nucleosides, sustaining proliferation and pro-inflammation and impairing traditional DHODH inhibitor efficacy. Human DHODH (hDHODH), the rate-limiting enzyme in de novo pyrimidine synthesis, is critical for activated lymphocyte proliferation, making it an ideal RA target.
In 2021, Zeng et al. designed new hDHODH inhibitors using structure-based methods (Fig. 2) [4]. Starting with lead compound 9 (45% inhibition at 10 µmol/L), they introduced an acrylamide bond to stabilize the naphthyl (hydrophobic tail) and benzoic acid (hydrophilic head), mimicking natural substrate interactions with the enzyme. Key changes included naphthyl substitution (compound 24, IC50 = 75 nmol/L), retaining a 2-methyl group on the acrylamide (boosting activity 10-fold), and adding a 5-fluorine to the benzoic acid (compound 54, IC50 = 32 nmol/L, 2.3-fold more potent than 24). Compound 54 showed strong in vitro activity (T cell proliferation half maximal effective concentration (EC50, 120 nmol/L)) and good pharmacokinetics (rat oral bioavailability 67%, t1/2 = 21.5 h). In CIA rats, 54 at 30 mg/kg reduced arthritis scores from 2.3 to 0.6 (P < 0.001) and cut synovial hyperplasia by 70%. Its "naphthyl-acrylamide-fluorinated benzoic acid" scaffold improves on leflunomide’s structure, making it a promising best-in-class candidate with potential applications in acute myeloid leukemia (AML) and viral infections.
TB remains a leading infectious killer (1.25 million deaths in 2023), with extensive drug-resistant TB (XDR-TB) and neurotoxic second-line drugs posing major challenges. Mycobacterium tuberculosis relies on de novo purine synthesis for survival, with Mycobacterium tuberculosis catalyzing the first committed step (PRPP → PRA). Due to structural differences from human PPAT, PurF is a highly selective target, though unvalidated prior to recent studies.
In 2025, Lamprecht et al. identified pyrrolidinopyrimidine derivative JNJ-6640 via phenotypic screening of 4924 compounds (Fig. 2) [5]. Genetic sequencing of resistant strains, structure-activity relationship (SAR) studies, and enzymatic assays confirmed PurF as the target: JNJ-6640 inhibits MtPurF with IC50 of 1 nmol/L, but human PPAT with IC50 14 µmol/L (selectivity > 10,000-fold). Stable isotope tracing (15N-glutamine) showed 100 nmol/L JNJ-6640 reduced purine incorporation by 80% and increased glutamine by 2.3-fold (P < 0.0001). Microfluidic imaging revealed 0.6 µmol/L JNJ-6640 blocked DNA replication within 6 hours, with 50% lysis by 144 h. In mouse models, a long-acting injectable (LAI) formulation achieved 1.8 log10[colony forming unit (CFU)] reduction in acute infection and 0.5 log10 in chronic infection. Notably, replacing linezolid with JNJ-6640 in bedaquiline-pretomanid regimens preserved efficacy, reducing neurotoxicity risks [5]. JNJ-6640’s potency (minimum inhibitory concentration inhibiting 90% of test isolates (MIC90, 8.6 nmol/L), selectivity, and activity against dormant bacteria (45-fold better in hypoxic models) validate PurF as a target, though metabolic stability (liver clearance 138 µL min−1 mg−1) and solubility need optimization.
Viral infections, especially RNA viruses, threaten global health, with traditional direct-acting antivirals (DAAs) prone to resistance. Targeting host nucleotide metabolic enzymes offers broad-spectrum activity and reduced resistance risk. DHODH, a key enzyme in pyrimidine synthesis and mitochondrial function, regulates viral replication and inflammation, becoming a focal target. ASLAN003 (2-(3,5-difluoro-3′-methoxybiphenyl-4-ylamino)nicotinic acid) is a quinolinamide DHODH inhibitor with IC50 35 nmol/L (2800-fold more potent than leflunomide) (Fig. 2). It inhibits severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with EC50 0.8122 µmol/L (therapeutic index (TI) > 12.3) and broad-spectrum activity against Dengue virus serotype 1-4 (DENV1-4, EC50 = 0.32–1.27 µmol/L), Zika virus (ZIKV, 0.48 µmol/L), and Chikungunya virus (CHIKV, 0.63 µmol/L). Time-course studies show efficacy from 1 h pre- to 4 h post-infection. ASLAN003 preferentially accumulates in metabolically active infected cells, sparing stationary-phase host nucleotide pools. It has high oral bioavailability (> 90%), t1/2 18 h, and stability (40℃ for 6 months, purity > 99%). Combined with dexamethasone, it synergistically inhibits IL-6 (78% vs. 52% monotherapy), potential for coronavirus disease 2019 (COVID-19) cytokine storms.
Favipiravir’s efficacy is limited by high doses and GTP-mediated reduced incorporation. Researchers combined it with inosine monophosphate dehydrogenase (IMPDH) inhibitor mycophenolate mofetil (MMF) to lower GTP pools (Fig. 2). MMF (10 µmol/L) reduced GTP by 62%, synergizing with favipiravir against influenza (IC50 1.829 µmol/L vs. 28.21 µmol/L alone, combination index (CI, 0.32) and increasing viral G → A mutations (2.1%→5.7%). In mice, the combination reduced lung viral load by 2.3 log10 and weight loss by 42%, with 73% less liver toxicity than DHODH inhibitors.
Foot-and-mouth disease virus (FMDV) depends on host-derived purines and pyrimidines for replication. Gong et al. demonstrated that the IMPDH inhibitor AVN-944 effectively inhibits FMDV serotype O with an EC50 of 12.02 µmol/L (selection index (SI) = 7.33) and serotype A with an EC50 of 3.95 µmol/L (SI = 22.30). In contrast, the DHODH inhibitor teriflunomide exhibited a much lower potency, with an EC50 of 294.1 µmol/L (SI = 1.84) (Fig. 2). Both inhibitors exert their antiviral effects during the early stages of infection (0–8 h post-infection), and their inhibitory activities can be reversed by exogenous supplementation with guanosine or uridine. In a suckling mouse model, administration of 60 µg of AVN-944 improved survival rates to 25% and significantly attenuated myocardial inflammation [6]. These findings suggest that targeting host nucleotide biosynthesis pathways represents a promising strategy for the control of FMDV and potentially other picornaviruses.
Recent years have seen remarkable progress in nucleotide metabolic enzyme-targeted drugs, a transformative therapeutic strategy across diseases. In oncology, DHODH inhibitor 416 shows promise for ATRA-resistant APL, while Brequinar-DT2216 combination addresses drug-resistant pancreatic cancer via synergistic "metabolism-apoptosis" effects. For RA, compound 54 (optimized structure) outperforms traditional DHODH inhibitors in activity/selectivity. JNJ-6640 validates PurF as a target for drug-resistant TB (especially XDR-TB). Antivirally, broad-spectrum agent ASLAN003 and favipiravir-MMF combination enhance efficacy and overcome resistance. Key challenges persist, including metabolic compensation with distinct disease-specific mechanisms (e.g., salvage pathway activation in cancer versus alternative metabolic pathway bypass in RA immune cells). Tissue-specific selectivity is crucial to spare healthy cells, particularly for the long-term treatment of chronic diseases. Additionally, systematic research is needed to identify optimal combination therapies that balance efficacy, safety, and pharmacokinetics. For anti-infective agents targeting host nucleotide metabolic enzymes, rigorous safety evaluation and comprehensive toxicological studies are essential, these enzymes are vital for normal cellular functions, and long-term inhibition may disrupt physiological processes.
Future innovations in this field will be driven by key frontier directions: dual-action molecules inhibiting both de novo nucleotide synthesis and salvage pathways (with strong potential to overcome metabolic redundancy, especially in tumors with flexible metabolic networks), cryo-electron microscopy-powered structural biology (revealing small molecule-target enzyme binding mechanisms at atomic resolution to guide precise drug design for improved potency, selectivity, and pharmacokinetics), and expanded applications of protein degradation strategies like PROTACs (e.g., BCL-XL PROTAC DT2216 in PDAC) enabling targeting of previously "undruggable" proteins; additionally, integration of AI-driven drug design and single-cell metabolomics will accelerate drug discovery (AI optimizes molecular interactions and chemical structures, while single-cell metabolomics uncovers disease metabolic heterogeneity for personalized therapies), and combining nucleotide metabolism-targeted therapies with immunotherapies or conventional drugs is expected to enhance anti-tumor immunity, reduce resistance, and improve outcomes. These therapies will extend beyond cancers, autoimmune disorders, TB, and viral infections to other infectious diseases, metabolic disorders, and neurodegenerative diseases linked to nucleotide metabolism imbalances, with multiple candidates in clinical development poised to deliver major breakthroughs in the next five years, establishing them as core components of modern precision medicine, while technological advances and deeper mechanistic insights drive the development of safer, more effective, and versatile treatments for a broad range of conditions.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Mei Wang: Writing – original draft, Conceptualization. Kai Song: Writing – original draft. Yuning Song: Writing – review & editing, Visualization. Haiyong Jia: Writing – review & editing, Visualization. Peng Zhan: Writing – review & editing, Visualization.
Acknowledgment
The authors are supported by the Key Research and Development Program, Ministry of Science and Technology of the People’s Republic of China (No. 2023YFC2606500).
Figure 1
Schematic diagram of the mechanism of action of nucleotide metabolism-targeted drugs. (A) Purine metabolism pathway: the core metabolic flow starting from PRPP, with key metabolites including PRA, IMP, GMP, AMP, GTP, and ATP. Key target enzymes are PurF and IMPDH; the corresponding targeted drug is JNJ-6640 (anti-drug-resistant TB). The "salvage pathway" is labeled to reflect alternative metabolic supplementation for purines. (B) Pyrimidine metabolism pathway: the core metabolic flow initiated by aspartic acid, with key metabolites including orotate, UMP, UDP, UTP, and CTP. The key target enzyme is DHODH; corresponding targeted drugs include ASLAN003 (broad-spectrum anti-viral), teriflunomide (anti-FMDV), Drug 416 (anti-APL), Brequinar (anti-PDAC), and compound 54 (anti-RA).
Figure 2
Chemical structures of representative compounds or drugs mentioned in this editorial, include: 416 (a selective DHODH inhibitor for APL resistant to ATRA); Brequinar (a DHODH inhibitor) and DT2216 (a BCL-XL PROTAC degrader) for combined treatment of PDAC; compounds 9, 24 and 54 (potent hDHODH inhibitors for RA); JNJ-6640 (a PurF inhibitor targeting drug-resistant Mycobacterium tuberculosis for TB treatment); ASLAN003 (a broad-spectrum antiviral DHODH inhibitor); AVN-944 (an IMPDH inhibitor) and teriflunomide (a DHODH inhibitor) for FMDV inhibition; MMF (an IMPDH inhibitor) used in combination with favipiravir against influenza. These compounds target key enzymes in nucleotide metabolism, providing novel therapeutic strategies for cancers, autoimmune diseases, TB, and viral infections.

 DownLoad:
DownLoad:

 下载:
下载:



 下载:
下载:
