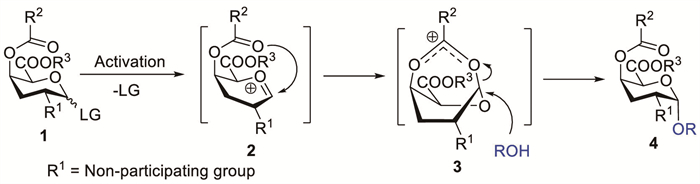
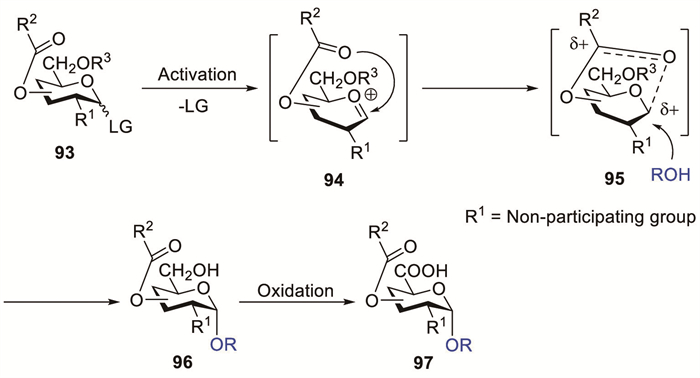
Scheme 1.
Proposed mechanism of O-4-acyl-mediated α-galacturonic acid glycosylation.
Chemical strategies for the stereoselective construction of 1,2-cis-galacturonic and aminogalacturonic acid glycosides
Juntao Cai , Qian Wang , Xu Shen , Lifeng Zhu , Shiqing Jiang , Jian Yin , Chunhong Dong

Carbohydrates are crucial for several biological processes, including cell–cell recognition, immune responses and pathogen–host interactions [1-3]. These glycans often exhibit unique biological activities, rendering them attractive targets for vaccine development [4], antimicrobial agents [3] and biomaterials [5]. In particular, 1,2-cis-glycosidic linkages, especially those involving galacturonic acid (GalA) and its amino derivatives, e.g., 2-amino-2-deoxy-galacturonic acid (GalNAcA), are structurally and functionally relevant [6-8], influencing molecular recognition and biochemical stability in bacterial polysaccharides [9], plant pectins [10], and glycosaminoglycans [11]. However, the paucity of structurally homogeneous oligosaccharides in quantities sufficient for biological studies remains a fundamental challenge in glycobiology research [12]. Although certain carbohydrates can be isolated from natural sources, the isolation of chemically pure and structurally well-defined samples of biologically relevant carbohydrates is impractical [13]. Therefore, the development of synthetic approaches for the precise construction of carbohydrates and their mimetics is indispensable for advancing glycobiology research [14-17]. Glycosylation reactions are vital for the synthesis of complex carbohydrates. Various parameters affect the glycosylation reaction selectivity and yield, such as the reaction temperature, solvent conditions, and activation strategy as well as the nature of protecting groups on the glycosyl donor and acceptor [18]. Unfortunately, current glycosylation methodologies remain limited in scope; in fact, there is no universal approach for efficiently constructing all glycosidic linkage types [13].
The stereoselective construction of 1,2-cis-GalA/GalNAcA glycosides remains a formidable challenge in synthetic carbohydrate chemistry owing to the interplay of electronic, steric and conformational factors [19]. Specifically, the C-5 carboxyl group in GalA/GalNAcA alters the reactivity of the glycosyl donor compared with neutral sugars, typically leading to competitive side reactions or poor stereocontrol [7]. Similarly, the electron-withdrawing properties of the C-5 carboxylate group deactivate neighbouring hydroxyl groups, endowing the C-4 hydroxyl moiety of GalA with poor nucleophilic behaviour [19]. In addition, the presence of a C-2-N-acyl or N-protecting group in aminogalacturonic acid derivatives further complicates the glycosylation by affecting neighbouring-group participation or introducing steric hindrance. For example, neighbouring-group participation at C-2 is essential for the highly stereoselective formation of 1,2-trans-glycosides via intermediate dioxolenium-ion formation because it effectively shields the α-face of the glycosyl donor [18]. In contrast, the synthesis of 1,2-cis-glycosides requires C-2-protecting groups without neighbouring-group participation (e.g., azide or benzyl ether) [20]. Although axial glycosylation products are typically thermodynamically preferred owing to the anomeric effect, these reactions frequently yield anomeric mixtures [20].
Multiple synthetic strategies have been developed for incorporating uronic acid moieties into oligosaccharide structures [6,20-23]. Two main methodologies can be adopted for uronic acid incorporation: pre-glycosylation, employing pre-formed glycuronic acid building blocks during oligosaccharide assembly (discussed in Section 2), and post-glycosylation, involving the oxidation of hydroxyl groups into carboxylates after oligomer construction (discussed in Section 3). This review summarises representative examples of both synthetic approaches, systematically and comparatively analysing their respective merits and limitations. By integrating theoretical and experimental perspectives, a comprehensive framework for the targeted assembly of these biologically critical glycoconjugates is provided.
In pre-glycosylation oxidation approaches, acidic oligosaccharides are assembled using appropriately protected glycuronic acid donors. Pre-glycosylation oxidation strategies enable the efficient assembly of α-GalA oligosaccharides by leveraging stereocontrolling elements, including remote participation effects, 4-O-glycosylated galacturonate donors, lactone-based donors and privileged donor architectures.
Although traditionally overshadowed by the dominant effects of neighbouring groups at C-2, remote substituent participation has emerged as a notable factor for stereocontrol [18]. Reportedly, the effectiveness of D-GalA derivatives in α-GalA synthesis was considerably improved when a participating functional group was introduced at C-4 [24]. The main step towards product formation is the stabilisation of the reactive oxacarbenium ion via the formation of the bicyclic acyloxonium intermediate 3 (Scheme 1). Subsequent nucleophilic attack by the glycosyl acceptor selectively occurs from the bottom face owing to top-face steric obstruction.
Vogel et al. reported the synthesis of homogalacturonan fragments using 4-O-acetyl-protected GalA donors [25,26]. Treatment of galacturonate bromide 5 with glycosyl acceptor 8 in the presence of silver triflate (AgOTf) and silver carbonate afforded disaccharide 9 in 35% yield (Scheme 2A). Subsequently, the effect of C-3 and C-4 substituents in trichloroacetimidate (TCAI) galacturonate donors 6 and 7 on the glycosylation stereoselectivity was assessed. The glycosylation of 3,4-di-O-acetyl derivative 6 with acceptor 8 via trimethylsilyl trifluoromethanesulfonate (TMSOTf) activation afforded disaccharide 9 in 72% yield and with good selectivity (α:β = 13.4:1). Contrary to previous cases, the more reactive glycosyl donor 7 reacted with acceptor 8 to afford disaccharide 10 in 59% yield with exclusive α-selectivity. Finally, the glycosylation of 8 using donor 7 under activation with boron trifluoride diethyl etherate (BF3·Et2O) afforded disaccharide 10 in 53% combined yield but with unsatisfactory stereocontrol (α:β = 1.6:1).
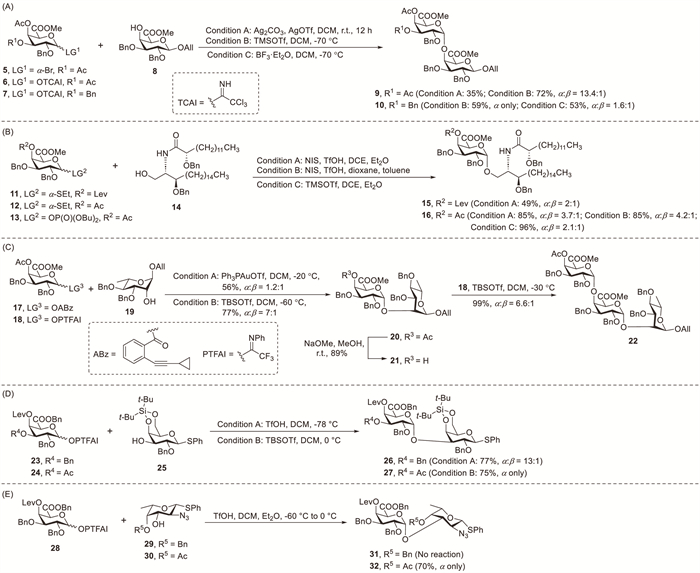
The synthesis of glycosphingolipids presents substantial synthetic challenges [21]. Seeberger et al. reported a streamlined synthesis of orthogonally protected D-GalA thioglycoside donors, and their application in assembling glycosphingolipids (Scheme 2B) [24]. Glycosylation using thioglycoside donors 11 and 12 with ceramide 14 in dichloroethane (DCE) and diethyl ether (Et2O) containing N-iodosuccinimide (NIS) and trifluoromethanesulfonic acid (TfOH) gave the corresponding products 15 (49%, α:β = 2:1) and 16 (85%, α:β = 3.7:1), respectively. Furthermore, employing phosphate donor 13 enhanced the glycosylation yield but reduced the anomeric selectivity (96%, α:β = 2.1:1). The low solubility of ceramide 14 in most solvents under cryogenic conditions limited these transformations. Systematic optimisation enabled an optimal balance between yield and stereoselectivity when using 4-O-acetyl protected thioglycoside 12 with NIS and TfOH activation in a dioxane–toluene (3:1) solvent mixture. This protocol afforded product 16 in 85% yield with good selectivity (α:β = 4.2:1). These studies confirmed the advantageous effects of ether solvents and the stereodirecting influence of C-4 esters in galacto-configured systems for achieving high α-selectivity.
In the chemical synthesis of pectin-derived oligosaccharides, the incorporation of α-GalA linkages is challenging [7,27]. Yu et al. demonstrated the synthesis of Nerium indicum rhamnogalacturonan oligosaccharides via direct α-GalA coupling using their glycosyl donors under various activation conditions (Scheme 2C) [28]. Coupling attempts between ortho-alkynylbenzoate donor 17 and acceptor 19 using triphenylphosphine gold(I) triflate (Ph3PAuOTf) as a catalyst produced disaccharide 20 in a moderate yield (56%) at −20 ℃ but with minimal stereocontrol (α:β = 1.2:1). Notably, the glycosylation efficiency considerably decreased at temperatures below −20 ℃ owing to the insufficient activation of donor 17. Under tert-butyldimethylsilyl triflate (TBSOTf) catalysis in dichloromethane (DCM), N-phenyl-2,2,2-trifluoroacetimidate (PTFAI) donor 18 reacted with acceptors 19 and 21 to afford disaccharide 20 (77%, α:β = 7:1) and trisaccharide 22 (99%, α:β = 6.6:1), respectively, with excellent yields and stereoselectivity.
The synthesis of Streptococcus pneumoniae serotype 1 (Sp1) polysaccharides encounters three principal obstacles: the incorporation of the rare 2-acetamido-4-amino-2,4,6-trideoxy-D-galactose, inherently poor reactivity of GalA donors in glycosylation reactions relative to their galactose analogues and stereochemical demand for establishing cis-glycosidic bonds throughout the oligosaccharide chain [29]. Codée et al. optimised a synthetic route for Sp1 oligosaccharides, achieving the stereocontrolled assembly from the trisaccharide to dodecasaccharide length (1–4 repeating units) [29,30]. The synthesis commenced with the challenging glycosylation of GalA 23 with galactose acceptor 25 (Scheme 2D). After an extensive condition screening, disaccharide 26 was obtained in 77% yield with outstanding α-selectivity (13:1), demonstrating scalability to multi-gram quantities. Next, under TBSOTf activation, the coupling of 3,4-di-O-acyl-GalA donor 24 and galactose acceptor 25 afforded disaccharide 27 in 75% yield and with complete α-selectivity.
Kuikarni et al. achieved the total synthesis of the tetrasaccharide repeating unit of Shewanella japonica type strain KMM 3299T [31]. The tetrasaccharide synthesis process was particularly challenging because it required three consecutive cis-linkages bridging unusual amino sugars and D-GalA units. Attempts to achieve the glycosylation by employing known trifluoroacetamidate donor 28 and L-fucosamine acceptor 29 using TfOH in DCM and Et2O at temperatures ranging from −60 ℃ to 0 ℃, failed to produce the desired disaccharide 31 (Scheme 2E). An aglycon transfer process occurred under the TfOH-catalyzed glycosylation conditions because of unmatched reactivities between the glycosyl donor and acceptor. Inspired by these observations, they employed a disarmed acceptor strategy comprising the coupling of donor 28 with L-fucosamine 30 under TfOH activation in DCM and Et2O as a cosolvent, successfully obtaining disaccharide 32 in 70% isolated yield with exclusive α-selectivity.
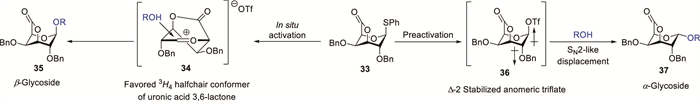
Uronic acid 3,6-lactones are valuable building blocks for oligosaccharide synthesis, exhibiting conformational rigidity, distinctive reactivity patterns, and enhanced stereocontrol in glycosylation reactions [32-34]. Glycosylation stereocontrol is particularly challenging because of the concurrent operation of competing SN1-like and SN2-like mechanisms generating opposite diastereomers and the inherent lack of stereoselectivity in the SN1-type pathway [35]. The pre-activation of GalA 3,6-lactone thioglycoside donor 33 generates reactive anomeric β-triflate intermediate 36, which has been characterised via low-temperature NMR spectroscopy (Scheme 3) [34]. This intermediate can be displaced in an SN2-like process to afford α-linked product 37. In the in situ activation process, β-linked product 35 is obtained via a reactive intermediate resembling a 3H4 oxocarbenium ion.
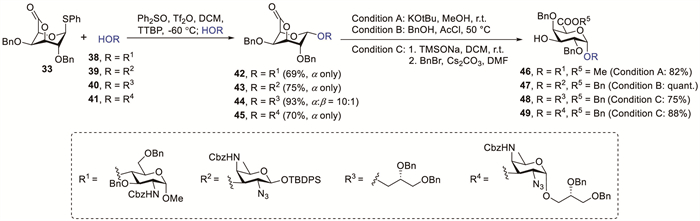
In 2005, Van der Marel et al. reported the synthesis of disaccharide 42 in 69% yield with complete α-selectivity via the pre-activation of donor 33 with diphenyl sulfoxide (Ph2SO) and trifluoromethanesulfonic anhydride (Tf2O) in the presence of 2,4,6-tri-tert-butylpyrimidine (TTBP) at −60 ℃ followed by glycosylation with acceptor 38 (Scheme 4). The selective opening of benzylated lactone 42 using potassium tert-butoxide (KOtBu) in anhydrous methanol (MeOH) gave disaccharide acceptor 46 in 82% isolated yield [32]. Codée et al. successfully prepared all three frame-shifted trisaccharide repeats of the zwitterionic polysaccharide Sp1 using 1-thiogalacturonic acid lactones as crucial glycosyl donors and acceptors [19]. The pre-activation of donor 33 with in situ formed β-triflate followed by coupling with acceptors 39, 40 and 41 afforded disaccharides 43 (75%, α only), 44 (93%, α:β = 10:1) and 45 (70%, α only), respectively. Treatment of lactone 43 with benzyl alcohol under acidic conditions provided disaccharide acceptor 47 in a quantitative yield. Unexpectedly, attempts to open lactone 44 and 45 using benzyl alcohol under acidic conditions did not yield the desired product. The lactone rings of 44 and 45 were cleaved using sodium trimethylsilanolate (TMSONa) to generate the free acid intermediates, which were benzylated with benzyl bromide (BnBr) and caesium carbonate (Cs2CO3) to yield disaccharide acceptors 48 (75%) and 49 (88%); these disaccharide acceptors function as pivotal synthetic intermediates for assembling the desired trisaccharide repeating motifs.
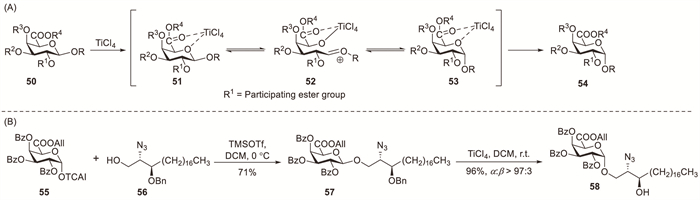
Murphy et al. circumvented the challenging direct α-glycosylation of GalA by developing innovative indirect synthetic strategies [36-38]. The synthesis involved two key steps: establishing 1,2-trans-β-glycosidic linkages via neighbouring 2-acyl group participation and subsequent construction of α-linked uronic acid glycolipids through TiCl4-mediated chelation-controlled anomerisation (Scheme 5A) [36]. The coupling of TCAI donor 55 and acceptor 56 using TMSOTf activation proceeded with exclusive β-selectivity to produce 57 in 71% yield (Scheme 5B). Subsequent TiCl4-mediated anomerisation efficiently converted β-glycoside precursor 57 to α-anomer 58 with excellent selectivity (α:β > 97:3). Notably, although TiCl4 cleaved the sphinganine benzyl group, anomerisation occurred faster, enabling the isolation of benzyl-protected α-anomers [38].
Numerous efficient synthetic routes enabling 1,2-cis-glycosidic bond formation have been reported. To modulate the stereochemical outcome of glycosylation reactions, steric hindrance can be introduced by attaching protecting groups to the donor [20]. For instance, the high steric hindrance effect of protecting groups at the C-4 position of completely protected oligogalacturonate donors can influence glycosylation stereoselectivity by hindering access to the β-face of the oxacarbenium ion, further promoting nucleophilic attack from the opposite, less hindered side (Scheme 6). Thus, glycosylation using galactonic acid donors containing C-4-attached oligosaccharides preferentially yields α-configured products.
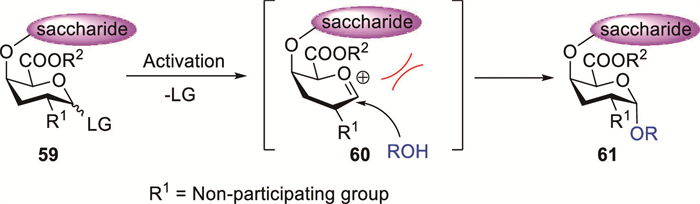
Oligosaccharide fragments of rhamnogalacturonan I (RG-Ⅰ) can be prepared via various synthetic approaches [7]. For instance, Reimer et al. developed a synthetic route to the tetrasaccharide fragment of the RG-Ⅰ backbone (Scheme 7A) [39]. The glycosylation step that converted disaccharide acceptor 64 into the target tetrasaccharide structure presented substantial challenges. By employing silver triflate (AgOTf) as a promoter, acceptor 64 was glycosylated with imidate donors 62 and 63 to form the target tetrasaccharide. The reaction, however, achieved low yields, providing 65 in 39% yield and 66 in the impure form. Further, multiple purification steps required for the removal of unreacted acceptor 64 from the desired tetrasaccharide 65 led to low yields. Despite numerous efforts to optimise the synthetic strategies, the efficiency of glycosyl acceptors conversion into tetrasaccharide products remains unsatisfactory. Vogel et al. utilised an analogous synthetic strategy to produce RG-Ⅰ oligosaccharide fragments [40]. TMSOTf-promoted glycosylation of acceptor 68 with an equimolar amount of donor 67 afforded tetrasaccharide 69 in 60% isolated yield after chromatographic purification (Scheme 7B). Meanwhile, glycosylation of acceptor 70 with donor 67 afforded hexasaccharide 71 in 59% isolated yield following HPLC purification. The β-anomers were detected (<5% NMR yield) in crude glycosylation mixtures, but the low isolated yields prevented their full characterisation. Glycosylation of acceptor 72 with rhamnogalacturonate donor 67 afforded tetrasaccharide 73 in 62% yield (Scheme 7C). A minor amount of β-linked isomer (<5%) was detected in the crude mixture by NMR spectroscopy.
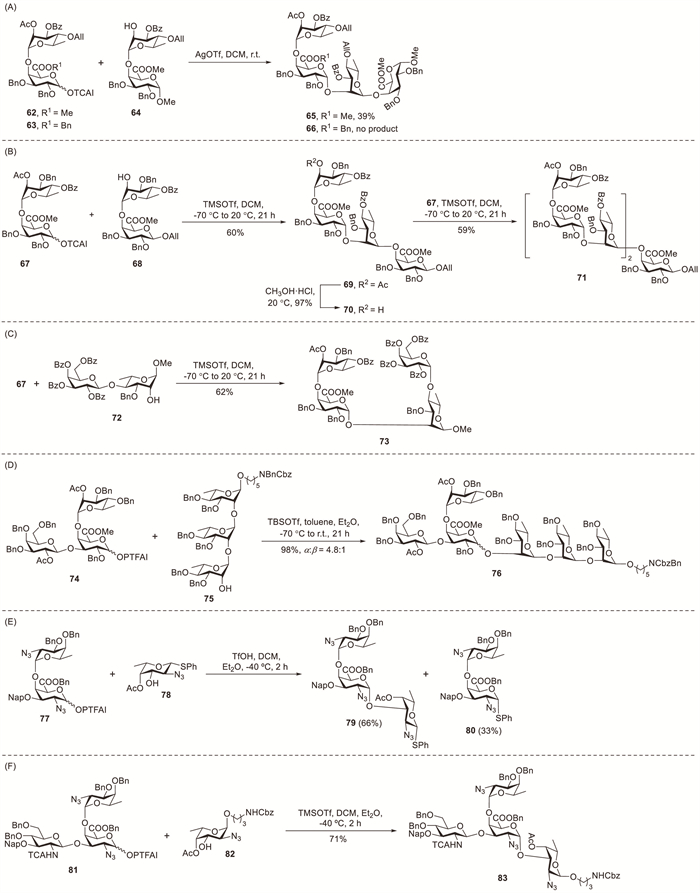
The PTFAI donors are the most extensively investigated for glycosylation reactions in contemporary carbohydrate chemistry [13,41]. Seeberger et al. reported a convergent [3 + 3] glycosylation strategy for assembling the hexasaccharide repeating unit of Klebsiella pneumoniae using a trisaccharide PTFAI donor (Scheme 7D) [42]. After optimisation, PTFAI donor 74 and acceptor 75 efficiently underwent glycosylation in toluene and Et2O at −70 ℃ to afford hexasaccharide 76 in 98% yield with good stereocontrol (α:β = 4.8:1). Kulkarni et al. reported the first chemical synthesis of Acinetobacter baumannii strains 34 and O5 tetrasaccharide units in a conjugation-ready form using oligosaccharide PTFAI donors to establish challenging 1,2-cis-linkages [43]. TfOH-promoted glycosylation of PTFAI donor 77 with L-fucosamine acceptor 78 in DCM and Et2O at 0 ℃ afforded trisaccharide 79 (50%) and aglycone-transfer product 80 (50%) (Scheme 7E). Notably, disaccharide side-product 80 could be successfully reutilised to prepare imidate 77. When the temperature was reduced to −40 ℃ as per established protocols, the yield of 79 increased to 66% and that of 80 decreased to 33%. PTFAI donor 81 underwent TMSOTf-promoted coupling with acceptor 82 at −40 ℃ to yield branched tetrasaccharide 83 with full functionality (Scheme 7F).
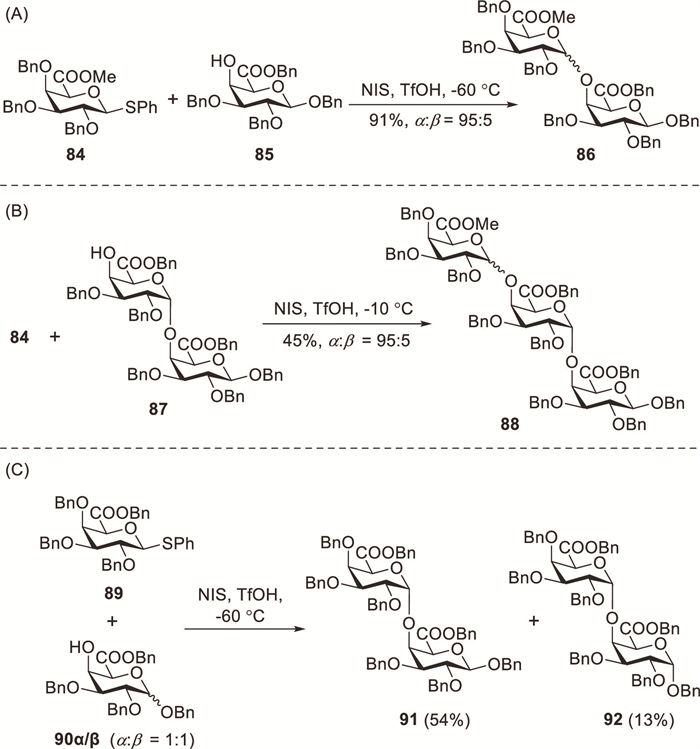
Literature reports indicate that glycosyl acceptors also play a critical role in determining glycosylation stereoselectivity [18]. Doutheau et al. demonstrated the highly stereoselective α(1→4) coupling of D-GalA esters using phenylthioglycoside donors, thereby achieving good yields [44,45]. The NIS–TfOH-promoted glycosylation of thioglycoside donor 84 with acceptor 85 at −60 ℃ afforded product 86 (91%, α:β = 95:5) with excellent stereoselectivity and high yield (Scheme 8A). TLC monitoring revealed that the reaction of disaccharide acceptor 87 with 2 equiv. of glycosyl donor 84 was sluggish at −60 ℃, whereas increasing the temperature to −10 ℃ afforded target trimer 88 (45%) along with 87 (50%) (Scheme 8B) [44]. Doutheau demonstrated that the reactivity of galactosiduronic acid acceptors bearing a free C-4 hydroxyl group is strongly influenced by their anomeric configuration (Scheme 8C) [46]. TLC monitoring showed faster consumption of 90β during the competitive glycosylation of acceptor mixture 90α/β (α:β, 0.5:0.5 equiv.) with donor 89 (0.6 equiv.) under standard conditions. The isolated product mixture contained 91 (54%) and 92 (13%). Theoretical calculations indicated that the higher reactivity of the β-anomer than that of the α-form could be attributed to the more robust hydrogen bond between the C-4 hydroxyl group and the ring oxygen.
The development of efficient glycosylation methodologies represents a critical challenge in α-GalA synthesis owing to the low reactivity of uronic acid derivatives, which typically leads to suboptimal coupling yields. The predominant indirect synthetic strategy for α-GalA oligosaccharides involves assembling the galactose oligosaccharide backbone followed by elective oxidation of specific primary hydroxyl groups to carboxylic acids. Protecting groups profoundly influence the reactivity and stereoselectivity; therefore, a comprehensive understanding of their effects is essential for rationalising and controlling the glycosylation outcome [20]. Numerous studies have reported stereoselective α-galactosylation reactions directed by remotely participating protecting groups at the C-3, C-4 and/or C-6 positions [47-51]. This through-space interaction leads to the stabilisation of the reactive intermediate and achieves stereochemical control, with acetyl groups shielding the β-face and promoting α-face selectivity in the glycosylation (Scheme 9) [52]. Notably, the C-4 substituent exerts greater stereocontrol than that the C-6 substituent in these glycosylation reactions [50]. Three effective approaches have been proposed for boosting 1,2-cis selectivity: (ⅰ) Supplementing donors having one remotely participating group with a second acyl substituent, (ⅱ) tuning remotely acyl groups to increase the nucleophilicity of the carbonyl oxygen and (ⅲ) appending sterically hindered side chains to participating acyl moieties [52]. However, the over-introduction of remotely participating acyl groups may reduce the 1,2-cis selectivity by stabilising the α-triflate intermediate, subsequently favouring β-product formation via nucleophilic substitution [52]. Primary alcohols can be oxidised to carboxylic acids using diverse methods, including the Dess–Martin/NaClO2 system, Swern–NaClO2 protocol, strongly acidic Jones reagent, milder pyridinium dichromate and pyridinium chlorochromate reagents or 2,2,6,6-tetramethylpiperidinooxy (TEMPO)-mediated catalysis, which offer distinct selectivity and compatibility [6].
4-O-Acylated galacto-configured donors can be used to produce complex GalA oligosaccharides, demonstrating their utility in natural product synthesis. Bundle et al. developed an indirect synthesis method of the trisaccharide repeat of a zwitterionic Sp1 capsular polysaccharide (Scheme 10A) [53]. The glycosylation between donor 98 and acceptor 99 was systematically evaluated under multiple activation conditions. Initial attempts using Ph2SO–Tf2O as the activator system afforded disaccharide 100 in suboptimal yields (≤30%), prompting investigation of alternative conditions. Although NIS–TfOH activation at −30 ℃ improved the yield to 57%, the optimal result was achieved using NIS–AgOTf, which afforded disaccharide 100 in 70% yield. Next, target trisaccharide 101 was isolated after sequential coupling reactions. Oxidation of diol 101 under TEMPO conditions generated the corresponding di-acid, which was totally de-protected via hydrogenolysis to obtain 102 in 53% overall yield.
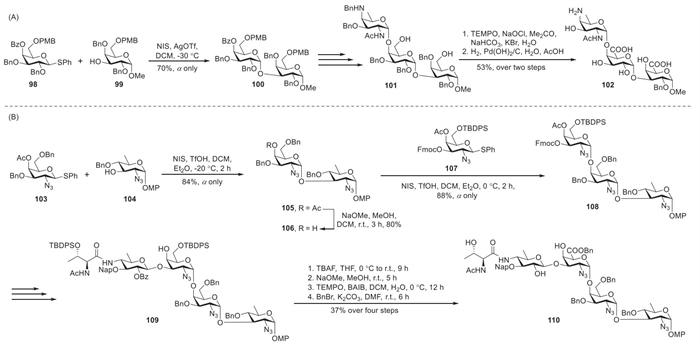
Kulkarni et al. reported the comprehensive total synthesis of the Vibrio cholerae O43 tetrasaccharide repeating unit using a 4-O-acetyl-protected galacturonate donor (Scheme 10B) [54]. Glycosylation of D-quinosamine acceptor 104 with D-galactosamine thioglycoside 103 using NIS and TfOH in DCM and Et2O (1:1) at −20 ℃ afforded α-linked disaccharide 105 in 84% yield as a single stereoisomer. Treatment of 105 with NaOMe in DCM and MeOH at room temperature afforded deacetylated acceptor 106 in 80% yield. Glycosylation of acceptor 106 with D-galactosamine thioglycoside 107 under NIS–TfOH activation at 0 ℃ exclusively provided α-linked trisaccharide 108 in 88% yield. The stereocontrol originated from the participation of the Et2O solvent in the reaction at low temperatures combined with 4-O-acetyl neighbouring-group assistance, guiding the acceptor to approach the α-face of the oxocarbenium ion. After the controlled regio- and stereoselective synthesis of tetrasaccharide 109, subsequent transformation into target derivative 110 was achieved. Selective removal of the tert-butyldiphenylsilyl (TBDPS) and benzyl protecting groups cleanly afforded the intended tetra-hydroxylated product. Furthermore, the primary alcohol was selectively oxidised using TEMPO–(diacetoxyiodo)benzene (BAIB) in a DCM–H2O system, followed by esterification with BnBr and sodium bicarbonate (NaHCO3) in N,N-dimethylformamide (DMF) to afford benzyl-protected derivative 110.
Bundle et al. synthesised an Sp1 capsular hexasaccharide using 6-O-acetyl-protected galactosyl donors [55]. Under NIS–TfOH activation at −30 ℃ in DCM, the coupling between disaccharide acceptor 112 and thiogalactoside donor 111 efficiently proceeded to afford target trisaccharide 113 in 73% isolated yield (Scheme 11A). 1H NMR analysis revealed the presence of only trace amounts of the β-anomeric galactose; this α-anomeric preference most likely resulted from the stereoelectronic influence exerted by the C-6 substituent (the acetyl group). The exposed hydroxymethyl groups, generated via deacetylation of 113, underwent simultaneous oxidation using TEMPO and sodium hypochlorite (NaOCl) to form uronic acid groups. Subsequent benzylation of these acidic groups enabled an efficient purification, affording target compound 114 in 51% overall yield. Hexasaccharide 117 was obtained in 85% yield via the TMSOTf-mediated glycosylation of trisaccharide building blocks 115 and 116 at room temperature (Scheme 11B). This high efficiency was achieved by carefully optimising the temperature and molar equivalents of the glycosyl donor and Lewis acid.
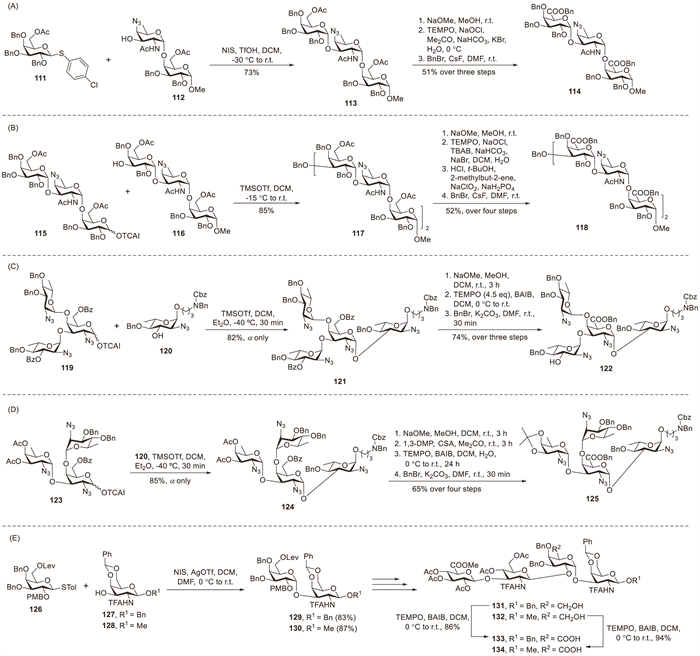
Gao et al. achieved the inaugural chemical synthesis of the structurally complex tetrasaccharide motifs present in the capsular polysaccharides of Vibrio vulnificus strains MO6-24 and BO62316 through post-glycosylation oxidation [56]. An optimised TEMPO–BAIB oxidation system was established for the direct transformation of α-D-galactosamine into α-D-aminogalacturonic acid residues, demonstrating the successful application to di-, tri- and tetra-saccharide substrates. The TMSOTf-mediated coupling between donor 119 and acceptor 120 in a DCM–Et2O mixture at −40 ℃ proceeded with unanticipated efficiency to exclusively afford tetrasaccharide 121 as the α-anomer in 82% isolated yield (Scheme 11C). Selectively oxidising specific hydroxyl moieties in complex oligosaccharide frameworks is highly challenging. Fortunately, the TEMPO-mediated oxidation (0.2 equiv.) of the 6′-hydroxyl group proceeded exceptionally well when using BAIB (4.5 equiv.) in a DCM–H2O system, affording target carboxylate 122 in excellent yield. Consistent with previous results, the coupling of donor 123 and acceptor 120 proceeded with complete α-selectivity, yielding tetrasaccharide 124 in 85% yield (Scheme 11D). After sequential acetylation and isopropylation steps, an optimised TEMPO–BAIB oxidation system efficiently generated target uronate 125 in excellent yield.
Li et al. established a productive semi-synthetic approach for preparing Colwellia psychrerythraea 34H capsular tetrasaccharide derivatives using disaccharide building blocks obtained via hyaluronic acid degradation [57]. The levulinoyl protecting group was strategically incorporated at C-6 to exploit its ability to stereocontrol 1,2-cis-linkages through remote anchimeric assistance. Donor 126 and acceptors 127 and 128 were prepared, after which established glycosylation methods were evaluated to optimise reaction yield and diastereoselectivity (Scheme 11E). The combination of NIS and AgOTf in DCM–DMF was selected as the optimal system, affording disaccharide 129 (83%) and its counterpart 130 (87%). From tetrasaccharides 131 and 132, carboxyl-bearing tetrasaccharides 133 and 134 were obtained via TEMPO-catalysed oxidation in 86% and 94% isolated yields, respectively.
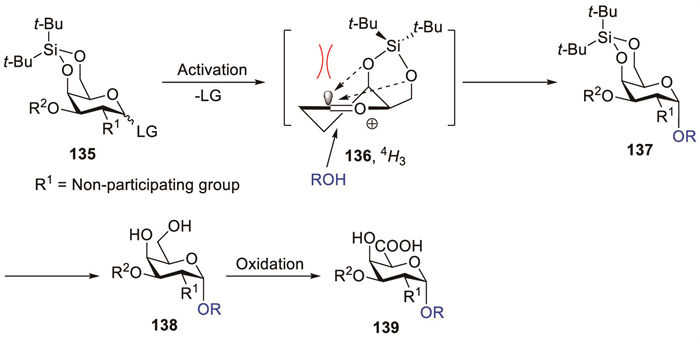
Sterically demanding protecting groups considerably affect the glycosylation stereochemistry and efficiency [20]. Kiso et al. demonstrated that the presence of the bulky 4,6-O-di-tert-butylsilylene (DTBS) group on the galactosyl donor promoted the α-selective galactosylation, overriding the influence of a participating substituent present at C-2 [58]. Notably, the DTBS moiety demonstrated exceptional and distinct stereocontrol capabilities compared with other cyclic protecting groups. A galactosyl donor featuring a six-membered cyclic protecting group between O-4 and O-6 predominantly adopts the 4H3 conformation when converted to the oxocarbenium ion (Scheme 12) [20]. Bulky alkyl groups within the DTBS protecting framework sterically hinder nucleophilic attack from the β-direction and influence the stability of transition state 136 via through-space charge delocalisation.
Kováč et al. described the chemical synthesis of an O-antigen pentasaccharide from Vibrio cholerae O139 [59]. They employed post-glycosylation oxidation to establish α-GalA linkages using a 4,6-O-di-tert-butylsilylidene galactopyranosyl donor 140 for stereocontrol. Nearly complete α-selectivity was achieved in the galactosylation of 141 with donor 140 (Scheme 13A). The challenging separation of β-anomeric impurities was avoided by performing purification after removing the 4,6-O-protecting groups, yielding essential α-product 142 with 73% efficiency. Target tetrasaccharide 143 was obtained through multiple synthetic steps. The carboxyl group generated via the TEMPO-mediated oxidation of 143 was subsequently esterified using methyl iodide (MeI) and NaHCO3, affording trisaccharide 144 in 90% overall yield.
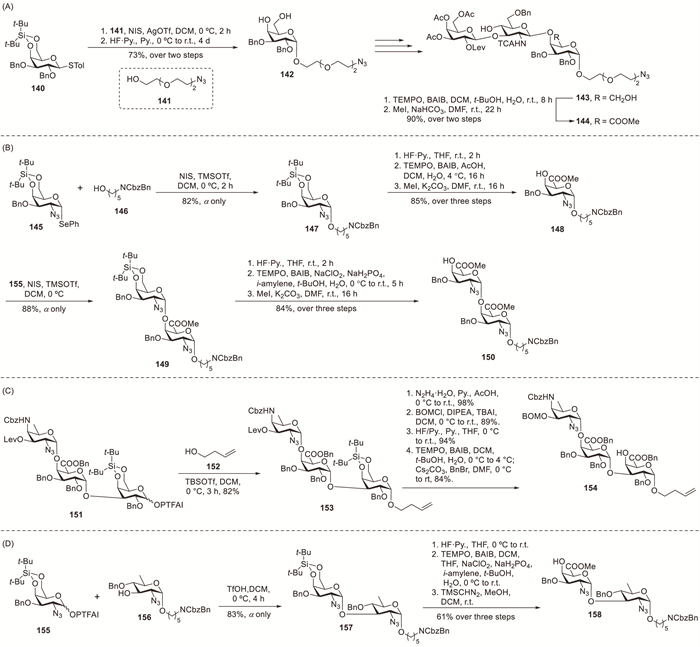
Codée et al. prepared the capsular polysaccharide repeating unit of Staphylococcus aureus strain M containing α-galactosaminuronic acid residues [60]. The coupling of donor 145 with acceptors 146 and 148 in the presence of NIS and TMSOTf smoothly proceeded to exclusively afford α-configured products 147 (82%) and 149 (88%), respectively (Scheme 13B). To prevent the glycosidic bond cleavage observed during TEMPO–BAIB oxidation of a disaccharide intermediate, a sequential approach using TEMPO–BAIB–Pinnick oxidation was successfully implemented. In 2019, the researchers established an efficient synthetic strategy for constructing extended Sp1 oligosaccharide structures [29]. The condensation of silylene-protected trisaccharide donor 151 with allylcarbinol 152 efficiently proceeded to afford trisaccharide 153 in 82% yield as a single stereoisomer, demonstrating the exceptional glycosylation capability of this donor structure (Scheme 13C). Trisaccharide 151 served as a versatile precursor for assembling higher oligomers ranging from hexasaccharides to dodecasaccharides. The TEMPO–BAIB system successfully oxidised trisaccharide 153; however, attempts to functionalise larger oligosaccharides failed, highlighting the inherent challenges in performing concurrent oxidations at multiple sites. Through systematic optimisation, alkaline conditions considerably enhanced the oxidation process. Consequently, extended oligosaccharides were successfully oxidised using TEMPO–BAIB in combination with NaHCO3.
Ragains et al. achieved the complete chemical synthesis of an all-1,2-cis-configured repeating segment from a Acinetobacter baumannii D78 capsular polysaccharide [61]. The reaction between DTBS-protected PTFAI 155 and acceptor 156 proceeded efficiently using TfOH activation, yielding disaccharide 157 in excellent yield (83%) with complete stereocontrol (Scheme 13D). The synthetic sequence involved cleavage of the DTBS group, carboxyl-group introduction via oxidation and TMSCHN2-mediated methylation, affording disaccharide 158 in 61% overall yield.
The torsional strain introduced by cyclic protecting moieties can profoundly affect anomeric selectivity during glycosylation [18]. This control is exemplified by the use of 4,6-O-benzylidene acetal protection to selectively generate β-mannosides [62-64], α-glucosides [65], and β-rhamnosides [66]. Furthermore, 4,6-O-benzylidene-protected galactosyl donors exhibit pronounced α-selectivity, as the cis-decalin conformation featuring an equatorially oriented phenyl group effectively blocks nucleophilic attack from the β-face [67-70]. Calenbergh et al. developed a synthetic strategy to access α-galacturonic-acid-modified KTHF RN7000 analogues (Scheme 14A) [71]. The Schmidt glycosidation method afforded compound 161 with excellent α-selectivity along with a small amount of β-anomeric by-products, which were easily removed via flash column purification [72]. Subsequently, the 6-hydroxy groups of 162 and 163 were selectively converted to carboxyl groups via TEMPO-mediated BAIB oxidation, furnishing galacturonates 164 and 165, respectively [71].
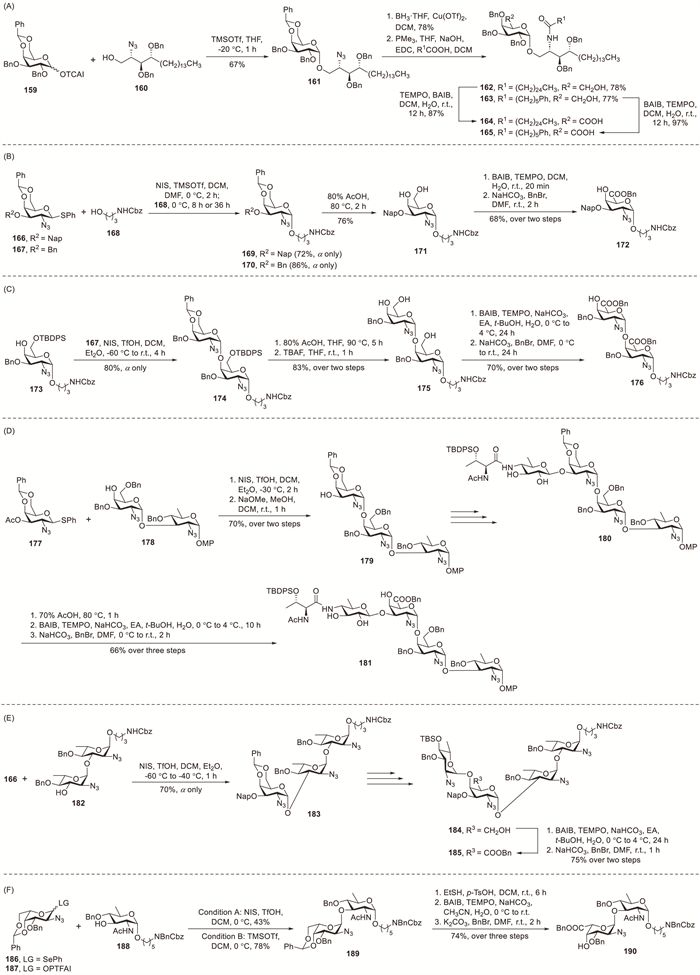
Kulkarni et al. developed an efficient synthetic route employing 4,6-O-benzylidene-protected D-galactosamine donors to access oligosaccharide repeating units of Staphylococcus aureus strain M [73], Proteus penneri 26 [74], Proteus vulgaris TG155 [74], Vibrio cholerae O43 [75] and Vibrio vulnificus MO6-24 [76]. Thiogalactoside donors 166 and 167 were activated with NIS–TMSOTf in DMF to generate intermediate imidates, followed by in situ addition of linker acceptor 168 to yield 1,2-cis-configured galactosamine derivatives 169 (72%) and 170 (86%), respectively, with complete stereocontrol (Scheme 14B). Acidic cleavage of the benzylidene acetal afforded diol 171, which was subjected to TEMPO-catalysed oxidation of its primary alcohol followed by benzyl ester formation to yield 172 in 68% overall yield for the two-step sequence. Applying this methodology, disaccharide 176 and tetrasaccharides 181 and 185 were efficiently prepared with high stereoselectivity and excellent yields (Schemes 14C–E).
Notably, natural L-galactosaminuronic acid residues have been exclusively observed in the α-anomeric form among all characterised bacterial glycans [8]. Yin et al. developed a synthetic route to conjugation-capable trisaccharide motifs of Pseudomonas aeruginosa serotype O10 and O19 O-antigens, featuring α-linked L-galactosaminuronic acid residues [77]. The glycosylation between selenoglycoside donor 186 and acceptor 188 mediated by NIS–TfOH in DCM at 0 ℃ afforded α-linked disaccharide 189 as the sole product, albeit in modest yield (43%). The pronounced α-selectivity stemmed from the synergistic combination of the conformational constraint of the 4,6-O-benzylidene group and inherent anomeric stabilisation. However, using selenophenyl as the leaving group for L-GalN3 donors reduced the glycosylation efficiency. Satisfactorily, the glycosylation between acceptor 188 and the more potent FTFAI donor 187 catalysed by sub-stoichiometric TMSOTf yielded α-linked product 189 in a markedly improved yield of 78%.
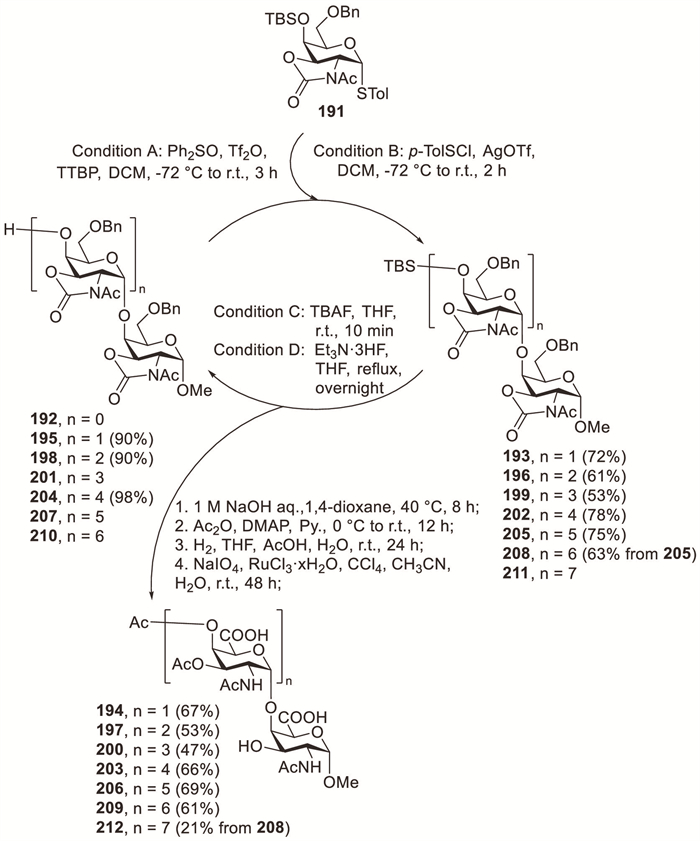
The 2,3-oxazolidinone protection strategy pioneered by Kerns et al. has enabled the highly stereoselective synthesis of diverse 1,2-cis-2-amino-2-deoxyglycosides [20,78]. For instance, Ye et al. developed an efficient method for the synthesis of α-(1→4)-linked GalNAcA repeating units characteristic of the Salmonella typhi capsular polysaccharide [79,80]. The combination of the 2,3-N,O-oxazolidinone protecting group strategy with pre-activation enabled glycosylations with complete α-anomeric control. Employing Ph2SO–Tf2O activation with sterically hindered TTBP as a base, disaccharide 193 was exclusively obtained as the α-anomer in 72% isolated yield (Scheme 15). Removal of the oxazolidinone and TBS protecting groups of 193 under basic conditions followed by acetylation efficiently produced an intermediate compound, which was subjected to Pd/C-mediated hydrogenolysis and ruthenium chloride (RuCl3)–sodium periodate (NaIO4) oxidative cleavage to afford target disaccharide 194. By performing iterative cycles of glycosylation, deprotection and oxidation, oligosaccharides ranging from tri- to octa-saccharides were successfully synthesised.
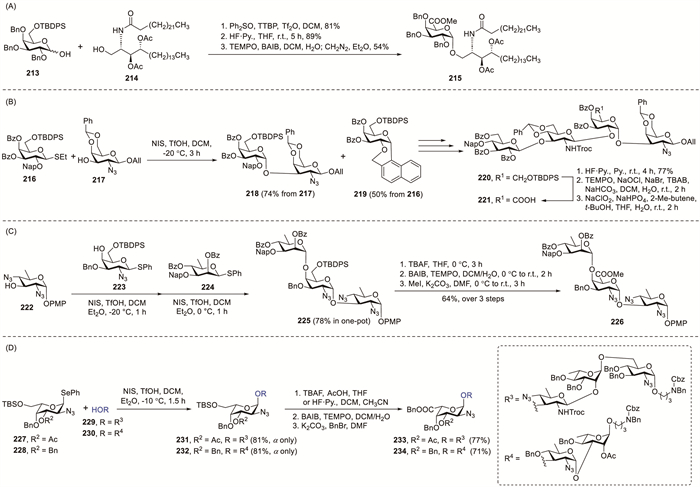
The steric effect of protecting groups on glycosyl donors can direct glycosylation stereochemistry by shielding one face of the molecule, promoting nucleophilic attack from the unhindered side [20]. One of the various α-directing galactosylation strategies includes using galactose donors containing bulky O-6 protecting groups such as TBDPS ethers [81]. The synthesis of glycolipid 215 was accomplished by Savage et al. in a moderate yield via glycosidic coupling followed by selective galactose oxidation to the corresponding uronic acid derivative (Scheme 16A) [82]. The synthetic sequence commenced with the Ph2SO–Tf2O-promoted coupling of donor 213 and acceptor 214, followed by sequential TBDPS deprotection, TEMPO-mediated oxidation and esterification to yield target compound 215. Bedini et al. developed a synthesis method for producing the capsular polysaccharide repeating unit of Colwellia psychrerythraea 34H using 6-O-TBDPS galactoside donors for controlled glycosylation [83]. Under NIS–TfOH activation, donor 216 generated intermolecular adduct 218 (74% yield from 217) and intramolecular C-glycoside 219 (50% yield from 216), indicating that the flexible oxonium species favoured a bimolecular reaction with the acceptor, except in the presence of excess donor (Scheme 16B). After sequential transformations, tetrasaccharide 220 was obtained. Subsequent HF–pyridine-mediated desilylation afforded an alcohol, which underwent TEMPO-catalysed oxidation to the corresponding carboxylic acid product 221. Kulkarni et al. reported the pioneering total synthesis of the trisaccharide repeating elements of Pseudomonas chlororaphis subsp. aureofaciens UCM B-306 via a convergent one-pot assembly of the key trisaccharide framework [84]. Donor 223 and acceptor 224 were first condensed under NIS–TfOH activation in DCM–Et2O at −20 ℃, generating a disaccharide intermediate within 1 h. In the same pot, D-rhamnosyl donor 224 was introduced with fresh NIS–TfOH at 0 ℃, affording α-trisaccharide 225 in 78% overall yield. After TBDPS cleavage, the resulting alcohol was oxidised to its carboxylate form using TEMPO–BAIB and then esterified with MeI and potassium carbonate (K2CO3) to yield protected trisaccharide 226 in 64% overall yield. Gao et al. reported the total synthesis of Pseudomonas aeruginosa O-antigen repeats containing α-N-acetyl-L-galactosaminuronic acid (α-L-GalpNAcA) units using 6-O-TBS-L-GalN3 donors for controlled glycosylation [85]. After establishing reliable α-L-GalN3 coupling conditions, they implemented this stereocontrolled glycosylation strategy in 233 and 234 synthesis. The glycosylation between 227 and 229 mediated by NIS–TfOH in DCM–Et2O proceeded to yield only α-linked tetrasaccharide 231 with 81% efficiency. Under the established conditions, coupling acceptor 228 with donor 230 afforded trisaccharide 232 in 81% isolated yield with exclusive α-anomeric configuration. After removing TBS from 231 and 232, the liberated primary alcohols underwent oxidation via the optimised TEMPO–BAIB protocol, and subsequent benzyl ester formation yielded products 233 and 234, respectively.
The n-pentenyl glycoside methodology was first established by Fraser-Reid et al. in 1988, introducing this alkyl group as an innovative anomeric substituent for glycosyl donors [86]. Glycosylation potentially initiated via the electrophile-induced cyclisation of 235 to produce bromonium intermediate 236, which was in equilibrium with oxonium cation 237 (Scheme 17A) [87]. After releasing the halomethyltetrahydrofuran by-product, the resulting oxocarbenium species 238 underwent nucleophilic capture by the alcohol acceptor to yield glycoside product 239. Clausen employed n-pentenyl glycosyl donors as a versatile method for oligogalacturonate assembly [88-90]. PMP-protected galactoside 240 was subsequently glycosylated with 241 in the presence of bromine gas (Br2) and tetrabutylammonium bromide (Et4NBr), affording the desired α-linked product 242 in 70% yield with excellent α-selectivity (α/β > 95:5). The activation of donor 240 generated a mixture of α- and β-glycosyl bromides, where the α-anomer was the major component. Owing to its superior reactivity, the transient β-glycosyl bromide was the sole participant in the coupling reaction, resulting in the formation of an α-linked product. Coupling of disaccharide donor 243 with acceptor 242 via activation using NIS and triethylsilyl triflate (TESOTf) yielded target α-configured trisaccharide 244 in 71% isolated yield. The PMP groups in compound 244 underwent CAN-catalysed oxidative cleavage followed by further oxidation and esterification. The oxidation sequence employed Dess–Martin periodinane followed by sodium chlorite to afford the carboxylic acids, which were immediately esterified in situ with (trimethylsilyl)diazomethane (TMSCHN2), thereby avoiding intermediate purification.
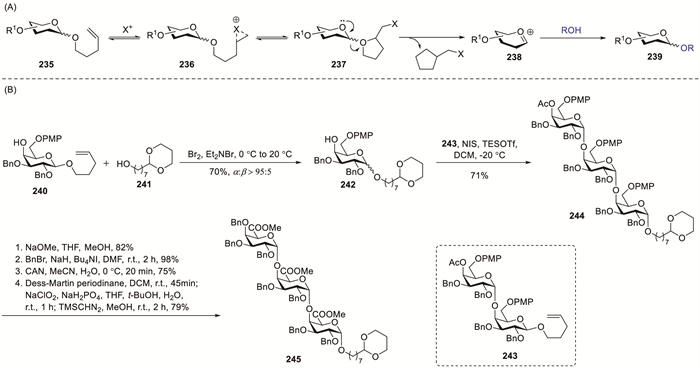
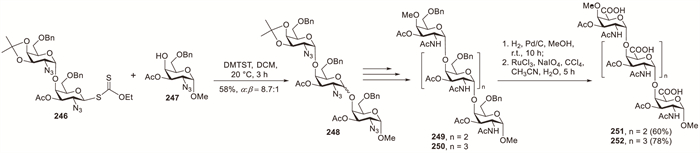
The immunodominant capsular polysaccharide of Salmonella typhi comprises of repeating α-(1→4)-linked N-acetyl-D-galactosaminuronic acid residues. Sinaӱ et al. demonstrated the synthesis of oligosaccharide fragments of Salmonella typhi containing repeating α-(1→4)-N-acetyl-D-galactosaminuronic acid polymers by employing 2-azido-2-deoxy-D-galactopyranosyl S-xanthates as key glycosyl donors (Scheme 18) [91]. The oligosaccharide construction commenced with DMTST-promoted glycosylation of acceptor 247 with donor 246, affording trisaccharide 248 in 58% yield with good selectivity (α:β = 8.7:1). Debenzylation of 249 and 250 via hydrogenation, followed by Sharpless oxidation of the primary alcohols, afforded oligogalacturonates 251 (60%) and 252 (78%), respectively.
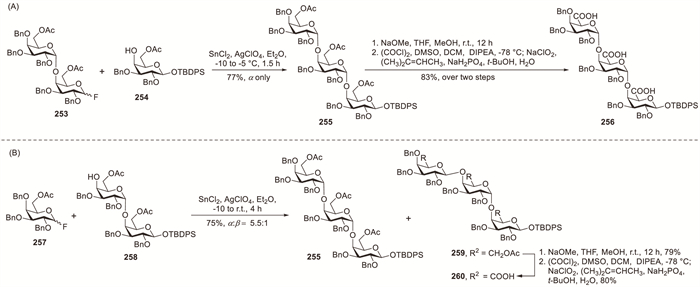
Ogawa et al. demonstrated the synthesis of α-(1→4)-linked GalA trisaccharides by employing D-galactopyranosyl fluoride donors in their glycosylation strategy (Scheme 19A) [92]. The stannous chloride (SnCl2)–silver perchlorate (AgClO4)-promoted glycosylation between 253 and 254 in Et2O proceeded with complete selectivity, yielding galactotrioside 255 as the sole product in 77% yield (Scheme 19A). Employing 1,2-dichloroethane as the solvent decreased the stereoselectivity, affording the desired product 255 in 68% yield along with 4% of the β-linked isomer. Under SnCl2–AgClO4 promotion, galactobiosyl acceptor 258 reacted with donor 257 to predominantly give trisaccharide 255 (63%) along with trisaccharide 259 (12%) as a minor side product (Scheme 19B). The α-D-galactopyranosyl moiety at C-4 in donor 253 demonstrated superior α-stereoselectivity compared with benzyl-protected analogue 257, suggesting that the bulky protecting group at C-4 considerably influences the glycosylation outcome.
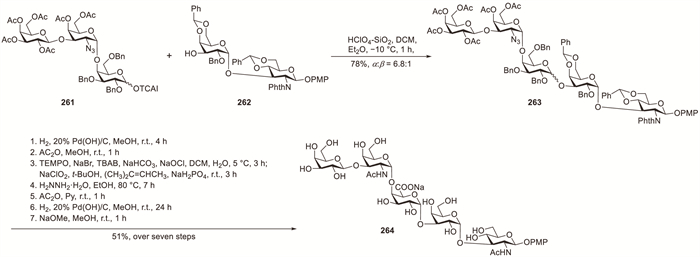
Misra et al. accomplished the convergent synthesis of an acidic pentasaccharide repeating unit that was structurally identical to the O-antigen polysaccharide of enterohemorrhagic Escherichia coli O113 by employing an efficient [3 + 2] block glycosylation strategy [93]. The HClO4–SiO2-promoted glycosylation of donor 261 with acceptor 262 in DCM–Et2O (3:1) proceeded stereoselectively, affording α-linked pentasaccharide derivative 263 in 68% yield along with a small amount of the stereoisomer (~10%), which was subsequently removed using silica-gel chromatography (Scheme 20). A selective TEMPO-catalysed oxidation reaction was employed in the final synthetic steps to incorporate the D-GalA moiety, demonstrating excellent functional-group compatibility.
Two synthetic strategies for incorporating α-linked galacturonic acid moieties into acidic oligosaccharides, i.e. pre- and post-glycosylation oxidation strategies, are described herein. The pre-glycosylation oxidation strategy for α-GalA/α-GalNAcA-containing oligosaccharides involves C-4 modification to control anomeric selectivity. Further, O-4-acyl protecting groups stabilise key intermediates, and bulky C-4 substituents direct α-selectivity via steric hindrance during glycosidic bond formation. Conformationally constrained 3,6-lactones enable the SN2-like displacement of anomeric triflates, yielding exclusive α-linkages. TiCl4-mediated anomerisation converts β-linked glycolipids into α-anomers via chelation control. Galactopyranosyl thioglycosides and phosphates are versatile donors achieving high α-selectivity after careful optimisation of the reaction conditions, while offering adjustable reactivity and selectivity for controlled oligosaccharide assembly. Premature oxidation at C-6 compromises the selectivity during protection of glycosyl donor hydroxyl groups. In addition, the electron-withdrawing character of the carboxylate ester moiety considerably reduces the reactivity of uronic acids. The challenging synthesis of 1,2-cis-α-GalA/α-GalNAcA derivatives can be achieved using a strategic approach involving initial glycosylation of 1,2-cis-Gal/α-GalNAc followed by subsequent oxidation. The presence of acyl protecting groups at C-3, C-4 and/or C-6 positions in galactopyranosyl donors facilitates the α-selective glycosylation by electronically stabilising the transient oxocarbenium ion intermediate. The 4,6-O-DTBS and 4,6-O-benzylidene protecting groups promote α-selectivity through dual mechanisms: (ⅰ) Steric shielding of the β-face and (ⅱ) conformational locking of the galactopyranosyl intermediate. The 2,3-N,O-oxazolidinone protecting group strategy based on the pre-activation protocol ensures exclusive α-selectivity. The strategic incorporation of sterically demanding 6-O-TBDPS or 6-O-TBS groups promotes α-selectivity by effectively shielding the β-face from nucleophilic attack during galactosylation. n-Pentenyl glycosides, galactosyl fluorides and S-xanthates offer unique activation pathways for α-control. Ultimately, achieving complete selectivity in the 1,2-cis-glycosylation hinges on three critical factors: strategic design of the protecting group pattern on the donor, precise control of the counterion and optimisation of the reaction conditions, including the use of modulating additives. Although TEMPO oxidation enables an efficient carboxyl-group incorporation, the post-glycosylation route presents synthetic complexity through necessary protecting group strategies and inherent risks of oxidative damage to pre-assembled oligosaccharides.
Despite these advances, some limitations persist, including moderate yields, competing side reactions and the need for extensive protecting group manipulations. Further development of catalytic activation methods, mechanistic studies and streamlined synthetic routes are essential to provide access to structurally diverse α-GalA/GalNAcA-containing glycans for biological and therapeutic applications. Recent efforts have also highlighted the limitations of conventional oxidation protocols in incorporating the C-5 carboxyl group of galacturonic acid derivatives. Traditional methods often suffer from poor functional group tolerance, over-oxidation or lack of stereocontrol at C-5, complicating downstream glycosylation. Emerging strategies such as electrochemical oxidation, enzyme-mimetic catalysis and organocatalytic dehydrogenation offer milder alternatives but require further optimisation for carbohydrate substrates.
Integrating multidisciplinary tools, e.g., machine-learning-guided reaction optimisation, bio-orthogonal chemistry and automated synthesis, can accelerate the development of robust protocols for these glycosides, unlocking their potential in glycoscience and materials science.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Juntao Cai: Writing – review & editing, Writing – original draft, Project administration, Investigation, Funding acquisition, Data curation, Conceptualization. Qian Wang: Investigation, Data curation. Xu Shen: Writing – review & editing. Lifeng Zhu: Writing – review & editing. Shiqing Jiang: Writing – review & editing, Supervision, Resources. Jian Yin: Writing – review & editing, Writing – original draft, Supervision, Software, Resources, Project administration, Methodology, Funding acquisition, Formal analysis, Conceptualization. Chunhong Dong: Writing – review & editing, Supervision, Resources, Project administration, Formal analysis, Conceptualization.
The authors are grateful to the National Natural Science Foundation of China (Nos. 22407042, 22325803, 22277042), the Natural Science Foundation of Henan Province (No. 232300421374) and the National Key R&D Program of China (No. 2023YFC2308000).
C.R. Bertozzi, L.L. Kiessling, Science 291 (2001) 2357–2364. doi: 10.1126/science.1059820
J. Finkelstein, Nature 446 (2007) 999 -999. doi: 10.1038/446999a
S.S. Shivatare, V.S. Shivatare, C.H. Wong, Chem. Rev. 122 (2022) 15603–15671. doi: 10.1021/acs.chemrev.1c01032
P.H. Seeberger, Chem. Rev. 121 (2021) 3598–3626. doi: 10.1021/acs.chemrev.0c01210
L. Su, Y.L. Feng, K.C. Wei, et al., Chem. Rev. 121 (2021) 10950–11029. doi: 10.1021/acs.chemrev.0c01338
L.J. van den Bos, J.D.C. Codée, R.E.J.N. Litjens, et al., Eur. J. Org. Chem. 2007 (2007) 3963–3976. doi: 10.1002/ejoc.200700101
C. Kinnaert, M. Daugaard, F. Nami, et al., Chem. Rev. 117 (2017) 11337–11405. doi: 10.1021/acs.chemrev.7b00162
C.J. Qin, H.L. Hou, M.R. Ding, et al., Chin. J. Nat. Med. 20 (2022) 387–392.
C.J. Qin, G.Z. Tian, J. Hu, et al., Curr. Opin. Chem. Biol. 78 (2024) 102424. doi: 10.1016/j.cbpa.2023.102424
S.T. Minzanova, V.F. Mironov, D.M. Arkhipova, et al., Polymers 10 (2018) 1407. doi: 10.3390/polym10121407
T. Ali, I. Murtaza, H.L. Guo, et al., Biochem. Biophys. Res. Commun. 765 (2025) 151861. doi: 10.1016/j.bbrc.2025.151861
W.L. Yao, D.C. Xiong, Y. Yang, et al., Nat. Synth. 1 (2022) 854–863. doi: 10.1038/s44160-022-00171-9
J.T. Cai, X. Yuan, Y.F. Kong, et al., Chin. J. Nat. Med. 21 (2023) 886–901.
T.J. Boltje, T. Buskas, G.J. Boons, Nat. Chem. 1 (2009) 611–622. doi: 10.1038/nchem.399
P.H. Seeberger, D.B. Werz, Nature 446 (2007) 1046–1051. doi: 10.1038/nature05819
X. Cao, X.J. Du, H. Jiao, et al., Acta Pharm. Sin. B 12 (2022) 3783–3821. doi: 10.1016/j.apsb.2022.05.020
R.D. Astronomo, D.R. Burton, Nat. Rev. Drug Discov. 9 (2010) 308–324. doi: 10.1038/nrd3012
S.S. Nigudkar, A.V. Demchenko, Chem. Sci. 6 (2015) 2687–2704. doi: 10.1039/C5SC00280J
A.E. Christina, L.J. van den Bos, H.S. Overkleeft, et al., J. Org. Chem. 76 (2011) 1692–1706. doi: 10.1021/jo102363d
R.A. Jeanneret, S.E. Johnson, M.C. Galan, J. Org. Chem. 85 (2020) 15801–15826. doi: 10.1021/acs.joc.0c02045
A. Wadouachi, J. Kovensky, Molecules 16 (2011) 3933–3968. doi: 10.3390/molecules16053933
S. Visansirikul, S.A. Kolodziej, A.V. Demchenko, Org. Biomol. Chem. 18 (2020) 783–798. doi: 10.1039/c9ob02546d
A.E. Christina, G.A. van der Marel, J.D.C. Codée, Recent developments in the construction of cis-glycosidic linkages, in: D.B. Werz, S. Vidal (Eds.), Modern Synthetic Methods in Carbohydrate Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2013, pp. 97–124.
P. Stallforth, A. Adibekian, P.H. Seeberger, Org. Lett. 10 (2008) 1573–1576. doi: 10.1021/ol800227b
S. Kramer, B. Nolting, A.J. Ott, et al., J. Carbohydr. Chem. 19 (2000) 891–921. doi: 10.1080/07328300008544125
B. Nolting, H. Boye, C. Vogel, J. Carbohydr. Chem. 20 (2001) 585–610. doi: 10.1081/CAR-100108276
S.A. Nepogodiev, R.A. Field, I. Damager, Approaches to chemical synthesis of pectic oligosaccharides, in: P. Ulvskov (Ed.), Annual Plant Reviews, Blackwell Publishing Ltd, Chicheste, 2010, pp. 65–92.
Y.Y. Ma, X. Cao, B. Yu, Carbohydr. Res. 377 (2013) 63–74. doi: 10.1016/j.carres.2013.05.008
Q.J. Zhang, A. Gimeno, D. Santana, et al., ACS Cent. Sci. 5 (2019) 1407–1416. doi: 10.1021/acscentsci.9b00454
Z. Wang, A. Gimeno, M.G. Lete, et al., Angew. Chem. Int. Ed. 62 (2023) e202211940. doi: 10.1002/anie.202211940
B.K. Ghotekar, S.S. Kulkarni, Org. Lett. 26 (2024) 4346–4350. doi: 10.1021/acs.orglett.4c01354
L.J. van den Bos, R.E.J.N. Litjens, R.J.B.H.N. van den Berg, et al., Org. Lett. 7 (2005) 2007–2010. doi: 10.1021/ol050491y
T. Furukawa, H. Hinou, K. Shimawaki, et al., Tetrahedron Lett. 52 (2011) 5567–5570. doi: 10.1016/j.tetlet.2011.08.024
A.E. Christina, J.A. Muns, J.Q.A. Olivier, et al., Eur. J. Org. Chem. 2012 (2012) 5729–5737. doi: 10.1002/ejoc.201200717
H. Elferink, R.A. Mensink, W.W.A. Castelijns, et al., Angew. Chem. Int. Ed. 58 (2019) 8746–8751. doi: 10.1002/anie.201902507
W. Pilgrim, P.V. Murphy, Org. Lett. 11 (2009) 939–942. doi: 10.1021/ol802915h
C. O’Reilly, P.V. Murphy, Org. Lett. 13 (2011) 5168–5171. doi: 10.1021/ol202042h
W. Pilgrim, C. O’Reilly, P.V. Murphy, Molecules 18 (2013) 11198–11218. doi: 10.3390/molecules180911198
D. Reiffarth, K.B. Reimer, Carbohydr. Res. 343 (2008) 179–188. doi: 10.1016/j.carres.2007.10.030
N. Nemati, G. Karapetyan, B. Nolting, et al., Carbohydr. Res. 343 (2008) 1730–1742. doi: 10.1016/j.carres.2008.03.020
B. Yu, H.C. Tao, Tetrahedron Lett. 42 (2001) 2405–2407. doi: 10.1016/S0040-4039(01)00157-5
P.H. Seeberger, C.L. Pereira, N. Khan, et al., Angew. Chem. Int. Ed. 56 (2017) 13973–13978. doi: 10.1002/anie.201700964
D. Rai, S.S. Kulkarni, Org. Lett. 25 (2023) 8332–8337. doi: 10.1021/acs.orglett.3c03417
D. Magaud, C. Grandjean, A. Doutheau, et al., Tetrahedron Lett. 38 (1997) 241–244.
D. Magaud, C. Grandjean, A. Doutheau, et al., Carbohydr. Res. 314 (1998) 189–199. doi: 10.1016/S0008-6215(98)00312-7
D. Magaud, R. Dolmazon, D. Anker, et al., Org. Lett. 2 (2000) 2275–2277. doi: 10.1021/ol006039q
J. Kalikanda, Z.T. Li, J. Org. Chem. 76 (2011) 5207–5218. doi: 10.1021/jo1025157
B.S. Komarova, N.E. Ustyuzhanina, Y.E. Tsvetkov, et al., Stereocontrol of 1, 2-cis-glycosylation by remote O-acyl protecting groups, in: D.B. Werz, S. Vidal (Eds.), Modern Synthetic Methods in Carbohydrate Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2013, pp. 125–159.
M. Shadrick, Y. Singh, A.V. Demchenko, J. Org. Chem. 85 (2020) 15936–15944. doi: 10.1021/acs.joc.0c01279
K. Greis, S. Leichnitz, C. Kirschbaum, et al., J. Am. Chem. Soc. 144 (2022) 20258–20266. doi: 10.1021/jacs.2c05859
K.X. Shou, Y.Q. Zhang, Y.J. Ji, et al., Chem. Sci. 15 (2024) 6552–6561. doi: 10.1039/d4sc01348d
B.S. Komarova, Y.E. Tsvetkov, N.E. Nifantiev, Chem. Rec. 16 (2016) 488–506. doi: 10.1002/tcr.201500245
I. Iynkkaran, D.R. Bundle, Carbohydr. Res. 378 (2013) 26–34. doi: 10.1016/j.carres.2013.05.005
K. Pradhan, A. Paul, D. Rai, et al., J. Org. Chem. 89 (2024) 4019–4030. doi: 10.1021/acs.joc.3c02886
X.Y. Wu, L. Cui, T. Lipinski, et al., Chem. Eur. J. 16 (2010) 3476–3488. doi: 10.1002/chem.200902460
H. Zhang, X.H. Wang, Y.H. Meng, et al., JACS Au 2 (2022) 97–108. doi: 10.1021/jacsau.1c00190
Z.T. Li, X.Y. Sun, Y.C. Wang, et al., CCS Chem. 6 (2024) 1698–1711. doi: 10.31635/ccschem.023.202303364
A. Imamura, H. Ando, S. Korogi, et al., Tetrahedron Lett. 44 (2003) 6725–6728. doi: 10.1016/S0040-4039(03)01647-2
X.W. Lu, P. Kovác, J. Org. Chem. 81 (2016) 6374–6394. ˇ doi: 10.1021/acs.joc.6b01019
B. Hagen, J.H.M. van Dijk, Q.J. Zhang, et al., Org. Lett. 19 (2017) 2514–2517. doi: 10.1021/acs.orglett.7b00747
D.K. Njeri, J.R. Ragains, Org. Lett. 24 (2022) 3461–3465. doi: 10.1021/acs.orglett.2c01034
D. Crich, S.X. Sun, J. Org. Chem. 61 (1996) 4506–4507. doi: 10.1021/jo9606517
D. Crich, Acc. Chem. Res. 43 (2010) 1144–1153. doi: 10.1021/ar100035r
M. Huang, G.E. Garrett, N. Birlirakis, et al., Nat. Chem. 4 (2012) 663–667. doi: 10.1038/nchem.1404
D. Crich, W.L. Cai, J. Org. Chem. 64 (1999) 4926–4930. doi: 10.1021/jo990243d
J.T. Cai, J. Hu, C.J. Qin, et al., Angew. Chem. Int. Ed. 59 (2020) 20529–20537. doi: 10.1002/anie.202007209
J.E. Yule, T.C. Wong, S.S. Gandhi, et al., Tetrahedron Lett. 36 (1995) 6839–6842. doi: 10.1016/00404-0399(50)1442K-
M. Moumé-Pymbock, T. Furukawa, S. Mondal, et al., J. Am. Chem. Soc. 135 (2013) 14249–14255. doi: 10.1021/ja405588x
X.Y. Liang, J. Yang, Z.H. Qiu, et al., Carbohydr. Res. 500 (2021) 108237. doi: 10.1016/j.carres.2021.108237
H.Y. Xue, Y. Zhou, K. Feng, et al., Org. Lett. 27 (2025) 6855–6861. doi: 10.1021/acs.orglett.5c02016
N. Pauwels, S. Aspeslagh, G. Vanhoenacker, et al., Org. Biomol. Chem. 9 (2011) 8413–8421. doi: 10.1039/c1ob06235b
S. Figueroa-Pérez, R.R. Schmidt, Carbohydr. Res. 328 (2000) 95–102. doi: 10.1016/S0008-6215(00)00092-6
A.A. Shirsat, D. Rai, B.K. Ghotekar, et al., Org. Lett. 25 (2023) 2913–2917. doi: 10.1021/acs.orglett.3c00997
A. Paul, S.S. Kulkarni, Org. Lett. 25 (2023) 4400–4405. doi: 10.1021/acs.orglett.3c01618
A. Paul, D. Rai, K. Pradhan, et al., Org. Lett. 25 (2023) 6413–6418. doi: 10.1021/acs.orglett.3c02430
A. Ghosh, S.S. Kulkarni, Org. Lett. 25 (2023) 7242–7246. doi: 10.1021/acs.orglett.3c02872
X.P. Zou, C.J. Qin, G.Z. Tian, et al., Org. Lett. 26 (2024) 9198–9202. doi: 10.1021/acs.orglett.4c03167
K. Benakli, C. Zha, R.J. Kerns, J. Am. Chem. Soc. 123 (2001) 9461–9462.
L. Yang, J.J. Zhu, X.J. Zheng, et al., Chem. Eur. J. 17 (2011) 14518–14526. doi: 10.1002/chem.201102615
G.L. Zhang, M.M. Wei, C.C. Song, et al., Org. Chem. Front. 5 (2018) 2179–2188. doi: 10.1039/c8qo00471d
D. van der Es, N.A. Groenia, D. Laverde, et al., Bioorg. Med. Chem. 24 (2016) 3893–3907. doi: 10.1016/j.bmc.2016.03.019
S.L. Deng, J. Mattner, Z. Zang, et al., Org. Biomol. Chem. 9 (2011) 7659–7662. doi: 10.1039/c1ob06276j
G. Vessella, A. Casillo, A. Fabozzi, et al., Org. Biomol. Chem. 17 (2019) 3129–3140. doi: 10.1039/c9ob00104b
B. Ghosh, N. Bhattacharjee, A.R. Podilapu, et al., Org. Lett. 24 (2022) 3696–3701. doi: 10.1021/acs.orglett.2c01318
X.Y. Yang, H. Zhang, Q.P. Zhao, et al., JACS Au 4 (2024) 2351–2362. doi: 10.1021/jacsau.4c00321
B. Fraser-Reid, P. Konradsson, D.R. Mootoo, et al., J. Chem. Soc., Chem. Commun. (1988) 823–825.
K.S. Kim, H.B. Jeon, S. Nakamura, et al., Glycoside synthesis from 1-oxygen substituted glycosyl donors: Sections 3.3 and 3.4, in: A.V. Demchenko (Ed.), Handbook of Chemical Glycosylation, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, pp. 185–259.
M.H. Clausen, M.R. Jørgensen, J. Thorsen, et al., J. Chem. Soc., Perkin Trans. 1 (2001) 543–551.
M.H. Clausen, R. Madsen, Chem. Eur. J. 9 (2003) 3821–3832. doi: 10.1002/chem.200204636
M. Clausen, R. Madsen, Carbohydr. Res. 339 (2004) 2159–2169.
L.K. Shi-Shun, J.M. Mallet, M. Moreau, et al., Tetrahedron 55 (1999) 14043–14068.
Y. Nakahara, T. Ogawa, Carbohydr. Res. 200 (1990) 363–375.
A. Santra, A.K. Misra, Glycoconj. J. 29 (2012) 181–188. doi: 10.1007/s10719-012-9383-4

Scheme 2 Synthesis of 1,2-cis-glycosides using O-4-actyl-protected galactouronates donors.

Scheme 3 Proposed glycosylation intermediates of uronic acid 3,6-lactone glycosyl donor 33.

Scheme 4 Proposed glycosylation intermediates of uronic acid 3,6-lactone glycosyl donor.

Scheme 5 Synthesis of α-linked galacturonic acid glycolipids via TiCl4-induced anomerisation.

Scheme 7 Synthesis of α-linked oligosaccharides using 4-O-glycosylated galacturonate donors.

Scheme 9 Preparation of α-galacturonic acid glycosides via the remote participation effect using a galactose donor.

Scheme 10 Synthesis of α-linked galacturonic acid oligosaccharides using 4-O-acyl-protected galactose donors.

Scheme 11 Synthesis of α-linked oligosaccharides using 6-O-acyl-protected galactosyl donors.

Scheme 12 Synthesis of α-galacturonic acid glycosides using 4,6-O-di-tert-butylsilylene-protected galactosyl donors.

Scheme 13 Synthesis of α-galacturonic acid glycoside using DTBS-protected galactosyl donors.

Scheme 14 Synthesis of α-galacturonic acid glycosides using 4,6-O-benzylidene-protected galactosyl donors.

Scheme 15 Synthesis of α-(1→4)-linked N-acetyl galactosaminuronic acids using 2,3-N,O-oxazolidinone-protected galactosyl donors.

Scheme 16 Synthesis of α-galacturonic acid glycosides using galactose donors containing bulky O-6 protecting groups.

Scheme 17 Synthesis of α-linked oligogalacturonates using n-pentenyl galactosyl donor.

Scheme 19 Synthesis of α-linked oligogalacturonates using D-galactosyl fluoride donors.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:




 下载:
下载: