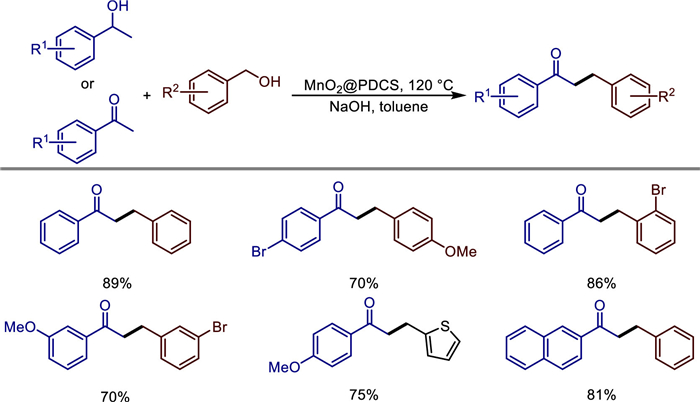
Scheme 1.
Alkylation of ketone with primary alcohols.
Recent advances of heterogeneous manganese catalysis in organic synthesis
Tianyi Zhou , Heng Yang , Guangbin Zhou , Feng Chen , Pan Gao

The relentless pursuit of sustainable and efficient methodologies defines modern organic synthesis. Catalysis stands as a cornerstone of modern chemical synthesis, enabling the efficient and selective construction of complex molecules that are integral to pharmaceuticals, agrochemicals, materials science, and energy technologies. The pursuit of catalytic systems that align with the principles of green and sustainable chemistry remains a primary driver of innovation in the field. The transition metal catalysts continue to play an indispensable role due to their ability to adopt multiple oxidation states and facilitate key redox processes. While noble metals have dominated advanced catalytic transformations, their high cost, limited abundance, and potential toxicity have spurred the search for earth-abundant and environmentally benign alternatives. Catalysis with earth-abundant first-row transition metals has become crucial in this endeavor, reducing reliance on precious and potentially toxic elements. Among these, manganese, Earth's second most abundant transition metal, commands exceptional attention due to its natural prevalence, low cost, benign toxicity profile, and remarkable redox flexibility. Capable of accessing oxidation states from MnⅡ to MnⅦ, manganese enables diverse mechanistic pathways for oxidations, reductions, C—H functionalizations, and cross-coupling reactions previously dominated by precious metals.
Catalysts are traditionally categorized as homogeneous or heterogeneous, each with distinct advantages and limitations [1]. Homogeneous catalysts typically offer superior activity, selectivity, and well-defined active sites due to high molecular dispersion. However, their industrial application is often hampered by formidable challenges in separation, recovery, and reuse, leading to catalyst loss and high operational costs. For example, reported manganese catalysis has been dominated by homogeneous systems, where the soluble catalyst offers high activity and tunability through ligand design [2–14]. However, these systems often face significant practical hurdles: Catalyst recovery and reuse are challenging, leading to potential contamination of products with metal residues; separation of the catalyst from the reaction mixture can be cumbersome; and ligand synthesis or catalyst decomposition. Conversely, heterogeneous catalysts, exist in a different phase than the reactants, usually as solid catalysts interacting with gaseous or liquid reactants, excel in easy separation, recyclability, and robustness under harsh conditions, making them the workhorses of large-scale industrial processes [15,16]. Nevertheless, they can suffer from lower efficiency due to mass transfer limitations and reduced accessibility to active sites [17]. The advent of nanotechnology has provided a paradigm shift, bridging the gap between homogeneous and heterogeneous catalysis [18]. Nanocatalysts, with their exceptionally high surface-area-to-volume ratio, offer a dramatic increase in active sites and unique size-dependent electronic properties that often lead to enhanced activity and selectivity. Their tunable size, shape, and surface chemistry allow for precise optimization toward specific reactions.
By immobilizing or integrating manganese species onto solid supports (e.g., metal oxides, carbon materials, zeolites, polymers, silica, MOFs) or utilizing inherently solid manganese-based materials (e.g., MnO2, Mn2O3, supported nanoparticles), these systems offer inherent advantages: Facile catalyst separation via simple filtration or centrifugation, excellent recyclability and reusability and potential for continuous flow processing. Driven by the sustainability and practicality, research into heterogeneous manganese catalysis has received increasing attention in recent years [19,20]. This growing field moves from simply anchoring homogeneous complexes to exploring novel material designs, nanostructuring, and leveraging unique properties arising from the solid-state environment to achieve high activity and selectivity. Advances in material science and characterization techniques have been instrumental in rationally designing and understanding these complex catalysts.
This review paper aims to provide a comprehensive and critical overview of the recent advances in heterogeneous manganese catalysis for organic synthesis. We will focus on the diverse strategies for immobilization and material design, including supported complexes, nanoparticles, and bulk manganese oxides. Since scholars have already conducted research and provided summaries on manganese-based magnetic catalysts, this paper will not elaborate on this aspect further [21]. The core of the review will systematically explore the application of these heterogeneous Mn catalysts across key reaction classes, such as: (Ⅰ) Coupling reactions (e.g., C—C bond formation, C—N bond formation, C—O bond formation); (Ⅱ) Oxidation reactions (e.g., alcohol oxidation, epoxidation, dehydrogention); (Ⅲ) Other reactions (e.g., hydrogen-deuterium exchange, hydrogenation). For each area, we will highlight the catalyst design principles, scope, mechanistic insights (where available), and performance metrics (activity, selectivity, recyclability). We will also address the ongoing challenges, including leaching control, understanding structure-activity relationships at the interface, achieving truly broad substrate scope, and scaling up processes. Finally, we will offer perspectives on the future directions of this dynamic field, emphasizing its tantalizing promise for developing robust, sustainable, and economically viable catalytic processes for the synthesis of complex organic molecules and fine chemicals.
The formation of carbon-carbon (C—C) bonds stands as a cornerstone of organic synthesis, enabling the construction of complex molecular architectures vital to pharmaceuticals, materials science, and agrochemicals. Traditional methodologies often rely on homogeneous catalysts, particularly those based on precious metals such as palladium, platinum, or rhodium. While effective, these systems face significant challenges, including catalyst separation, recyclability, and environmental concerns due to metal toxicity and scarcity. In response, the field has increasingly shifted toward heterogeneous catalysis, which offers enhanced stability, facile recovery, and reusability—attributes critical for sustainable industrial applications. Amid this transition, heterogeneous manganese catalysts have emerged as compelling alternatives, distinguished by its earth abundance, cost-effectiveness, and recyclable properties.
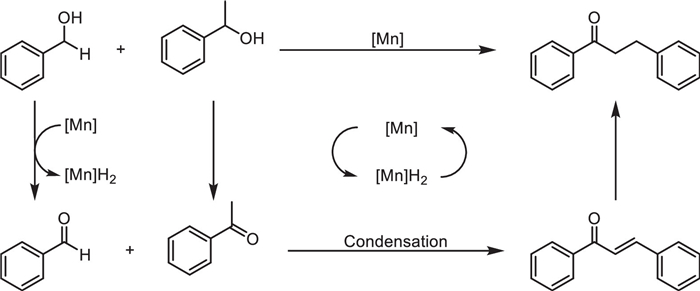
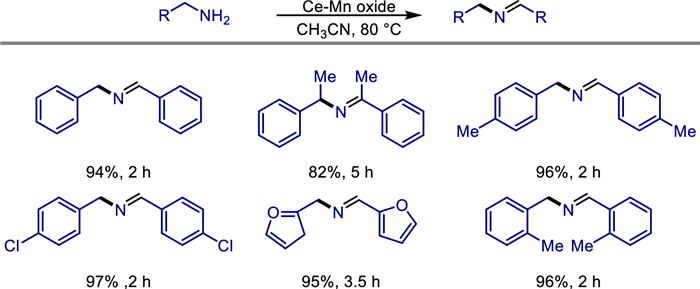
The conversion of alcohols by borrowing hydrogen (BH) method has attracted widespread attention due to its high atomic economy and substrate utilization efficiency [22–24]. In 2019, Wang et al. prepared manganese dioxide nanoparticles and immobilized onto the surface of modified poly(2,4-dichlorophenyl) microspheres (PDCS) [25]. This α-MnO2@PDCS catalyst, characterized by SEM, TEM, XRD, and XPS, demonstrated exceptional catalytic efficiency in borrowing hydrogen reactions, enabling direct alkylation of ketones with alcohols (Scheme 1). The catalyst retained high activity over five cycles, attributed to the stabilizing role of the PDCS support. Mechanistic studies ruled out radical pathways and highlighted sequential dehydrogenation-condensation-hydrogenation steps (Scheme 2). Notably, the system's scalability was validated through gram-scale synthesis, underscoring its potential for industrial applications. This work advances the use of earth-abundant manganese in recyclable heterogeneous catalysis, offering an economical and eco-friendly alternative to noble-metal systems for C—C bond formation and bioactive heterocycle synthesis.
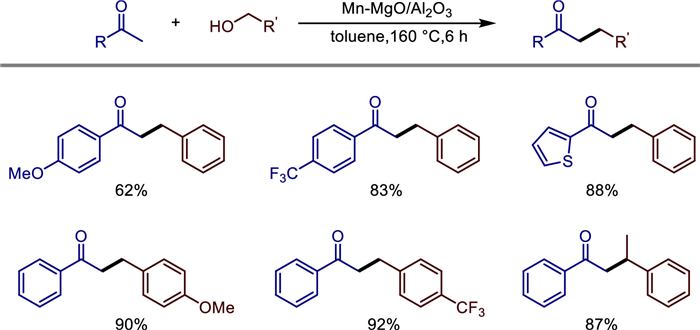
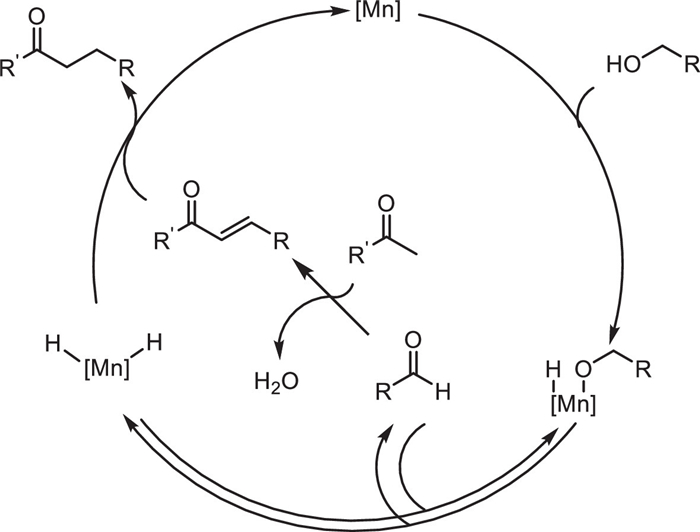
Non-precious metal heterogeneous catalysts have been widely studied in recent years. However, the difficulty in observing the types of active metals hinders the study of mechanisms. In 2022, Michikazu Hara and co-workers reported the use of Al2O3 supported low-valent Mn catalyst to construct C—C bonds through α-alkylation of ketones and alcohols using the BH method (Scheme 3) [26]. The catalyst is a Mn2+-MgO mixture deposited on an Al2O3 support (Mn-MgO/Al2O3), which exhibits catalytic performance for the corresponding reaction with a yield of 50%-92%. The characterization of the Mn-MgO/Al2O3 catalysts disclosed that Mn oxide and MgO nanoparticles are strongly contacted onto Al2O3 particles catalyst and Mn oxide mainly consists of Mn2+. The alcohol could adsorb on the catalyst surface and Mn alkoxides undergo reversible dehydrogenation to generate Mn—H species and generate aldehyde intermediate. Then aldehyde condenses with a ketone to give the α,β-unsaturated ketone, which is hydrogenated by Mn—H species to form the desired product (Scheme 4). The catalyst can be recycled three times without a decrease in activity, and its structural stability is proven by characterizations (XRD, ICP-AES). This provides an efficient and sustainable non-noble metal catalytic solution for the green synthesis of C—C bonds and functional molecules.
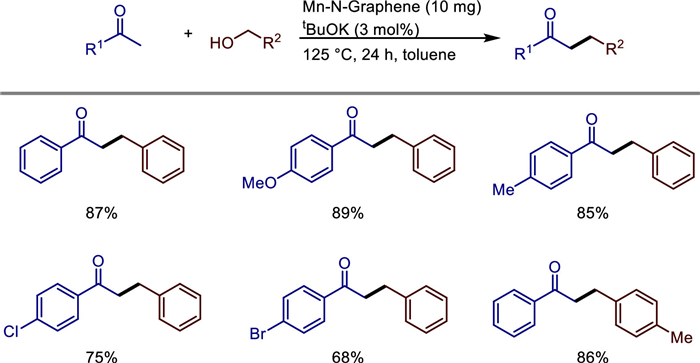
In the same year, Tetsuya Kida and colleagues discovered a new method to convert difficult-to-recover molecular homogeneous catalysts into heterogeneous catalysts [27]. Specifically, by performing pyrolysis on pre-formed crystalline polymer based on manganese ethylene diamine, they successfully transformed homogeneous manganese complexes into heterogeneous manganese-based catalysts. The resulting catalysts not only exhibit high stability and recyclability but also provide excellent control over reactions at a molecular level. These catalysts were applied to C—C and C—N bond-forming reactions, such as the alkylation of ketones or amines using primary alcohols as alkylating agents, which proceeded under mild conditions with water as the sole by product (Scheme 5). They combined XPS, TEM, and XRD characterizations to confirm that Mn atoms in the catalyst are anchored on the surface of nitrogen-doped graphene in a monodispersed form, forming stable Mn-N active sites. This catalyst exhibits excellent substrate universality and is compatible with sensitive functional groups such as aromatic ketones, aliphatic alcohols, halogenated substances, and cyano groups. The highest yield can reach 93%, and the cross-alkylation of secondary alcohols and ketones has been successfully achieved. Meanwhile, this catalyst can be recycled five times without a decrease in activity. This study provides a new strategy for replacing noble metal catalysts and achieving green and efficient C—C/C—N bond formation. It combines atomic economy (with water being the only by-product) and has great potential for industrial applications.
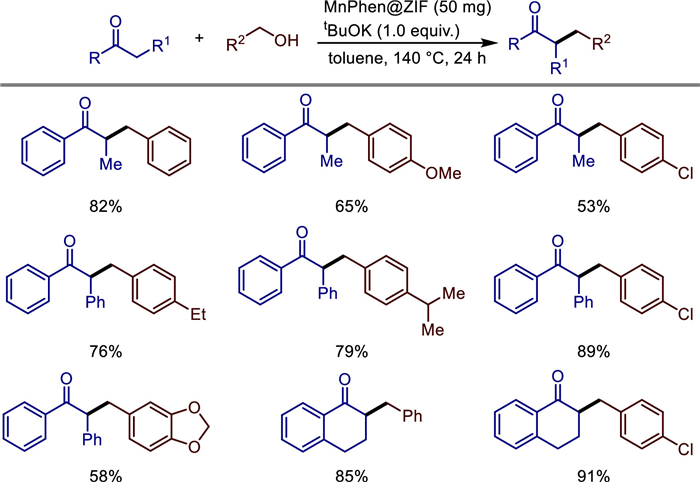
With the deepening of research, Arshad Aijaz et al. reported for the first time that metal-organic frameworks (MOFs) can be used as catalysts for the α-alkylation of ketones with alcohols in 2022 [28]. The research team developed a manganese-based heterogeneous catalyst (MnPhen@ZIF) based on metal-organic frameworks (MOFs). By encapsulating the homogeneous manganese-phenanthroline complex (MnPhen) into the pores of ZIF-8, they achieved a highly selective α-alkylation reaction between ketones and alcohols to synthesize β,β-disubstituted branched ketones (Scheme 6). Its innovative points include: (1) For the first time, MOFs were used in manganese-catalyzed C—C bond formation. The nanoconfinement effect of ZIF-8 was utilized to stabilize the active sites, and SEM, TEM, XRD, and XPS characterizations confirmed that MnPhen was uniformly distributed within the pores of the MOF. (2) Without the need for additional homogeneous strong bases, the reaction only requires a small amount of t-BuOK, and water is the only by-product, which is in line with the principles of green chemistry. (3) It has high selectivity and substrate universality, and is suitable for aromatic ketones, aliphatic alcohols, halogenated substances, and heterocyclic alcohols, with the highest yield reaching 91%. (4) It has excellent cycle stability. The catalyst can be reused five times without a significant decrease in activity, and there is no manganese leaching. (5) The reaction mechanism was revealed. The hydrogen borrowing pathway was confirmed through deuterium-labeling experiments and intermediate detection, and it was found that defects and Lewis acid sites were formed in the pores of the MOF during the reaction, which improved the catalytic efficiency. This work provides a new strategy for the development of non-noble metal catalysts supported by MOFs and the efficient synthesis of branched ketones.
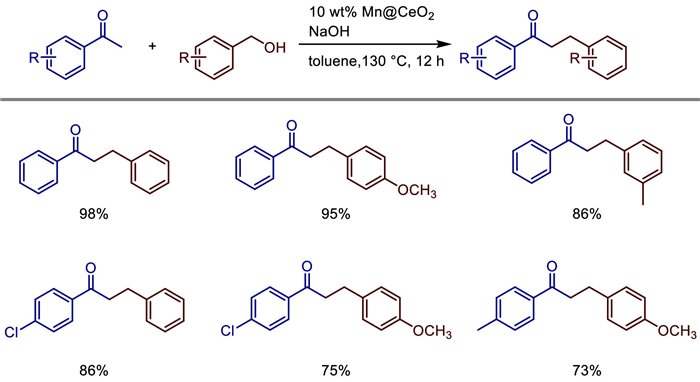
In 2025, Rajagopal Swaathy et al. reported a heterogeneous Mn@CeO2 catalyst (with 10 wt% Mn doping) synthesized via a simple co-precipitation method [29]. This catalyst is used for the α-alkylation of ketones with alcohols as the alkyl source through a hydrogen-borrowing strategy. The catalyst exhibits excellent crystallinity and thermal stability (up to 800 ℃). Structural characterizations (XRD, FE-SEM, HR-TEM) reveal that 19 nm MnO2 nanoparticles are uniformly distributed on 33 nm rod-shaped CeO2. XPS and EPR analyses indicate that the introduction of Mn4+ species creates oxygen vacancies and surface defects, thereby enhancing catalytic activity. Under optimized reaction conditions (toluene as solvent, 130 ℃, 12 h, 50 mol% NaOH), the catalyst achieves yields of 70%–98% for the α-alkylation of various aromatic ketones with alcohols (Scheme 7). It can also be recycled for 6 runs without significant loss of activity. ICP-MS confirms no leaching of manganese ions, demonstrating its good heterogeneous nature. Being environmentally friendly and highly atom-economical, this catalyst only produces water as a by-product, making it suitable for sustainable C—C bond construction.
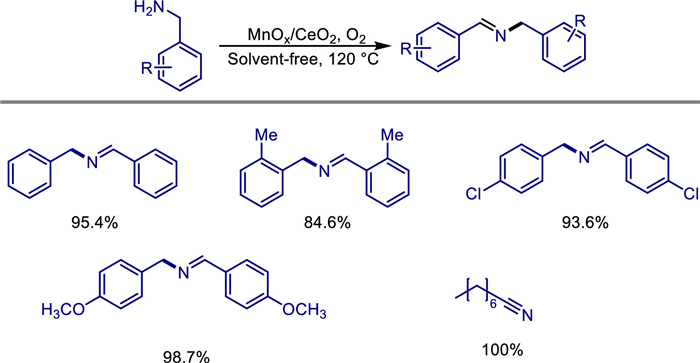
In the research field of catalytic synthesis of imines, the structural design and performance control of catalysts have always been the core direction to achieve green and efficient reactions. Suresh K. Bhargava et al. studied the structure activity properties of MnOx/CeO2 nanorods and MnOx/CeO2 nanoparticles in 2016, as well as the importance of CeO2 morphology for oxygen as a green oxidant for solvent-free amine oxidation [30]. The research team prepared CeO2 nanorods with different morphologies and nanoparticles by hydrothermal method and precipitation method respectively, and loaded 10% manganese oxide (MnOx) by wet impregnation method. The influence of the morphology of the support on the structure of the catalyst and the performance of the selective oxidation of amines was systematically studied. Through characterizations such as HRTEM, XPS, and Raman, it was found that due to the exposure of highly active crystal planes, CeO2 nanorods have a higher concentration of Ce3+ and oxygen vacancies, and a multivalent state coupling of Mn2+/Mn3+/Mn4+ is formed after loading, while only Mn3+/Mn4+ exist on the nanoparticle support. In the oxidation reaction of amines with oxygen as the oxidant in the absence of a solvent, the activity of the MnOx/CeO2 nanorod catalyst is twice that of the nanoparticles, and the selectivity for dibenzylamine reaches 99%, and it is applicable to a variety of aliphatic amine substrates (Scheme 8). Its innovation lies in revealing the mechanism by which the morphology of CeO2 synergistically enhances the catalytic performance by regulating the surface defect structure (Ce3+/oxygen vacancies) and the valence state distribution of manganese species, providing a theoretical basis for the design of efficient non-noble metal catalysts.
In 2019, Fushan Chen et al. synthesized three manganese oxide catalysts (M-3, M-4, M-5) via a template-free oxalate route at different calcination temperatures (350, 450, 550) for the efficient preparation of imines through the oxidative self-coupling of primary amines [31]. Among them, M-4 (calcined at 450 ℃) catalyzed the reaction of benzylamine under mild conditions (toluene as solvent, 110 ℃, air as oxidant, no alkaline additives) for 2 h, achieving 100% conversion and 96.2% imine selectivity. Its performance was significantly superior to most noble metal or complex catalyst systems reported in existing literature.
Catalyst characterization (XRD, XPS, N2 adsorption-desorption, etc.) showed that M-4 is mainly composed of cubic phase Mn2O3 with a micro-rod morphology. Its high selectivity is attributed to the synergistic effect between the surface Mn3+/Mn4+ ratio (0.486/0.254) and the water molecule content in surface oxygen species (0.143). Specifically, Mn3+ is the main active site for oxidizing amines to imines, while Mn4+ tends to cause excessive oxidation of imines to by-product nitriles. Surface water molecules promote the hydrolysis of imine intermediates to aldehydes, which then condense with amines to form the target imines. In comparative experiments, M-3 (350 ℃) exhibited high activity but low selectivity (77.2%) due to its high specific surface area (304.7 m2/g) and Mn4+ content (0.386), easily generating benzonitrile. M-5 (550 ℃) showed significantly reduced activity due to its low specific surface area (19.4 m2/g) and lattice oxygen mobility.
The authors proposed the reaction pathway: amines undergo dehydrogenation oxidation to form imine intermediates (not detected), part of which is hydrolyzed to aldehydes and then condensed with another molecule of amine to form imines. A small amount of imine intermediates is over-oxidized to nitriles, which are further hydrolyzed to amides. This pathway explains the reason for the decrease in imine selectivity under high conversion rates (accumulation of by-products). This work provides an efficient and low-cost non-noble metal catalyst scheme for the green synthesis of imines (Scheme 9).
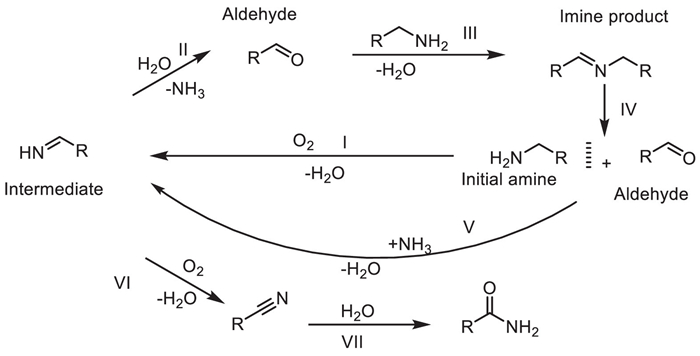
Organic modifiers have shown great potential in regulating the activity and selectivity of heterogeneous catalysts by adjusting their surface properties. Despite the increasing application of organic modification techniques in regulating metal oxide redox acid catalysis, controlling the acidity of metal oxide catalysts to improve reaction selectivity without sacrificing their redox activity remains a major challenge. Qianqian Hao et al. demonstrated in 2021 the successful control of metal oxide redox acid catalysis using non proton tertiary amine modifiers [32]. They carried out surface modification of the manganese dioxide (MnO2) catalyst by using non-protic tertiary amines (such as N,N-dimethylcyclohexylamine, DMCHA), selectively blocking its Lewis acid sites, thus regulating the reaction pathway of amine oxidation. Experiments show that the MnO2 modified by DMCHA can increase the selectivity of imines from 42% (for the unmodified catalyst) to over 90% in the aerobic oxidation reaction of benzylamine and its derivatives, and significantly inhibit the formation of nitrile by-products. Through characterizations such as FT-IR and adsorption experiments, it is confirmed that DMCHA is stably adsorbed on the surface of the catalyst through acid-base interactions, effectively covering the acid active sites, while not affecting the oxidation active sites of MnO2. Thus, selectivity regulation is achieved while maintaining a high reaction rate (the TOF is comparable to that of the unmodified catalyst). This method breaks through the limitation that traditional protic modifiers (such as carboxylic acids) lead to a significant decrease in activity, and the catalyst can be recycled more than three times. Its innovation lies in the first use of non-protic amine modifiers to precisely regulate the acid-oxidation synergistic catalytic performance of metal oxides, providing a new strategy for designing highly active and highly selective heterogeneous catalysts, especially suitable for the reaction system of efficiently synthesizing imines from aryl amine compounds (Scheme 10).
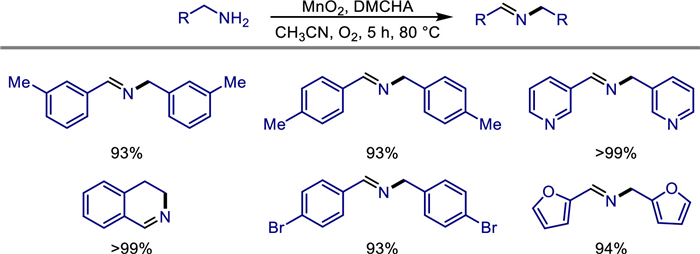
In 2021, Murugan Subaramanian developed a nitrogen-doped graphene-supported manganese-based nanocatalyst (Mn@NrGO) for the selective N-alkylation of alcohols and amines under mild conditions [33]. This heterogeneous catalyst was prepared by the pyrolysis of an in-situ formed manganese-phenanthroline complex at 800 ℃. Characterized by multiple techniques including PXRD, XPS, and TEM, its structure was confirmed to contain MnOx nanoparticles and a nitrogen-doped graphene support. Under the optimized reaction conditions (8 mol% catalyst, 1 equiv. KOtBu, n-octane as solvent, 140 ℃, oxygen-free environment), the catalyst efficiently catalyzed the N-alkylation of various anilines with benzyl alcohols, yielding secondary amine products with moderate to excellent yields and demonstrating good functional group tolerance (Scheme 11). Mechanistic studies revealed that the reaction follows a "hydrogen-borrowing" mechanism, which involves three steps: dehydrogenation of alcohols to form aldehydes, condensation of aldehydes with amines to generate imines, and hydrogenation of imines. Water is the only by-product in this process.
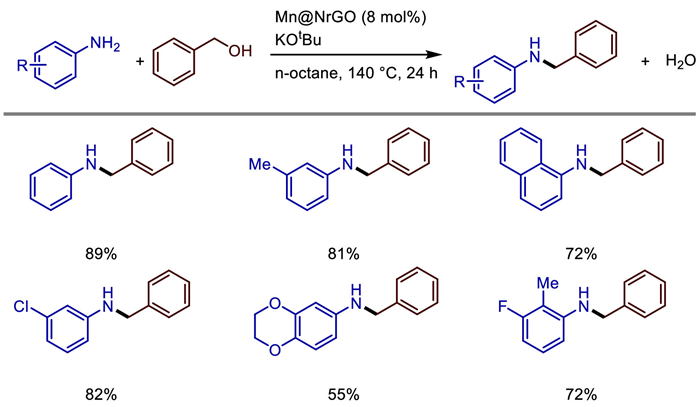
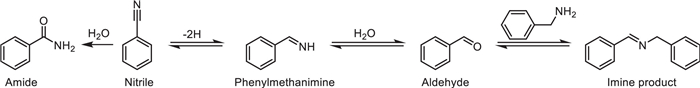
The catalyst exhibits excellent recyclability and reusability, maintaining high activity even after six consecutive uses. Moreover, hot filtration experiments confirmed that it is a genuine heterogeneous catalyst. Additionally, this catalytic system can be applied to the synthesis of pharmaceutical intermediates, highlighting its potential application value in green synthesis. Primary amine self-coupling is important in imine synthesis, but its thermal catalysis usually requires higher temperatures or the addition of expensive oxidants. Hepeng Zhang et al. prepared Ce doped MnOx catalysts using three different methods and thoroughly characterized their surface properties [34]. They prepared cerium-doped manganese dioxide (Ce-MnO2) catalysts through three methods, namely redox coprecipitation, acid-assisted calcination, and coprecipitation, and systematically studied their performance in the catalytic self-coupling oxidation of benzylamine for imine synthesis. It was found that the CeMn-RO catalyst prepared by the redox coprecipitation method, due to its high concentration of Mn3+ and Ce3+, abundant surface oxygen species and oxygen vacancies, as well as the coexistence of acidic/basic sites, exhibited excellent catalytic performance under mild conditions (80 ℃, air as the oxidant, solvent-free). The conversion rate of benzylamine reached 92%, and the selectivity of imines reached 80%. Through characterizations such as H2-TPR and XPS, the mechanism by which Ce doping enhances the oxidation ability by regulating the manganese oxidation state and oxygen vacancies was revealed. Combined with the in-situ DRIFTS technique to track the intermediates, the reaction pathway was proposed as follows: dehydrogenation of benzylamine → hydrolysis of NH-imine into benzaldehyde → condensation of aldehyde and amine to form imine (Scheme 12).
The oxidation self-coupling of amines and the cross-coupling with alcohols are important reactions for the synthesis of imines; however, the catalytic systems for these reactions typically require expensive oxidants, complex catalysts, or additives such as bases or co-catalysts. In 2022, Jonggol Tantirungrotechai et al. developed an amorphous cerium-manganese oxide (Ce-Mn) microsphere catalyst, which was prepared by a simple coprecipitation method [35]. It is used to efficiently catalyze the oxidative self-coupling of amines and the cross-coupling reaction between amines and alcohols. Using air as a green oxidant, a variety of imines can be synthesized under mild conditions. The innovation of this catalyst lies in the fact that its hierarchical microsphere structure significantly improves the catalytic activity and stability. There is no need for expensive oxidants or additives, and it can be recycled more than ten times through filtration and calcination without deactivation. Experiments show that the catalyst exhibits good universality for substrates such as benzylamine derivatives, heterocyclic amines, and benzyl alcohol. Moreover, conditions such as the Ce/Mn ratio, solvent, and temperature have been systematically optimized. This method is both economical and environmentally friendly, providing a new strategy for the green synthesis of imines (Scheme 13).
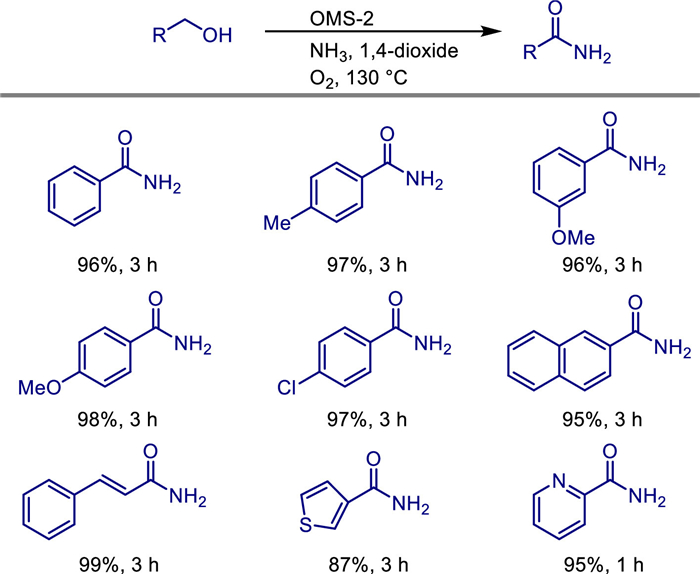
Amides are a very important class of compounds in chemistry and biology, widely used as intermediates in the synthesis of peptides and proteins, fragrance enhancers, anti-blocking agents, pigments for inks, detergents, and lubricants. The most common process for amide synthesis is the reaction of activated carboxylic acid derivatives, such as acid chlorides, anhydrides, and esters, with amines (including ammonia). Beckmann rearrangement, Aube-Schmidt rearrangement, and Staudinger ligation are also commonly used procedures. However, these procedures require stoichiometric amounts of (hazardous) reagents and produce at least equivalent amounts of by-products. Therefore, developing new environmentally friendly methods for amide synthesis is a very important topic in modern organic synthesis. For example, homogeneous manganese catalysts have been successfully developed for the synthesis of amides [36] and urea [37] from alcohols and amines successfully. In parallel, the development of heterogeneous manganese catalysts for amide synthesis has also garnered growing interest among chemists.
In 2012, Noritaka Mizuno and colleagues demonstrated that manganese oxide-based octahedral molecular sieves (KMn8O16; OMS-2), which possess a 2 × 2 hollandite structure with one-dimensional pores, can facilitate this transformation [38]. They developed a heterogeneous catalytic system based on manganese oxide molecular sieve (OMS-2). For the first time, they achieved the direct and efficient synthesis of primary amides under mild conditions (130 ℃, 3 atm O2) using primary alcohols and ammonia water as raw materials and oxygen as the oxidant (Scheme 14). They utilized the triple catalytic functions of OMS-2: (1) Dehydrogenation of alcohols to aldehydes, (2) Condensation of aldehydes with ammonia to form imines and subsequent dehydrogenation to nitriles, and (3) hydration of nitriles to amides, with only water as the by-product. This avoids the dependence of traditional methods on dangerous reagents (such as acyl chlorides and acid anhydrides) and noble metal catalysts. Experiments show that OMS-2 is applicable to substrates such as benzyl alcohol derivatives (yield ≥95% for those with substituents), allyl alcohols, heterocyclic alcohols (such as thiophene methanol and pyridine methanol), and aliphatic alcohols. Moreover, the catalyst can be recovered by simple filtration and reused more than 11 times without a decrease in activity. Through hot filtration experiments and ICP-AES analysis, it was confirmed that the catalytic process follows a heterogeneous mechanism, and there is no dissolution of manganese species. In addition, this method can be extended to the direct synthesis of amides from aldehydes or nitriles, and the products are easy to purify (by washing with n-hexane). This work breaks through the limitations of traditional primary amide synthesis, such as low substrate universality, the need for additives, and the non-recyclability of catalysts, providing a new strategy for green and sustainable amide synthesis.
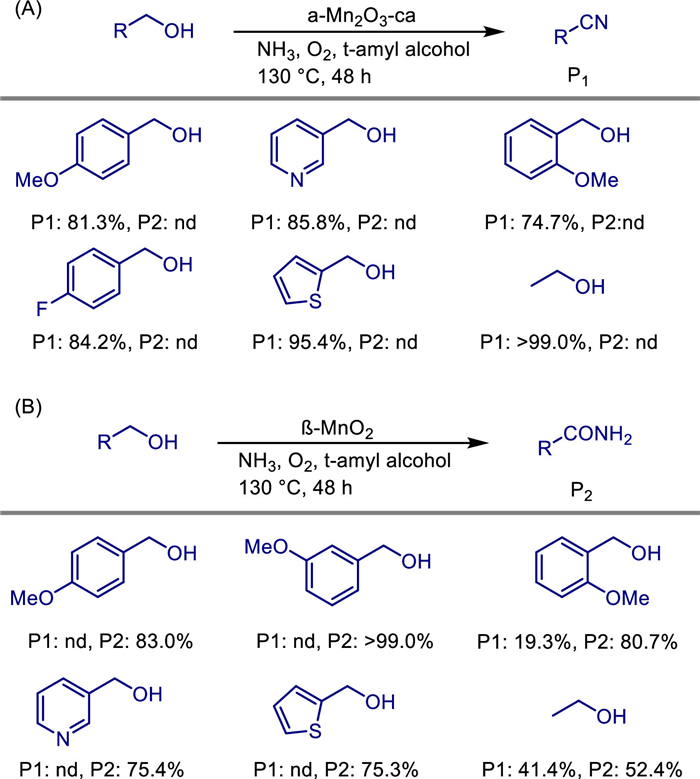
In 2021, Feng-Shou Xiao and his team demonstrated that the performance of heterogeneous catalysts can be effectively tuned by altering their structure [39]. They achieved efficient selective control of nitrile and amide products in the ammoxidation reaction of alcohols by regulating the crystal structures of manganese oxides. It was found that the α-Mn2O3 catalyst can selectively catalyze the direct synthesis of nitrile compounds from primary alcohols and ammonia water under mild conditions (130 ℃, in an oxygen atmosphere) with a yield of > 99%, while catalysts with crystal phases such as β-MnO2 tend to produce amides through a tandem hydration reaction (Scheme 15). Its innovation lies in revealing the unique mechanism by which α-Mn2O3 blocks the hydration side reaction of nitriles by inhibiting the activation of water molecules and the adsorption of nitriles, while the strong water decomposition ability of the β-MnO2 crystal accelerates the conversion of nitriles to amides. Experiments combined with kinetics and in-situ infrared spectroscopy confirmed that the surface of α-Mn2O3 cannot effectively dissociate water and has weak adsorption of nitriles, thus exhibiting high selectivity and stability (it can be recycled more than 10 times) for substrates such as benzyl alcohol, aliphatic alcohol, and heterocyclic alcohol. This work breaks through the bottleneck of uncontrollable product selectivity in traditional ammoxidation reactions and provides a new strategy for the green synthesis of high-value-added nitrile compounds.
The sequential cleavage and functionalization of C—C bonds in alcohols have rapidly developed into an emerging field for discovering new transformations. However, due to the inherent inertness of C—C bonds, achieving this in a direct and selective manner remains challenging. In 2022, Zehui Zhang, Wen Dai, and colleagues reported a novel and efficient approach for the direct synthesis of amides through sequential C—C bond cleavage and amidation of alcohols, catalyzed by heterogeneous manganese oxide [40]. The catalyst is prepared by the coprecipitation method, featuring a high specific surface area (201 m2/g), abundant oxygen vacancies, and moderate acidic sites. Characterizations such as XRD and XPS reveal that the oxygen vacancies of the catalyst can activate oxygen and reduce the energy barrier for C—H bond cleavage (1.15 eV). Experiments show that this system applies to primary alcohols, secondary alcohols, vicinal diols, and lignin model compounds (Scheme 16). The highest yield of amides can reach 96%. By switching the solvent (acetonitrile) and adjusting the temperature, sterically hindered nitriles can be selectively produced (with a yield of 69%-91%). The mechanism confirms that phenylacetaldehyde (a) generated after oxidation can be efficiently converted to phenylacetonitrile (b) in an NH3 and O2 atmosphere. Subsequently, part of b undergoes a hydration reaction to form 2-phenylacetamide (c). The remaining b and the generated c can be further oxidized to benzoyl cyanide (d) and 2-oxo-2-phenylacetamide (e). Similar to b, part of d can also form e through a hydration reaction. Finally, the α-carbonyl carbon of d and e is subjected to a nucleophilic attack by NH3, directly yielding the target product benzamide (Scheme 17). The catalyst maintains high activity in gram-scale reactions, shows no deactivation after being recycled 5 times, and has been successfully applied to the late-stage functionalization of bioactive molecules (such as δ-tocopherol derivatives), providing an efficient and recyclable heterogeneous catalytic solution for green synthesis and biomass conversion.
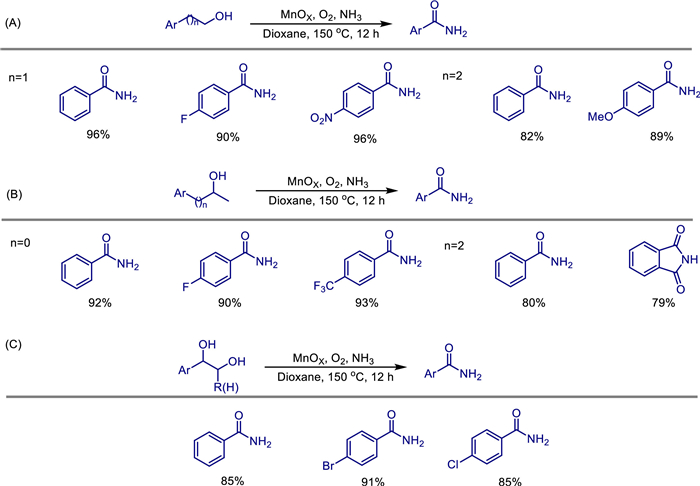
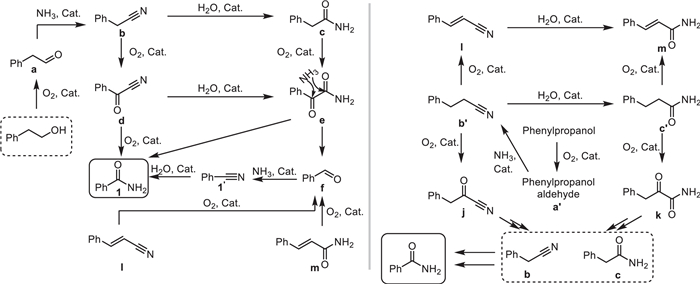
Recently, Zehui Zhang and his team reported a novel heterogeneous manganese catalyst, nitrogen-doped carbon-supported manganese carbodiimide (MnNCN/NC-600), which was first used for the efficient and highly selective synthesis of nitriles from alcohols under the conditions of aqueous ammonia (aq. NH3) and oxygen (O2) [41]. The catalyst was prepared by pyrolyzing a solid mixture of manganese acetate and dicyandiamide. Characterization results (XRD, XPS, TEM, FT-IR, etc.) confirmed the formation of MnNCN nanoparticles and their uniform dispersion on the nitrogen-doped carbon support. Compared with traditional manganese oxides (MnOx), MnNCN/NC-600 has stronger O2 adsorption and activation ability, a larger specific surface area, and weaker Lewis acidity. These properties endow it with broad substrate applicability in reactions such as ammoxidation of primary alcohols to nitriles, oxidative C—C bond cleavage and ammoxidation of secondary alcohols, and ammoxidation of phenyl-substituted aliphatic alcohols and diols to nitriles, effectively inhibiting side reactions such as over-oxidation of aldehydes (to form carboxylic acids) and hydrolysis of nitriles (to form amides) (Scheme 18). This study provides new insights into manganese-catalyzed aerobic oxidation and serves as an important complement to the manganese oxide catalytic system.
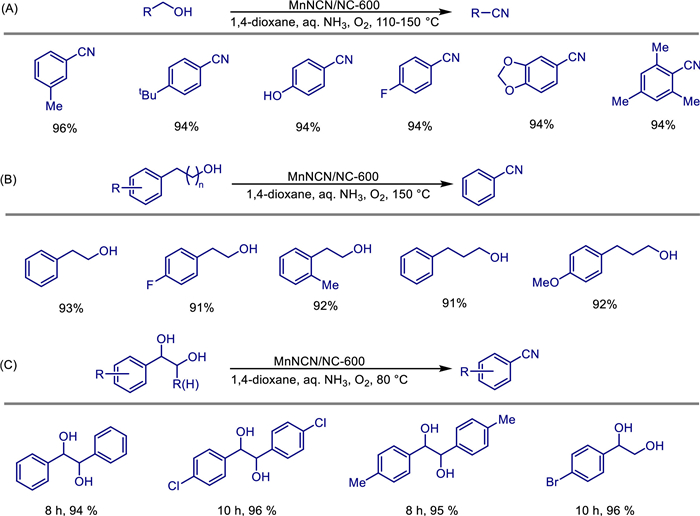
In 2018, Bo Li et al. reported a method for the heterogeneous oxidation synthesis of quinazoline compounds catalyzed by octahedral molecular sieve OMS-2 under ligand-free conditions [42]. The OMS-2-U catalyst prepared with urea as an additive (specific surface area 113 m2/g, crystallinity better than conventional OMS-2 and OMS-2-H) can efficiently catalyze the oxidation/cyclization reaction of alcohols and amidines in toluene solvent under an oxygen atmosphere at 110 ℃ to synthesize a series of substituted quinazolines (Scheme 19). The structural stability of the catalyst was confirmed by XRD, BET, SEM and other characterizations, and it can be recycled 4 times without significant activity decline. Substrate applicability studies have shown that benzyl alcohols with electron-withdrawing groups react better than those with electron-donating groups, but steric hindrance (such as 2-chlorobenzyl alcohol with a yield of 56%) and strong electron-donating groups (such as 3,4-dimethoxybenzyl alcohol without product) will limit the reaction. This system provides a recyclable heterogeneous catalytic strategy for the green synthesis of nitrogen heterocycles.
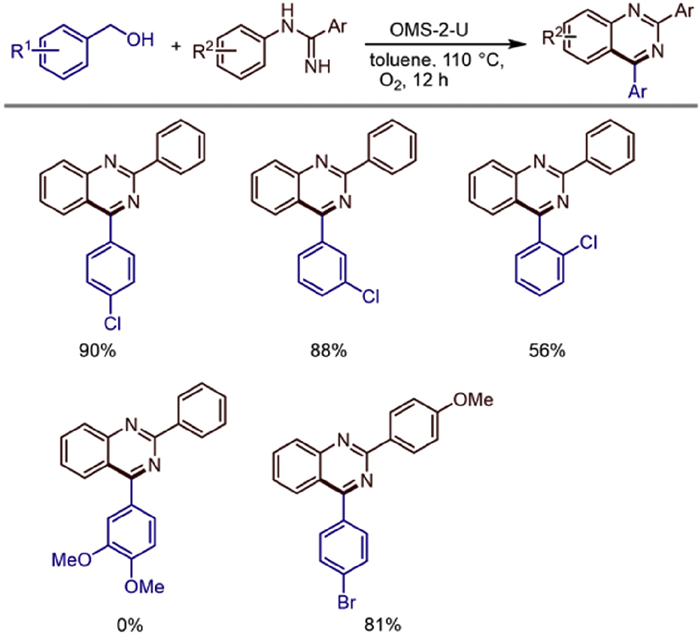
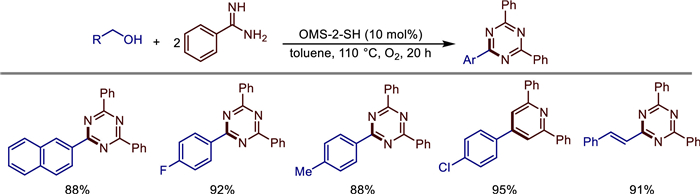
Following closely on this, in 2019, Jian Shen's team achieved new breakthroughs in the preparation of OMS-2. They successfully prepared manganese oxide octahedral molecular sieves (OMS-2) with increased specific surface area and mixed valence states using 4Na2SO4·2H2O2·NaCl as a reducing agent [43]. OMS-2 exhibited excellent catalytic performance for the aerobic oxidation synthesis of 1,3,5-triazines from benzyl alcohol and benzamidine (Scheme 20). Toluene, DMF, and DMSO could also serve as substrates to react with benzimidine under heterogeneous conditions to form triazines. This catalytic system features base-free conditions, a wide substrate range, high chemoselectivity, simple operation, catalyst recyclability, and the use of O2 as a green oxidant.
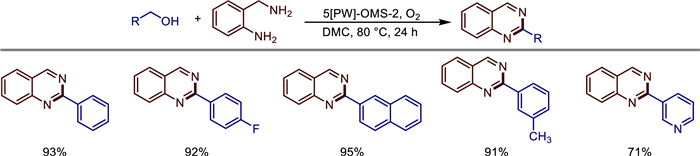
The exploration of the performance optimization of OMS-2 materials does not stop. In 2021, Xiang Liu et al. reported a sodium phosphotungstate-modified octahedral molecular sieve OMS-2 nanocomposite ([PW]-OMS-2), which was used for the efficient synthesis of quinazoline compounds through a one-pot reaction of benzyl alcohol and 2-aminobenzylamine using O2 as the oxidant and dimethyl carbonate (DMC) as the solvent [44]. Catalysts prepared by the reflux method were confirmed by TEM, XRD, EDS and other characterizations: sodium phosphotungstate decomposed into phosphotungstic acid and sodium tungstate during the doping process, in which phosphotungstic acid was attached to the surface of OMS-2, and W ions were successfully doped into the OMS-2 framework. Compared with the phosphotungstic acid/OMS-2 prepared by the impregnation method and pure OMS-2, the optimized 5[PW]-OMS-2 (molar ratio of sodium phosphotungstate to KMnO4 is 5 mol%) had the highest catalytic activity, with a specific surface area of 169.1 m2/g and an average oxidation state of Mn of 3.82. Under the optimal conditions (80 ℃, O2 atmosphere, 24 h), the yield of 2-phenylquinazoline catalyzed by 5[PW]-OMS-2 reached 93%. Substrate expansion showed that aryl methanol containing electron-donating groups (such as -CH3, -OCH3) or electron-withdrawing groups (such as -F, -Cl, -Br, -CF3) and heterocyclic methanol (pyridine/thiophene) could be efficiently converted (yield 71%–95%), but fatty alcohols were ineffective. Kinetic studies showed that the reaction was the rate-limiting step (apparent activation energy 87.73 kJ/mol), and the reaction rate constant of 5[PW]-OMS-2 (0.1635 min−1) was significantly higher than that of pure OMS-2 (0.0482 min−1). Mechanistic analysis indicated that the synergistic effect of the mixed crystal phases of phosphotungstic acid and OMS-2 and the enhanced redox ability by W doping were the key to the high performance of the catalyst. Hot filtration experiments confirmed that it was a heterogeneous catalytic process (Scheme 21).
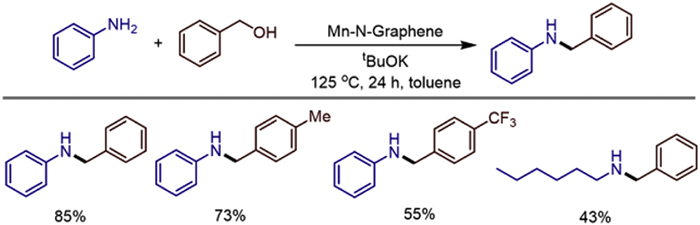
We all know that a series of organic transformations are catalyzed by molecularly well-defined homogeneous catalysts, but they are difficult to recycle. Meanwhile, heterogeneous catalysts are required in many industrial processes due to their stability and recyclability, but controlling them at the molecular level is challenging. In 2022, Tetsuya Kida and colleagues converted homogeneous manganese complexes into heterogeneous manganese-based catalysts by pyrolyzing pre-formed crystalline polymers [27]. The resulting catalysts were then used in C—C/C—N bond-forming reactions, such as the alkylation of ketones and/or amines, utilizing primary alcohols as alkylating agents and releasing water as the only by product under nearly mild conditions. The author's team used characterization techniques such as XPS, TEM, and XRD to confirm that the catalyst is composed of a graphene layer structure. Manganese is anchored in the nitrogen-dopedcarbon matrix in an atomically dispersed form without forming nanoparticles orclusters, and there are active sites such as Mn—N and Mn—Cl on the surface. This catalyst exhibits high catalytic activity under mild conditions (using toluene as the solvent, at 125 ℃, with 3 mol% BuOK). It can use alcohols as alkylation reagents to achieve the C-alkylation of ketones and the N-alkylation of amines through the "hydrogen borrowing" mechanism, with only water as the by-product (Scheme 22). Experiments show that the catalyst has good compatibility with substrates containing electron-donating or electron-withdrawing groups (such as halogenated, methoxy, trifluoromethyl), and is applicable to both primary and secondary alcohols, with primary alcohols showing higher reactivity. In addition, the catalyst maintains stable activity after five cycles without obvious deactivation. The innovation lies in the first construction of a manganese-based heterogeneous catalyst through the molecular precursor pyrolysis strategy, breaking through the limitation that traditional homogeneous manganese catalysts cannot be recycled, and avoiding the use of noble metals at the same time. It provides a low-cost, sustainable, and scalable solution for the formation of C—C/C—N bonds. This work fills the gap of manganese-based heterogeneous catalysts in hydrogen borrowing reactions, and verifies the synergistic effect of manganese active centers in dehydrogenation, condensation, and hydrogen transfer through mechanistic experiments, providing a reference for the design of new single-atom metal catalysts.
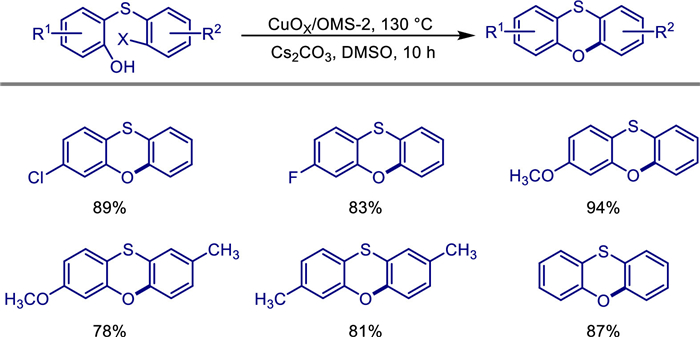
Phenoxathiin and its derivatives have garnered extensive attention due to their unique chemical and physical properties, as well as their applications in fluorescent materials, antifungal activity, and selective inhibitors. In 2019, Xiang Liu and colleagues achieved the heterogeneous catalytic synthesis of phenoxathiin derivatives via intramolecular arylation reactions with aryl bromides or aryl chlorides, using a copper-loaded manganese oxide octahedral molecular sieve (OMS-2) catalyst (CuOx/OMS-2) [45]. The author's team prepared the CuOx/OMS-2 catalyst by the wet impregnation method. Its structure was characterized as short nanorods by TEM and XRD, and the copper species on the surface were highly dispersed. Under the optimized conditions (using DMSO as the solvent, Cs2CO3 as the base, at 130 ℃), this catalyst successfully catalyzed the intramolecular arylation reaction of phenols and aryl halides, efficiently synthesizing a series of substituted benzothiophene derivatives with a maximum yield of up to 94% (Scheme 23). Experiments show that the reaction has a wide compatibility with the substituents of the substrates, and it can well adapt to both electron-withdrawing groups (such as -Cl, -F) and electron-donating groups (such as -CH3, -OCH3). The catalyst still maintains stable activity after being reused 8 times, and there is no significant change in its structure after recovery. XRD and TEM analyses confirm its high stability. The innovation lies in the first application of the CuOx/OMS-2 heterogeneous catalytic system in the intramolecular arylation reaction, breaking through the limitation that traditional homogeneous catalysts cannot be recycled. This work not only provides a green and scalable method for the synthesis of benzothiophene compounds but also expands the application scope of OMS-2-based catalysts in organic synthesis, especially showing potential application values in fields such as fluorescent materials and antifungal drugs.
Coincidentally, aiming at the problems existing in the traditional synthesis methods of another important class of phenoxazin derivatives, 2-amino-3H-phenoxazin-3-one (APXO), with significant antiproliferative activity, such as reliance on precious metals, alkaline additives, and hydrogen peroxide with potential risks, Yu Long's team proposed a greener and more economical solution in 2022 [46]. They developed a cerium-doped manganese oxide catalyst (e.g., Mn0.9Ce0.1Oy-350) via a co-precipitation method. This catalyst enabled the green synthesis of APXO using atmospheric oxygen as the oxidant in an air environment, with solvents such as 1,4-dioxane and without the need for additional additives (Scheme 24).
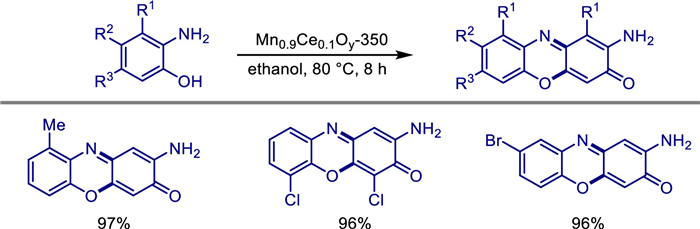
Characterization revealed that the catalyst consists of irregular nanoparticles with uniform distribution of cerium, manganese, and oxygen. Manganese exists as Mn2+, Mn3+, and Mn4+, while cerium includes Ce3+ and Ce4+. The catalyst exhibits a high specific surface area, and cerium doping significantly enhances its oxidative capacity, oxygen activation ability, and alkalinity. The abundant alkaline sites on the surface promote the Michael addition and oxidative dehydrogenation steps. Under optimized conditions (80 ℃, 1000 rpm, 8-h reaction), the conversion rate of 2-aminophenol reached 98%, with APXO selectivity at 99% and a yield as high as 97%. The catalyst also demonstrated high applicability for 2-aminophenol derivatives containing electron-donating or electron-withdrawing groups. After five cycles of reuse, the GC yield and isolated yield remained at 92% and 88%, respectively. ICP analysis indicated extremely low metal leaching (Mn < 0.4 ppm, Ce < 0.1 ppm), highlighting its excellent stability.
Compared to other catalysts reported in the literature, this system uses air as the oxidant and requires no additives, resulting in a lower E-factor and process mass intensity (PMI), making it more environmentally sustainable. Mechanistic studies showed that surface oxygen on the catalyst oxidizes 2-aminophenol to an o-quinone imine intermediate, which undergoes intermolecular Michael addition, oxidative dehydrogenation, and cyclization to form APXO (Scheme 25). Notably, even under a nitrogen atmosphere, cerium doping enhances the catalytic oxidation capability.
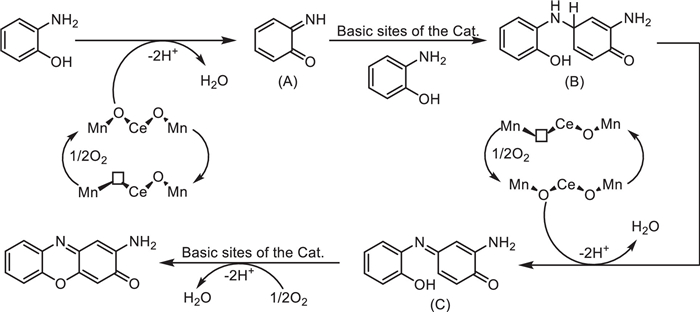
Additionally, the synthesized 2-amino-1,9-dimethyl-3H-phenoxazin-3-one exhibited excellent fluorescence properties in low-polarity solvents, making it suitable as a fluorescent molecular probe for detection and labeling studies. This research provides an environmentally friendly and cost-effective strategy for the industrial synthesis of APXO, expands the application of cerium-doped manganese oxide catalysts in oxidative coupling reactions, and offers new directions for the development of fluorescent molecular probes.
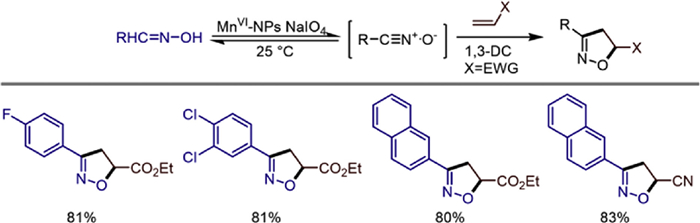
The high-valent species of manganese have unique advantages in selective oxidation. In the same year, Saikat Khamarui studied the versatility and efficiency of MnⅥ-NPs as catalysts in the synthesis of nitrile oxides from aldoximes and subsequent 1,3-dipolar cycloaddition reactions [47]. The author's team prepared the MnⅥ-NPs catalyst by thermally decomposing manganese oxalate and combining a surfactant self-assembly strategy. The highly active manganese species on its surface can mildly oxidize aldoximes to form nitrile oxides, avoiding the use of acids/bases or excessive metal oxidants in traditional methods and effectively suppressing the dimerization side reaction. Under optimized conditions (using CH2Cl2 as the solvent, with 5 mol% catalyst, NaIO4 as the co-oxidant, at room temperature), this catalytic system achieved stereoselective cycloaddition reactions with alkenes or alkynes, with a yield of up to 85%. It also has wide compatibility with functional groups such as electron-withdrawing groups (-Cl, -F), electron-donating groups (-CH3, -OCH3), ester groups, and cyano groups. The innovation lies in the first application of MnⅥ-NPs in the transformation of glycosyl aldoximes, successfully achieving the synthesis of glycosyl isoxazolines with 100% enantioselectivity and breaking through the limitation of glycosidic bond destruction by traditional HCl or strong oxidants. In addition, this catalyst can be recycled and reused. This work provides a green, efficient, and stereocontrolled new strategy for the synthesis of bioactive drug molecules and glycosyl functional materials, especially showing potential application value in the synthesis of bioactive molecules for antidepressant and anticancer purposes (Scheme 26).
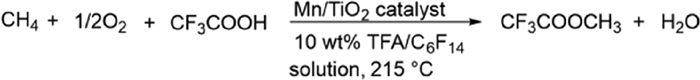
Compared with direct methane conversion without product protection, the strategy of protecting methanol through in-situ esterification during methane conversion can achieve higher yields, but most of these high-yield methods operate under harsh conditions. At present, research on developing heterogeneous catalysts for the conversion of methane to methyl esters is extremely limited, and studies on the activity of manganese in methane conversion are also scarce. In 2023, Jeroen A. van Bokhoven and his team developed a heterogeneous manganese-based catalyst for the aerobic conversion of methane to methyl trifluoroacetate (MTFA) under mild conditions (10 wt% TFA/C6F14 solution, 215 ℃) (Scheme 27) [48]. Manganese catalysts supported on SiO2, TiO2, and ZrO2 were prepared by the wet impregnation method. It was found that although the SiO2 support achieved high activity (up to 1068 μmol g-1sat h-1) and manganese dispersion, there was severe manganese leaching and homogeneous catalytic contribution. In contrast, TiO2 and ZrO2-supported catalysts (such as 10 wt% Mn/TiO2) achieved an activity of 613 μmol g-1sat h-1 while maintaining heterogeneous characteristics (no leaching, verified by hot filtration). Characterization confirmed that manganese on TiO2 mainly existed in the form of low-dispersion Mn3O4 phase, and its deactivation was due to the formation of inactive MnF2 phase during the reaction. Although the manganese utilization rate was low at high loadings, the manganese-based turnover frequency (TOF) reached 1.23 mol mol−1Mn h−1 at low loadings (< 1 wt%). This study first demonstrated the potential of simply synthesized supported manganese catalysts for heterogeneous conversion of methane in diluted acids, providing new ideas for the development of economical and efficient methane-methyl ester conversion systems.
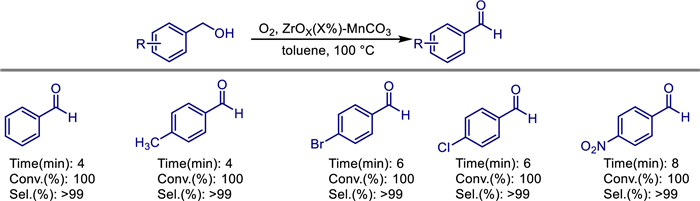
In 2017, Muhammad Nawaz Tahir and colleagues proposed an efficient green process for the selective oxidation of benzyl alcohol derivatives to the corresponding aldehydes [49]. They utilized a ZnOx-doped MnCO3 catalyst supported on highly reduced graphene oxide (HRG), using molecular O2 as an environmentally friendly oxidant under base free conditions (Scheme 28). The research team synthesized ZrOx(1%)–MnCO3/(X%HRG) nanocatalysts with different HRG ratios (0-7%) through the coprecipitation method, and systematically investigated the effects of parameters such as the doping amount of HRG, calcination temperature (300 and 500 ℃), catalyst dosage, and reaction temperature on the catalytic performance. Experiments showed that the catalyst ZrOx(1%)–MnCO3/(1%HRG) containing 1% HRG and calcined at 300 ℃ exhibited the best performance. It could achieve a 100% conversion rate of benzyl alcohol and a selectivity for benzaldehyde of > 99% within only 4 min. Its specific activity was as high as 60.0 mmol g-1 h-1, significantly superior to most of the reported graphene-based catalysts. The introduction of HRG not only enhanced the dispersibility of the active components and the charge transfer efficiency through its high specific surface area (211.03 m2/g) and defect sites, but also promoted the redox cycle of manganese species, thereby improving the catalytic activity. This catalyst showed excellent universality for a variety of substituted benzyl alcohols (including derivatives with electron-donating groups, electron-withdrawing groups, and steric hindrance groups). Although the oxidation of fatty alcohols (such as citronellol) required a longer time, complete conversion could still be achieved.
Palladium nanoparticles are effective catalysts for the selective oxidation of benzyl alcohol, a fundamental reaction in fine chemical production. In 2020, Rong Chen and colleagues reported an atomic-level MnOx-modified palladium nanoparticle catalyst (MnOx/Pd/Al2O3) prepared via atomic layer deposition (ALD) for the selective oxidation of benzyl alcohol to benzaldehyde [50]. By precisely controlling the deposition of MnOx on the Pd surface, this catalyst achieved selective passivation of the Pd(111) crystal plane, completely inhibiting the decarbonylation side reaction (eliminating toluene by-product). Meanwhile, the strong interaction between MnOx and Pd promoted electron transfer, increased the concentration of metallic Pd0 species (XPS confirmed Pd0 accounted for 51%), and enhanced interfacial oxygen migration capacity (O2-TPD showed desorption peaks shifted to lower temperatures). Catalytic performance was significantly improved: at 120 ℃ under solvent-free conditions, benzyl alcohol conversion reached 84.7%, benzaldehyde selectivity was 90.3%, and yield was 76.5%; the TOF value was as high as 31,561 h-1, 8.7 times that of the unmodified Pd/Al2O3. This work highlights the advantages of ALD technology in the directional regulation of active sites and interfacial structures, providing a new strategy for the design of efficient catalysts.
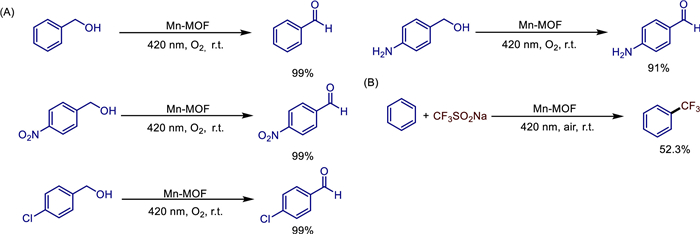
The multifunctional metal-organic frameworks (MOFs) have garnered widespread attention, although they also present certain challenges. In 2022, Shixiong Li and colleagues successfully synthesized and designed a three-dimensional Mn-MOF ([Mn4(L)4(DMF)3]n, L = 4,4′-(anthracene-9,10-diyl)dibenzoic acid) [51]. They introduced anthracene groups with π-conjugated structures into the MOF framework through ligand design, endowing the material with remarkable photo response properties (absorption edge at 403 nm, bandgap of 2.82 eV) and high porosity (specific surface area of 58.55 m2/g). Structural analysis shows that Mn(Ⅱ) ions are bridged by ligands to form a one-dimensional chain structure, which is further expanded into a three-dimensional framework with rhombic channels, providing an ideal environment for the mass transfer of reactants and the exposure of active sites. Under the drive of visible light (420 nm), this material exhibits dual catalytic functions: Firstly, it can efficiently catalyze the aerobic oxidation of benzyl alcohol and its derivatives by generating superoxide radicals (O2⁻), achieving a 100% conversion rate and an aldehyde selectivity of > 99% within 6 h, and it is universal for substrates containing electron-donating/electron-withdrawing groups. Secondly, it can utilize trifluoromethyl radicals (CF3·) to mediate the direct trifluoromethylation of the C—H bonds in arenes, with a yield of 52.3% within 12 h in DMSO solvent and it can tolerate a variety of substituents (Scheme 29).
In 2022, Michikazu Hara and his team reported a novel method for synthesizing high specific surface area β-MnO2 nanoparticles (β-MnO2-4) by using inexpensive and readily available KMnO4 instead of NaMnO4 [52]. This was achieved through the crystallization of a layered manganese oxide precursor, resulting in a specific surface area of 124 m2/g. Under mild conditions (50–80 ℃, with O2 as the sole oxidant), this material efficiently and selectively catalyzes the aerobic oxidation of aromatic, heteroaromatic, and allylic alcohols to the corresponding carbonyl compounds (aldehydes/ketones), with conversion rates reaching 84%–99% (Scheme 30). The catalyst can be recycled three times without significant deactivation.
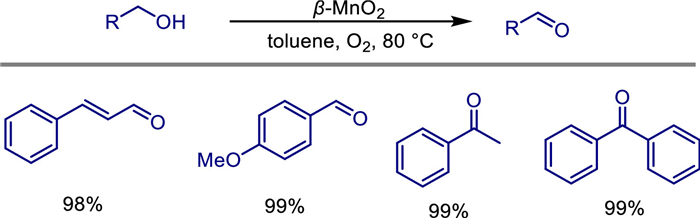
Additionally, β-MnO2 enables one-pot tandem oxidation reactions of alcohols with ammonia: in toluene solvent, it selectively synthesizes nitriles (e.g., benzyl alcohol to benzonitrile with a 95% yield), while in 1,4-dioxane, it produces amides. Compared to α-MnO2-based OMS-2 catalysts, β-MnO2 exhibits superior selectivity for nitrile synthesis due to its higher oxidation activity and lower nitrile hydration activity. Mechanistic studies indicate that alcohol oxidation follows the Mars-van Krevelen mechanism (involving lattice oxygen), while the difference in nitrile/amide selectivity stems from the disparity in acid sites (OMS-2 contains Brønsted acid sites, whereas β-MnO2 only has Lewis acid sites) and crystal structures (β-MnO2 features planar μ3-oxygen, while OMS-2 has both planar and bent μ3-oxygen), affecting their substrate activation capabilities.
In 2023, Behrooz Maleki's team studied the synthesis of a crown ether-containing Schiff base manganese complex and immobilized it on nanocellulose (MnL2@nanocellulose) [53]. The research team synthesized a Schiff base ligand containing a crown ether (L2) and a ligand without a crown ether (L1) through ligand design. These ligands were respectively complexed with manganese and immobilized on a nanocellulose carrier, and the catalytic performances of the two were systematically compared. Experiments showed that MnL2@nanocellulose containing a crown ether could achieve an aldehyde yield of 95% for benzyl alcohol within 30 min under mild conditions (60 ℃, 0.5 mmol Oxone), which was significantly superior to MnL1@nanocellulose without a crown ether (requiring 80 ℃ and 1 mmol Oxone). The crown ether promotes the contact between HSO5+ and the manganese active center by chelating K+ in Oxone, accelerating the oxidation cycle from Mn(Ⅲ) to Mn(Ⅴ), thus improving the catalytic efficiency. This catalyst shows high universality for benzyl alcohol derivatives containing electron-donating groups (such as methoxy groups) and electron-withdrawing groups (such as nitro groups), and its activity only decreases by 9% after being recycled 5 times (Scheme 31).

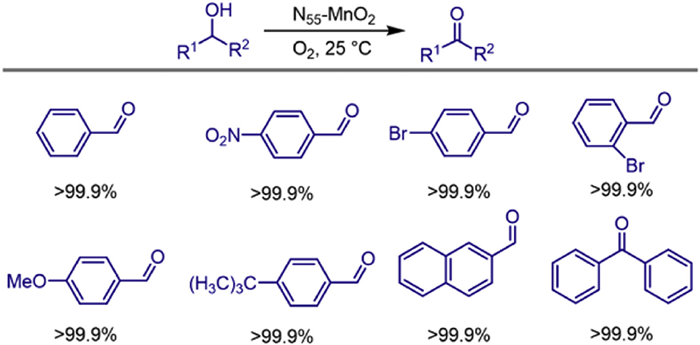
The development of innovative catalysts capable of efficiently activating oxygen (O2) into singlet oxygen (1O2) represents a frontier field with the potential to revolutionize green chemical synthesis. Despite its great promise, practical implementation still faces significant challenges. In 2023, Yusuke Yamauchi and colleagues designed a series of nitrogen (N)-doped manganese dioxide (Ny-MnO2, where y denotes the molar amount of N precursor used) nanocatalysts via partitioned microemulsion crystallization combined with post-calcination [54]. These nanocatalysts exhibited the remarkable ability to directly generate 1O2 at room temperature without the need for external field assistance. Through strategic introduction of defect engineering and interstitial N, the concentration of surface oxygen atoms (Os) near oxygen vacancies (Ov) in the N55-MnO2 nanocatalyst reached 51.1%. Density functional theory (DFT) calculations showed that the increased Ov content and N doping reduced the O2 adsorption energy from -1.24 eV (ε-MnO2) to -2.18 eV, thereby driving the excitation of O2 into 1O2. In the aerobic oxidation of alcohols at room temperature, N55-MnO2 achieved a benzaldehyde yield of > 99.9%, with a turnover frequency (TOF) of 0.14 molcat-1 h-1, representing a 6.7-fold improvement over undoped ε-MnO2, and maintained > 94% activity after 10 cycles. This study introduces a method for the spontaneous activation of oxygen for the green synthesis of fine chemicals (Scheme 32).
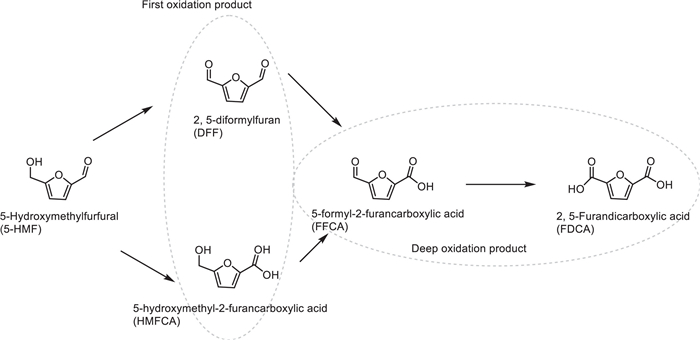
In 2022, the team led by Yongdan Li developed a heterogeneous catalyst of manganese oxide (MnOx) supported on hydroxyapatite (HAP) (MnOx/HAP) for the selective oxidation of the biomass platform molecule 5-hydroxymethylfurfural (HMF) to the high-value chemical 2,5-diformylfuran (DFF) using oxygen as the oxidant under alkali-free conditions [55]. The Ca/P molar ratio of the HAP support was regulated by the precipitation method (pH = 7.5–11.0), and the catalyst with a manganese loading of 10 wt% was prepared by the deposition method. Characterizations showed that when pH = 10.0, Ca/P = 1.65 (close to the theoretical value of HAP, 1.67), and the MnOx/HAP-10.0-400 catalyst calcined at 400 ℃ had the highest Mn4+ content (34%) and lattice oxygen ratio (32%), and exhibited a mesoporous structure (specific surface area 76 m2/g). Under the conditions of toluene solvent, 120 ℃, 1.0 MPa O2, and reaction for 12 h, the catalyst achieved 86.4% HMF conversion and 90.9% DFF selectivity (Scheme 33). Performance optimization showed that the Ca/P ratio affected the distribution of acid-base sites on the catalyst, and the basic sites at pH = 10.0 were most suitable for activating the O—H bond of HMF; calcination at 400 ℃ could stabilize high-valent manganese species (Mn4+/Mn3+), whose redox cycle directly participated in the oxidation reaction through lattice oxygen (the lattice oxygen content decreased from 32% to 17% after the reaction), and oxygen vacancies were supplemented by gaseous O2 (Scheme 34). Solvent screening confirmed that toluene could inhibit side reactions, while high temperature (> 120 ℃) led to ring-opening decomposition of DFF. The catalyst showed no significant activity decline after 4 cycles (HMF conversion 84.9%, DFF selectivity 86.4%), demonstrating its environmental friendliness and reusability, and providing a new strategy for the green synthesis of biomass-derived chemicals.
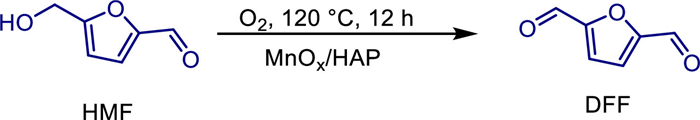
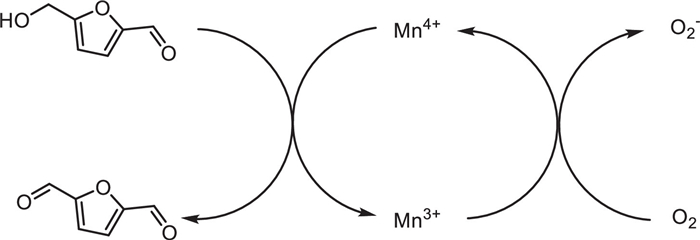
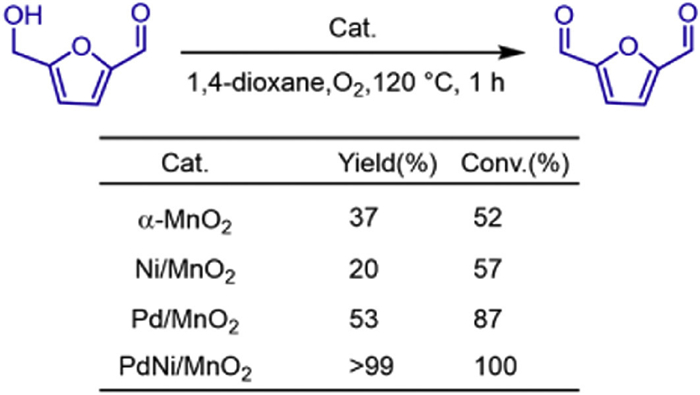
In 2022, Xiaopeng Li and his colleagues reported a synergistic bimetallic single-atom (Pd and Ni) promotion method, which regulates the catalytic activity and selectivity of α-MnO2 for the oxidation of HMF (Scheme 35). Pd and Ni have different spatial doping distributions and catalytic functions [56]. Pd single atoms are embedded in the lattice of α-MnO2, activating the lattice oxygen and generating abundant oxygen vacancies. Ni single atoms, mainly anchored on the surface of α-MnO2, improve the ability to decompose molecular oxygen to repair the oxygen vacancies. Pd and Ni work synergistically to accelerate the consumption and recovery of active lattice oxygen during the oxidation of HMF, increasing the production rate by three times. Compared with traditional single-atom catalysts that require a metal loading of 1-10 wt%, single-atom cocatalysts with a tiny metal loading of less than 0.5 wt% can significantly enhance the activity of α-MnO2 (Scheme 36).
In 2017, Paul T. Anastas and colleagues developed a novel catalyst, EcoMnOx, using waste biomass from hyperaccumulator plants rich in manganese [57]. This valorization of manganese-rich biomass provides an alternative to its costly conventional disposal and offers a new source of manganese. The extracted metal ions, including Mn2−, were oxidized under mild conditions using H2O2 /NaOH to yield EcoMnOx, a polymetallic oxide material containing 8.9-14.1 wt% Mn. Spectroscopic studies indicated the presence of layered mixed manganese oxides in the material, rich in Mn(Ⅳ) as well as Mn(Ⅲ) and Mn(Ⅱ) species.
The catalytic performance of EcoMnOx was evaluated in the aerobic oxidative cleavage of 1,2-diols under atmospheric pressure of O2 or air. At a Mn concentration of only 10 mol%, the catalyst achieved high conversion rates for active benzylic and allylic diols, with excellent selectivity for aldehydes or ketones (98%-99%). Additionally, due to its heterogeneous nature, the catalyst could be easily removed by filtration and reused at least six times without any loss of activity. Finally, E-factor analysis revealed that EcoMnOx generated 4 to 17 times less waste compared to traditional reagents such as NaIO4 and Pb(OAc)4, highlighting the sustainable attributes of this novel heterogeneous catalyst (Scheme 37).
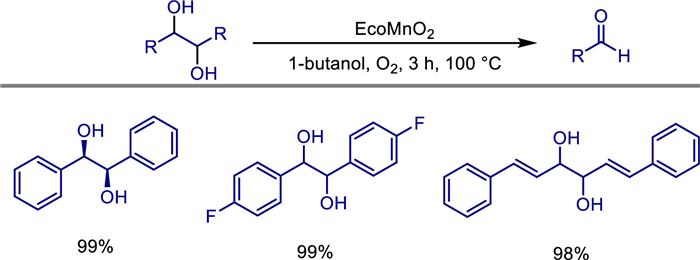
In the same year, Paul T. Anastas and colleagues first reported the use of a heterogeneous catalyst based on the rare-earth metals manganese and sodium for the aerobic oxidative cleavage of 1,2-diols [58]. They prepared the Na-Mn layered mixed oxide (LMO) catalyst by oxidizing MnCl2·4H2O with H2O2 under alkaline conditions. Its structural characterization showed that it was a layered calcium-manganese ore-type oxide (Na0.27MnO2·6H2O), with an average oxidation state of manganese being +3.7, and sodium ions intercalated between the layers. This catalyst efficiently catalyzes the oxidative cleavage of activated 1,2-diols such as benzyl, allyl, and heterocyclic ones under mild conditions (atmospheric pressure O2 or air, 100 ℃, 1-butanol as the solvent). The conversion rate reaches 92%-99%, the selectivity for aldehydes/ketones is > 99%, and only water is generated as a byproduct. Through kinetic experiments and mechanism studies, a monodentate two-electron oxidative cleavage mechanism was proposed, which involves the Mn4+/Mn3+ redox cycle, and oxygen participates in the reoxidation step of manganese. The heterogeneous nature of the catalyst supports simple filtration for recovery, and there is no loss of activity after at least six cycles, and its surface reaction mechanism was verified through filtration experiments. For low-activity substrates (such as unsubstituted diols), adding Na2CO3 can improve the efficiency. This catalyst shows good compatibility in domino one-pot reactions and has been successfully applied to complex synthesis. Compared with traditional periodic acid and lead reagents, this method abandons toxic reagents and solvents, uses cheap metals and green oxidants, providing an environmentally friendly and scalable new strategy for the oxidative cleavage of 1,2-diols (Scheme 38).
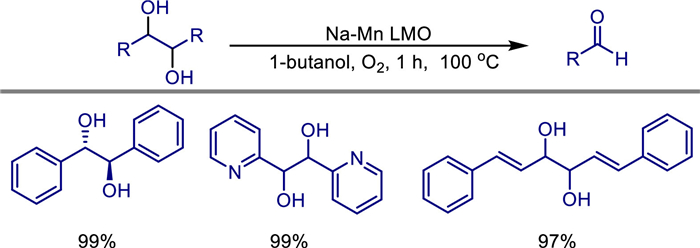
Medium-chain furan compounds have significant application potential, particularly in pharmaceuticals and polymers. In 2022, Yao Fu and colleagues described an environmentally friendly and efficient heterogeneous sodium-doped porous manganese oxide catalyst (Na-MnOx) for the oxidative cleavage of furan 1,2-diols into medium-chain furan aldehyde compounds. They prepared the Na-MnOx catalyst through the coprecipitation method [59]. Characterization shows that it has a layered calcium-manganese ore structure, and manganese exists in a mixed valence state of Mn2−, Mn3+, and Mn4+, among which the proportion of Mn4+ is directly related to the catalytic activity (optimal when the concentration of NaOH is 1.8 mol/L). This catalyst efficiently catalyzes the oxidative cleavage of biomass substrates such as (E)-5-(furan-2-yl)pent-4-ene-2,3-diol (FPED) under mild conditions (atmospheric pressure O2, 1-butanol as the solvent, 90 ℃). The yield of furan-2-acrolein reaches 91%, and it has been successfully extended to 5-hydroxymethylfurfural (HMF) and dialdehyde furan (DFF) derivatives to synthesize a variety of medium-chain furan aldehyde compounds. By adding the weak base Na2CO3, the efficiency of low-activity substrates can be improved, and the aldehyde products can be further converted into high-value-added chemicals such as diacids, esters, and alcohols, demonstrating its potential in the fields of medicine and biodegradable materials (Scheme 39). Mechanistic studies show that the reaction follows a two-electron oxidative cleavage pathway, and the Mn4+-O2−; acid-base pair drives the cleavage of the substrate, and the regeneration of Mn2+; by oxygen is the key rate-determining step. The catalyst can be recycled five times while maintaining 87% of its activity, and it has both stability and heterogeneous recyclability.
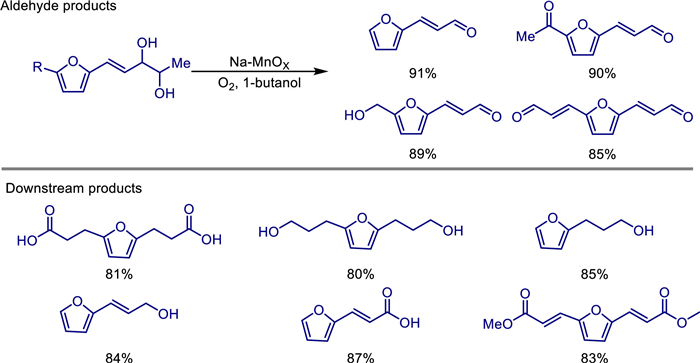
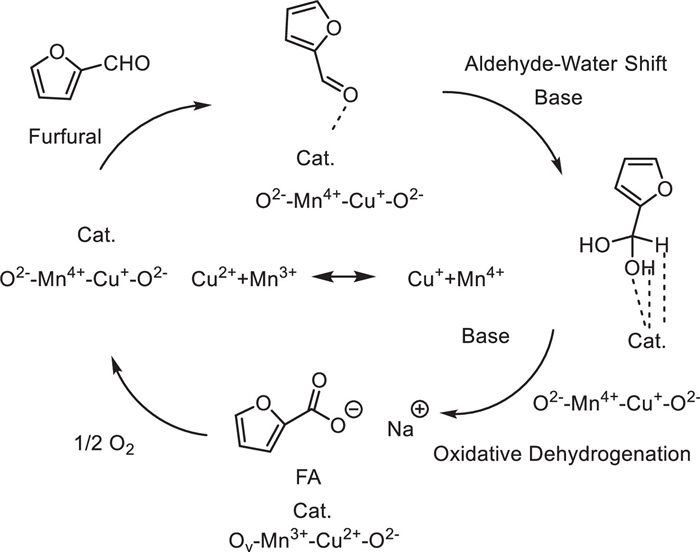
In 2023, Lu Lin and his team reported an efficient, sustainable, and noble-metal-free Mn-Cu bimetallic oxide catalytic system (prepared by hydrothermal-redox method) for the selective oxidation of furfural to furoic acid (FA) [60]. It was found that among catalysts with different Mn/Cu molar ratios, Mn2Cu1Ox exhibited the most excellent catalytic performance: under the conditions of 120 ℃, 1 MPa O2 pressure, and the addition of 2 equiv. of NaOH, a 99% conversion of furfural and quantitative FA selectivity (i.e., selectivity approaching 100%) could be achieved after a 3-h reaction. The catalyst still maintained an FA yield of more than 90% after 5 cycles of use, showing good stability. Characterization analyses (XRD, SEM, TEM, BET, XPS, Raman, O2-TPD, H2-TPR, O2-TPO, CO2-TPD, FTIR) showed that the introduction of CuO formed a Cu1.5Mn1.5O4 spinel structure, which significantly weakened the strength of the Mn-O bond and increased the proportion of lattice oxygen (O1), reaction activity (O1 mobility and oxygen coordination ability), and reducibility on the catalyst surface. This is the key reason why its catalytic activity is significantly higher than that of single MnO2 or CuO. In addition, the strong surface basic sites of Mn2Cu1Ox promote furfural to bind in a way of vertical adsorption of the aldehyde group (with the furan ring away from the catalyst surface), which is conducive to the high-selectivity generation of FA and inhibits side reactions. The reaction follows the Mars-van Krevelen mechanism, involving the direct participation of lattice oxygen in oxidation and the redox cycle of Cu−/Mn4+ ↔ Cu2−/Mn3+. This study provides a scientific basis and practical strategy for the development of low-cost, efficient, and environmentally friendly aerobic oxidation catalysts for biomass-derived molecules (Scheme 40).
In 2018, Steven L. Suib and colleagues reported a one-step peroxide-mediated heterogeneous catalytic oxidation of amides to imines using a series of manganese oxides [61]. The author team used N-benzylbenzamide as a model substrate to screen various manganese oxide catalysts, among which cesium-promoted mesoporous manganese oxide (meso Cs/Mn2O3) and amorphous manganese oxide (AMO) showed the best performance. By optimizing reaction conditions (such as using tert-butyl hydroperoxide (TBHP) as the oxidant, N-hydroxyphthalimide (NHPI) as the promoter, acetonitrile as the solvent, and combining with molecular sieves for water removal), efficient selective partial oxidation was achieved, with the conversion of the target product diphenyl imide reaching up to 90% and the selectivity reaching 95%. This method can be extended to various aromatic, heterocyclic, and lactam substrates, showing good yields for amides with substituents such as chlorine and methoxy groups on the benzene ring, as well as thiophene amides, but it is ineffective for aliphatic amides and sterically hindered ortho-chlorinated substrates (Scheme 41).
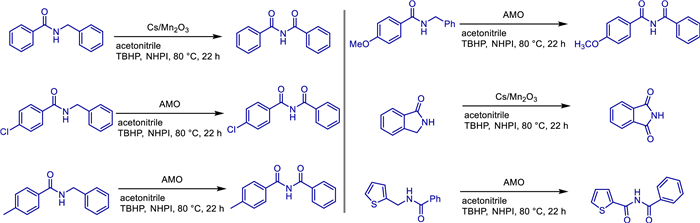
The team compared different manganese oxide catalysts (such as K-OMS-2, commercial MnO2) and found that the cesium-ion-promoted mesoporous manganese oxide significantly enhanced catalytic activity due to increased surface basicity and lattice oxygen activation; amorphous manganese oxide (AMO) exhibited excellent performance due to its high specific surface area and abundant active sites. Kinetic studies showed that the reaction followed first-order kinetics with respect to the amide, and NHPI accelerated the cleavage of the α-C—H bond by generating PINO radicals; Hammett analysis indicated that the transition state carried a partial negative charge, supporting a reaction mechanism involving polar effects. Catalyst stability tests showed that meso Cs/Mn2O3 had no obvious loss of activity after 3 cycles, and X-ray diffraction confirmed that its amorphous structure remained stable. The heterogeneous nature of the catalyst supports simple filtration and recovery, and combined with its tolerance to various functional groups, it provides an efficient and sustainable new strategy for the functional group transformation from amides to imides, showing potential applications in pharmaceutical synthesis, polymer materials, and other fields.
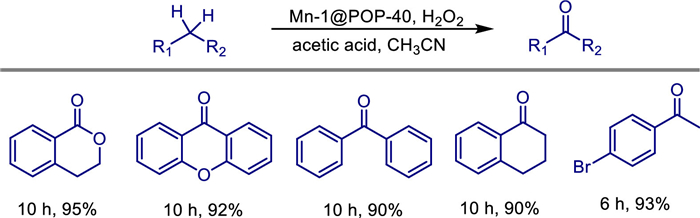
In recent decades, bio-inspired manganese complexes have emerged as attractive catalysts for various selective oxidation reactions. However, these catalysts still suffer from oxidative degradation. In 2021, Wei Sun and colleagues reported the preparation of porous organic polymer (POP)-supported single-site manganese catalysts (such as Mn-1@POP-40) by copolymerizing vinyl-functionalized Mn-N4 complexes (mimicking the active centers of non-heme enzymes) with divinylbenzene [62]. The catalyst features a hierarchical pore structure (BET specific surface area up to 750 m2/g) and atomically dispersed manganese active sites (confirmed by HAADF-STEM and EDS mapping), effectively suppressing the oxidative degradation common in homogeneous catalysts.
In aliphatic C—H oxidation reactions (using cyclohexane as a model substrate, H2O2 as the oxidant, and acetic acid as an additive), Mn-1@POP-40 achieved a ketone yield of 82%, outperforming the homogeneous Mn-1 catalyst (74%). Prolonging the reaction time significantly increased the yield of sterically hindered substrates (such as dibenzofuran) to 92% (no improvement in the homogeneous system). The catalyst can be recycled 5 times without significant loss of activity or selectivity. This design achieves efficient, stable, and recyclable applications of single-site manganese catalysts by mimicking the confined environment of enzymes (Scheme 42).
Under mild conditions, heterogeneous catalytic oxidation for C—H functionalization represents a direct chemical approach to obtain target oxygenated products. In 2022, Keigo Kamata and Michikazu Hara reported the successful synthesis of nanoscale Murdochite-type oxide Mg6MnO8 (Mg6MnO8-MA) with a high specific surface area (104 m2/g) via a malic acid-assisted sol-gel method [63]. Its specific surface area is approximately 7 times higher than that of samples synthesized by traditional methods (2–15 m2/g).
This material efficiently catalyzes the C—H oxidation of alkylarenes (e.g., fluorene) with molecular oxygen (O2) as the sole oxidant under mild conditions (40 ℃, 1 atm O2) without additives, outperforming other Mn-based oxides with Mn-O-Mn active sites (e.g., SrMnO3 and β-MnO2) and Mg-based oxides. The catalyst can be recovered by simple filtration and reused at least 5 times without significant activity loss. It shows broad substrate applicability, including mono/multi-substituted fluorenes, heteroatom-containing arenes (e.g., xanthene, thianthrene), and non-fused ring compounds (e.g., diphenylmethane, benzylpyridine), with high yields (32%–99%) of target oxygenated products for various substrates (Scheme 43).
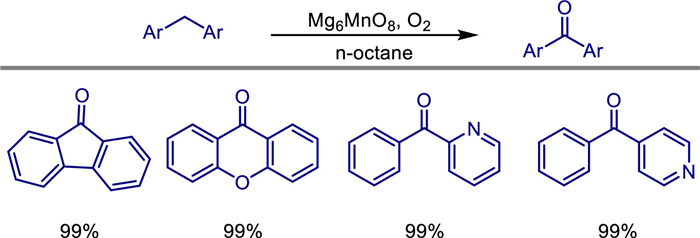
Mechanistic studies indicate that the reaction follows a basicity-controlled hydrogen atom transfer mechanism (reaction rate shows good linear correlation with substrate pKa, with small kinetic isotope effects, KIE = 1.2–2.0). 18O labeling experiments, kinetic analysis, and inhibition tests confirm that the reaction involves an O2 activation pathway (73%–85% of oxygen in products originates from gaseous O2), rather than the Mars-van Krevelen mechanism. The synergistic effect of isolated Mn4+ species in the MgO matrix and basic sites on the material surface is crucial for high activity (CO2-TPD shows significantly more basic sites than reference catalysts, and CHCl3 adsorption IR confirms its basicity is similar to MgO). This work first achieves the synthesis of high-surface-area Mg6MnO8 nanoparticles and their application in liquid-phase selective oxidation with O2 as the sole oxidant, providing a new strategy for developing efficient heterogeneous oxidation catalysts.
From an environmental and economic perspective, strategies for converting toxic petroleum ethylbenzene into high-value-added products through hydrogen atom transfer processes, along with the design of related catalysts, have recently regained attention.
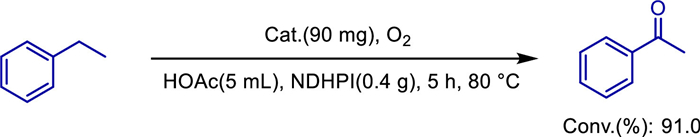
In 2022, Ali Reza Faraji and colleagues successfully synthesized a novel manganese-based dendritic catalyst supported on a polymeric magnetic core-shell [64]. This supported dendritic macromolecule-coated manganese nanoparticle catalyst offers significant advantages: combined with N-hydroxyphthalimide derivatives (NDHPI), it enables efficient and selective oxidation of ethylbenzene under mild conditions (80 ℃, using O2 as the oxidant), achieving a 91% conversion of ethylbenzene and 98.3% selectivity for acetophenone within 5 h. The optimized conditions are: 90 mg catalyst, 0.4 g NDHPI, and 5 mL acetic acid as the solvent (Scheme 44). Meanwhile, the catalyst efficiently oxidizes oxime derivatives to the corresponding carbonyl compounds (e.g., > 99% conversion of acetophenone oxime with 100% selectivity for acetophenone), with the optimal conditions being: 100 mg catalyst, 5 mmol benzaldehyde, toluene as the solvent, and 50 ℃ (Scheme 45). Additionally, the study deeply explores the influence of various operational parameters on the catalytic efficiency of the synthesized dendritic catalyst, including catalyst dosage, solvent properties, temperature, reaction time, and the roles of several N-hydroxyimides. The results indicate that the magnetic dendritic structure, high Mn(Ⅱ) content, hydrophobic arms of the dendritic macromolecules, and the interaction between Mn and the dendritic framework collectively contribute to the improved catalytic efficiency of the catalyst.
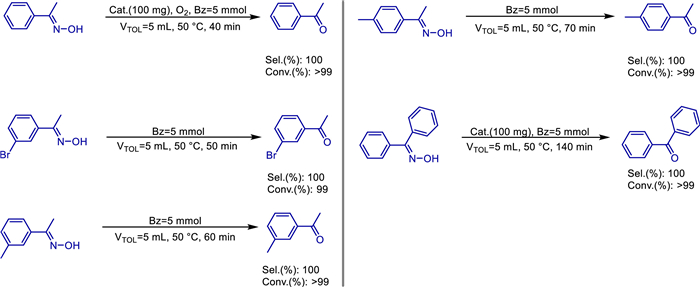
In the realm of organic chemical synthesis, achieving the conversion of ethylbenzene to acetophenone under solvent-free conditions with high conversion and selectivity is of great significance for both academic research and industrial applications. In 2019, the research team led by Hong Jiang delved into relevant research topics. Through pyrolysis and calcination methods, they successfully developed a unique catalyst composed of nitrogen-doped carbon and manganese (Mn) supported on mesoporous alumina [65]. Research data indicates that in the solvent-free oxidation reaction system, where molecular oxygen serves as the oxidant and ethylbenzene is the reaction substrate, this newly developed catalyst exhibits extremely excellent performance. Its catalytic activity reaches 27.8%, and the selectivity for the target product acetophenone exceeds 99% (Scheme 46). Moreover, in eight consecutive cycles of use, no significant decrease in various performance indicators is observed, demonstrating good stability.
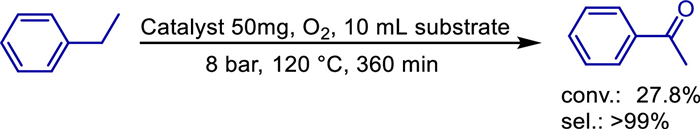
In the entire heterogeneous catalytic system, phenanthroline plays an indispensable dual-role. On one hand, as a heterogeneous chelating ligand, phenanthroline can bind tightly to manganese elements, dispersing manganese elements evenly in the catalyst, thereby enhancing the catalytic activity of manganese. On the other hand, phenanthroline also acts as a nitrogen source, providing 9.4% of doped pyridinic-N to the catalyst. These doped pyridinic-N act like a "booster" for the catalyst. They can not only promote the adsorption of molecular oxygen on the catalyst surface but also assist molecular oxygen in breaking bonds smoothly, strongly facilitating the subsequent oxidation reaction.
In-depth research reveals that the content of active manganese in the bulk phase of this catalyst is significantly higher than that on the surface. This characteristic is an important guarantee for the stability and reusability of the catalyst. During heterogeneous catalytic reaction processes, the active manganese in the bulk phase can continuously participate in the reaction, maintaining the activity of the catalyst. As a result, the catalyst can still maintain good performance after multiple cycles of use. At the same time, mesoporous alumina also plays an irreplaceable role in the entire catalytic system. It provides a stable support structure for the active components, preventing the loss or agglomeration of active ingredients during the reaction. Its rich pore structure also provides a large number of adsorption sites for reactant molecules, enabling reactant molecules to quickly come into contact with the active components, thus accelerating the reaction rate and significantly improving the catalytic efficiency.
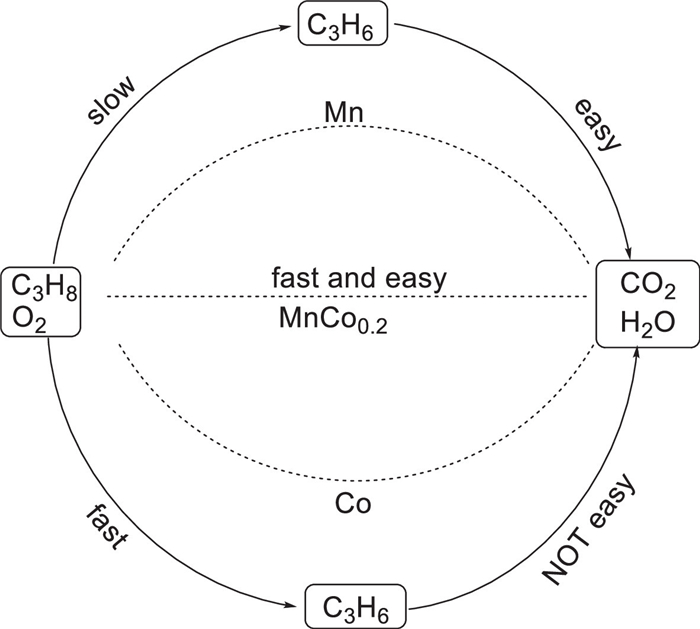
In 2019, Xiping Song and his team reported the synthesis of cobalt-doped manganese dioxide (MnO2) nanofiber catalysts and their application in the deep oxidation of propane [66]. The researchers adopted a host-guest strategy to introduce cobalt (Co) into the α-MnO2 framework via a one-step hydrothermal method, successfully constructing MnCox nanofibers with dual active sites (Mn sites and Co sites). Characterization results (XRD, TEM, SEM) showed that when the actual Co/Mn molar ratio was as high as 0.15, doping did not significantly change the tetragonal crystal structure and nanofiber morphology of α-MnO2, and the specific surface area (73.9–81.3 m2/g) was also similar to that of undoped MnO2 (77.6 m2/g). XPS and ICP-OES analyses indicated that at low doping levels (MnCo0.05), Co was mainly enriched on the surface, while at high doping levels (MnCo0.2, MnCo0.5), Co entered the framework and possibly partially replaced K− ions in the tunnels. Meanwhile, cobalt doping increased the average oxidation state of manganese and the proportion of surface adsorbed oxygen.
Catalytic performance tests showed that cobalt doping significantly reduced the ignition temperature (T50) for propane oxidation. The optimized sample MnCo0.2 (Co/Mn = 0.2) exhibited the best activity: At a space velocity of 90 L g-1 h-1, the temperatures corresponding to 50% (T50) and 90% (T90) propane conversion were 205 ℃ and 223 ℃, respectively. Comparative studies revealed that cobalt species mainly promoted the activation of propane at low temperatures (reducing T5n), while manganese species were more conducive to complete oxidation to CO2 (reducing T90). In-situ DRIFTS studies elucidated the reaction mechanism: Co sites dominated the direct dehydrogenation pathway of propane (C3H8 + 2O → C3H6 + 2OH), while Mn sites dominated the oxidative dehydrogenation pathway (C3H8 + O* → C3H6 + H2O). The synergistic effect of the two promoted C—H bond breaking and water generation, thus significantly improving the overall catalytic efficiency. This work confirms the synergistic effect of Mn-Co dual active sites and provides new ideas for the development of efficient catalysts for low-carbon alkane oxidation (Scheme 47).
The selective oxidation of C(sp2)-H bond of polycyclic aromatic hydrocarbons to corresponding ketones is an idea method to prepare 1,2-dicarbonyl compound. In 2024, Rafatosadat Badihi and her team synthesized manganese oxide octahedral molecular sieve (OMS-2) nanorods in the presence of nitric acid, followed by preparation through Ru3+ ion exchange [67]. Structural characteristics were confirmed using multiple techniques including XRD, FTIR, SEM, EDS, XPS, and EDS mapping, demonstrating successful and uniform modification of Ru3+ while maintaining the OMS-2 crystal framework (ionic radius ratio Ru3+/K+ = 0.49).
The catalyst efficiently promotes the selective oxidation of polycyclic aromatic hydrocarbons in aqueous solutions under mild conditions (30–45 ℃), using NaIO4 as the oxidant. Excellent yields of 55%–99% were achieved for few substrates, alongside high selectivity: polycyclic systems preferentially yielded ketones, small molecules like biphenyl mainly formed benzoic acid. This catalyst also could be used for the oxidation of sulfur-containing heterocyclic compounds (such as olanzapine and benzothiophene) to sulfoxides (e.g., olanzapine converted to olanzapine sulfoxide with 99% yield). This catalyst could be recycled 10 times with only a 7.5% activity decline, primarily attributed to partial pore blockage after cycling (BET specific surface area decreased from 54.48 m2/g to 25.20 m2/g). Mechanistic studies showed that Ru3+ was in-situ oxidized to RuO2 by NaIO4, with RuO2 serving as the active site to drive the oxidation (Scheme 48).
The oxidation and cleavage of unsaturated C—C bonds are key processes for producing carbonyl compounds from hydrocarbon feedstocks. So far, no method has been reported for the direct amidation of unsaturated hydrocarbons using molecular oxygen as a green oxidant for the oxidative cleavage of unsaturated C—C bonds. In 2023, Wen Dai and his team introduced a novel self-tandem catalytic strategy involving manganese oxide catalysis, enabling the direct formation of amides from unsaturated hydrocarbons through a combination of oxidative cleavage and amidation coupling [68]. The author's team screened out an amorphous MnOx catalyst, which exhibited excellent performance towards various mono- and multi-substituted alkenes and alkynes (including aromatic, aliphatic, and heterocyclic substrates) under the conditions of 130 ℃, 0.5 MPa O2, and NH3 (Scheme 49). It could efficiently convert substrates such as styrene into the corresponding amides (with a maximum yield of 88%), and by adjusting the reaction conditions, sterically hindered nitriles could be selectively synthesized (with yields ranging from 51% to 68%). Characterizations show that the high activity of MnOx stems from its large specific surface area (302 m2/g), abundant oxygen vacancies, and moderate acidic sites. Its amorphous structure and the Mn3+/Mn4+ redox couple promote the activation of oxygen species and the adsorption of substrates. Mechanistic studies, combined with GC-MS, in-situ DRIFTS, and DFT calculations, reveal that the reaction proceeds through different pathways: α-substituted alkenes generate ketones via a dioxetane intermediate, while β-substituted alkenes are transformed via an epoxide intermediate, and both ultimately form amides through ammoxidation, with superoxide radicals being the key reactive species. This system does not require noble metals and has substrate universality (supporting late-stage functionalization of complex molecules), gram-scale synthesis ability (with a yield of 86%), and catalyst recyclability (showing no significant loss of activity after 8 cycles). It provides an efficient and sustainable new route for the green transformation of unsaturated hydrocarbons, breaking through the dependence of traditional methods on high-pressure CO or noble metals and having application potential in the fields of drug synthesis and material intermediates.
In 2017, Mircea Dinca and co-workers reported the successful construction of mononuclear Mn2+ sites with weak ligand fields (MnⅡ-MOF-5) by partially substituting Zn2+ with Mn2+ in Zn4O-terephthalate (MOF-5), whose structure was confirmed by Mn K-edge X-ray absorption spectroscopy [69]. The Mn2+ sites can undergo redox reactions with various oxidants. Notably, when reacting with tBuSO2PhIO, they are proposed to generate highly active, high-spin MnⅣ–oxo intermediates, which can capture ambient hydrogen to form stable MnⅢ–hydroxyl species (MnⅢ(OH)-MOF-5).
In the presence of tBuSO2PhIO, the MnⅢ-MOF-5 precatalyst efficiently catalyzes the selective epoxidation of cyclic olefins (e.g., cyclopentene, norbornene) with selectivity > 99%. The catalytic activity originates from the high-valent MnⅣ–oxo intermediates formed due to the weak carboxylic acid ligand field, whose unique oxygen atom transfer (OAT) capability avoids common side reactions (e.g., C—H bond oxidation).
Characterizations such as X-ray absorption spectroscopy (XAS) and electron paramagnetic resonance (EPR) confirmed the local structure and valence state changes of Mn sites. The study also found that the catalytic activity is positively correlated with the ring strain of substrates (e.g., norbornene exhibits the highest activity), and the reaction primarily occurs within the pores of MOF-5 (Scheme 50). Although the catalyst gradually deactivates during continuous reactions, this work achieves highly selective oxidation in MOF weak oxygen ligand fields for the first time, providing new insights for mimicking enzymatic catalysis and designing novel selective oxidation catalysts.
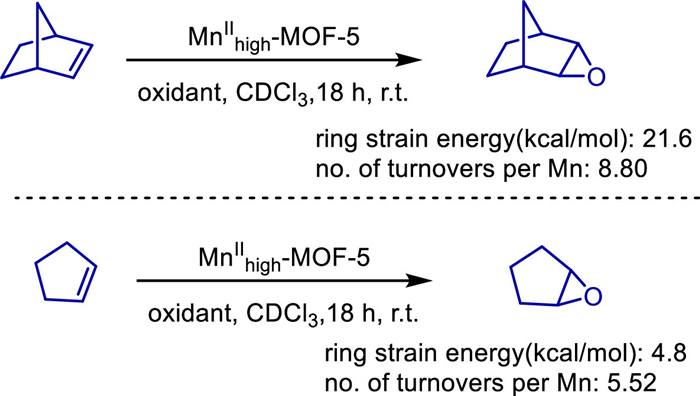
In 2021, Arumugam Pandurangan and colleagues reported the synthesis of an SBA-16 support with a three-dimensional cubic cage-like mesoporous structure via a hydrothermal method, and the preparation of MnWO4/SBA-16 catalysts by dispersing MnWO4 bifunctional metal oxides with different loadings (1 wt%, 3 wt%, 5 wt%) onto SBA-16 using the incipient wet impregnation method [70]. The catalyst was applied to the epoxidation of styrene with environmentally friendly tert-butyl hydroperoxide (TBHP) as the oxidant. Characterization results (XRD, BET, TEM, XPS, TGA) showed that MnWO4 was uniformly dispersed on the surface of SBA-16, forming strong metal-metal interactions (synergistic effects), and the catalyst exhibited a high specific surface area (277 m2/g for the optimal 3 wt% sample), ordered mesoporous structure, and good thermal stability.
Under optimized conditions (60 ℃, 24 h, acetonitrile solvent, 50 mg catalyst, styrene/TBHP molar ratio 1:1), 3 wt% MnWO4/SBA-16 demonstrated the best performance, achieving a styrene conversion of 90% and a styrene oxide selectivity of 75%. The performance advantages were attributed to the dual redox sites of MnWO4 and the 3D mesoporous structure of SBA-16, which facilitated reactant diffusion and efficient utilization of active sites. Additionally, the catalyst showed higher activity toward electron-rich olefins (such as styrene and trans-stilbene) and could be recycled four times without significant loss of activity (Scheme 51).
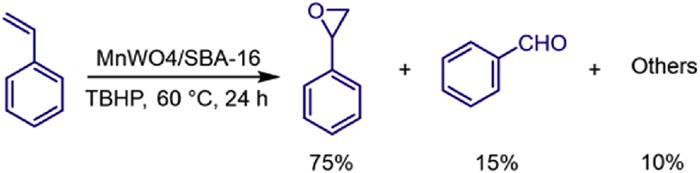
The study also explored the reaction mechanism, proposing that TBHP was activated at Mn2+ sites to form a peroxo-manganotungstate intermediate, which then nucleophilically attacked the C═C bond of the olefin to generate the epoxide. Compared with other catalysts reported in the literature, this material achieved high activity and selectivity at lower temperatures, catalyst dosages, and oxidant ratios (Scheme 52).
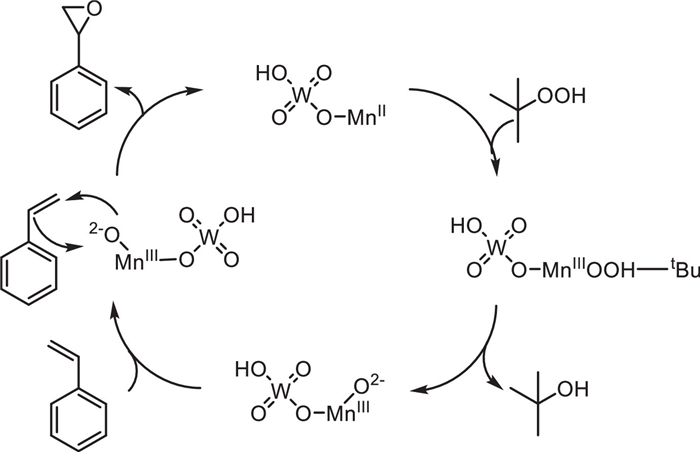
In 2023, Yuehui Li and his team successfully synthesized a highly dispersed manganese-supported carbon nitride catalyst (Mn/g-C3N4, with a Mn loading of 2.56 wt%) via an impregnation-calcination method, enabling bifunctional catalysis for alkene epoxidation and CO2 cycloaddition [71]. In the epoxidation of aromatic alkenes (using styrene as the model substrate), the catalyst, with a dosage of 0.65 mol%, at 100 ℃, and tert-butyl hydroperoxide (TBHP) as the oxidant, achieved a 91% conversion and 93% selectivity after 8 h of reaction. It also showed universality for monosubstituted styrene derivatives containing electron-withdrawing/donating groups, but exhibited lower activity towards aliphatic alkenes (Scheme 53).
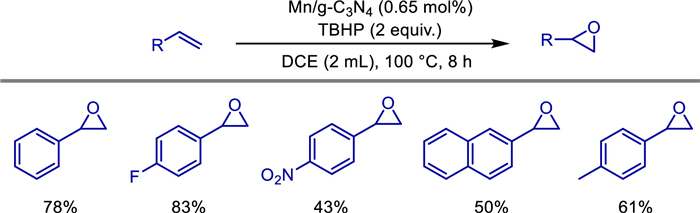
Characterization analyses (XRD, HRTEM, XPS) revealed that manganese is anchored to the pyrrolic nitrogen sites of g-C3N4 in an atomically dispersed form, forming a Mn-Nx structure. This structure generates Mn-O active oxygen species through the heterolysis of TBHP, realizing efficient oxygen atom transfer to alkenes. Furthermore, the catalyst can further catalyze the cycloaddition reaction of styrene oxide with CO2, achieving 88% conversion and 89% selectivity to cyclic carbonate in the presence of an ammonium salt co-catalyst (Scheme 54). Mechanistic studies indicated that its bifunctional property stems from the synergistic effect between the basic sites of the g-C3N4 support and the Lewis acidity of the Mn-Nx sites, which co-activate CO2 and epoxides.
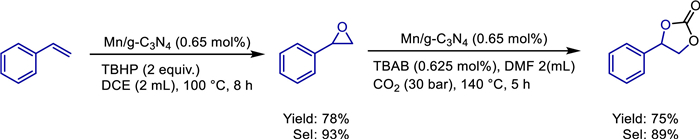
The catalyst showed no significant activity decline after four cycles (leached Mn < 0.3 ppm), and poisoning experiments (KSCN inhibiting activity) confirmed that the Mn-Nx sites are the active centers of the reaction. This work provides a new idea for designing efficient heterogeneous catalysts mimicking cytochrome P450 enzyme catalytic systems, and also establishes a green synthetic pathway from alkenes to epoxides and then to cyclic carbonates.
Oxidative dehydrogenation reactions, especially for saturated nitrogen heterocyclic compounds, are a key step in the construction of heterocyclic aromatic hydrocarbons such as quinoline and indole, the core structures of many drugs. For a long time, efficient catalysis of this process often relied on precious metals or harsh additives, limiting its prospects for green applications. In 2017, Steven L. Suib and his team reported the use of mesoporous manganese oxide (meso MnOx) prepared by a reverse micelle template method as a heterogeneous catalyst for the aerobic, additive-free oxidative aromatization of saturated nitrogen heterocyclic compounds [72]. The catalyst efficiently converts 1,2,3,4-tetrahydroquinoline to quinoline in air (with a conversion of 72% and selectivity > 99% under optimal conditions), and demonstrates broad applicability to tetrahydroquinoline derivatives with electron-donating groups (e.g., -OMe) or electron-withdrawing groups (e.g., -NO2), 2-methylindoline, 1,2,3,4-tetrahydroquinoxaline, and Hantzsch esters (with conversions > 99% for some substrates) (Scheme 55). The catalyst calcined at 350 ℃ features a high specific surface area (226 m2/g) and weak crystallinity, which are crucial for its excellent performance.
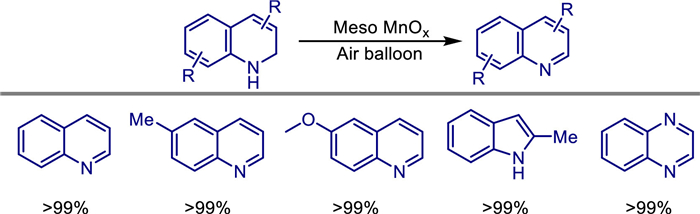
Mechanistic studies indicate that the reaction proceeds via electron transfer from the nitrogen heterocycle to the Mn3+ center, forming an amine radical intermediate, followed by two dehydrogenation steps (involving α-C—H activation) to achieve aromatization. Lattice oxygen participates in the multi-electron redox cycle, while atmospheric oxygen replenishes oxygen vacancies to maintain catalytic activity. The catalyst can be recovered by simple centrifugation, reused four times without activity decay after activation, and shows no metal leaching (confirmed by hot filtration experiments).
The innovation of this method lies in the first use of non-noble metal mesoporous manganese oxide for the aromatization of nitrogen heterocycles, breaking through the limitations of traditional noble metal catalysts. With broad substrate applicability (including compounds with electron-donating/withdrawing groups and steric hindrance), environmental friendliness (additive-free, air as the oxidant), and operational simplicity (easy product separation and catalyst recyclability), this approach provides an efficient new strategy for the green synthesis of intermediates for drugs and bioactive molecules.
In 2018, Mingyang He and colleagues proposed a reaction mechanism involving an imine intermediate. The key catalytic site for the dehydrogenation reaction is Mn3+, which can be stabilized by Ni2+ within the layered double hydroxide structure [73]. They prepared LDHs with different Ni/Mn ratios via the coprecipitation method, among which Ni2Mn-LDH exhibited the best performance. Without additives and using air as the oxidant, it showed high dehydrogenation activity towards various N-heterocycles such as tetrahydroquinoline derivatives, tetrahydroquinoxaline, and dihydroindole (with the highest conversion > 99% and selectivity up to 92%). Characterization techniques including XRD and XPS confirmed the hydrotalcite structure of the catalyst and the synergistic effect between Mn3+ and Ni2+—Ni2+ stabilized the Mn3+ active sites in the hydrotalcite structure, promoting the oxidative dehydrogenation reaction. Kinetic studies indicated that the dehydrogenation of tetrahydroquinoline was a first-order reaction with an apparent activation energy of 113 kJ/mol. Mechanistic analysis, combined with radical trapping experiments and XPS valence state changes, proposed that the reaction involved the formation of an imine intermediate and the redox cycle of Mn3+/Mn⁴+. This catalytic system overcomes the problems of traditional homogeneous catalysts such as difficult separation, dependence on noble metals, or additives. It combines low cost, environmental friendliness (with only water as a by-product), and substrate universality, providing an efficient and sustainable new strategy for the green synthesis of N-heterocycles, especially in the preparation of intermediates for pharmaceuticals and bioactive molecules (Scheme 56).
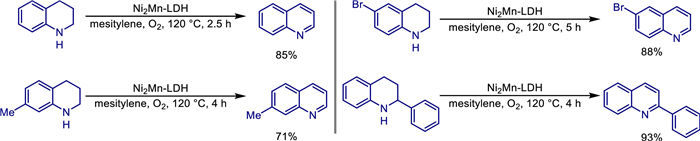
In 2020, Peiqing Zhao and colleagues first prepared a tungsten-doped OMS-2 nanocomposite catalyst ([PW]-OMS-2) for the aerobic oxidative dehydrogenation of N-heterocyclic compounds [74]. The catalyst prepared by the reflux method (with 2 mol% tungsten-doped 2[PW]-OMS-2 exhibiting the best performance) features a mixed crystal phase of cryptomelane (OMS-2) and Mn2O3 hydrate, with the specific surface area significantly increased to 204 m2/g (67 m2/g for undoped OMS-2). XPS characterization shows that the Mn3+ content reaches 63.9%, and the lattice oxygen activity is enhanced.
In the green solvent dimethyl carbonate (DMC) under an oxygen atmosphere, the catalyst efficiently catalyzes the conversion of tetrahydroquinoline to quinoline (99% conversion, > 99% selectivity, 80 ℃, 6 h) and demonstrates broad substrate applicability, covering 20 types of N-heterocyclic derivatives such as tetrahydroquinazoline and dihydroindole (conversion rates of 46%–99%). Kinetic studies indicate that the dehydrogenation of tetrahydroquinoline is a first-order reaction with an apparent activation energy of 29.66 kJ/mol. The catalyst shows excellent stability: after five cycles, the activity is maintained (39% conversion). XRD and BET confirm the structural stability, while hot filtration experiments and ICP-AES analysis (W/Mn leaching rate in the filtrate < 0.0001%) verify its heterogeneous nature. The performance improvement is attributed to the synergistic effect of the mixed crystal phase induced by tungsten doping, high specific surface area, abundant Mn3+ sites, and labile lattice oxygen (Scheme 57).
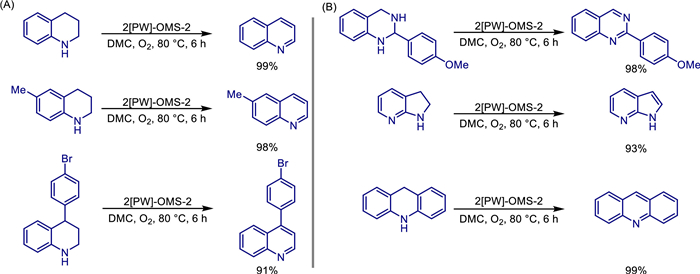
In the same year, Peiqing Zhao and his team developed a synergistic catalytic system composed of sodium-doped amorphous manganese oxide (Na-AMO) and 4-tert-butylcatechol (Q-2), which efficiently achieved aerobic oxidative dehydrogenation of various N-heterocyclic compounds in aqueous phase at room temperature using oxygen as the oxidant. Na-AMO was prepared by coprecipitation of potassium permanganate and manganese sulfate in the presence of sodium nitrate [75]. Characterizations (XRD, XPS, BET, H2-TPR, etc.) showed that it had an amorphous structure, abundant surface-adsorbed active oxygen species, and basic sites. Sodium doping enhanced the concentration of oxygen vacancies and catalytic activity.
Optimization of reaction conditions determined the optimal system: Na-AMO (10 mol%), Q-2 (10 mol%), H2O as the solvent, O2 atmosphere, and reaction at room temperature for 20 h. The system exhibited broad applicability, including tetrahydroquinolines (up to 95% yield), tetrahydroquinazolines (60%–80%), dihydroindoles (e.g., 90% for 7-azaindole), and Hantzsch ester derivatives (65%–95% in ethanol/water mixed solvent at 40 ℃), and enabled the synthesis of key intermediates of bioactive molecules (such as building blocks for the antiparasitic agent TAS-103) (Scheme 58).
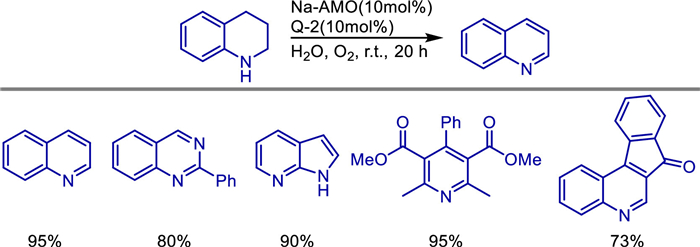
Mechanistic studies showed that Na-AMO and Q-2 formed a redox synergistic cycle: Q-2 reoxidized the reduced Mn species to the active state, while being regenerated by O2. The catalyst could be recycled 8 times (activity stabilized from 95% to 87%). This system combines the advantages of environmental friendliness (water solvent, O2 oxidant), mild conditions (room temperature), non-noble metal catalyst, and recyclability.
The oxygen evolution reaction (OER) is a kinetic process involving four-electron transfer, which is a major bottleneck in improving the efficiency of hydrogen production by water electrolysis. Therefore, extensive research efforts have been focused on identifying single-atom electrocatalysts with excellent OER performance. Although a comprehensive understanding of the OER mechanism has been achieved, its application in other valuable synthetic reactions has been restricted.
In 2023, the team of Dingsheng Wang reported a single-atom manganese catalyst anchored on a nitrogen-doped carbon support (Mn-SA@NC), which enables the efficient electrochemical oxidation of silanes to silanols by utilizing the key intermediate Mn-OOH in the oxygen evolution reaction (OER) (Scheme 59) [76]. The catalyst exhibits a high turnover number (TON) of up to 9132 under low loading (600 ppm), and demonstrates excellent stability—it can be recycled for more than 5 times and is compatible with flow chemistry systems. Experimental and theoretical calculations show that the reaction proceeds through a non-radical pathway: Mn-OOH directly performs electrophilic attack on the Si-H bond of silane, avoiding the high-energy OH* radical pathway, with an energy barrier of only +15.95 kcal/mol. This work for the first time extends the OER mechanism of single-atom catalysts to the field of organic electrosynthesis, addressing the problems of severe pollution from strong oxidants and low electrosynthesis efficiency in traditional silanol synthesis, and providing a new strategy for the clean synthesis of high-value-added chemicals.
It is well-known that nitrile hydration is widely used in amide production, typically employing homogeneous catalysts such as acids, bases, and metal complexes. In 2019, the team led by Fengshou Xiao reported a heterogeneous nanorod α-MnO2 (NR-MnO2) catalyst synthesized via a simple precipitation method (with a diameter of ~3 nm) [77]. This catalyst efficiently promotes the hydration of aromatic, heterocyclic, and aliphatic nitriles to afford corresponding amides under mild conditions (75–120 ℃, atmospheric air).
For benzonitrile, both the conversion and benzamide selectivity exceed 99.5%, significantly outperforming commercial Pd/C, Ru/C catalysts, and reported homogeneous/heterogeneous catalysts. The catalyst demonstrates broad substrate applicability, including substituted benzonitriles with electron-withdrawing/donating groups, furan-2-carbonitrile, thiophene-3-carbonitrile, nicotinonitrile (all yields > 99.5%), and challenging aliphatic nitriles. Recycling experiments show excellent stability: No activity/selectivity loss after 5 cycles, with negligible Mn leaching (Scheme 60).
Mechanistic studies indicate that the rate-determining step involves nucleophilic attack of hydroxyl species (OH⁻), generated by surface oxygen sites activating water, on the nitrile group to form a negatively charged transition state (Hammett slope ρ = 1.2), rather than water dissociation (kinetic isotope effect rH/rD = 1.1). Using earth-abundant manganese, this catalyst combines high activity, selectivity, and recyclability, offering a new pathway for green amide synthesis.
The demand for deuterium-labeled products is increasing in various applications in life sciences and other fields. In 2022, Kathrin Junge and Matthias Beller, along with their team, reported a new class of heterogeneous catalysts for the practical incorporation of deuterium into aniline, phenol, and heterocyclic substrates [78]. This catalyst is prepared by pyrolyzing manganese salts with starch, forming Mn/MnOx core-shell nanoparticles supported on a carbon carrier. Using inexpensive D2O as the deuterium source, it enables efficient hydrogen isotope exchange at 120 ℃ and 10 bar H2. The author's team optimized the catalyst preparation conditions and found that a pyrolysis temperature of 1000 ℃ and starch as the carbon source can significantly enhance the catalytic activity. At a low catalyst loading (10 mol%), the deuterium incorporation rate at the ortho- and para-positions of aniline substrates can reach up to 98%. The catalyst has good tolerance to functional groups such as halogens, thioethers, and amine groups, and is also applicable to the deuterium labeling of nitrogen-containing heterocycles (such as imidazoles and indoles), phenols, natural products, and commercially available drugs (such as fluconazole and ketoconazole) (Scheme 61). Mechanistic studies indicate that the reaction may proceed through a radical pathway. The Mn3+ active centers on the catalyst surface are involved in the homolytic cleavage of D2O to generate deuterium radicals, thereby achieving hydrogen isotope exchange. This method breaks through the limitations of traditional homogeneous catalysis that require noble metals or strong acid-base conditions, solves the problem of catalyst separation, and has advantages such as easy operation, a wide range of substrates, and inexpensive and readily available deuterium sources. It provides an efficient and sustainable heterogeneous catalytic strategy for the synthesis of deuterium-labeled compounds, is particularly suitable for the late-stage functionalization labeling of drugs, and promotes the practical application of manganese-based catalysts in isotope exchange reactions.
In 2022, Feng Shen et al. synthesized the MnOx catalyst under hydrothermal conditions, which was used to convert glucose into formic acid (FA) in water [79]. The aim was to develop a vanadium-free reaction system. When using the MnOx catalyst and glucose as the substrate in water at 160 ℃, a FA yield of 81.1% was obtained, which was almost twice the value obtained using the vanadium-based heterogeneous catalyst. The manganese oxide material prepared under hydrothermal conditions at 100 ℃ has a higher Mn2+/Mn3+ ratio and more adsorbed oxygen species compared with those prepared at higher temperatures. Among all the evaluated catalysts, this material achieved the highest formic acid yield. Studies on the reaction mechanism showed that there are two parallel pathways for glucose conversion: (1) The α-cleavage pathway (main pathway): Arabinose intermediate is generated through the cleavage of the C1—C2 bond (verified by HPLC and 13C NMR), and finally FA is produced; (2) The β-cleavage pathway: Glyoxylic acid intermediate is generated through the cleavage of the C2—C3 bond (a 73.4% FA yield can be obtained with glyoxylic acid as the substrate), accompanied by CO2 by-product (accounting for approximately 11% of glucose carbon) (Scheme 62).
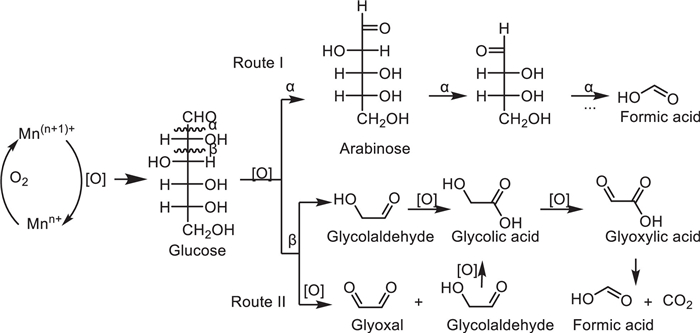
In 2022, Ahmed Alzamly and co-workers successfully synthesized a novel manganese-based metal-organic framework (MnMOF), denoted as UAEU-50, which is assembled from benzenedicarboxylate linkers (BDC) and trinuclear manganese clusters [80]. Prepared via a solvothermal method using terephthalic acid (BDC) ligands and trinuclear manganese clusters, this material exhibits a hexagonal layered structure with a pore size of 3.5 Å, featuring excellent thermal stability (decomposition temperature > 500 ℃) and chemical stability (resistant to various organic solvents).
Characterizations by single-crystal X-ray diffraction, scanning electron microscopy, and UV-visible diffuse reflectance spectroscopy confirm that UAEU-50 has a band gap of 3.04 eV, enabling efficient visible-light absorption. Experiments show that under visible-light irradiation with tetrabutylammonium bromide as a co-catalyst, UAEU-50 efficiently converts 1,2-epoxy-2-methylpropane and CO2 into cyclic carbonates with a conversion rate of 90%, and demonstrates moderate to good catalytic activity toward other epoxides (e.g., styrene oxide, cyclohexene oxide) (Scheme 63). The innovation of this work lies in the first application of a two-dimensional manganese-based MOF for photocatalytic CO2 cycloaddition: through the synergistic effect of trinuclear manganese clusters and the photoresponse properties of BDC ligands, combined with the cooperative effect of the co-catalyst, high-efficiency catalysis is achieved under mild conditions (room temperature and atmospheric pressure), overcoming the limitations of poor photostability in traditional MOFs and their reliance on noble metals or harsh reaction conditions. Control experiments confirm that the photocatalytic process requires visible-light excitation, and the catalyst can be recycled over 4 times with no significant activity loss, demonstrating its practical application potential. Mechanistic studies reveal that Lewis acid sites at manganese nodes activate epoxides, while photogenerated electrons promote CO2 insertion, synergistically enabling the green synthesis of cyclic carbonates. This study provides a novel manganese-based photocatalytic platform for CO2 resource utilization and offers a new strategy for low-cost and sustainable CO2 resource utilization (Scheme 64).
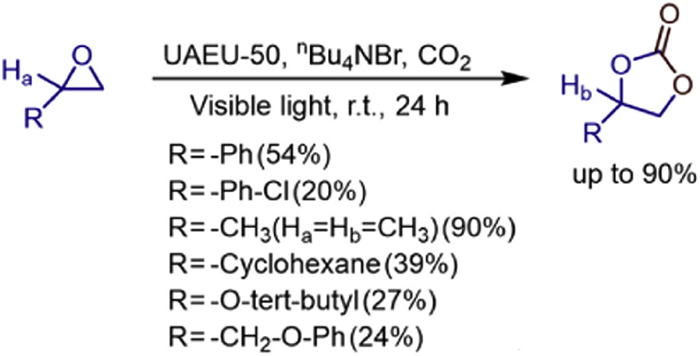
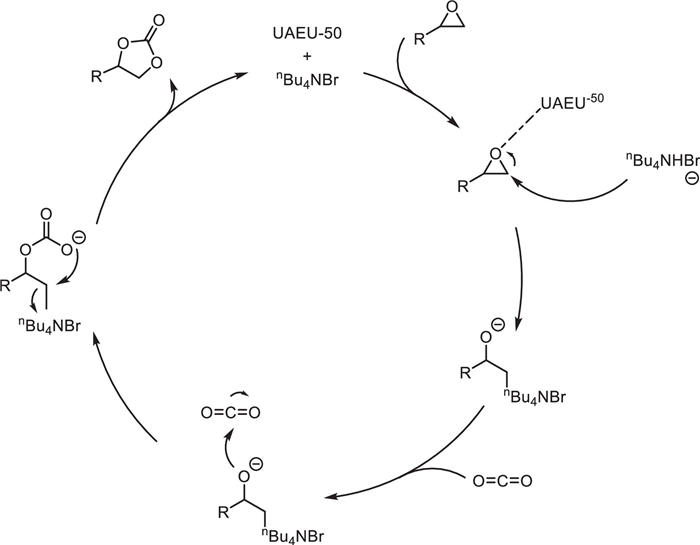
It is well-known that the development of sustainable catalytic systems for producing fuels and chemicals from biomass remains a crucial yet highly challenging process. In this context, the selective hydrogenation of 5-hydroxymethylfurfural (HMF), a biomass-derived molecule, to 5-methylfurfural (MF) using molecular hydrogen is one of the most important conversion reactions. However, this is a difficult task because the reduction of the C═O bond in HMF is far more favorable than that of the C—OH bond.
In 2024, Tetsuya Kida and his colleagues reported a manganese-doped graphene oxide (Mn_N_Graphene) heterogeneous catalyst for the highly selective hydrogenation of HMF, a biomass-derived platform compound, to high-value-added chemical MF under molecular hydrogen conditions [81]. The catalyst was synthesized via a wet impregnation method: manganese precursor (Mn(acac)3) and 1,10-phenanthroline were deposited on graphene oxide (GO) support at a 1:1 molar ratio, followed by pyrolysis at 900 ℃ under an argon atmosphere. Characterization results showed that Mn exists as +4 valence state (MnOx) nanoparticles (4–70 nm) uniformly dispersed in the nitrogen-doped graphene framework, with a BET specific surface area of 220 m2/g. XPS confirmed that nitrogen exists in the form of pyridinic nitrogen and graphitic nitrogen, providing anchoring sites for MnOx. Raman spectroscopy (ID/IG = 1.61) and HAADF-STEM revealed that the embedding of MnOx causes distortion in the graphene structure, facilitating charge transfer.
In the reaction performance test (160 ℃, 1.0 MPa H2, THF solvent, 12 h), Mn_N_Graphene achieved complete conversion of HMF (> 99%) and MF selectivity > 99%, while pure GO, nitrogen-doped rGO (NrGO), and nitrogen-free manganese catalyst (Mn_Graphene) showed no significant activity. FT-IR confirmed that the catalyst selectively adsorbs the C—OH groups of HMF (methanol adsorption forms methoxy species at 1155 cm-1), while the C═O groups (butyraldehyde) were not adsorbed, explaining its high selectivity for C—OH hydrogenolysis. MF cannot be further hydrogenated over this catalyst, which confirms the stability of the product. The MF selectivity remained unchanged after the catalyst was recycled 4 times. XPS showed that the Mn valence state did not change after recycling, and removing the catalyst after 3 h of reaction could stop the conversion at 67%, proving its heterogeneous nature and low leaching property.
This study is the first to use an earth-abundant metal manganese-based catalyst to directly achieve the selective hydrogenolysis of HMF to MF under mild conditions, breaking through the limitations that non-noble metals are difficult to activate H2 and traditional processes rely on noble metals (such as Pt, Pd, Re-Ni) or toxic reagents (such as phosgene). It provides a green, efficient, and recyclable catalytic system for the high-value utilization of biomass resources (Scheme 65).
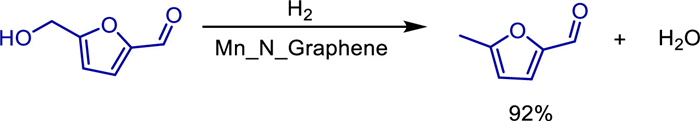
Heterogeneous manganese catalysis holds significant promise in organic synthesis, leveraging manganese's natural abundance, low toxicity, and environmental compatibility. The synergy between different oxidation states and manganese compounds within heterogeneous systems enables highly efficient and selective catalytic transformations. Rational catalyst design further enhances reaction regioselectivity and stereoselectivity, facilitating access to complex molecular architectures. As a non-precious metal catalyst, Mn aligns with green chemistry principles. These systems effectively catalyze diverse reactions, including coupling reactions, oxidations, and C—H functionalizations, expanding the synthetic toolbox.
Despite these advances, several challenges remain, which also define the future direction of this field. (a) Advanced material design and nanostructuring: Future efforts should focus on rationally designing manganese catalysts with well-defined active sites (e.g., single-atom catalysts, defect-rich surfaces, and hybrid nanostructures). Incorporating dopants, heteroatoms, or secondary metals could further enhance redox flexibility and tune activity/selectivity; (b) Mechanistic insights and operando studies: While many catalytic pathways have been proposed, direct evidence for structure–activity relationships remain limited. Advanced in situ/operando characterization (XPS, DRIFTS, XAFS, TEM) combined with computational modelling will be essential for uncovering the dynamic changes in oxidation states and surface intermediates during catalysis; (c) Expanding scope to sustainable feedstocks: Beyond traditional organic substrates, heterogeneous manganese catalysis has great potential in biomass valorization (e.g., lignin depolymerisation, HMF oxidation), CO2 utilization, and late-stage functionalization of pharmaceuticals. These directions would highlight the role of manganese catalysis in green and circular chemistry. (d) Integration with modern catalytic platforms: The synergy between heterogeneous manganese catalysis and emerging methodologies—such as photocatalysis, electrocatalysis, mechanochemistry, and flow chemistry—could lead to new reactivity modes, improved atom economy, and scalable processes [74]; (e) Stability, scalability, and industrial translation: For real-world applications, challenges such as catalyst deactivation, leaching, and recyclability must be addressed. Developing robust catalysts with long-term stability under continuous-flow or industrially relevant conditions will be crucial. Furthermore, life-cycle analysis (LCA) and techno-economic assessments should be undertaken to evaluate the true sustainability benefits of manganese-based systems compared to existing technologies.
In summary, heterogeneous manganese catalysis offers a sustainable and versatile approach to organic synthesis. Its efficiency, selectivity, and environmental profile solidify its importance (Table 1). While current limitations exist, ongoing research and development hold strong potential for Mn-based heterogeneous systems to become pivotal in advancing green chemistry and sustainable chemical synthesis.
 DownLoad:
CSV
DownLoad:
CSV
| Catalyst | Introduction | Substrate | Number of substrates | Yield (%) | Sel. (%) | TOF | Catalytic reactions | Ref. |
| MnO2@PDCS | Manganese dioxide nanoparticles immobilized on the surface of modified poly(2,4-dichlorophenyl) microspheres (PDCS) | Primary alcohol, ketone | 27 | 69-89 | - | - | C—C bond formation | [25] |
| Mn-MgO/Al2O3 | A mixture of divalent manganese ions (Mn2+) and magnesium oxide (MgO) deposited on an Al2O3 support | Primary alcohol, ketone | 16 | 50-92 | - | - | C—C bond formation | [26] |
| Mn-N-Graphene | A heterogeneous catalyst prepared by pyrolyzing the manganese-ethylenediamine precursor, which has well-defined Mn–N active sites and a graphene support structure | Primary alcohol, ketone | 20 | 42-93 | - | - | C—C bond formation | [27] |
| MnPhen@ZIF | A homogeneous manganese-phenanthroline catalyst encapsulated in the pores of ZIF-8 MOF | Primary alcohol, amine | 4 | 43-85 | - | - | C—C bond formation | [28] |
| Primary alcohol, ketone | 16 | 28-91 | - | - | C—C bond formation | [28] | ||
| Mn@CeO2 | A heterogeneous catalyst with CeO2 as the support and doped with 10% manganese | Primary alcohol, ketone | 24 | 5-98 | - | - | C—C bond formation | [29] |
| MnOx/CeO2 | A heterogeneous catalyst with 10 wt% manganese supported on two types of CeO2 supports (with Ce(NO3)3·6H2O and NH4Ce(NO3)4·6H2O as the precursors respectively) | Amine | 10 | - | 69.5-100 | - | C—N bond formation | [30] |
| M-4 | A heterogeneous catalyst mainly composed of cubic-phase Mn2O3 and containing a small amount of metastable Mn5O8 | Amine | 7 | - | 96.1-100 | - | C—N bond formation | [31] |
| MnO2/DMCHA | A heterogeneous catalyst in which N,N-dimethylcyclohexanamine (DMCHA) selectively covers the Lewis acid sites on the surface of MnO2 through associative adsorption | Amine | 13 | 14-99 | - | - | C—N bond formation | [32] |
| Mn@NrGO | A heterogeneous catalyst in which the complex (Mn-Phen) formed by Mn(acac)3 and 1,10-phenanthroline at a 1:1 ratio is deposited on reduced graphene oxide (rGO) | Amine | 31 | 52-89 | - | - | C—N bond formation | [33] |
| CeMn-RO | A heterogeneous catalyst with a structure featuring high defects and oxygen vacancies, formed via a redox reaction using Mn(NO2)2·4H2O and Ce(NO3)3·6H2O as precursors | Benzylamine | 1 | 52 | - | - | C—N bond formation | [34] |
| Ce-Mn | A heterogeneous catalyst prepared using cerium nitrate (Ce(NO3)3) and manganese nitrate (Mn(NO3)2) as precursors | Amine | 11 | 28-97 | - | - | C—N bond formation | [35] |
| OMS-2 | A cryptomelane-type manganese oxide with a porous 2×2 tunnel structure | Primary alcohol, ammonia | 10 | 65-99 | - | - | C—N bond formation | [38] |
| α-Mn2O3-ca | A heterogeneous catalyst prepared by the citric acid-assisted sol-gel method using manganese nitrate (Mn(NO3)2) as the precursor | Primary alcohol | 14 | 6.2-99 | - | - | C—N bond formation | [39] |
| β-MnO2 | β-MnO2 | Primary alcohol | 14 | 1.1-99 | - | - | C—N bond formation | [39] |
| MnOx | Heterogeneous manganese oxide featuring a high specific surface area, abundant oxygen vacancies, and moderate acidic sites | Primary alcohol | 24 | 19-96 | - | - | C—N bond formation | [40] |
| Secondary alcohol | 18 | 54-93 | - | - | C—N bond formation | [40] | ||
| 1,2-Diol | 9 | 85-97 | - | - | C—N bond formation | [40] | ||
| MnNCN/NC-600 | A heterogeneous catalyst prepared using manganese acetate (Mn(OAc)2) and dicyandiamide (DCDA) as precursors | Primary alcohol | 35 | 68-96 | - | - | C—N bond formation | [41] |
| Alcohol | 30 | 79-96 | - | - | C—N bond formation | [41] | ||
| 1,2-Diol | 19 | 27-96 | - | - | C—N bond formation | [41] | ||
| OMS-2-U | OMS-2(prepared using urea as a template agent) | Primary alcohol | 10 | 75-95 | - | - | C—N bond formation | [42] |
| OMS-2-SH | OMS-2(prepared using H2O2 as the reducing agent under acidic conditions) | Primary alcohol, phenyl amidine | 15 | 55-95 | - | - | C—N bond formation | [43] |
| 5[PW]-OMS-2 | OMS-2 modified by sodium phosphotungstate | Primary alcohol | 16 | 57-95 | - | - | C—N bond formation | [44] |
| CuOx/OMS-2 | Copper supported on manganese oxide octahedral molecular sieve | 2-((2-Halogenated phenyl)thio)phenol | 5 | 82-95 | - | - | C—O bond formation | [45] |
| Mn0.9Ce0.1Oy-350 | Cerium-doped manganese oxide | 2-Aminophenol | 8 | 83-98 | - | - | C—O bond formation | [46] |
| MnⅥ-NPs | The high-valent manganese nanoparticles (MnV-NPs) modified with surface bromine/siloxy groups | Nitrile oxide | 8 | 71-85 | - | - | C—O bond formation | [47] |
| Mn/TiO2 | A heterogeneous catalyst with titanium dioxide (TiO2) as the support and manganese (Mn) as the active component | Methane | 1 | - | - | 1.23 (h-1) | C—O bond formation | [48] |
| ZrOx(X%)-MnCO3/(X%)HRG | A heterogeneous catalyst with a core of zirconium oxide-doped manganese carbonate nanocomposites supported on highly reduced graphene oxide (HRG) | Primary alcohol | 21 | - | 99 | - | Oxidation of alcohol | [49] |
| MnOx/Pd/Al2O3 | A heterogeneous catalyst with atomic-level modification of MnOx on Pd nanoparticles via atomic layer deposition (ALD) technology | Benzyl alcohol | 1 | 76.5 | 90.3 | - | Oxidation of alcohol | [50] |
| Mn-MOF | Mn-MOF ([Mn4(L)4(DMF)3]n, L = 4,4′-(anthracene-9,10-diyl)dibenzoic acid) | Primary alcohol | 6 | 0-99 | - | - | Oxidation of alcohol | [51] |
| β-MnO2 | β-MnO2 | Primary alcohol, secondary alcohol | 14 | 14-98 | - | - | Oxidation of alcohol | [52] |
| MnL1@Cellulose | A crown ether-free Schiff base manganese complex supported on nanocellulose | Primary alcohol | 9 | 52-85 | - | - | Oxidation of alcohol | [53] |
| MnL2@Cellulose | A crown ether-modified Schiff base manganese complex is supported on nanocellulose | Primary alcohol | 9 | 85-96 | - | - | Oxidation of alcohol | [53] |
| N55-MnO2 | Nitrogen-doped manganese dioxide | Primary alcohol, secondary alcohol | 12 | 30.5-99.9 | 0-99.9 | - | Oxidation of alcohol | [54] |
| MnOx/HAP | Manganese oxide supported on hydroxyapatite | HMF | 1 | - | 90.9 | - | Oxidation of alcohol | [55] |
| PdNi/MnO2 | A heterogeneous catalyst in which α-MnO2 is synergistically promoted by trace amounts of Pd and Ni single atoms | HMF | 1 | 99 | - | - | Oxidation of alcohol | [56] |
| EcoMnOx | A heterogeneous catalyst extracted and prepared from manganese hyperaccumulator plant biomass | 1,2-Diol | 10 | - | 0-99 | - | Oxidation of glycol | [57] |
| Na-Mn LMO | The layered oxides based on sodium and manganese | 1,2-Diol | 14 | 15-99 | - | - | Oxidation of glycol | [58] |
| Na-MnOx | The sodium-doped porous sodium manganese oxide | FPED | 4 | 85-91 | - | - | Oxidation of glycol | [59] |
| Mn2Cu1Ox | Mn-Cu bimetallic oxide with different Mn/Cu molar ratios | Furfural | 1 | 83 | - | - | Oxidation of aldehyde | [60] |
| AMO | Amorphous manganese oxide | Amide | 8 | - | 0-98 | - | Oxidation of C—H bond | [61] |
| Cs/Mn2O3 | Cesium-promoted mesoporous manganese oxide | Amide | 2 | - | 95-100 | - | Oxidation of C—H bond | [61] |
| Mn-1@POP-40 | Porous organic polymers supporting single-site manganese active site | Cyclohexane | 13 | 35-82 | - | - | Oxidation of C—H bond | [62] |
| Mg6MnO8-MA | Murdochite-type Mg6MnO8 nanoparticles with a high specific surface area, composed of isolated Mn4+ species embedded in an alkaline MgO matrix | Aromatic hydrocarbon | 16 | 32-99 | - | - | Oxidation of C—H bond | [63] |
| Mn(Ⅱ)@G3.0-P(4-VT/VS/GMA)-MB | A dendritic manganese structure supported on a polymeric magnetic core-shell | Alkane, oxime | 22 | - | 69-100 | - | Oxidation of C—H bond | [64] |
| Mn/N-C/Al2O3 | A heterogeneous catalyst using nitrogen-doped carbon and mesoporous alumina as supports, with highly dispersed manganese as the active site |
Phenylethane | 1 | - | 99 | - | Oxidation of C—H bond | [65] |
| MnCo0.2 | Cobalt-doped MnO2 nanofiber structure with Mn–Co dual active sites | Propane | 1 | - | - | - | Oxidation of C—H bond | [66] |
| Ru-OMS-2 | Ruthenium-modified octahedral manganese oxide molecular sieve | Polycyclic aromatic hydrocarbons | 10 | 55-99 | - | - | Oxidation of C—H bond | [67] |
| MnOx | Manganese oxide | Olefin | 34 | 18-87 | - | - | Oxidation of C—H bond | [68] |
| Alkyne | 16 | 29-88 | - | - | Epoxidation | [68] | ||
| MnⅡ-MOF-5 | Manganese-exchanged MOF-5 | Cyclic olefin | 1 | - | 99 | - | Epoxidation | [69] |
| MnWO4/SBA-16 | The three-dimensional cubic mesoporous SBA-16 supported MnWO4 active component | Styrene | 7 | - | 27-75 | - | Epoxidation | [70] |
| Mn/g-C3N4 | Mn supported on g-C3N4 | Olefin | 11 | 39-83 | 0-99 | - | Epoxidation | [71] |
| Meso MnOx | Mesoporous manganese oxide | N-Heterocycle | 9 | - | 0-99 | - | Dehydrogenation of N-heterocycles | [72] |
| Ni2Mn-LDH | A heterogeneous catalyst based on NiMn layered double hydroxide (LDH) | Tetrahydroquinoline derivative | 16 | - | 39-93 | - | Dehydrogenation of N-heterocycles | [73] |
| 2[PW]-OMS-2 | OMS-2-based heterogeneous catalyst with a sodium phosphotungstate doping amount (2 mol%) | N-Heterocycle | 20 | - | 82-99 | - | Dehydrogenation of N-heterocycles | [74] |
| Na-AMO | The sodium-doped amorphous manganese oxide (Na-AMO) | N-Heterocycle | 28 | 50-95 | 82-99 | - | Dehydrogenation of N-heterocycles | [75] |
| Mn-SA@NC | The manganese single atoms anchored on nitrogen-doped carbon | Silane | 16 | 65-90 | - | - | Oxidation of silane | [76] |
| NR-MnO2 | A well-defined α-MnO2 crystal structure with a specific nanorod morphology | Nitrile | 10 | - | 99.5 | - | Hydration of nitriles | [77] |
| Mn@Starch-1000 | A heterogeneous manganese catalyst with pyrolyzed starch carbon as the support and a Mn/MnOx core-shell structure | Aniline and N-heterocycle | 24 | 61-99 | - | - | Deuteration | [78] |
| MnOx-100 | A vanadium-free manganese-based heterogeneous catalyst | Glucose | 1 | 81.1 | - | - | Conversion of glucose to formic acid (FA) | [79] |
| UAEU-50{Mn3(BDC)3} | A heterogeneous Mn-MOF catalyst constructed from trinuclear Mn clusters and phthalate (BDC) ligands | Epoxide | 1 | 90 | - | - | Reaction of epoxides and CO2 to cyclic carbonates | [80] |
| Mn_N_Graphene | MnOx nanoparticles supported on nitrogen-doped graphene oxide | HMF | 1 | 92 | - | - | Cleavage of C—O bond | [81] |
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Tianyi Zhou: Writing – original draft. Heng Yang: Writing – original draft. Guangbin Zhou: Writing – original draft. Feng Chen: Writing – review & editing, Conceptualization. Pan Gao: Writing – review & editing, Conceptualization.
M. Kazemi, M. Ghobadi, Nanotechnol. Rev. 6 (2017) 549–571. doi: 10.1515/ntrev-2016-0113
J.R. Carney, B.R. Dillon, S.P. Thomas, Eur. J. Org. Chem. 2016 (2016) 3912–3929. doi: 10.1002/ejoc.201600018
J. Wang, Y. Zhang, Y. Zhou, et al., Org. Chem. Front. 11 (2024) 6850–6917. doi: 10.1039/d4qo01499e
L.J. Li, Y. He, Y. Yang, et al., CCS Chem. 6 (2023) 537–584.
C. Wang, H. Chen, M Mi, et al., Chin. J. Org. Chem. 45 (2025) 2326–2334. doi: 10.6023/cjoc202412002
R. Sharma, S. Pandey, M. Mohit, D.S. Rawat, Tetrahedron 171 (2025) 134416. doi: 10.1016/j.tet.2024.134416
S. Kostera, L. Gonsalvi, ChemCatChem 16 (2024) e202301391. doi: 10.1002/cctc.202301391
R.V. Ottenbacher, A.A. Bryliakova, V.I. Kurganskii, et al., Chem. Eur. J. 29 (2023) e202302772. doi: 10.1002/chem.202302772
D.C. Lacy, P.C. Abhyankar, Chem. Eur. J. 29 (2023) e202300518. doi: 10.1002/chem.202300518
K. Das, S. Waiba, A. Jana, B. Maji, Chem. Soc. Rev. 51 (2022) 4386–4464. doi: 10.1039/d2cs00093h
G. Anusree, P.S. Devi, G. Anilkumar, Adv. Synth. Catal. 366 (2024) 3963–3999. doi: 10.1002/adsc.202400611
N. Ahmed, J. Organometal. Chem. 1009 (2024) 123071. doi: 10.1016/j.jorganchem.2024.123071
M.F. Ansari, A.K. Maurya, A. Kumar, S. Elangovan, Beilstein J. Org. Chem. 20 (2024) 1111–1166. doi: 10.3762/bjoc.20.98
Y. Wang, M. Wang, Y. Li, Q. Liu, Chem 7 (2021) 1180–1223. doi: 10.1016/j.chempr.2020.11.013
C.M. Friend, B. Xu, Acc. Chem. Res. 50 (2017) 517–521. doi: 10.1021/acs.accounts.6b00510
F. Zaera, Chem. Rev. 122 (2022) 8594–8757. doi: 10.1021/acs.chemrev.1c00905
A.N. Fajer, H.A. Al-Bahrani, A.A.H. Kadhum, M. Kazemi, J. Mol. Struct. 1296 (2024) 136800. doi: 10.1016/j.molstruc.2023.136800
L. Liu, A. Corma, et al., Chem. Rev. 118 (2018) 4981–5079. doi: 10.1021/acs.chemrev.7b00776
J. Wang, Y. Zhang, X. Guo, et al., Green Chem. 26 (2024) 2365–2383. doi: 10.1039/d3gc04117d
M. Kazemi, R. Ali, V. Jain, et al., RSC Adv. 15 (2025) 23054–23088. doi: 10.1039/d5ra02493e
M. Kazemi, R. Ali, V. Jain, et al., RSC Adv. 15 (2025) 23054–23088. doi: 10.1039/D5RA02493E
B.G. Reed-Berendt, D.E. Latham, M.B. Dambatta, L.C. Morrill, ACS Cent. Sci. 7 (2021) 570–585. doi: 10.1021/acscentsci.1c00125
K.R. Rohit, S. Radhika, S. Saranya, G. Anilkumar, Adv. Synth. Catal. 362 (2020) 1602–1650. doi: 10.1002/adsc.201901389
S. Mullick, A. Ghosh, D. Banerjee, Chem. Commun. 60 (2024) 4002–4014. doi: 10.1039/d4cc00003j
Y. Qiu, Y. Zhang, L. Jin, et al., Org. Chem. Front. 6 (2019) 3420–3427. doi: 10.1039/c9qo00892f
Y. Kita, M. Kuwabara, K. Kamata, M. Hara, ACS Catal. 12 (2022) 11767–11775. doi: 10.1021/acscatal.2c03085
M.S. Ahmad, Y. Inomata, T. Kida, Mol. Catal. 526 (2022) 112390.
G. Dey, S. Saifi, M. Sk, et al., Dalton Trans. 51 (2022) 17973–17977. doi: 10.1039/d2dt02629e
R. Swaathy, S. Karthikeyan, ACS Omega 10 (2025) 9649–9660. doi: 10.1021/acsomega.4c10938
P. Sudarsanam, B. Hillary, M.H. Amin, S.B.A. Hamid, S.K. Bhargava, Appl. Catal. B: Environ. 185 (2016) 213–224. doi: 10.1016/j.apcatb.2015.12.026
F. Chen, T. Yang, S. Zhao, et al., Chin. Chem. Lett. 30 (2019) 2282–2286. doi: 10.1016/j.cclet.2019.09.007
Q. Hao, X. Jia, J. Ma, et al., Chem. Asian J. 16 (2021) 1388–1391. doi: 10.1002/asia.202100264
M. Subaramanian, P.M. Ramar, G. Sivakumar, et al., ChemCatChem 13 (2021) 4334–4341. doi: 10.1002/cctc.202100635
C. Wu, J. Bu, W. Wang, et al., Ind. Eng. Chem. Res. 61 (2022) 5442–5452. doi: 10.1021/acs.iecr.2c00311
T. Chutimasakul, W. Tirdtrakool, P. Na Nakhonpanom, et al., ChemistrySelect 7 (2022) e202203028. doi: 10.1002/slct.202203028
A. Kumar, N.A. Espinosa-Jalapa, G. Leitus, et al., Angew. Chem. Int. Ed. 56 (2017) 14992–14996. doi: 10.1002/anie.201709180
J. Guo, J. Tang, H. Xi, S.Y. Zhao, W. Liu, Chin. Chem. Lett. 34 (2023) 107731. doi: 10.1016/j.cclet.2022.08.011
K. Yamaguchi, H. Kobayashi, T. Oishi, N. Mizuno, Angew. Chem. Int. Ed. 51 (2012) 544–547. doi: 10.1002/anie.201107110
H. Wang, Q. Luo, L. Wang, et al., Chin. J. Catal. 42 (2021) 2164–2172. doi: 10.1016/S1872-2067(21)63803-2
P. He, B. Chen, L. Huang, et al., Chem 8 (2022) 1906–1927. doi: 10.1016/j.chempr.2022.02.021
X. Liu, B. Han, C. Wu, et al., Angew. Chem. Int. Ed. 64 (2024) e202413799.
B. Li, C. Li, L. Tian, et al., New J. Chem. 42 (2018) 15985–15989. doi: 10.1039/c8nj02551g
J. Shen, X. Meng, Catal. Commun. 127 (2019) 58–63. doi: 10.1016/j.catcom.2019.05.005
N. Yao, X. Bi, L. Zhang, et al., Mol. Catal. 504 (2021) 111499.
N. Liu, F. Chao, Y. Huang, et al., Tetrahedron Lett. 60 (2019) 151259. doi: 10.1016/j.tetlet.2019.151259
F. Sun, J. Qin, X. Cao, et al., ACS Sustain. Chem. Eng. 10 (2022) 9087–9095. doi: 10.1021/acssuschemeng.2c01703
S. Khamarui, Y. Saima, SynOpen 06 (2022) 173–178. doi: 10.1055/a-1896-3987
Y. Ji, A.N. Blankenship, J.A. van Bokhoven, ACS Catal. 13 (2023) 3896–3901. doi: 10.1021/acscatal.2c06292
M.E. Assal, M.R. Shaik, M. Kuniyil, et al., RSC Adv. 7 (2017) 55336–55349. doi: 10.1039/C7RA11569E
J. Yang, K. Cao, M. Gong, B. Shan, R. Chen, J. Catal. 386 (2020) 60–69. doi: 10.1016/j.jcat.2020.03.029
B. Liao, S. Li, J. Catal. 414 (2022) 294–301. doi: 10.1016/j.jcat.2022.09.021
K. Kamata, N. Kinoshita, M. Koutani, et al., Catal. Sci. Technol. 12 (2022) 6219–6230. doi: 10.1039/d2cy01476a
B. Maleki, R. Sandaroos, S. Naderi, S. Peiman, J. Organometal. Chem. 990 (2023).
J. Tang, J. Chen, Z. Zhang, et al., Chem. Sci. 14 (2023) 13402–13409. doi: 10.1039/d3sc04418a
H. Chen, Y. Li, L. Yu, et al., Catal. Lett. 152 (2022) 3716–3724. doi: 10.1007/s10562-022-03945-0
X. Liao, M. Guo, W. Tang, et al., Green Chem. 24 (2022) 8424–8433. doi: 10.1039/d2gc01769e
V. Escande, C.H. Lam, C. Grison, P.T. Anastas, ACS Sustain. Chem. Eng. 5 (2017) 3214–3222. doi: 10.1021/acssuschemeng.6b02979
V. Escande, C.H. Lam, P. Coish, P.T. Anastas, Angew. Chem. Int. Ed. 56 (2017) 9561–9565. doi: 10.1002/anie.201705934
Q.S. Kong, X.L. Li, H.B. Shen, H.J. Xu, Y. Fu, Green Energy Environ. 7 (2022) 957–964. doi: 10.1016/j.gee.2020.12.007
X. Yu, T. Jin, H. Wang, et al., Green Energy Environ. 8 (2023) 1683–1692. doi: 10.1016/j.gee.2022.03.016
S. Biswas, H.S. Khanna, Q.A. Nizami, et al., Sci. Rep. 8 (2018) 13649. doi: 10.1038/s41598-018-31729-3
B. Wang, J. Lin, Q. Sun, C. Xia, W. Sun, ACS Catal. 11 (2021) 10964–10973. doi: 10.1021/acscatal.1c02738
E. Hayashi, T. Tamura, T. Aihara, K. Kamata, M. Hara, ACS Appl. Mater. Interfaces 14 (2022) 6528–6537. doi: 10.1021/acsami.1c20080
S. Movahedian, A.R. Farahani, A.R. Faraji, J. Alloys Compd. 908 (2022) 164585. doi: 10.1016/j.jallcom.2022.164585
W.F. Xu, W.J. Chen, D.C. Li, B.H. Cheng, H. Jiang, Ind. Eng. Chem. Res. 58 (2019) 3969–3977. doi: 10.1021/acs.iecr.8b05328
L. Chen, J. Ding, J. Jia, et al., ACS Appl. Nano Mat. 2 (2019) 4417–4426. doi: 10.1021/acsanm.9b00818
A. Khorshidi, M. Panahdar, R. Badihi, Catal. Lett. 154 (2024) 3309–3322. doi: 10.1007/s10562-023-04557-y
B. Chen, L. Zhang, H. Luo, et al., JACS Au 3 (2023) 476–487. doi: 10.1021/jacsau.2c00608
A.W. Stubbs, L. Braglia, E. Borfecchia, et al., ACS Catal. 8 (2017) 596–601.
S. Manimaran, K. Subramanian, R. Tschentscher, A. Pandurangan, J. Porous Mat. 29 (2021) 357–369.
M. Du, Y. Sun, J. Zhao, et al., Chin. Chem. Lett. 34 (2023) 108269. doi: 10.1016/j.cclet.2023.108269
K. Mullick, S. Biswas, A.M. Angeles-Boza, S.L. Suib, Chem. Commun. 53 (2017) 2256–2259. doi: 10.1039/C6CC09095H
W. Zhou, Q. Tao, F. a. Sun, et al., J. Catal. 361 (2018) 1–11.
X. Bi, T. Tang, X. Meng, et al., Catal. Sci. Technol. 10 (2020) 360–371. doi: 10.1039/c9cy01968e
T. Tang, X. Bi, X. Meng, et al., Tetrahedron Lett. 61 (2020) 151425. doi: 10.1016/j.tetlet.2019.151425
H.T. Tang, H.Y. Zhou, Y.M. Pan, et al., Angew. Chem. Int. Ed. 63 (2023) e202315032.
H. Wang, Y. Wang, H. Xu, et al., Ind. Eng. Chem. Res. 58 (2019) 17319–17324. doi: 10.1021/acs.iecr.9b04128
F. Bourriquen, N. Rockstroh, S. Bartling, K. Junge, M. Beller, et al., Angew. Chem. Int. Ed. 61 (2022) e202202423. doi: 10.1002/anie.202202423
J. Li, R.L. Smith, S. Xu, et al., Green Chem. 24 (2022) 315–324. doi: 10.1039/D1GC03637H
L.A. Siddig, R.H. Alzard, H.L. Nguyen, et al., ACS Omega 7 (2022) 9958–9963. doi: 10.1021/acsomega.2c00663
M.S. Ahmad, Y. Nagata, K. Masumoto, et al., Mol. Catal. 553 (2024) 113787.

Scheme 2 The proposed possible mechanism for alkylation of ketone with primary alcohols catalyzed by MnO2@PDCS.

Scheme 4 Proposed mechanism for alkylation reaction of ketones and alcohols over Mn-MgO/Al2O3.

Scheme 8 Solvent-free oxidation of benzylamine over CeO2 supports and MnOx/CeO2 nanocatalysts.

Scheme 10 The catalytic performance of DMCHA modified MnO2 catalyst in the amine aerobic oxidation reaction.

Scheme 11 Mn@NrGO catalyzed N-alkylation of amines with benzyl alcohol: Scope of amines.

Scheme 13 Ce-Mn oxides catalyze the self-coupling reaction of benzylamine derivatives.

Scheme 15 Catalytic ammoxidation of various alcohols over α‐Mn2O3 and β‐MnO2 catalysts.

Scheme 16 (A) Oxidation of primary alcohols. (B) Oxidation of secondary alcohols. (C) Oxidation of 1,2-diols.

Scheme 18 (A) Production of nitriles from ammoxidation of alcohols. (B) Synthesis of nitriles from alcohols via oxidative cleavage of C-/C(O)-C bonds. (C) Synthesis of nitriles from the ammoxidation of 1,2-diols.

Scheme 19 The reaction of alcohols with substituted N-phenylformamidines catalyzed by OMS-2-U.

Scheme 20 OMS-2-based catalytic systems for the oxidative synthesis of heterocycles using amidines as substrates.

Scheme 23 The reaction of 2-((2-halogenated phenyl)thio)phenol with different substituents.

Scheme 26 Generation of nitrile oxides and their regioselective cyclization to isoxazolines.

Scheme 28 Aerobic oxidation of benzyl alcohol derivatives using ZrOx(1%)-MnCO3/(1%)HRG nanocomposite.

Scheme 29 (A) Photocatalytic results of Mn-MOF in the aerobic oxidation of different benzyl alcohols. (B) Photocatalytic results of Mn-MOF in the trifluoromethylation of benzene.

Scheme 31 The oxidation reaction of benzyl alcohols in the presence of MnL1@Cellulose and MnL2@Cellulose.

Scheme 39 Oxidative cleavage of FPED catalyzed by Na-MnOx and multiple derivatives of biomass sources.

Scheme 40 Plausible catalytic mechanism for the Mn2Cu1Ox-catalyzed aerobic oxidation of furfural to FA.

Scheme 44 The aerobic oxidation of EB Catalyzed by Mn(Ⅱ)@G3.0-P(4-VT/VS/GMA)-MB. nanocomposite.

Scheme 45 Aerobic oxidation of oximes derivatives catalyzed by Mn(Ⅱ)@G3.0-P(4-VT/VS/GMA)-MB nanocomposite.

Scheme 47 Synergistic effect of Mn−Co dual sites on the MnCo0.2 sample on pro-pane complete oxidation.

Scheme 48 Oxidation of various arenes and S-heterocyclic aromatic compounds catalyzed by Ru-OMS-2.

Scheme 49 Formation of amides from aromatic olefins and aromatic alkynes catalyzed by MnOx.

Scheme 51 Epoxidation of various alkenes over MnWO4 (3 wt%)/SBA-16 catalysts at 60 ℃ in 24 h.

Scheme 56 Oxidative dehydrogenation of tetrahydroquinoline derivatives catalyzed by Ni2Mn-LDH.

Scheme 57 (A) Oxidative dehydrogenation of tetrahydroquinoline derivatives catalyzed by 2[PW]-OMS-2. (B) Oxidative dehydrogenation of other N-containing compounds by 2[PW]-OMS-2.

Scheme 58 N-Heterocyclic compounds catalyzed by Na-AMO in aerobic oxidative dehydrogenation reaction.

Scheme 61 Manganese catalyzed actual doping of deuterium into aniline, phenol, and heterocyclic substrates.

Scheme 62 Proposed oxidation pathways of glucose to FA in water for the MnOx-100 catalyst.
Table 1. Summary of heterogeneous manganese catalysts.
| Catalyst | Introduction | Substrate | Number of substrates | Yield (%) | Sel. (%) | TOF | Catalytic reactions | Ref. |
| MnO2@PDCS | Manganese dioxide nanoparticles immobilized on the surface of modified poly(2,4-dichlorophenyl) microspheres (PDCS) | Primary alcohol, ketone | 27 | 69-89 | - | - | C—C bond formation | [25] |
| Mn-MgO/Al2O3 | A mixture of divalent manganese ions (Mn2+) and magnesium oxide (MgO) deposited on an Al2O3 support | Primary alcohol, ketone | 16 | 50-92 | - | - | C—C bond formation | [26] |
| Mn-N-Graphene | A heterogeneous catalyst prepared by pyrolyzing the manganese-ethylenediamine precursor, which has well-defined Mn–N active sites and a graphene support structure | Primary alcohol, ketone | 20 | 42-93 | - | - | C—C bond formation | [27] |
| MnPhen@ZIF | A homogeneous manganese-phenanthroline catalyst encapsulated in the pores of ZIF-8 MOF | Primary alcohol, amine | 4 | 43-85 | - | - | C—C bond formation | [28] |
| Primary alcohol, ketone | 16 | 28-91 | - | - | C—C bond formation | [28] | ||
| Mn@CeO2 | A heterogeneous catalyst with CeO2 as the support and doped with 10% manganese | Primary alcohol, ketone | 24 | 5-98 | - | - | C—C bond formation | [29] |
| MnOx/CeO2 | A heterogeneous catalyst with 10 wt% manganese supported on two types of CeO2 supports (with Ce(NO3)3·6H2O and NH4Ce(NO3)4·6H2O as the precursors respectively) | Amine | 10 | - | 69.5-100 | - | C—N bond formation | [30] |
| M-4 | A heterogeneous catalyst mainly composed of cubic-phase Mn2O3 and containing a small amount of metastable Mn5O8 | Amine | 7 | - | 96.1-100 | - | C—N bond formation | [31] |
| MnO2/DMCHA | A heterogeneous catalyst in which N,N-dimethylcyclohexanamine (DMCHA) selectively covers the Lewis acid sites on the surface of MnO2 through associative adsorption | Amine | 13 | 14-99 | - | - | C—N bond formation | [32] |
| Mn@NrGO | A heterogeneous catalyst in which the complex (Mn-Phen) formed by Mn(acac)3 and 1,10-phenanthroline at a 1:1 ratio is deposited on reduced graphene oxide (rGO) | Amine | 31 | 52-89 | - | - | C—N bond formation | [33] |
| CeMn-RO | A heterogeneous catalyst with a structure featuring high defects and oxygen vacancies, formed via a redox reaction using Mn(NO2)2·4H2O and Ce(NO3)3·6H2O as precursors | Benzylamine | 1 | 52 | - | - | C—N bond formation | [34] |
| Ce-Mn | A heterogeneous catalyst prepared using cerium nitrate (Ce(NO3)3) and manganese nitrate (Mn(NO3)2) as precursors | Amine | 11 | 28-97 | - | - | C—N bond formation | [35] |
| OMS-2 | A cryptomelane-type manganese oxide with a porous 2×2 tunnel structure | Primary alcohol, ammonia | 10 | 65-99 | - | - | C—N bond formation | [38] |
| α-Mn2O3-ca | A heterogeneous catalyst prepared by the citric acid-assisted sol-gel method using manganese nitrate (Mn(NO3)2) as the precursor | Primary alcohol | 14 | 6.2-99 | - | - | C—N bond formation | [39] |
| β-MnO2 | β-MnO2 | Primary alcohol | 14 | 1.1-99 | - | - | C—N bond formation | [39] |
| MnOx | Heterogeneous manganese oxide featuring a high specific surface area, abundant oxygen vacancies, and moderate acidic sites | Primary alcohol | 24 | 19-96 | - | - | C—N bond formation | [40] |
| Secondary alcohol | 18 | 54-93 | - | - | C—N bond formation | [40] | ||
| 1,2-Diol | 9 | 85-97 | - | - | C—N bond formation | [40] | ||
| MnNCN/NC-600 | A heterogeneous catalyst prepared using manganese acetate (Mn(OAc)2) and dicyandiamide (DCDA) as precursors | Primary alcohol | 35 | 68-96 | - | - | C—N bond formation | [41] |
| Alcohol | 30 | 79-96 | - | - | C—N bond formation | [41] | ||
| 1,2-Diol | 19 | 27-96 | - | - | C—N bond formation | [41] | ||
| OMS-2-U | OMS-2(prepared using urea as a template agent) | Primary alcohol | 10 | 75-95 | - | - | C—N bond formation | [42] |
| OMS-2-SH | OMS-2(prepared using H2O2 as the reducing agent under acidic conditions) | Primary alcohol, phenyl amidine | 15 | 55-95 | - | - | C—N bond formation | [43] |
| 5[PW]-OMS-2 | OMS-2 modified by sodium phosphotungstate | Primary alcohol | 16 | 57-95 | - | - | C—N bond formation | [44] |
| CuOx/OMS-2 | Copper supported on manganese oxide octahedral molecular sieve | 2-((2-Halogenated phenyl)thio)phenol | 5 | 82-95 | - | - | C—O bond formation | [45] |
| Mn0.9Ce0.1Oy-350 | Cerium-doped manganese oxide | 2-Aminophenol | 8 | 83-98 | - | - | C—O bond formation | [46] |
| MnⅥ-NPs | The high-valent manganese nanoparticles (MnV-NPs) modified with surface bromine/siloxy groups | Nitrile oxide | 8 | 71-85 | - | - | C—O bond formation | [47] |
| Mn/TiO2 | A heterogeneous catalyst with titanium dioxide (TiO2) as the support and manganese (Mn) as the active component | Methane | 1 | - | - | 1.23 (h-1) | C—O bond formation | [48] |
| ZrOx(X%)-MnCO3/(X%)HRG | A heterogeneous catalyst with a core of zirconium oxide-doped manganese carbonate nanocomposites supported on highly reduced graphene oxide (HRG) | Primary alcohol | 21 | - | 99 | - | Oxidation of alcohol | [49] |
| MnOx/Pd/Al2O3 | A heterogeneous catalyst with atomic-level modification of MnOx on Pd nanoparticles via atomic layer deposition (ALD) technology | Benzyl alcohol | 1 | 76.5 | 90.3 | - | Oxidation of alcohol | [50] |
| Mn-MOF | Mn-MOF ([Mn4(L)4(DMF)3]n, L = 4,4′-(anthracene-9,10-diyl)dibenzoic acid) | Primary alcohol | 6 | 0-99 | - | - | Oxidation of alcohol | [51] |
| β-MnO2 | β-MnO2 | Primary alcohol, secondary alcohol | 14 | 14-98 | - | - | Oxidation of alcohol | [52] |
| MnL1@Cellulose | A crown ether-free Schiff base manganese complex supported on nanocellulose | Primary alcohol | 9 | 52-85 | - | - | Oxidation of alcohol | [53] |
| MnL2@Cellulose | A crown ether-modified Schiff base manganese complex is supported on nanocellulose | Primary alcohol | 9 | 85-96 | - | - | Oxidation of alcohol | [53] |
| N55-MnO2 | Nitrogen-doped manganese dioxide | Primary alcohol, secondary alcohol | 12 | 30.5-99.9 | 0-99.9 | - | Oxidation of alcohol | [54] |
| MnOx/HAP | Manganese oxide supported on hydroxyapatite | HMF | 1 | - | 90.9 | - | Oxidation of alcohol | [55] |
| PdNi/MnO2 | A heterogeneous catalyst in which α-MnO2 is synergistically promoted by trace amounts of Pd and Ni single atoms | HMF | 1 | 99 | - | - | Oxidation of alcohol | [56] |
| EcoMnOx | A heterogeneous catalyst extracted and prepared from manganese hyperaccumulator plant biomass | 1,2-Diol | 10 | - | 0-99 | - | Oxidation of glycol | [57] |
| Na-Mn LMO | The layered oxides based on sodium and manganese | 1,2-Diol | 14 | 15-99 | - | - | Oxidation of glycol | [58] |
| Na-MnOx | The sodium-doped porous sodium manganese oxide | FPED | 4 | 85-91 | - | - | Oxidation of glycol | [59] |
| Mn2Cu1Ox | Mn-Cu bimetallic oxide with different Mn/Cu molar ratios | Furfural | 1 | 83 | - | - | Oxidation of aldehyde | [60] |
| AMO | Amorphous manganese oxide | Amide | 8 | - | 0-98 | - | Oxidation of C—H bond | [61] |
| Cs/Mn2O3 | Cesium-promoted mesoporous manganese oxide | Amide | 2 | - | 95-100 | - | Oxidation of C—H bond | [61] |
| Mn-1@POP-40 | Porous organic polymers supporting single-site manganese active site | Cyclohexane | 13 | 35-82 | - | - | Oxidation of C—H bond | [62] |
| Mg6MnO8-MA | Murdochite-type Mg6MnO8 nanoparticles with a high specific surface area, composed of isolated Mn4+ species embedded in an alkaline MgO matrix | Aromatic hydrocarbon | 16 | 32-99 | - | - | Oxidation of C—H bond | [63] |
| Mn(Ⅱ)@G3.0-P(4-VT/VS/GMA)-MB | A dendritic manganese structure supported on a polymeric magnetic core-shell | Alkane, oxime | 22 | - | 69-100 | - | Oxidation of C—H bond | [64] |
| Mn/N-C/Al2O3 | A heterogeneous catalyst using nitrogen-doped carbon and mesoporous alumina as supports, with highly dispersed manganese as the active site |
Phenylethane | 1 | - | 99 | - | Oxidation of C—H bond | [65] |
| MnCo0.2 | Cobalt-doped MnO2 nanofiber structure with Mn–Co dual active sites | Propane | 1 | - | - | - | Oxidation of C—H bond | [66] |
| Ru-OMS-2 | Ruthenium-modified octahedral manganese oxide molecular sieve | Polycyclic aromatic hydrocarbons | 10 | 55-99 | - | - | Oxidation of C—H bond | [67] |
| MnOx | Manganese oxide | Olefin | 34 | 18-87 | - | - | Oxidation of C—H bond | [68] |
| Alkyne | 16 | 29-88 | - | - | Epoxidation | [68] | ||
| MnⅡ-MOF-5 | Manganese-exchanged MOF-5 | Cyclic olefin | 1 | - | 99 | - | Epoxidation | [69] |
| MnWO4/SBA-16 | The three-dimensional cubic mesoporous SBA-16 supported MnWO4 active component | Styrene | 7 | - | 27-75 | - | Epoxidation | [70] |
| Mn/g-C3N4 | Mn supported on g-C3N4 | Olefin | 11 | 39-83 | 0-99 | - | Epoxidation | [71] |
| Meso MnOx | Mesoporous manganese oxide | N-Heterocycle | 9 | - | 0-99 | - | Dehydrogenation of N-heterocycles | [72] |
| Ni2Mn-LDH | A heterogeneous catalyst based on NiMn layered double hydroxide (LDH) | Tetrahydroquinoline derivative | 16 | - | 39-93 | - | Dehydrogenation of N-heterocycles | [73] |
| 2[PW]-OMS-2 | OMS-2-based heterogeneous catalyst with a sodium phosphotungstate doping amount (2 mol%) | N-Heterocycle | 20 | - | 82-99 | - | Dehydrogenation of N-heterocycles | [74] |
| Na-AMO | The sodium-doped amorphous manganese oxide (Na-AMO) | N-Heterocycle | 28 | 50-95 | 82-99 | - | Dehydrogenation of N-heterocycles | [75] |
| Mn-SA@NC | The manganese single atoms anchored on nitrogen-doped carbon | Silane | 16 | 65-90 | - | - | Oxidation of silane | [76] |
| NR-MnO2 | A well-defined α-MnO2 crystal structure with a specific nanorod morphology | Nitrile | 10 | - | 99.5 | - | Hydration of nitriles | [77] |
| Mn@Starch-1000 | A heterogeneous manganese catalyst with pyrolyzed starch carbon as the support and a Mn/MnOx core-shell structure | Aniline and N-heterocycle | 24 | 61-99 | - | - | Deuteration | [78] |
| MnOx-100 | A vanadium-free manganese-based heterogeneous catalyst | Glucose | 1 | 81.1 | - | - | Conversion of glucose to formic acid (FA) | [79] |
| UAEU-50{Mn3(BDC)3} | A heterogeneous Mn-MOF catalyst constructed from trinuclear Mn clusters and phthalate (BDC) ligands | Epoxide | 1 | 90 | - | - | Reaction of epoxides and CO2 to cyclic carbonates | [80] |
| Mn_N_Graphene | MnOx nanoparticles supported on nitrogen-doped graphene oxide | HMF | 1 | 92 | - | - | Cleavage of C—O bond | [81] |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




































 下载:
下载: