Different natural iron-bearing minerals activated peroxymonosulfate for in-situ chemical oxidation process: Insights into the active Fe sites and activation mechanism
Citation:
Jinyan Yang, Ying Chen, Minghan Yu, Lan Zhu, Yilin Wang, Jingsong Chen, Hui Ma, Shengyan Pu. Different natural iron-bearing minerals activated peroxymonosulfate for in-situ chemical oxidation process: Insights into the active Fe sites and activation mechanism[J]. Chinese Chemical Letters,
2026, 37(7): 112210.
doi:
10.1016/j.cclet.2025.112210
Different natural iron-bearing minerals activated peroxymonosulfate for in-situ chemical oxidation process: Insights into the active Fe sites and activation mechanism
English
Different natural iron-bearing minerals activated peroxymonosulfate for in-situ chemical oxidation process: Insights into the active Fe sites and activation mechanism
State Key Laboratory of Geohazard Prevention and Geoenvironment Protection, Chengdu University of Technology, Chengdu 610059, China
b.
State Environmental Protection Key Laboratory of Synergetic Control and Joint Remediation for Soil&Water Pollution, Chengdu University of Technology, Chengdu 610059, China
Received Date:
11 June 2025 Accepted Date:
03 December 2025 Revised Date:
26 November 2025 Available Online:
15 July 2026
Abstract:In situ chemical oxidation (ISCO) technology using peroxymonosulfate (PMS) and natural iron-bearing minerals for groundwater remediation has received increasing interest. The interaction between PMS and active Fe sites in minerals significantly influences the effectiveness of groundwater remediation. Nevertheless, there has been limited research investigating the relationship between the minerals active Fe sites and PMS activation. Herein, we distinguished and quantified the active Fe sites of common natural iron-bearing minerals in groundwater aquifers. Lewis acid sites (Fe-OH) were confirmed as the reaction sites in iron oxide/hydroxide/bearing clay minerals. The activation performance of minerals is positively correlated with their Lewis acid content. In iron sulfide minerals, Fe-S sites act as electron transfer mediators, facilitating PMS adsorption and activation. The activation of PMS by Lewis acid and Fe-S sites free radical both led to the generation of free radicals (SO4·- and ·OH) for CPs removal. Moreover, typical ferrihydrite/PMS and pyrite/PMS systems exhibited resistance to environmental interference and broad pH adaptability. A one-dimensional sand column experiment further proved their feasibility and long-term applicability in saturated porous media. These findings highlight the critical influence of active Fe sites of natural iron-bearing minerals and provide technical support for the application of PMS-ISCO strategies for groundwater remediation.
Chlorophenols (CPs) are one of the most prevalent groundwater contaminants because of their widespread use in industry and agriculture as wood preservatives, herbicides, and fungicides [1,2]. In recent years, CPs pollutions have been commonly detected in groundwater [2,3]. For example, 2,4-dichlorophenol (2,4-DCP) has been detected in groundwater and aquifers of some contaminated sites at levels up to 10 mg/L [4]. Owing to their high toxicity, carcinogenicity, and persistence, CPs pose serious threats to both the environment and humans [5,6]. CPs have recently been designated as priority pollutants by the European Union, the United States, and China [7]. However, conventional physical, chemical, and biological methods are generally ineffective for the removal of CPs. Therefore, the development of efficient, sustainable, and economically viable technologies for the remediation of CPs-contaminants in groundwater has emerged as a critical focus in current environmental research.
In situ chemical oxidation (ISCO) is an economic and sustainable approach for remediating CPs-contaminated groundwater due to its rapid implementation, small environmental disturbance, and short restoration cycle [8,9]. Peroxymonosulfate (PMS) receives increasing attention owing to the formation of highly reactive radicals (SO4·- (E0 = 2.6–3.1 V) and ·OH (E0 = 1.9–2.7 V)). In addition, PMS exhibits wide pH adaptability and groundwater transport capability, where generated SO4·- and ·OH radicals mineralize CPs via bond cleavage to small molecule intermediates or end products (CO2 and H2O) [10,11]. In recent years, extensive research has demonstrated that carbon-based and iron-based materials can effectively activate PMS for the degradation of CPs pollutants [12-16]. Due to their low cost and environmentally friendly, iron-based materials have been considered as good catalysts for groundwater remediation [17-19]. Zero-valent iron (ZVI), as a PMS activator [20], has been applied for the degradation of CPs in groundwater. However, the passivation layer may form on the ZVI surface during storage and transport, which may restrict its reactivity. Nature iron-bearing minerals (such as iron (hydro) oxide mineral, iron-bearing clay minerals, and iron sulfide minerals) are widely distributed in geological formations, which can be utilized directly. Therefore, this eliminates the need for mining, storage, and transportation. Additionally, these natural iron-bearing minerals have the characteristics of abundant reserves, environmental friendliness, and stable catalytic performance. Moreover, these iron-bearing minerals exhibit greater resistance to inorganic constituents in groundwater compared to external Fe-based catalysts [21]. Therefore, exploring the potential of natural iron-bearing minerals in aquifers as PMS activator for in-situ remediation of polluted groundwater has become a hot research topic in environmental protection.
It is important to note that the activation of PMS by natural iron-bearing minerals primarily proceeds via heterogeneous catalysis [20,22], wherein the reaction predominantly occurs at the mineral-oxidant interface. This means that the provision of sufficient active sites of natural iron-bearing minerals plays a key role in the reaction. Fe sites are generally recognized as the primary reactive centers in the context of PMS activation by natural iron-bearing minerals. Therefore, it is crucial to identify the types of active Fe sites in natural iron-bearing minerals and investigate the mechanism of PMS interaction with various types of natural iron-bearing minerals. In general, Fe sites can be categorized into two types due to structural differences among natural iron-bearing minerals. The first type includes Lewis acid sites, such as Fe-OH [23,24]. Fe-OH structure primarily originates from two primary forms: Adsorbed water molecules on the mineral surface, and inherent hydroxyl groups bound within the mineral lattice. The corresponding reaction mechanism aligns with prior literature findings [25]. Specifically, PMS forms a surface complex with the mineral by displacing the hydroxyl group of the Fe-OH structure on the mineral surface. Then PMS accepts electrons from the Fe(Ⅱ)/Fe(Ⅲ) redox cycle within the complex, facilitating O—O bond cleavage and the generation of reactive oxygen species (ROS). The second type involves Fe-S sites, where the iron donates electrons and the sulfur accepts electrons [3,26]. According to previous studies [27], the ≡Fe(Ⅱ)···HSO5- is formed when the interaction between Fe(Ⅱ) and PMS. The ≡Fe(Ⅱ)···HSO5- could trigger the broken of O—O bond of PMS, accompanied by the generation of ≡Fe(Ⅲ) and SO4·-. Then, the sulfur sites of mineral accepts electrons to promote the Fe(Ⅲ)/Fe(Ⅱ) cycle. However, comparative studies on Fe sites-mediated PMS activation across different classes of natural iron-bearing minerals remain limited. Moreover, the active site content of different types of minerals has also been insufficiently explored. Therefore, it is essential to systematically investigate the influence of type of Fe site on the remediation of CPs-contaminated groundwater. Such an investigation will help to elucidate the underlying relationship between natural iron-bearing mineral structure and PMS activation efficiency, thereby advancing the rational design and application of natural iron-bearing minerals in ISCO for groundwater remediation.
In this study, the in situ activation of PMS by four common types of natural-bearing minerals (including iron oxide minerals, iron hydroxide minerals, iron-bearing clay minerals and iron sulfide minerals) for the removal of 2,4,6-trichlorophenol (2,4,6-TCP, a typical CPs compound) was investigated. Meanwhile, the catalytic PMS oxidation mechanism was explored by identifying the active Fe sites and quantifying the active site amount of different minerals. Additionally, the reactive species, the possible degradation pathways of 2,4,6-TCP, and the toxicity of the intermediate products were evaluated. Finally, the remediation capability of mineral-based ISCO for CPs in actual groundwater and porous media was investigated. In summary, our work provides a deeper understanding of PMS activation mechanisms by natural iron-bearing minerals and presents a scientific basis for material selection in the remediation of organically contaminated groundwater through PMS-ISCO technology.
All chemical reagents used in the experiment are shown in Texts S1 and S2 (Supporting information). All experimental methods are shown in Texts S3-S6 (Supporting information).
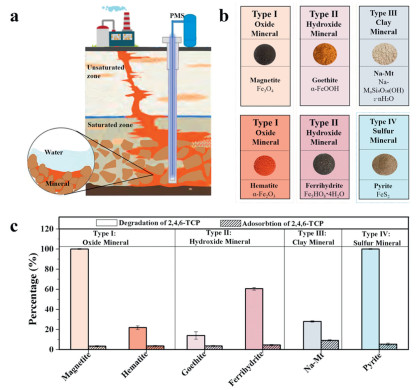
The schematic diagram of ISCO technology based on PMS and natural iron-bearing minerals for the remediation of polluted groundwater is shown in Fig. 1a. Four types of abundant natural iron-bearing minerals in aquifers were investigated. Photographs of the typical examples of these natural iron-bearing minerals (including iron oxide minerals (such as magnetite, hematite), iron hydroxide minerals (such as ferrihydrite, goethite), iron-bearing clay minerals (such as Na-Mt) and iron sulfide minerals (such as pyrite)) were shown in Fig. 1b. Natural iron sulfide minerals primarily include pyrite, mackinawite, and pyrrhotite. Mackinawite is unstable and readily transforms into pyrite, while pyrite constitutes the dominant form of iron sulfides in aquifers [28]. Therefore, research on iron sulfide minerals primarily focuses on pyrite. As shown in the physical image, different types of natural iron-bearing minerals exhibited distinct color variations. These samples exhibited their expected crystal structure and phase composition (Fig. S1 in Supporting information).
Figure 1
Figure 1.
(a) Schematic representation of the degradation of 2,4,6-TCP by different minerals. (b) Physical drawings of different minerals. (c) Adsorption and degradation of 2,4,6-TCP by different minerals/PMS system. Experimental conditions: T0 = 25 ± 2 ℃, [Minerals]0 = 1.0 g/L, 2,4,6-TCP = 8 mg/L, PMS = 2 mmol/L, pH 3.5.
The catalytic performance of the mentioned-above minerals activated PMS was further evaluated. As shown in Fig. S2a (Supporting information), the adsorption capacity of Na-Mt toward 2,4,6-TCP was about 10%, which was higher than that of other minerals (< 4%). Approximately 4% of 2,4,6-TCP was removed only in the presence of PMS within 8 h (Fig. S2b in Supporting information), suggesting that the self-decomposition capacity of PMS was incapable of effectively removing 2,4,6-TCP. The results suggest that both the removal of 2,4,6-TCP are negligible when only PMS or natural iron-bearing mineral exist in the reaction system. Notably, all minerals had a certain capacity for the removal of 2,4,6-TCP in the coexistence of 1.0 g/L natural iron-bearing minerals and 2.0 mmol/L PMS (Fig. 1c and Fig. S2b). For iron oxide minerals, magnetite/PMS system attained 100% degradation efficiency of 2,4,6-TCP within 5 h, whereas the hematite/PMS system was 21.9% within 8 h. For iron hydroxide minerals, the degradation efficiency of 2,4,6-TCP in goethite/PMS system was 14.1% within 8 h, while that of ferrihydrite/PMS system was 60.6% during the same period. For iron-bearing clay minerals, the degradation efficiency of 2,4,6-TCP in Na-Mt/PMS system was 28.1% within 8 h. For iron sulfide minerals, it was worth noting that pyrite/PMS system reached 100% degradation efficiency within 2 h. The catalytic performance and the responding pseudo-first-order kinetics showed that the reaction rate constant (kobs) value for magnetite/PMS, hematite/PMS, goethite/PMS, ferrihydrite/PMS, Na-Mt/PMS and pyrite/PMS systems were 9.56 × 10–3, 0.378 × 10–3, 0.275 × 10–3, 1.87 × 10–3, 0.633 × 10–3 and 18.08 × 10–3 min-1, respectively (Fig. S2c in Supporting information). Overall, pyrite exhibited the highest PMS activation capacity, followed by magnetite, and ferrihydrite. These three types of natural iron-bearing minerals show high effectiveness in removing 2,4,6-TCP pollution. In contrast, Na-Mt demonstrates a relatively lower PMS activation capacity.
Previous studies indicate that the released Fe ions from natural iron-bearing minerals may synergistically enhance activation of PMS through homogeneous reactions [29]. Therefore, it was essential to quantify the concentration of dissolved Fe in the solution (Table S1 in Supporting information). The results demonstrated that only pyrite and Na-Mt released measurable amounts of Fe2+. In contrast, no Fe2+ or Fe3+ was detected from goethite, only Fe3+ was released from the other minerals. For magnetite, the released Fe2+ may have been re-adsorbed onto the its surface due to its strong magnetic properties [30]. The homogeneous catalytic activation of PMS by different natural iron-bearing minerals was further evaluated based on the concentrations of released Fe2+ and Fe3+ (Fig. S3 in Supporting information). The results indicated that only 10% - 20% of 2,4,6-TCP was removed. These findings suggest that PMS activation by different natural iron-bearing minerals predominantly occurs through heterogeneous catalytic mechanism. Previous studies have reported that structural differences in natural iron-bearing minerals affect PMS activation [31]. Therefore, the observed differences in PMS activation performance among natural iron-bearing minerals mentioned-above may be attributed to type and content of active Fe sites.
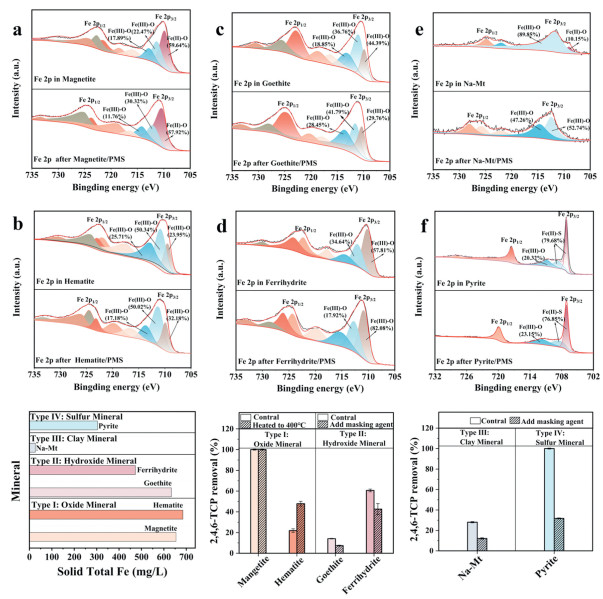
According to the previous study, Fe(Ⅱ) on iron-bearing minerals has stronger activity and ability to activate PMS than Fe(Ⅲ) [28]. Hence, X-ray photoelectron spectroscopy (XPS) was utilized to gain a deeper understanding of the role of Fe species in different natural iron-bearing minerals (Figs. 2a-f). The results showed that, goethite, ferrihydrite, and hematite primarily existed in the form of Fe(Ⅲ). Na-Mt and magnetite existed in the form of Fe(Ⅱ) and Fe(Ⅲ). Pyrite primarily existed in the form of Fe(Ⅱ), and the presence of Fe(Ⅲ) was likely attributed to oxidation during the analytical process [32]. Thus, pyrite exhibits the highest catalytic ability, which can be attribute to its higher proportion of Fe(Ⅱ) compared to magnetite and Na-Mt. Meanwhile, the iron content may also influence the catalytic capacity of minerals, the total Fe content in each mineral was quantified (Fig. 2g). The results showed that, iron concentration followed the order: hematite > magnetite > goethite > ferrihydrite > pyrite > Na-Mt. Note that although the proportion of Fe(Ⅱ) in ferrihydrite is relatively lower than that in Na-Mt, ferrihydrite has stronger ability to activate PMS than Na-Mt. This suggests that additional factors, such as types of reactive site, may also influence the catalytic activity of natural iron-bearing minerals.
Figure 2
Figure 2.
(a-f) XPS patterns of Fe 2p before and after the reaction. (g) Solid Total Fe of minerals. (h, i) Active site exploration experiment for different systems. Experimental conditions: T0 = 25 ± 2 ℃, [Minerals]0 = 1.0 g/L, 2,4,6-TCP = 8 mg/L, PMS = 2 mmol/L, pH 3.5. Magnetite and hematite were heated up to 400 ℃ for 12 h; [PO43-]0 = 4 mmol/L for goethite and ferrihydrite; [pyrrole]0 = 0.1 mol/L for Na-Mt; [S-]0 = 2 mmol/L for pyrite.
Multiple characterization techniques were employed to further investigate the active sites of natural iron-bearing minerals (Figs. 2h and i and Figs. S4a-e in Supporting information). The degradation rate and efficiency of 2,4,6-TCP in iron oxide minerals/PMS system was significantly enhanced after calcination at 400 ℃. This results are probably because thermal treatment would remove oxygen to cause the re-exposure of Fe sites of iron oxide minerals, which is available to adsorb more PMS [24]. Note that the degradation rate and efficiency of 2,4,6-TCP declined both in the goethite/PMS and ferrihydrite/PMS systems when phosphate was added. This decline in degradation performance was likely due to phosphate coordinating with Fe-OH groups, thereby inhibiting their function as Lewis acid sites [33]. Moreover, the degradation rate and efficiency of 2,4,6-TCP decreased upon the addition of pyrrole in the Na-Mt/PMS system. This decline is attributed to the competitive binding of pyrrole with PMS at Fe sites on the surface of Na-Mt [34]. Meanwhile, as shown in the XPS analyses (Figs. 2a-e), the Fe-O ratios in the iron oxide, iron hydroxide and iron-bearing clay minerals decreased significantly after the reaction. The result hints that the catalytic reaction predominantly occurred on the Fe-O sites of the three types of minerals. Furthermore, the degradation rate and efficiency of 2,4,6-TCP decreased in the pyrite/PMS system upon the addition of Na2S·9H2O. This decline was attributed to the incorporation of Na2S·9H2O, which led to the competitive adsorption of H2S onto the surface of pyrite via molecular interactions. In contrast, as shown in the XPS analyses (Fig. 2f), the Fe-S ratios in pyrite decreased significantly after activation reaction, indicating that Fe-S sites of pyrite were the main reactive sites. Additionally, previous studies have confirmed that Fe(Ⅱ) in pyrite can act as an electron donor and undergo oxidation to Fe(Ⅲ), while sulfur species (e.g., S22-) serve as electron acceptors, achieving iron cycle process during reaction. This dynamic establishes a sustainable Fe(Ⅱ)/Fe(Ⅲ) redox cycle that drives PMS activation [35]. Thus, the different catalytic performance of natural iron-bearing minerals toward PMS activation can be attributed to different types of their active sites. The degradation capacity is primarily associated with Lewis acid sites of iron oxide minerals, iron hydroxide minerals, and iron-bearing clay minerals. In contrast, the catalytic activity of sulfur-containing minerals (such as pyrite) is mainly governed by Fe-S sites.
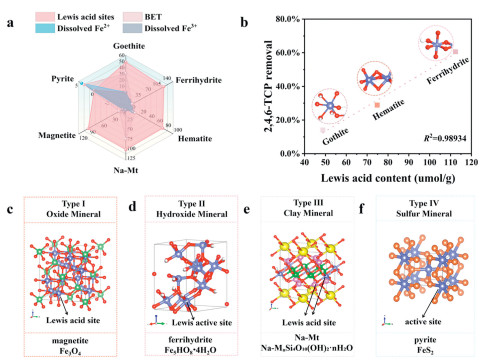
We established that the active sites of natural iron-bearing minerals (including iron oxide minerals, iron hydroxide minerals, and iron-bearing clay minerals) were primarily assigned to Lewis acid sites (Fig. 3a). The structural diagrams of Lewis acid sites within minerals mentioned-above were illustrated in Figs. 3c-f and Figs. S5a and b (Supporting information). Considering the potential link between degradation efficiency of 2,4,6-TCP and the Lewis acidity of natural iron-bearing minerals, Py-IR (Pyridine-infrared spectroscopy) technique was used to analyze the surface acid properties of these minerals [24]. As shown in Figs. S6a-c (Supporting information), the peaks at 1445 cm-1 and 1596 cm-1 corresponded to the chemisorption of pyridine at Lewis acid sites, while the peak at 1490 cm-1 corresponded to Brønsted acid sites [36,37]. These results suggested that the acidity of the minerals mentioned-above is predominantly attributed to Lewis acid sites because of relatively low Brønsted acid content.
Figure 3
Figure 3.
(a) Radar charts of different nature iron-bearing minerals. (b) Linear correlation plot between Lewis acid content and degradation capacity. (c-f) Schematic diagram of active sites of different nature iron-bearing minerals.
According to Table S2 (in Supporting information), the Lewis acid content of minerals followed the order: ferrihydrite > Na-Mt > magnetite > hematite > goethite. Higher Lewis acid content means that more PMS can be adsorbed on the surface of those minerals. This observation explained why ferrihydrite exhibited higher PMS activation capacity than Na-Mt, hematite, and goethite. As shown in Fig. 3b, a significant linear correlation (R2 = 0.98934) was observed between the Lewis acid content and the 2,4,6-TCP degradation efficiency in goethite/PMS, hematite/PMS, and ferrihydrite/PMS systems. This finding further supports the conclusion that higher Lewis acid content enhances the removal efficiency of 2,4,6-TCP. These three minerals contain only Fe(Ⅲ) based on the XPS analysis, indicating that the observed linear relationship between Lewis acid content and PMS activation capacity was primarily attributable to the properties of Fe(Ⅲ) minerals. Na-Mt has no linear correlation with catalytic activity, this might be related to physical adsorption mechanism. Furthermore, there is no linear correlation between the catalytic activities of magnetite and Lewis acid content. The finding is probably because its strong magnetic properties make the particles prone to agglomeration [30,38], thus affecting the exposure of Lewis acid sites on its surface. Thus, except for magnetite and Na-Mt, the catalytic activity of iron oxide/hydroxide/bearing clay minerals showed a significant linear correlation with the Lewis acid content and PMS activation capacity.
In summary, the variation in degradation performance of 2,4,6-TCP among natural iron-bearing minerals is primarily attributed to differences in Lewis acid sites and Fe-S sites. In other words, the catalytic activity of Fe(Ⅲ)-dominant minerals (e.g., ferrihydrite) is primarily governed by Lewis acid content. Whereas, Fe-S sites play the predominant role for Fe(Ⅱ)-dominant minerals (e.g., pyrite).
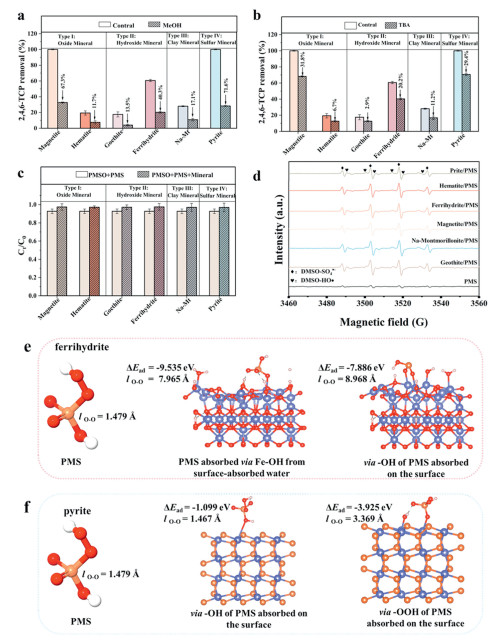
To explore the mechanism of PMS activation by the four types of natural iron-bearing minerals mentioned above, quenching experiment was initially adopted to identify the contribution of potential ROS to 2,4,6-TCP degradation. Previous research has identified the potential ROS, including sulfate radicals (SO4·-), hydroxyl radicals (·OH), superoxide radicals (O2·-), and singlet oxygen (1O2), within mineral-activated PMS systems [30,39]. MeOH is regarded to be a quencher of ·OH (9.7 × 108 L mol-1 s-1) and SO4·- (2.5 × 107 L mol-1 S-1) [40], while TBA (tert-butyl alcohol) is considered as a quencher of ·OH (6.0 × 108 L mol-1 s-1) [40-42]. FFA (furfuryl alcohol), as a specific probe detecting 1O2 (1.2 × 108 L mol-1 s-1), is used to identify the 1O2 generation [43-45]. While p-BQ is used to be a quencher of O2·- (0.9 × 109 L mol-1 s-1) [40,46,47]. Accordingly, MeOH (0.5 mol/L), TBA (0.5 mol/L), FFA (0.5 mmol/L), and p-BQ (0.5 mmol/L) were selected in each quenching experiment. As shown in Figs. 4a and b, the degradation of 2,4,6-TCP was significantly inhibited when the addition of MeOH and TBA in the mineral/PMS system. These results confirm that SO4·- and ·OH are the dominant reactive species in the mineral/PMS system. Importantly, there was no significant change in 2,4,6-TCP removal efficiency with the addition of FFA and p-BQ (Figs. S7a and b in Supporting information), suggesting that O2·- and 1O2 were scarcely present in the iron-bearing minerals/PMS system. Previous studies have reported that the activity of the process mainly comes from the generated reactive oxygen species (ROS), such as SO4·-, ·OH, O2·-, and 1O2, via the breakage of the peroxide (O—O) bond in the PMS structure. SO4·- (k = 3.0 × 104 L mol-1 s-1) and ·OH (k = 1.6 × 105 L mol-1 s-1) are suggested to be the predominant reactive oxygen species responsible for pollutant degradation under acidic conditions. It is generally accepted that the low reactivity of O2·- toward organic pollutants. In addition, 1O2 production associated with PMS decay under alkaline conditions (HSO5- + SO52− → HSO4- + SO42- + 1O2) [30,39]. However, our system maintains a nearly acid environment. Therefore, there is negligible contributions form O2·- and 1O2 in the mineral/PMS systems. In summary, it is shown that SO4·- and ·OH, but not O2·- and 1O2, play an important role in the natural iron-bearing minerals/PMS system.
Figure 4
Figure 4.
(a) Effect of MeOH, (b) TBA, (c) PMSO on 2,4,6-TCP degradation of different systems. (d) EPR characterization results of different systems. (e-f) DFT configurations on PMS before and after adsorption onto ferrihydrite and pyrite. Experimental Conditions: T0 = 25 ± 2 ℃, PMS = 2 mmol/L, [Minerals]0 = 1.0 g/L, 2,4,6-TCP = 8 mg/L, [tert-butanol]0 = [Methanol]0 = 0.5 mol/L, [PMSO]0 = 4.0 mmol/L, pH 3.5.
According to previous reports, ferryl ion species (Fe(Ⅳ)) may be produced in the minerals/PMS system, thus affecting the removal of 2,4,6-TCP [48]. To examine the existence of Fe(Ⅳ), PMSO probing test was performed because PMSO reacted with Fe(Ⅳ) in the mineral/PMS system. As shown in Fig. 4c, approximately 10% consumption of PMSO when PMS was present alone. However, the consumption of PMSO decreased rather than increased when minerals were added. The consumption of PMSO would been enhanced through Fe(Ⅳ)-mediated oxidation if Fe(Ⅳ) were indeed present. However, this phenomenon was not observed, indicating the absence of Fe(Ⅳ). This was because the production of Fe(Ⅳ) was negligible under acidic conditions. This conclusion aligned with previous research findings [49], further supporting the above-mentioned mechanistic insights into PMS activation by natural iron-bearing minerals.
To further elucidate the generation of reactive oxygen species (ROS), the DMPO and TEMP trapped electron paramagnetic resonance (EPR) tests were conducted. As shown in Fig. 4d, weak signals of DMPO-·OH and DMPO-SO4·- adducts were detected in the EPR spectrum in the PMS solution alone. This result was aligned with the observation that PMS alone exhibits weak degradation capability. In contrast, obvious signals of DMPO-·OH and DMPO-SO4·- adducts were detected in the different natural iron-bearing minerals/PMS system, confirming that both ·OH and SO4·- were generated. Notably, no signal of DMPO—O2·- and TEMP-1O2 were observed, suggesting the absence of O2·-and 1O2 was absent in the mineral/PMS system.
The formation mechanisms of SO4·- and ·OH can be attributed to the reaction pathways outlined in Eqs. 1–15. In summary, the findings provide critical insights into the ROS involved in the mineral/PMS system from both the quenching experiments and EPR spectroscopy. The results demonstrate that four types of minerals-activated PMS systems are predominantly governed by SO4·- and ·OH, with negligible contributions from O2·-, 1O2, and Fe(Ⅳ). Consequently, the degradation of 2,4,6-TCP in the natural iron-bearing systems primarily proceeds via free radical pathways. To elucidate the types of active Fe sites and the mechanism of PMS interaction with different natural iron-bearing minerals, density functional theory (DFT) calculations were performed. The adsorption energy (Ead) of a PMS molecule onto the active Fe site of mineral was calculated (Figs. 4e and f). Ferrihydrite, characterized by high Lewis acidity, and pyrite, dominated by Fe-S active sites, were selected for comparative analysis. The results revealed that ferrihydrite formed ≡Fe(Ⅲ)-(OH)SO4 complexes with PMS through Fe-OH groups derived from surface-adsorbed water, rather than from the -OH moiety of PMS. (-9.535 eV for -OH on the mineral surface and -7.886 eV for -OH of PMS). Pyrite preferentially bonded with -OOH rather than -OH of PMS molecule, forming ≡Fe(Ⅱ)···HSO5- complexes (-3.925 eV for -OOH on the PMS and -1.099 eV for -OH on the PMS). Furthermore, to elucidate the difference in PMS activation between the two minerals, the O—O bond length (lO—O) of PMS before and after adsorption was calculated.
The lO—O of free PMS molecule was 1.479 Å, whereas it significantly increased upon adsorption onto both minerals, indicating effective PMS activation. Notably, PMS adsorbed on ferrihydrite exhibited longer lO—O value (7.965 Å for -OH on the mineral surface, 8.968 Å for -OH on the PMS) than those on pyrite (1.467 Å for -OOH of PMS, 3.369 Å for -OH of PMS), suggesting stronger bond weakening by ferrihydrite. This finding explains the strong catalytic ability of ferrihydrite even though it is a trivalent iron mineral. Based on the above theoretical investigations, it can be inferred that both ferrihydrite and pyrite enhance the adsorption and activation ability of Fe sites toward PMS, thereby boosting catalytic activity.
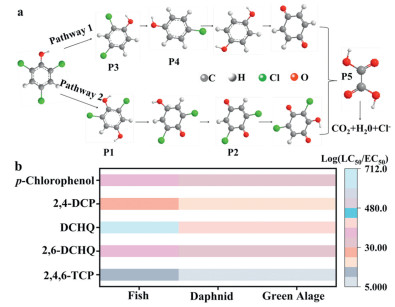
To identify the possible degradation pathways of 2,4,6-TCP in the mineral/PMS system, the intermediates were monitored via GC–MS. As shown in Fig. 5a and Fig. S8 (Supporting information), five intermediates of 2,4,6-TCP degradation were recognized. In Pathway 1, the Cl- in the adjacent position of 2,4,6-TCP was lost to form 2,4-dichlorophenol (P3) after 2,4,6-TCP attacked by ROS (SO4·- and ·OH) [50]. Subsequently, 2,4-dichlorophenol was further dechlorinated to generate 4-chlorophenol (P4) [51]. In Pathway 2, ROS (SO4·- and ·OH) induce hydroxylation of 2,4,6-TCP, resulting in the formation of 2,6-dichloro-1,4-benzenediol (P1). At the same time, one hydroxyl group on the benzene ring was readily oxidized to a carbonyl group to generate the intermediate product P2 [52]. These intermediate products were gradually converted to oxalic acid (P5), followed by the C—C bond breaking and ring-opening reactions [53]. Eventually, these intermediate products were mineralized into CO2 and H2O, with a mineralization and dechlorination efficiency of approximately 50% (Figs. S9a and b in Supporting information). These findings confirm the feasibility of the PMS-ISCO approach for effective mineralization of 2,4,6-TCP.
Figure 5
Figure 5.
(a) Possible degradation pathways of 2,4,6-TCP. (b) Toxicological evaluation of intermediates.
Evaluating the residual toxicity of degradation intermediates was critical to ensuring the environmental safety of the applied remediation technology. The 2,4,6-TCP toxicity and its degradation intermediates were analyzed using the Ecological Structure Activity Relationship Model (Ecosar v2.2) published by the United States Environmental Protection Agency [54]. Fig. 5b showed that degradation intermediate P1-P4 demonstrated harmful effects (LC50/EC50: 10–100 mg/L range). However, P5 as the other degradation intermediate, was relatively harmless (LC50/EC50 > 100 mg/L). In other words, almost all the intermediates show lower toxicity than 2,4,6-TCP. Consequently, the mineral/PMS system has proved to be an efficient and safe technology for 2,4,6-TCP degradation. This approach not only effectively eliminated 2,4,6-TCP but also reduced the potential secondary pollution of groundwater environments by minimizing the biotoxicity of the degradation intermediates. The results further highlight the potential of this technique as a viable PMS-ISCO strategy for sustainable groundwater remediation.
Based on the analysis of active Fe sites, ferrihydrite, the mineral with the highest Lewis acid catalytic ability, and pyrite, the mineral based on Fe-S active sites, were selected as comparative analysis. The effect of the mineral dosage and PMS concentration on the degradation of 2,4,6-TCP in the ferrihydrite/PMS and pyrite/PMS systems were investigated. As shown in the Fig. S10a (Supporting information), the degradation efficiency of 2,4,6-TCP increased significantly with ferrihydrite dosage, which was attributed to the fact that higher ferrihydrite dosage could provide more Lewis acid sites for PMS activation. Conversely, nearly 100% degradation of 2,4,6-TCP was achieved across a wide range of dosage (0.5–5.0 g/L) in the pyrite/PMS system, indicating the exceptional catalytic efficiency of pyrite. The effects of the amount of initial PMS were systematically investigated as well, and the result was shown in Fig. S10b (Supporting information). The results clearly indicated that the degradation efficiency of 2,4,6-TCP both in the ferrihydrite/PMS and pyrite/PMS systems were significantly enhanced with the increase of PMS concentration. This phenomenon indicates that increasing the PMS concentration enhances the generation of radicals (SO4·- and ·OH), thus leading to accelerated degradation of 2,4,6-TCP. In addition, it was observed that the pyrite/PMS system showed better degradation ability. Previous studies have reported that, ≡Fe2+ transfers an electron to PMS to generate SO4·-, ·OH and ≡Fe3+ (k = 3.0 × 104 L mol-1 s-1). However, due to the rate-limiting reaction between PMS and ≡Fe3+ (k = 2–5 L mol-1 s-1), the generation of ROS is sluggish and the degradation of pollutants is limited Fe(Ⅱ) [55]. Therefore, pyrite exhibits better catalytic ability relative to ferrihydrite.
The pH conditions at groundwater contamination sites may affect the degradation efficiency of 2,4,6-TCP [56]. Therefore, the 2,4,6-TCP degradation at different solution pH (3.0–9.0) in the ferrihydrite/PMS and the pyrite/PMS system was investigated. As shown in Fig. S10c (Supporting information), the degradation efficiency of 2,4,6-TCP in acidic solutions is lower than that in alkaline solutions both in the ferrihydrite/PMS and pyrite/PMS systems. The surface charge of minerals depends on their pHzpc and the solution pH [29]. The pHzpc of ferrihydrite, pyrite and other minerals mentioned above was negatively charged as shown in Table S3 (Supporting information). PMS is negatively charged at pH (3.0–9.0) because the pKa2 is about 9.4 [57]. Therefore, under acidic conditions, the reduced electrostatic repulsion between ferrihydrite and PMS decreased their effective contact. As a result, ROS (SO4·- and ·OH) generation was inhibited. Furthermore, previous studies demonstrated that pyrite can effectively buffers solution pH, explaining its stable 2,4,6-TCP degradation performance across a wide pH ranges [20]. These findings indicate that both the ferrihydrite/PMS and the pyrite/PMS system exhibit effective degradation capabilities for 2,4,6-TCP over a wide pH range.
Environmental groundwater is always a complex system, including HCO3-, Cl-, NO3- and SO42- [58].These anions may ultimately affecting PMS activation [59,60]. As shown in Fig. S10d (Supporting information), the degradation efficiency of 2,4,6-TCP had negligible effects in both systems upon the addition of SO42- and NO3- respectively. In contrast, the presence of HCO3- and Cl- led to 100% removal of 2,4,6-TCP in both systems. This enhancement may be attributed to pH modification by HCO3- and the formation of highly oxidative hypochlorous acid via the reaction between Cl- and PMS [61]. The relevant reaction equations are shown in Eqs. 16–19. These results demonstrate that both ferrihydrite/PMS and pyrite/PMS systems maintain effective 2,4,6-TCP degradation in the presence of major co-existing anions, highlighting their potential for groundwater remediation.
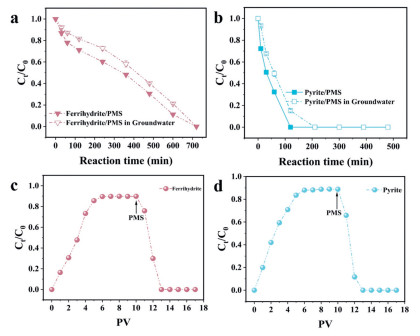
It is essential to evaluate the effectiveness of these systems in degrading 2,4,6-TCP in real groundwater. Therefore, the real groundwater samples were collected from a certain automobile waste recycling station in Neijiang City, Sichuan Province. The water quality parameters were shown in Table 1. The 2,4,6-TCP degradation capabilities of both systems for actual contaminated groundwater were shown in Figs. 6a and b. The degradation rate and efficiency of 2,4,6-TCP in the ferrihydrite/PMS and pyrite/PMS system was weakened compared to the laboratory results. This reduced performance likely resulted from competitive PMS consumption by co-existing organic pollutants (such as petroleum) in the groundwater matrix (Table S4, Fig. S10d, Fig. S11a and b in Supporting information). Both the two mineral/PMS system still showed a more satisfactory degradation performance despite the removal efficiency of 2,4,6-TCP decreased in the real groundwater. In addition, after the nearly 100% removal of 2,4,6-TCP, the ferrihydrite/PMS system exhibited lower PMS consumption compared to the pyrite/PMS system. This difference can be attributed to the fast decomposition of PMS and radical quenching each other at high dosage of pyrite. Hence, the ferrihydrite/PMS system is more suitable than the pyrite/PMS system for the remediation of organic pollutants in groundwater by PMS-ISCO technology. In addition, one-dimensional column simulations were performed to evaluate the degradation of 2,4,6-TCP in saturated porous media. The one-dimensional column simulation experiment setup is shown in Fig. S12 (Supporting information). As shown in Figs. 6c and d, the concentration of 2,4,6-TCP increased after pumping 2,4,6-TCP alone and reached a stable state at 6 pore volumes (PV) in the quartz sand/ferrihydrite and quartz sand/Pyrite system.
Figure 6.
(a, b) The degradation rate and efficiency of 2,4,6-TCP in real groundwater for ferrihydrite/PMS and pyrite/PMS system. (c, d) Degradation of 2,4,6-TCP in one-dimensional column experiment. Experimental conditions: T0 = 25 ± 2 ℃, [Minerals]0 = 1.0 g/L, 2,4,6-TCP = 8 mg/L, PMS = 2 mmol/L, pH 3.5.
The concentration of 2,4,6-TCP decreased rapidly after pumping 2 mmol/L PMS at the same flow rate. Both the ferrihydrite/PMS and pyrite/PMS systems accomplished 100% 2,4,6-TCP degradation at 13 PVs. This phenomenon was attributed to the efficient activation of PMS by ferrihydrite and pyrite, generating SO4·- and ·OH radicals. This was consistent with the phenomena observed in the laboratory. In summary, the ferrihydrite/PMS and pyrite/PMS system exhibit remarkable degradation effects on 2,4,6-TCP in saturated porous media through the one-dimensional column degradation experiments. Hence, the ferrihydrite/PMS and pyrite/PMS system possess extensive application in the groundwater remediation by PMS-ISCO technology.
In this study, four different types of natural iron-bearing minerals (iron oxide minerals, iron hydroxide minerals, iron-bearing clay minerals, and iron sulfide minerals) were employed to activate PMS for 2,4,6-TCP degradation. The variation in catalytic performance of natural iron-bearing minerals in PMS-mediated degradation of 2,4,6-TCP was primarily governed by differences in Lewis acid sites and Fe-S sites, which arise from variations in Fe site characteristics. Among them, Fe(Ⅲ) minerals (e.g., ferrihydrite) were dominated by Lewis acid, and the higher the Lewis acid content, the stronger the ability to catalyze PMS. Iron sulfide minerals, such as pyrite, are governed by Fe-S sites. Additionally, the degradation pathway of 2,4,6-TCP and the toxicity of its intermediates were evaluated. The intermediates were significantly less toxic, with mineralization and dechlorination efficiencies reaching approximately 50%. The degradation performance of 2,4,6-TCP was further investigated under real groundwater and saturated porous media conditions using the ferrihydrite/PMS and pyrite/PMS systems. Both systems demonstrated excellent removal efficiency, confirming their practical applicability in field-relevant environments. Building on the above findings, this study not only advanced the mechanistic understanding of PMS activation by natural iron-bearing minerals but also offers robust technical support for the application of PMS-ISCO technology in remediating 2,4,6-TCP-contaminated groundwater.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This research was supported by the National Natural Science Foundation of China (No. U22A20591), the National Key Research and Development Program of China (No. 2024YFC3712700), and the Research Fund of State Key Laboratory of Geohazard Prevention and Geoenvironment Protection (No. SKLGP2020Z002).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.112210.
[1]
A. Peng, J. Gao, Z. Chen, et al., Environ. Sci. Tech. 52 (2018) 5208–5217. doi: 10.1021/acs.est.7b06664
Figure 1
(a) Schematic representation of the degradation of 2,4,6-TCP by different minerals. (b) Physical drawings of different minerals. (c) Adsorption and degradation of 2,4,6-TCP by different minerals/PMS system. Experimental conditions: T0 = 25 ± 2 ℃, [Minerals]0 = 1.0 g/L, 2,4,6-TCP = 8 mg/L, PMS = 2 mmol/L, pH 3.5.
Figure 2
(a-f) XPS patterns of Fe 2p before and after the reaction. (g) Solid Total Fe of minerals. (h, i) Active site exploration experiment for different systems. Experimental conditions: T0 = 25 ± 2 ℃, [Minerals]0 = 1.0 g/L, 2,4,6-TCP = 8 mg/L, PMS = 2 mmol/L, pH 3.5. Magnetite and hematite were heated up to 400 ℃ for 12 h; [PO43-]0 = 4 mmol/L for goethite and ferrihydrite; [pyrrole]0 = 0.1 mol/L for Na-Mt; [S-]0 = 2 mmol/L for pyrite.
Figure 3
(a) Radar charts of different nature iron-bearing minerals. (b) Linear correlation plot between Lewis acid content and degradation capacity. (c-f) Schematic diagram of active sites of different nature iron-bearing minerals.
Figure 6
(a, b) The degradation rate and efficiency of 2,4,6-TCP in real groundwater for ferrihydrite/PMS and pyrite/PMS system. (c, d) Degradation of 2,4,6-TCP in one-dimensional column experiment. Experimental conditions: T0 = 25 ± 2 ℃, [Minerals]0 = 1.0 g/L, 2,4,6-TCP = 8 mg/L, PMS = 2 mmol/L, pH 3.5.

 DownLoad:
DownLoad:

 下载:
下载:







 下载:
下载:
