Citation:
Meng Zhang, Junjuan Shi, Yu Feng, Boyan Liu, Jie Jiang, Daqian Song, Yanxiao Jiang. Advancements in pretreatment strategies: A comprehensive overview of desalination techniques for enhanced mass spectrometry analysis[J]. Chinese Chemical Letters,
2026, 37(7): 112208.
doi:
10.1016/j.cclet.2025.112208

Advancements in pretreatment strategies: A comprehensive overview of desalination techniques for enhanced mass spectrometry analysis
English
Advancements in pretreatment strategies: A comprehensive overview of desalination techniques for enhanced mass spectrometry analysis
Abstract:
Mass spectrometry is a highly sensitive and precise analytical technique widely utilized in biomedical research, drug development, and environmental monitoring. Nonetheless, before mass spectrometry analysis, complex matrix samples frequently harbor numerous salts, which may compromise the stability and accuracy of mass spectrometry signals. Hence, it is imperative to employ effective desalting techniques, which serve as critical pretreatment steps in sample preparation for mass spectrometry analysis. While numerous scholars have developed desalting techniques, a comprehensive overview of such techniques is lacking. This paper provides a summary of desalination methods for mass spectrometry analysis developed since the twenty-first century (including extraction techniques, chromatography, ion exchange, crystallography, ambient ionization mass spectrometry and desalting agents, etc.), along with their principles, advantages, and disadvantages. Furthermore, recent research advancements and technological enhancements are discussed to introduce novel ideas and methodologies for sample desalting as a pretreatment step before mass spectrometry analysis. Against the backdrop of the growing importance of precision medicine and environmental monitoring, the continuous improvement of desalination technology will promote its wide application in areas such as clinical diagnosis, food safety testing, and environmental pollution assessment.
-
1. Introduction
Mass spectrometry (MS) is a crucial analytical technology due to its outstanding sensitivity, high resolution, and broad application range [1]. Its sensitivity allows for the detection of trace compounds in samples, thereby fulfilling pivotal roles in biomedicine, environmental science, food safety, and beyond [2]. Simultaneously, its exceptional resolution accurately discriminates minute differences in compounds, providing a reliable approach for drug development, pathological diagnosis, environmental monitoring, and more [3]. From drug analysis to protein identification, from environmental pollution monitoring to food safety detection, its significance is importance [4].
However, there are still several challenges on the road to MS research that affect the development and application of the field to some extent [5]. More generally, the presence of salts in a sample can lead to interfering ion signals with the molecule to be measured, and high salt concentrations lead to ion suppression and adduct formation, which masks or complicates the ion signal [6]. Removal of salts reduces these interfering signals and improves the reliability of the analyzed data. In addition, the accumulation of salts negatively affects the performance and stability of the mass spectrometer, which can be protected and prolonged by desalting [7]. Therefore, effective desalting prior to MS analysis is essential for maintaining instrument stability and improving data quality [8].
Over the years, numerous desalting techniques have been developed and refined to cater to the diverse requirements of mass spectrometry applications [9,10]. Presently, high-performance liquid chromatography (HPLC) stands as the predominant and extensively utilized technique, owing to its desalting proficiency and distinctive capability to segregate the target analytes from numerous co-existing interferences [11]. Various LC-related devices have been developed to suit different matrix and material types, including precolumns for sample enrichment and desalting, as well as analytical columns for separation [12]. Furthermore, a range of offline desalting techniques, including ion exchange, utilization of desalting agents, crystallization, ambient ionization mass spectrometry (AIMS) and application of hydrophobic materials, have been employed for rapid salt removal, owing to their broad applicability and versatility [13–19].
In recent years, although numerous innovative techniques have emerged, including new materials and ambient ionization methods, a systematic summary of these advancements is lacking [20,21]. In this review, we provide a comprehensive overview of desalting techniques developed for MS sample preparation since the 21st century. We focus on their working principles, advantages, limitations, and recent technological developments. Methods designed for non-MS purposes, such as dialysis, ultrafiltration, reverse osmosis, and large-scale thermal desalination, are beyond the scope of this article. By critically evaluating the performance and application scenarios of different desalting approaches, we aim to guide researchers in selecting and optimizing suitable pretreatment strategies.
2. Extraction techniques
2.1 Liquid-liquid extraction (LLE)
LLE is one of the most widely used sample preparation methods for pre-mass spectrometry desalination. Conventional LLE desalting relies on a significant difference in the partitioning behavior of the target analyte and the salt ions between two immiscible phases. However, conventional LLE usually requires relatively large amounts of organic solvent and sample volume, and the extraction is often followed by a solvent evaporation and concentration step. To overcome the problems of traditional LLE, some new technologies have emerged, such as liquid-phase microextraction (LPME) and dispersive liquid-liquid microextraction (DLLME).
The core of LPME lies in the dramatic reduction of the volume of the extraction solvent to micro- or even nano-volumes. In single-drop microextraction mode, a small drop of organic solvent is suspended from the tip of a micro-syringe needle and immersed into a stirred aqueous solution of a salt-containing sample or placed on top of the sample solution for extraction [22]. Hollow fiber liquid phase microextraction utilizes a porous hydrophobic hollow fiber as support, whose lumen is saturated with an organic solvent to form a "liquid film", and a small amount of the receiving phase can be injected into the lumen of the fiber [23]. The target analytes in the sample cross the sample-organic solvent interface by diffusion and are ultimately enriched in the microfilmed organic solvent droplets or the receiver phase inside the fiber lumen [24]. The selectivity of the LPME can be achieved by selecting the extraction solvents, adjusting the pH of the samples, and adding chelating agents or ion-pairing reagents [25].
DLLME, as another highly efficient microextraction technique, has been rapidly developing in the field of desalination for mass spectrometry sample pretreatment in recent years. The most important advantages of DLLME are its extremely high enrichment factor, fast extraction kinetics, and simplicity of operation [26]. For desalting applications, the DLLME process itself effectively excludes salts from the trace organic extraction phase that is ultimately enriched with the target. It is worth noting that the high salt concentration in the sample solution can produce a "salting-out effect", which reduces the solubility of organic compounds in the aqueous phase [27]. DLLME has been successfully applied to the desalting and enrichment of organic pollutants in environmental water samples [28], as well as drugs and their metabolites in biological fluids [29]. However, for strongly hydrophilic or charged compounds, which is a common problem of LLE technology, the partition coefficients in conventional organic solvents are low, and more specific extraction systems need to be explored.
2.2 Solid-phase extraction (SPE)
SPE is a sample preparation technique that separates analytes from complex matrices through adsorption onto a solid sorbent, followed by elution with a solvent [30]. The choice of stationary phase material significantly impacts SPE performance. Traditional materials, such as C18 reversed-phase media and cation exchange resins, effectively remove matrix interferences but offer limited selectivity [31]. To address this limitation, novel materials like graphitized carbon black [32], molecularly imprinted polymers [33] and nanomaterials [34] have been developed. These advanced materials enhance target enrichment by leveraging improved hydrophobicity, electrostatic interactions, or specific molecular recognition. For instance, a study using graphitized carbon for solid-phase extraction enabled the sensitive and rapid analysis of paralytic shellfish toxins in highly saline seawater, demonstrating the potential of high selectivity, cost-effective, efficient desalting [35].
Solid-phase microextraction (SPME), a miniaturized branch of SPE, is particularly notable for its minimal sample requirements and high efficiency in handling micro-volume samples [36]. At the core of SPME is the use of miniature adsorbent materials tailored to capture specific target molecules. Common materials include agarose beads [37], C4 or C18 particles [38], polymer nanobeads [39], and carbon nanotubes [40]. Selection of these materials is based on the chemical properties of the analytes to optimize separation efficiency further. Among these, poly(methyl methacrylate) (PMMA) nanospheres have gained considerable attention for their excellent desalting and peptide enrichment capabilities. PMMA nanospheres exhibit weak interactions with inorganic salts, enabling high-quality MS signals even in high-salinity matrix. This capability enhances flexibility in sample handling and significantly reduces experimental costs [41].
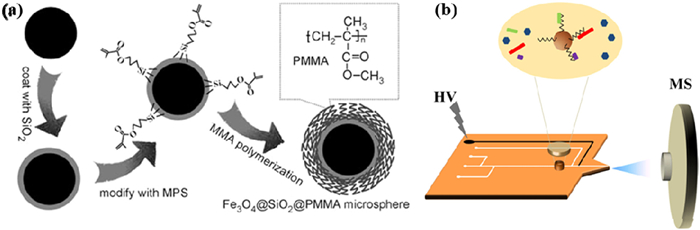
The integration of magnetic materials into SPE has significantly expanded its capabilities, leading to the development of magnetic solid-phase microextraction (MSPME). This innovative approach combines efficient analyte enrichment with convenient separation by utilizing magnetic beads coated with functional materials, such as PMMA-coated Fe3O4@SiO2 magnetic beads (Fig. 1a). These magnetic microspheres can rapidly capture target analytes from complex matrix, and their separation and recovery are facilitated by an applied magnetic field. Compared to traditional SPE techniques, MSPME offers simplicity, speed, and enhanced methodological flexibility. For example, the use of magnetic beads for peptide enrichment has proven effective in excluding salt interference, thus improving the sensitivity of MS analysis while also reducing experimental time [42].
Figure 1

Building on MSPME, researchers have developed innovative hybrid techniques, such as microwave-assisted extraction [43] and micropipette solid-phase extraction tip technology [44]. These advancements enhance the speed and efficiency of sample processing by novel energy sources and microfluidic techniques. For instance, microwave-assisted extraction employs magnetite beads for the selective trapping of oligonucleotides, followed by the rapid release of target molecules via microwave heating [43]. Meanwhile, microtube tip technology, utilizing polypropylene C–CP fiber media, achieves highly sensitive MS detection by minimizing matrix interference while enriching target analytes [44]. In addition, SPE techniques have been combined with microfluidic technology to achieve online solid-phase extraction-gradient elution-MS analysis (Fig. 1b) [45]. This integrated approach simplifies the sample handling process by using magnetic beads for desalting and enrichment, followed by stepwise elution directly coupled with MS detection.
Although SPE offers high selectivity and compatibility with various matrices, its desalting efficiency can be compromised when handling samples with extremely high salt concentrations, leading to potential analyte losses. Furthermore, traditional offline SPE workflows are labor-intensive and not easily scalable for high-throughput applications.
3. Chromatography
3.1 Liquid chromatography-mass spectrometry (LC–MS)
Another pivotal advancement in modern MS pretreatment is the implementation of online precolumn enrichment and purification techniques [46]. Among these, reversed-phase LC is particularly prominent. Its stationary phase typically comprises hydrophobic alkyl chains (e.g., C18, C8) that adsorb target molecules through nonpolar interactions. Reversed-phase LC is widely favored for its high efficiency, flexibility, resolution, reproducibility, stability, and automation, making it a cornerstone for desalting in analytical workflows.
C18 reversed-phase columns are among the most widely used chromatographic tools for desalting and purifying diverse compounds [47]. Applications include phosphopeptides [48], guanidinium- and urea-based compounds from human thyroid tissues [49], bioactive proteins such as FIP-fve and Flammutoxin from enoki mushrooms [50], Aβl-1 tyrosine kinase [51], total proteins from human serum [52], neuropeptides from crustacean hemolymph [53], and nucleotides [54]. They are also effective for small molecules, including heparin, acetylheparin sulfate, and their derived unsaturated disaccharides [55], pharmacologically active compounds from mouse hepatocyte samples [56], lipophilic marine toxins in shellfish [57], microcystins in surface water [58], and alkaloids such as oxidized picrotoxin and quinazoline-like compounds in rat blood and skin samples [59]. Other examples include the antiretroviral agent emtricitabine [60], caffeine [61] and theobromine in human fluids [62]. For complex samples, gradient elution with aqueous and organic phases enhances desalting and separation efficiency.
Two-dimensional liquid chromatography (2D–LC) is widely used in pharmaceutical analysis for metabolite profiling, drug residue detection, and quantification in complex biological matrices. As an extension of reversed-phase technology, it combines different separation mechanisms, offering advantages for desalting and resolving complex samples [63]. A common setup pairs a first-dimensional cation-exchange (CEX) column with a second-dimensional reversed-phase column. Stoll et al. used 2D–LC with TOF–MS to analyze rituximab isomers, employing a C4 reversed-phase column with gradient elution to desalt samples and address CEX–MS compatibility. This enhances peak capacity and resolution, providing a cost-effective alternative by lessening reliance on advanced MS [64]. Moreover, Luo et al. confirmed 2D–LC–TOF–MS feasibility for synthetic peptides using reversed-phase chromatography in both dimensions [65]. Overall, 2D–LC improves peptide detection sensitivity and accuracy for MS.
ZipTip® Micro Pipette Tip, a micro-tipped desalting and enrichment tool developed by Millipore Sigma (formerly Millipore), use reversed-phase C18 or C4 particles, or cation exchange resins, as packing materials [66]. This column leverages capillary action to adsorb small sample volumes, followed by salt removal through appropriate elution steps. Recent advances include functionalizing C18 particles with conductive poly pyrrole or polyacrylamide, enhancing the adsorption of charged analytes [67]. Such modifications have significantly improved ZIPTIP's efficiency in enriching phosphorylated peptides and cleaning up complex biological matrix [68,69].
SEC columns, also known as gel permeation chromatography columns, gel filtration columns, or molecular sieve columns, are widely used for desalting large molecular samples, such as proteins and oligomers. SEC columns are typically packed with porous polymer materials, enabling separation based on molecular size. The underlying principle of SEC is the separation of molecules by differences in size or molecular weight. For instance, Duivelshof et al. developed a workflow integrating non-denaturing chromatography with MS for analyzing daratumumab. This method employed rapid desalting via SEC as an alternative to manual desalting using spin filters. Before ESI–MS analysis, short isocratic online desalting was performed using an Advance Bio SEC column. This approach offered a more generalized and efficient solution compared to traditional offline buffer desalting methods [70].
HiTrap desalting columns, another type of SEC column, are pre-packed with gel filtration resins designed for high-efficiency desalting. These columns operate by separating molecules based on size, utilizing the pores within the gel matrix. When integrated with automated systems, HiTrap columns have demonstrated high efficiency in desalting and purifying biological proteins [71] and seawater samples [72]. The development of the column technologies, particularly their integration with MS, has significantly enhanced the accuracy of analyzing complex samples.
3.2 Gas chromatography-mass spectrometry (GC–MS)
GC–MS is a core technology for analyzing volatile compounds [73]. Conventional desalting strategies for GC–MS rely mainly on the retention of salts in the aqueous phase or their precipitation upon heating to avoid their entry into the gas phase system. In recent years, the development of new adsorbent materials has significantly contributed to the advancement of desalting techniques for GC samples. Hydrophilic-lipophilic balanced (HLB) polymeric adsorbents have shown outstanding advantages [74], with their unique three-dimensional structure combining hydrophilic groups (e.g., N-vinylpyrrolidone) and lipophilic groups (e.g., divinylbenzene), which can maintain good solubility at high ionic strength, effectively overcoming the bottleneck of the "salting-out effect" of traditional reversed-phase packing materials. For example, using Oasis HLB columns to treat high-salt seawater samples, endocrine-disrupting compounds were almost completely removed from NaCl while maintaining excellent detection limits (2 ng/mL) and elution recoveries (> 98%) [75].
Online and automated desalting techniques are another important development. The seamless integration of online SPME and GC systems is particularly prominent [76]. Researchers have innovatively prepared coatings with desalination capabilities (e.g., polyacrylates with ion-exchange resin particles) directly on SPME fibers. The fiber can be directly immersed into high-salt samples, and the coating can both adsorb target compounds and capture salt ions by ion exchange or adsorption during stirred extraction. After the extraction is completed, the fiber is directly thermal desorbed and fed to GC, which realizes the full automation of desalting, enrichment, and feeding and avoids the loss and contamination of offline operation [77].
Although the novel adsorbent materials and online coupling techniques have significantly improved the desalting efficiency of GC–MS analysis, the existing methods are generally insufficient for salt-saturated brines, high-viscosity samples, or hyperpolar/thermally unstable compounds, and it is difficult to cope with the complex interference environments of multiple ions coexisting in the actual samples.
3.3 Electrophoresis
Electrophoresis is a commonly used analytical method for the separation, enrichment, analysis, and detection of ionic compounds [78]. The principle of electrophoresis lies in the migration of charged particles under an electric field, where particles move towards the electrode of opposite polarity based on their charge [79]. The migration rate depends on factors such as the molecule's size, shape, charge, and the characteristics of the medium. Typically, smaller and more highly charged molecules migrate faster, enabling separation [80].
To overcome the low throughput and efficiency of traditional electrophoresis, Harkins et al. developed an automated system enabling parallel electrophoresis of 96 samples for MS analysis. This 96-well cartridge integrates electrophoresis, concentration, fractionation, and desalting. Each well contains a polyacrylamide gel for separation and a reversed-phase C8 capture layer for analyte binding. Target molecules migrate through the gel under electric current and are captured on a hydrophobic slide, allowing direct MALDI–MS analysis. This system significantly enhances desalting, protein concentration, and detection sensitivity while maintaining high throughput [81].
Capillary electrophoresis (CE), one of the most commonly used electrophoretic techniques, was developed in the 1980s. Chalcraft et al. proposed a newborn screening method combining CE and ESI–MS to analyze amino acids, acylcarnitine, and their stereoisomers directly from dried blood spot extracts. Using electrokinetic focusing, the method enhances sensitivity for low-abundance metabolites [82]. However, in the context of biological sample analysis, ESI–MS often encounters challenges such as sensitivity issues and ionization suppression caused by salts. To overcome this, Rahman et al. introduced a current-limited polarity-reversed nano electrospray ionization MS method. By employing a 10 GΩ series resistance and reversing the polarity, this technique efficiently drives salt cations toward the electrode while generating negatively charged sprays. Subsequent application of a positive high voltage (+1.7 kV) at the nESI tip produces protonated analytes free from salt interference [83].
Electrophoresis-based desalting is advantageous for its low cost, ease of implementation, and minimal sample consumption. However, its throughput remains limited, and it is less effective for small molecule desalting or highly complex biological matrices. Moreover, it requires highly pure samples, and its application in desalting remains limited, primarily focusing on proteins, nucleic acids, enzymes, viruses, and cells.
4. Ion exchange
Ion exchange technology, which originated in the early 20th century, was widely used to water treatment, chemical separation, and analytical chemistry [84]. Ion exchange columns, packed with resins bearing specific functional groups, achieve desalination by selectively adsorbing target analytes while releasing exchangeable ions into the sample, thereby reducing salt ion concentrations. Similarly, ion exchange membranes have been employed for sample cleanup and interference removal, particularly for small sample volumes or specialized applications [85].
High-performance anion exchange chromatography (HPAEC) eluents (e.g., NaOH, NaOAc) are incompatible with ESI. Membrane desalting devices address this by using cation exchange membranes to swap alkali cations for hydrated H+ ions, converting eluents to ESI-compatible solvents [86]. These nanoporous polymer membranes regulate ion/water transport via charged pores, replacing Na+ with H3O+ while regenerating via electrolyzed water acid. Micro-processing units enable rapid desalination (95% salt removal in 1 s) by exploiting diffusion differences between salts and analytes (e.g., peptides), preserving MS-suitable concentrations when coupled to ESI tips [87]. Additionally, microfluidic ion suppression modules based on anion exchange chromatography have been introduced for pre-desalination treatment. Microfluidic suppressors based on anion exchange use sulfonated PTFE films for acid-base reactions, inhibiting basic mobile phase ions. Dilute H2SO4 regenerant replaces K+ with H+, neutralizing OH– and removing potassium. These membrane suppressors generate counter-ions via water electrolysis, avoid continuous regenerant needs, and enhance ion transfer via electric fields—though localized heating increases noise [88].
Ion exchange membranes efficiently desalt samples by selectively separating ions. While sufficient for low-salt samples, they struggle with high-salt oligosaccharides. To overcome this, researchers developed a capillary-scale HPAEC system combining pulsed amperometric detection and capillary-scale post-desalination ESI ion trap MS (sub-picomolar sensitivity). Its desalination unit uses a Nafion cation exchange capillary within a PEEK column. Dilute sulfuric acid saturates the capillary with H3O+, enabling efficient transfer of neutral/anionic compounds to MS while converting eluent to water/acetic acid. This successfully analyzed carbohydrates in chicory inulin and food products [86,89]. However, the application of HPAEC with online MS has been somewhat limited by the availability of commercially viable small-caliber LC columns. Microporous HPAEC–MS/MS effectively addresses this, enabling characterization of phosphopeptides [90], small molecules like galacturonic acid [91], and heterogeneous N–glycan mixtures in biomacromolecule protein digests [92].
Ion exchange technology offers efficient salt removal, capable of eliminating large quantities of salt ions rapidly. Nonetheless, the relatively high cost of ion-exchange resins, limited resin reusability, and the need for careful pH control during operation constrain their applicability in large-scale or cost-sensitive workflows. Additionally, the limited lifespan of ion exchange resins or membranes, coupled with the potential generation of wastewater, poses challenges regarding environmental sustainability.
5. Crystallography
Crystallization desalting is a straightforward approach for rapid salt removal. The process involves two key steps: (1) Inducing salt crystallization by drying the sample solution, and (2) dissolving the analyte in a suitable spray solvent for MS analysis. This technique, when coupled with an ion source, facilitates rapid analysis of complex samples [93]. For instance, Gong et al. developed an efficient method for desalting trace samples by leveraging the spontaneous separation of biomolecules from salt during crystallization. Biomolecules are deposited at the tip of a quartz pipette as the solvent evaporates. This approach enabled the detection of proteins and peptides at concentrations as low as 50 amol [94].
Desalination relies on crystallization/resolubilization, where target molecules spontaneously separate from salts and redissolve in MS-compatible solvents. To minimize the "coffee-ring effect" (uneven solute distribution during evaporation), Wang et al. used hydrophobic silicone coatings to inhibit droplet fixation. Enriched samples were analyzed via vibrating sharp-edged spray ionization, detecting macrolide antibiotics in physiological buffers/PBS (LOD: 1 nmol/L) [93]. In addition, Chen et al. developed online crystallization/solvent evaporation ionization MS [95], enabling spontaneous salt separation for high-salt LC eluates with simplified maintenance. However, crystallization rates are hard to control, risking reproducibility. To address this, heat-assisted methods were introduced. Chen et al.'s dual-spray HADSI uses sample and organic solvent sprays (e.g., methanol) with a heating plate, rapidly crystallizing salt while depositing targets on crystals [96]. Dual-spray tuning challenges prompted a simpler design: thermally-assisted recrystallization ionization MS. This places a heating plate between the electrospray unit and MS inlet, leveraging spontaneous target-salt separation during crystallization [97].
Crystalline desalting effectively separates biomolecules from salts through a process of salt crystallization and re-dissolution. However, controlling the crystallization rate is challenging, which can affect reproducibility and analyte recovery. Additionally, the method may be less effective for volatile or unstable compounds that degrade during the drying process.
6. Hydrophobic materials
Hydrophobic materials are extensively utilized in sample pre-treatment [98]. These materials operate based on the differential hydrophilicity and hydrophobicity of molecules, enabling efficient sample purification and enrichment through selective adsorption and separation of target analytes. In MS, hydrophobic coatings are employed not only for the enrichment and separation of biomolecules such as proteins [99,100] but also for addressing the challenges associated with low-abundance analytes [101].
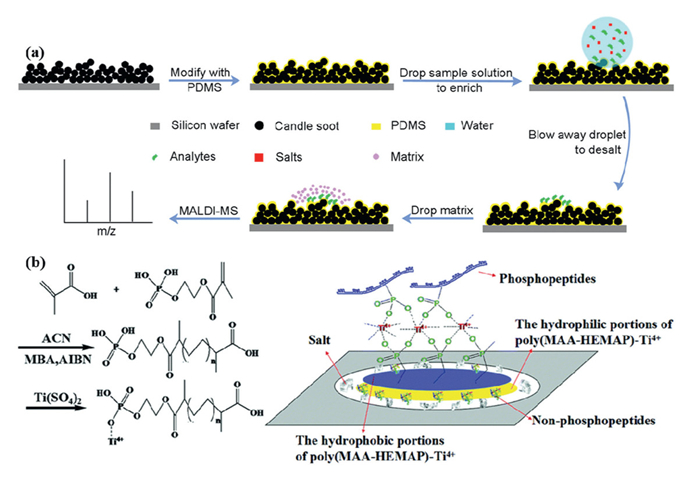
In recent years, various desalination methods based on polymeric hydrophobic materials have emerged. These methods extract and separate analytes from sample solutions through hydrophobic interactions with materials such as carbon materials [102], polytetrafluoroethylene [103], paraffin wax [104], nylon [105], nafion-coated sponges [106], PMMA [41], self-assembled layers of Vmh2 hydrophobic veins [107], octadecyl trichlorosilane monolayers [108], poly(glycidyl propargyl ether/divinyl benzene) [109], poly(N–isopropylacrylamide) [110]. These materials achieve desalination by adsorbing analytes and removing salts through washing. Building on this, Wang et al. introduced a superhydrophobic candle soot/PDMS substrate for one-step peptide enrichment and desalting in MALDI–MS analysis. This method utilizes the strong confinement effect of the superhydrophobic surface and the selective trapping capability of PDMS, enabling peptide enrichment within a confined area while efficiently removing residual salt contamination. Unlike conventional hydrophobic surfaces, superhydrophobic surfaces exhibit weaker interactions with aqueous solutions, enhancing their ability to remove salts. By integrating candle soot coating with PDMS, a superhydrophobic C/PDMS substrate was developed and successfully applied to detect angiotensin Ⅲ in real samples, achieving a detection sensitivity three orders of magnitude higher than that of traditional MALDI steel plates. This method also demonstrated excellent performance in the mass spectrometric analysis of myoglobin and bovine serum albumin digests (Fig. 2a) [111].
Figure 2

Polymeric hydrophobic materials offer advantages in sample desalination. However, they face challenges such as low selectivity and complex preparation. Among the various enrichment strategies, immobilized metal affinity chromatography (IMAC) remains the most widely used method for phosphopeptide separation, leveraging the strong affinity between phosphate groups and immobilized metal ions on the substrate. Recent advances in IMAC adsorbents, particularly those employing metal ions like Ti4+ and Zr4+, have exhibited remarkable selectivity and specificity. These adsorbents form stable metal-phosphate monolayers due to the strong coordination of each Ti4+ or Zr4+ ion with multiple phosphate molecules, providing well-defined metal sites for selective phosphopeptide enrichment while minimizing non-specific binding to carboxylic acid groups or other peptides [112]. Such properties are essential for phosphopeptide enrichment, especially under conditions where the sample is loaded at low pH and ionized at high pH. Building on this, researchers have developed a novel strategy using circular hydrophobic-hydrophilic-Ti4+ immobilized phosphate polymer-patterned silica sheets as sample carriers. When a sample solution is applied dropwise, the outer hydrophobic coating concentrates the sample into a small circular area (1800 μm in diameter), while the Ti4+-immobilized polymer captures phosphopeptides. Concurrently, non-phosphopeptides, proteins, and salts are segregated into the hydrophobic and hydrophilic regions of the silica carrier, facilitating simultaneous enrichment and desalination (Fig. 2b) [113].
Additionally, synthetic hydrophobic membranes have been employed in desalination. Unlike ion exchange membranes, synthetic hydrophobic membranes are suitable for retaining the integrity of biomolecules by rejecting salts primarily through physical hydrophobicity and removing impurities in conjunction with a washing step, and are widely used for direct analysis of complex biological samples such as MALDI–IMS. The main difference between the two is that hydrophobic membranes provide passive desalination through physical surface properties, making them flexible and suitable for a wide range of biological samples, while ion exchange membranes actively separate ions through chemical selectivity, making them suitable for applications such as high purity water treatment. Studies have shown that surfaces like polyethylene, polypropylene, and C8 or C18 coatings effectively remove salts and contaminants [114]. However, this process can be time-consuming and complex. Using commercial hydrophobic coatings and picosecond laser micromachining, hydrophilic surface energy traps are created on glass substrates. These devices rapidly concentrate droplets through directional evaporation, enhancing analyte drying uniformity and reducing analysis time, making them particularly suitable for MALDI–IMS analysis [115]. Another innovative approach is surface-assisted laser desorption ionization (SALDI), which, in conjunction with fluorocarbon coatings, selectively separates ionic or hydrophilic analytes from biofluid electrolytes for quantitative MS analysis. This technique relies on nanoporous silicon films, prepared via grazing angle deposition and coated with fluorocarbons like perfluorooctyl trichlorosilane, achieving contact angles of 105°−120° [116].
Overall, hydrophobic materials offer significant advantages in desalination, such as reducing salt-water contact, enhancing evaporation efficiency, and preventing salt crystal accumulation, which helps extend equipment lifespan. However, limitations such as low adsorption capacity, regeneration challenges, susceptibility to contaminants, material-dependent treatment efficiency, and potential environmental impacts require further research to optimize their applications. Moreover, these materials often suffer from limited binding capacities, potential sample loss during washing steps, and difficulty in achieving complete removal of strongly bound salt residues.
7. Ambient ionization mass spectrometry (AIMS)
7.1 Paper spray ionization (PSI)
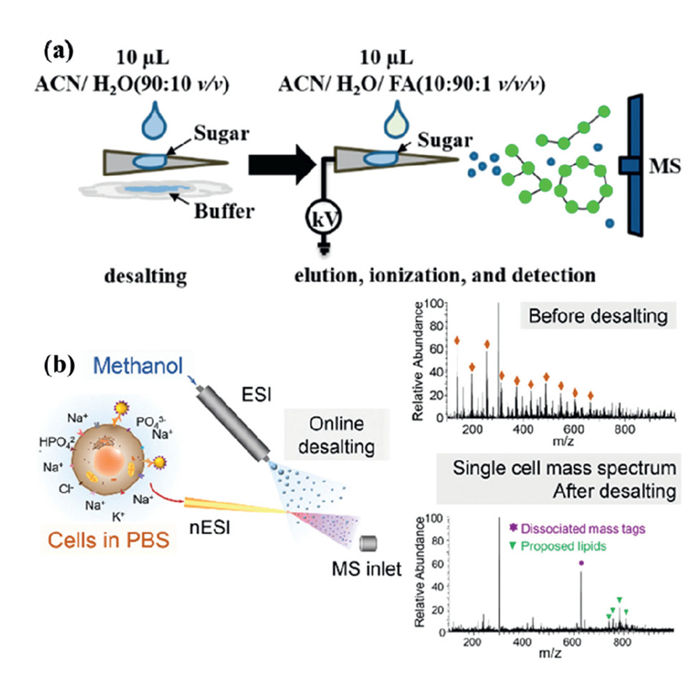
Recent advances in MS highlight the benefits of simplifying or eliminating sample preparation and reducing analysis time, thereby streamlining analytical workflows [117]. AIMS is a representative MS-based technique that enables rapid and straightforward analysis of complex matrix samples, often requiring little to no sample pretreatment. Among the most popular environmental ionization techniques developed in recent years is PSI [117]. The PSI involves cutting a paper substrate into a triangular shape with a pointed tip, loading the sample onto the substrate, and applying a high voltage, spray solvent to facilitate simultaneous sample elution and ionization. First introduced by Ouyang, Cooks, and colleagues in 2010 [118], PSI has since gained widespread use across various fields [119]. Many researchers have modified paper substrates to improve enrichment and desalting efficiency. Modified substrates, such as polystyrene [120], polystyrene microspheres [121], polyvinylidene fluoride molecularly imprinted membranes [122], organ siloxane polymers [123], zirconia-coated papers [124], have shown excellent desalting effects. In addition to substrate modifications, innovations in MS ionization devices have further improved. For example, Riboni et al. developed Solvent-assisted paper spray ionization, a device that integrates a power supply and fluidic system to extend the analysis time for proteins, peptides, and N-glycans. It also discriminates between protein species characterized by different post-translational modifications and adducts of electrophilic compounds [125]. Traditionally, MS analysis of small carbohydrate fragments (oligosaccharides with a degree of polymerization between 2 and 12) has required offline sample preparation, which is time-consuming. To address this, Wang et al. introduced a new paper spray ionization technique called Desalting paper spray (DPS). DPS utilizes triangular filter paper for both desalting and ionization. Unlike conventional PSI and nano ESI, DPS–MS enables the rapid and sensitive detection of oligosaccharides in complex biological matrix, such as tris, PBS, HEPES buffers, and urine. This method is fast (approximately < 5 min per sample), simple, and highly sensitive, and has been successfully applied to detect various oligosaccharides in non-volatile buffers (e.g., tris, phosphate, and HEPES buffers) (Fig. 3a) [126]. The cellulose-based paper substrate interacts strongly with oligosaccharides through hydrogen bonding, serving as an effective stationary phase for desalting. Building on this, DPS–MS has been extended to detect small amounts of complex glycans and glycopeptides in non-volatile buffers [127]. Paper spray MS offers significant advantages, such as the elimination of complex sample pretreatment, suitability for complex samples, and the ability to perform both qualitative and quantitative analyses. However, the paper spray technique has certain limitations, such as limited resolution and lower sensitivity compared to traditional multistage MS techniques [118].
Figure 3

7.2 Extractive electrospray ionization MS (EESI–MS)
EESI–MS is a widely used technique for the rapid detection of natural proteins in a variety of matrix, including untreated biological samples. It has been successfully applied for the direct determination of macromolecules in complex matrix. EESI is particularly beneficial because it can tolerate highly complex matrix, enabling the direct analysis of untreated biological samples for molecular-level characterization under natural conditions [128]. Some researchers have developed a straightforward method for online EESI–MS analysis of in vivo metabolites, allowing for desalting and real-time analysis without the need for sample pretreatment [129]. Improved ESI-based ionization techniques effectively address challenges such as ion suppression and interference from non-volatile salts. These techniques are easy to implement and are not compromised by the rapid, high-throughput nature of single-cell screening in MS. However, desalting ionization methods are generally limited to the analysis of high-abundance free targets. To enable organic mass flow cytometry in cellular non-denaturing buffers, online desalting ionization techniques with higher sensitivity and greater compatibility with in situ cell sampling and intact cell screening are necessary. Single-cell MS remains a challenge in natural environments due to the presence of heavily ionized suspensions of non-volatile salts. To address this, Xu et al. developed a dual-spray ionization method using nano ESI and ESI emitters. This method enables cells to be directly injected into PBS buffer and undergo online, simultaneous desalting and ionization analysis during the dual-spray process (Fig. 3b) [130]. Online desalting maintains the physiological environment of the sample for as long as possible, making simultaneous desalting and ionization ideal for preserving the natural environment and eliminating salt interference.
7.3 Solvent–fragment microextraction–nESI
Online ionization based on solvent-fragment microextraction (SFME) is a recent advancement in AIMS and serves as a powerful online sample preparation technique for direct MS analysis of ultra-low-volume biological liquid samples. Our team recently improved SFME by employing a nano tip for online microextraction and analysis in SFME–MS, successfully applying it for the trace detection of seawater pollutants [20,131]. The SFME–nESI–MS is simple to operate: The nano tip is filled with a high-salt sample and extraction solvent, and a platinum wire is inserted at the other end to serve as the conductive electrode. Results demonstrate that high voltage on the platinum wire facilitates rapid chemical exchange between the extraction solvent and the sample. This technique enables online desalting pretreatment and assay analysis, providing rapid results in less than two minutes, with detection limits comparable to those of other assays (as low as 0.06 ng/mL).
7.4 Ambient electric arc ionization (AEAI)
AEAI has demonstrated significant advantages in mass spectrometry of high-salt samples. Studies have shown that AEAI–MS can maintain excellent signal intensity at salt concentrations up to 1000 mmol/L, whereas conventional electrospray ionization fails completely due to the salt inhibition effect. The core mechanism is that the thermal desorption effect generated by the arc effectively reduces salt deposition at the entrance of the mass spectrometer and avoids the formation of sodium adducts, thus improving the reliability of analysis in complex matrices [132]. The further developed tip-assisted ambient arc ionization technique combines microinjection tip and arc ionization to directly analyze organophosphorus pesticide residues in complex matrices such as strawberry juice without cumbersome pre-treatment. The method demonstrated high sensitivity (LOD 0.0124–0.0245 μg/g), high precision (RSD < 9.2%) and excellent quantification (R2 > 0.995), and the recoveries in real samples were in the range of 82.8%−116% [133]. Its fast analytical characteristics and resistance to matrix interference provide an efficient solution for direct mass spectrometric detection of high-salt or complex biological samples.
The introduction of ambient ionization has significantly advanced analytical chemistry, enabling faster, simpler, and more efficient sample analysis. Nevertheless, they often face limitations in quantification accuracy, lower resolution compared to conventional MS methods, and susceptibility to matrix effects when dealing with highly complex biological samples.
8. Desalting agents
Protein-metal interaction studies using ESI–MS are often complicated by the tendency of metal ions to undergo non-specific binding. This makes it challenging to determine whether the detected level of metalation accurately reflects the bulk solution conditions without additional data. For instance, ESI mass spectra recorded in the presence of Na+ frequently exhibit significant sodium adduction, even for proteins with no inherent metal affinity in bulk solution. This non-specific sodium adduction is especially prevalent in the commonly used positive ion mode, where salt-free solutions yield 'clean' mass spectra dominated by multi protonated [M+nH]n+ ions. However, in the presence of sodium, one or more protons can be replaced by Na+, resulting in heterogeneous species denoted as [M+mNa++(n-m)H]n+, where m varies from 0 to n. Similar behavior is observed with many other inorganic cations. Various desalting techniques, such as on-line dialysis, reversed-phase HPLC, and size-exclusion chromatography, have been developed to address non-specific metal binding. However, they are often unsuitable for studies focused on characterizing protein metalation in liquid phase, as desalting can disrupt the corresponding binding equilibria [134]. In contrast, the use of specific desalting agents offers an effective solution by mitigating the adverse effects of metal ion salts. This approach significantly reduces non-specific sodium ion adducts and enhances the abundance of gaseous protein and protein complex ions generated by ESI [135].
In some cases, desalting agents alone can effectively achieve the desired results. For instance, cerebrospinal fluid (CSF) samples, when unsalted, do not show clear signs of GM1 in MALDI–TOF–MS spectra. However, pretreatment with desalting agents can improve the analysis. One method involves applying two microliters of cold 0.1% trifluoroacetic acid (TFA) to a dry CSF sample on the sample plate prior to MALDI–TOF–MS analysis [135]. In addition, pressurized reagent desalting is an emerging technique that can help reduce interference from sodium ion adducts in MS. Recently, Loo and colleagues demonstrated that, in desorption electrospray ionization of HPLC column effluent containing TFA, the presence of m-NBA or cyclobutene sulfone in the spray solvent decreased the intensity of TFA cluster ions and improved the signal-to-noise (S/N) ratio of protein MS signals. This effect is likely due to the pressurizing reagent binding to the TFA anion, which inhibits the formation of TFA clusters or prevents the dissociation of TFA into anions and free protons in solution, thereby reducing the concentration of free TFA anions [136]. Similarly, Cassou et al. used m-NBA and cyclobutene sulfone as pressurized reagents to desalinate proteins in natural MS. These reagents use sodium chelation to facilitate desalting. Cyclobutene sulfone was found to be more effective than m-NBA in reducing sodium adduction while maintaining non-covalent protein-protein and protein-ligand interactions. Notably, desalting protein ions with m-NBA resulted in a seven-fold increase in protein ion S/N due to fewer clusters and reduced chemical noise [137].
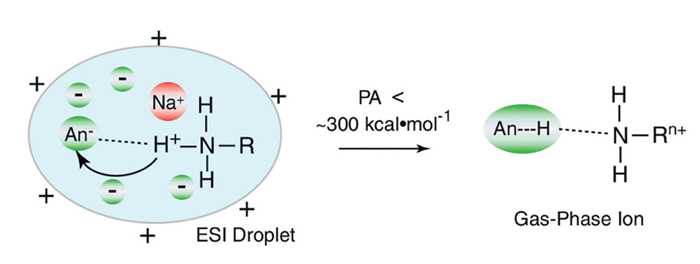
Several alternative methods have been developed for desalting protein samples prior to ESI while preserving specific salts essential for maintaining protein structure and function. These strategies include buffer up sampling with high concentrations of ammonium acetate [138], the addition of low concentrations of reagents, desalination of protein ions during ESI [139,140], and ion reactions with organic vapors [141,142]. Among them, the addition of volatile buffers like ammonium acetate has been widely used to mitigate sodium ion adduction without extensive sample pretreatment. For example, studies have demonstrated that adding 20 mmol/L ammonium acetate to an aqueous solution containing 7 mmol/L NaCl can increase the S/N ratio of cytochrome c and ubiquitin by 6-fold and over 11-fold, respectively [138,143]. Other ammonium salts can also achieve similar results at lower concentrations. For example, the addition of 25 mmol/L ammonium bromide to a ubiquitin solution with 1 mmol/L NaCl reduced the average number of sodium ions in the most abundant charge state of ubiquitin from 6.0 to 0.4, resulting in a 66-fold improvement in the S/N ratio for this ion (Fig. 4) [139,144]. The desalting efficiency of these salts is largely dependent on the proton affinity of the anion, with anions of lower proton affinity being more effective at reducing sodium ion adduction. However, these low-proton-affinity anions may bind to proteins as acid molecules, leading to the formation of ionic clusters [144], which can negatively impact protein ion signal quality. Furthermore, some salts, such as citrate and tartrate, can chelate metal ions (e.g., calcium ions) in solution, which may non-specifically bind to proteins. In ESI droplets, these metal cations may influence protein behavior through ion-pairing with free anions and could also form metal complexes with negatively charged carboxylic acid groups in proteins. Such non-specifically bound metal cations contribute to the total number of cations associated with proteins due to liquid-phase interactions [134].
Figure 4
 Figure 4. Diagram of the interaction between anions with basic sites with lower PA values. Copied with permission [144]. Copyright 2011, Springer.
Figure 4. Diagram of the interaction between anions with basic sites with lower PA values. Copied with permission [144]. Copyright 2011, Springer.Sodium adduction in ESI-generated protein ions reduces ionization efficiency and disperses signal across Multiple species. Desalting agents can mitigate this effect by decreasing non-specific sodium binding, thereby enhancing ion abundance. Their efficacy largely depends on the associated anion. While widely used in protein analysis, conventional methods like SPE, HPLC, and electrophoresis remain standard for desalting in non-ESI workflows.
9. Conclusions and prospects
Desalting pretreatment techniques before MS analysis have witnessed rapid development over the past decades. Despite various reported offline desalting methods like SPE and ion exchange, LC–MS-based online desalting remains the predominant and recommended approach. Moreover, driven by ongoing advancements in sample preparation and separation techniques, novel desalination techniques have emerged from traditional solid-phase extraction, encompassing desalting agents, crystallization methods, and hydrophobic coating materials. Nevertheless, each method or technique possesses distinct advantages and limitations (refer to Table S1 in Supporting information). The selection of the method may vary depending on the specific requirements of the research application, Table S2 (Supporting information) provides the specific performance of these methods.
However, existing methods often lose analytes and suffer residual ion suppression when processing very high-salt samples leading to 30%−50% declines in ionization efficiency despite extensive pretreatment. Moreover, most processes are labor-intensive and do not have high-throughput capabilities. The development of automated microfluidic systems shows promise in addressing this bottleneck. Beyond technical performance, sustainability considerations are becoming increasingly crucial, as many high-performance desalination materials suffer from high production costs and limited reusability. As a result, research is shifting toward greener sorbents (e.g., biodegradable polymers) and energy-efficient processes. Looking ahead, the integration of smart responsive materials with artificial intelligence-assisted method optimization appears particularly promising for developing next-generation desalination strategies.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Meng Zhang: Writing – review & editing, Writing – original draft, Methodology, Conceptualization. Junjuan Shi: Methodology, Conceptualization. Yu Feng: Conceptualization. Boyan Liu: Methodology. Jie Jiang: Supervision, Funding acquisition. Daqian Song: Methodology, Conceptualization. Yanxiao Jiang: Writing – review & editing, Writing – original draft, Supervision, Methodology, Funding acquisition, Conceptualization.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (Nos. 2023YFF0614201 and 2023YFF0614202), the National Natural Science Foundation of China (Nos. 22304038 and 22401107), the Natural Science Foundation of Shandong Province (No. ZR2022QB248) and the Scientific Research Foundation of Harbin Institute of Technology at Weihai (No. HIT (WH) 2022).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:
10.1016/j.cclet.2025.112208 .
-
-
[1]
B. Domon, R. Aebersold, Science (1979) 312 (2006) 212–217. doi: 10.1126/science.1124619
-
[2]
B.F. Cravatt, G.M. Simon, J.R. Yates Iii, Nature 450 (2007) 991–1000. doi: 10.1038/nature06525
-
[3]
M. Fessenden, Nature 540 (2016) 153–155. doi: 10.1038/540153a
-
[4]
B.C. Collins, C.L. Hunter, Y. Liu, et al., Nat. Commun. 8 (2017) 291. doi: 10.1038/s41467-017-00249-5
-
[5]
G. Huang, G. Li, R.G. Cooks, Angew. Chem. Int. Ed. 50 (2011) 9907–9910. doi: 10.1002/anie.201103687
-
[6]
X. Song, M. Mofidfar, R.N. Zare, Front. Chem. 9 (2022) 807244. doi: 10.3389/fchem.2021.807244
-
[7]
L. Han, F. Ma, P. He, et al., Food Chem. 450 (2024) 139195. doi: 10.1016/j.foodchem.2024.139195
-
[8]
W. Su, S. Liu, Q. Zhang, et al., Chin. Chem. Lett. 36 (2025) 110237. doi: 10.1016/j.cclet.2024.110237
-
[9]
B. Wu, N. Wang, Y. Shen, et al., J. Membr. Sci. 666 (2023) 121144. doi: 10.1016/j.memsci.2022.121144
-
[10]
R.A. Zubarev, A. Makarov, Anal. Chem. 85 (2013) 5288–5296. doi: 10.1021/ac4001223
-
[11]
B.T. Veach, T.K. Mudalige, P. Rye, Anal. Chem. 89 (2017) 3256–3260. doi: 10.1021/acs.analchem.6b04889
-
[12]
Y. Qi, C. Ma, S. Chen, et al., ACS. ES. T. Water. 1 (2021) 1975–1982. doi: 10.1021/acsestwater.1c00162
-
[13]
J. Xu, T. Li, Z. Yu, et al., Chin. Chem. Lett. 35 (2024) 108578. doi: 10.1016/j.cclet.2023.108578
-
[14]
Q. Zhang, H. Xiao, J. Zhan, et al., Chin. Chem. Lett. 33 (2022) 4746–4749. doi: 10.1016/j.cclet.2022.01.004
-
[15]
Y. Li, J. Han, X. Wang, et al., Chin. Chem. Lett. 36 (2024) 110708.
-
[16]
M. Wang, L. Zheng, B. Wang, et al., Chin. Chem. Lett. 33 (2022) 3484–3487. doi: 10.1016/j.cclet.2022.03.098
-
[17]
R. Sekar, S.K. Kailasa, Y. Chen, et al., Chin. Chem. Lett. 25 (2014) 39–45. doi: 10.1016/j.cclet.2013.10.012
-
[18]
Z. Su, W. Hu, L. Ye, et al., Chin. Chem. Lett. 34 (2023) 107790. doi: 10.1016/j.cclet.2022.107790
-
[19]
H. Lu, Y. Yin, J. Sun, et al., Chin. Chem. Lett. 32 (2021) 3457–3462. doi: 10.1016/j.cclet.2021.05.074
-
[20]
M. Zhang, R. Shang, Z. Hong, et al., J. Hazard. Mater. 469 (2024) 134039. doi: 10.1016/j.jhazmat.2024.134039
-
[21]
L. Huang, J. Wang, H. Jiang, et al., Chin. Chem. Lett. 36 (2025) 109896. doi: 10.1016/j.cclet.2024.109896
-
[22]
G. Li, K.H. Row, J. Sep. Sci. 45 (2022) 883–895. doi: 10.1002/jssc.202100682
-
[23]
A. Alsharaa, C. Basheer, M. Sajid, J. Chromatogr. B 1007 (2015) 43–48. doi: 10.1016/j.jchromb.2015.11.004
-
[24]
P. Vinoth Kumar, J. Jen, Chemosphere 83 (2011) 200–207. doi: 10.1016/j.chemosphere.2010.12.041
-
[25]
C. Huang, K.F. Seip, A. Gjelstad, et al., Anal. Chem. 87 (2015) 6951–6957. doi: 10.1021/acs.analchem.5b01610
-
[26]
I. Tarazona, A. Chisvert, Z. León, et al., J. Chromatogr. A 1217 (2010) 4771–4778. doi: 10.1016/j.chroma.2010.05.047
-
[27]
C.V. Ramos-Dorta, P. Verónica, A.M. Afonso, Environ. Technol. 34 (2013) 607–616. doi: 10.1080/09593330.2012.710255
-
[28]
S. Erarpat, G. Özzeybek, D.S. Chormey, et al., Chemosphere 189 (2017) 180–185. doi: 10.1016/j.chemosphere.2017.09.072
-
[29]
M. Rezaee, H.A. Mashayekhi, Anal. Methods 4 (2012) 2887–2892. doi: 10.1039/c2ay25460c
-
[30]
X. Han, J. Chen, Z. Li, et al., Anal. Chim. Acta 1078 (2019) 78–89. doi: 10.1016/j.aca.2019.06.022
-
[31]
X. Han, J. Chen, H. Qiu, et al., Microchim. Acta 186 (2019) 375. doi: 10.1007/s00604-019-3498-2
-
[32]
F. Baroudi, J. Al-Alam, S. Chimjarn, et al., Microchem. J. 154 (2020) 104593. doi: 10.1016/j.microc.2019.104593
-
[33]
A.T. Silveira, A.M.d.C. Barbosa, H.D. de Faria, et al., Analyst 147 (2022) 2779–2792. doi: 10.1039/d2an00446a
-
[34]
G. Han, Q. Zeng, Z. Jiang, et al., Talanta 164 (2017) 355–361. doi: 10.1016/j.talanta.2016.11.044
-
[35]
M.J. Boundy, A.I. Selwood, D.T. Harwood, et al., J. Chromatogr. A 1387 (2015) 1–12.
-
[36]
Y. Wang, J. Chen, H. Ihara, et al., TrAC, Trends Anal. Chem. 143 (2021) 116421. doi: 10.1016/j.trac.2021.116421
-
[37]
Y. Zhao, B.T. Chait, Anal. Chem. 66 (1994) 3723–3726. doi: 10.1021/ac00093a029
-
[38]
R.L. Winston, M.C. Fitzgerald, Anal. Biochem. 262 (1998) 83–85. doi: 10.1006/abio.1998.2754
-
[39]
W. Jia, X. Chen, H. Lu, et al., Angew. Chem. Int. Ed. 45 (2006) 3345–3349. doi: 10.1002/anie.200503485
-
[40]
C. Pan, S. Xu, H. Zou, et al., J. Am. Soc. Mass Spectrom. 16 (2005) 263–270. doi: 10.1016/j.jasms.2004.11.005
-
[41]
W. Shen, H. Xiong, Y. Xu, et al., Anal. Chem. 80 (2008) 6758–6763. doi: 10.1021/ac801001b
-
[42]
H. Chen, C. Deng, X. Zhang, Angew. Chem. Int. Ed. 49 (2010) 607–611. doi: 10.1002/anie.200904885
-
[43]
W. Chen, Y. Chen, Anal. Chem. 79 (2007) 8061–8066. doi: 10.1021/ac0709450
-
[44]
D.S. Fornea, Y. Wu, R.K. Marcus, Anal. Chem. 78 (2006) 5617–5621. doi: 10.1021/ac060447b
-
[45]
N. Gasilova, L. Qiao, D. Momotenko, et al., Anal. Chem. 85 (2013) 6254–6263. doi: 10.1021/ac400171e
-
[46]
H. Liu, P. Jin, F. Zhu, et al., TrAC, Trends Anal. Chem. 134 (2021) 116132. doi: 10.1016/j.trac.2020.116132
-
[47]
J. Chen, Z. Gong, W. Tang, et al., TrAC, Trends Anal. Chem. 134 (2021) 116135. doi: 10.1016/j.trac.2020.116135
-
[48]
J. Wang, Y. Zhang, H. Jiang, et al., J. Proteomics. 6 (2006) 404–411. doi: 10.1002/pmic.200500223
-
[49]
R. Fan, F. Zhang, X. Chen, et al., Anal. Chim. Acta 961 (2017) 82–90. doi: 10.1016/j.aca.2017.01.036
-
[50]
C. Tung, C. Lin, C. Tung, et al., J. Food Drug Anal. 26 (2018) 1045–1053. doi: 10.1016/j.jfda.2017.12.004
-
[51]
H. Chen, E. Adams, A. Van Schepdael, J. Chromatogr. B 897 (2012) 17–21. doi: 10.1016/j.jchromb.2012.04.001
-
[52]
A. Boichenko, N. Govorukhina, A.G.J. van der Zee, et al., Anal. Bioanal. Chem. 405 (2013) 3195–3203. doi: 10.1007/s00216-013-6749-9
-
[53]
R. Chen, M. Ma, L. Hui, et al., J. Am. Soc. Mass Spectrom. 20 (2009) 708–718. doi: 10.1016/j.jasms.2008.12.007
-
[54]
J. Klawitter, V. Schmitz, J. Klawitter, et al., Anal. Biochem. 365 (2007) 230–239. doi: 10.1016/j.ab.2007.03.018
-
[55]
F. Galeotti, N. Volpi, Anal. Chem. 83 (2011) 6770–6777. doi: 10.1021/ac201426e
-
[56]
M. Goel, E. Larson, C.J. Venkatramani, et al., J. Chromatogr. B 1084 (2018) 89–95. doi: 10.1016/j.jchromb.2018.03.029
-
[57]
J. Regueiro, A.E. Rossignoli, G. Álvarez, et al., Food Chem. 129 (2011) 533–540. doi: 10.1016/j.foodchem.2011.04.054
-
[58]
G. Munoz, S. Vo Duy, A. Roy-Lachapelle, et al., J. Chromatogr. A 1516 (2017) 9–20. doi: 10.1016/j.chroma.2017.07.096
-
[59]
H. Zheng, G. Chen, L. Shi, et al., J. Pharm. Biomed. Anal. 49 (2009) 427–433. doi: 10.1016/j.jpba.2008.11.032
-
[60]
M. Pendela, D.A. Mamade, J. Hoogmartens, et al., Talanta 82 (2010) 125–128. doi: 10.1016/j.talanta.2010.04.004
-
[61]
A.S. Ptolemy, E. Tzioumis, A. Thomke, et al., J. Chromatogr. B 878 (2010) 409–416. doi: 10.1016/j.jchromb.2009.12.019
-
[62]
M.O. Magnusson, R. Sandström, Rapid. Commun. Mass Spectrom. 18 (2004) 1089–1094. doi: 10.1002/rcm.1450
-
[63]
A.K. Mallik, H. Qiu, M. Takafuji, et al., TrAC, Trends Anal. Chem. 108 (2018) 381–404. doi: 10.1016/j.trac.2018.09.003
-
[64]
D.R. Stoll, D.C. Harmes, J. Danforth, et al., Anal. Chem. 87 (2015) 8307–8315. doi: 10.1021/acs.analchem.5b01578
-
[65]
H. Luo, W. Zhong, J. Yang, et al., J. Pharm. Biomed. Anal. 137 (2017) 139–145. doi: 10.1016/j.jpba.2016.11.012
-
[66]
C. Shi, H. Li, X. Shi, et al., Chin. Chem. Lett. 33 (2022) 3613–3622. doi: 10.1016/j.cclet.2021.12.010
-
[67]
S. Zheng, X. Wang, J. Fu, et al., Anal. Chim. Acta 724 (2012) 73–79. doi: 10.1016/j.aca.2012.02.043
-
[68]
J.R. Avasarala, M.R. Wall, G.M. Wolfe, J. Mol. Neurosci. 25 (2005) 119–125. doi: 10.1385/JMN:25:1:119
-
[69]
M. Wan, Y. Wang, L. Zhan, et al., Eur. J. Mass Spectrom. 26 (2019) 55–62.
-
[70]
B.L. Duivelshof, S. Fekete, D. Guillarme, et al., Anal. Bioanal. Chem. 411 (2019) 4615–4627. doi: 10.1007/s00216-018-1561-1
-
[71]
S. Murko, J. Ščančar, R. Milačič, J. Anal. At. Spectrom. 26 (2011) 86–93. doi: 10.1039/C0JA00088D
-
[72]
N. García-Otero, P. Bermejo-Barrera, A. Moreda-Piñeiro, Anal. Chim. Acta 760 (2013) 83–92. doi: 10.1016/j.aca.2012.11.024
-
[73]
M. Khan, C. Manes, C. Aubry, et al., Environ. Sci. Technol. 47 (2013) 10884–10894. doi: 10.1021/es402138e
-
[74]
Y. Fang, W. Wang, Y. Xu, et al., Talanta 281 (2025) 126823. doi: 10.1016/j.talanta.2024.126823
-
[75]
M. Rocha, C. Cruzeiro, E. Rocha, J. Water. Health 11 (2013) 281–296. doi: 10.2166/wh.2013.021
-
[76]
P. González-Hernández, H. Manuel, P. Verónica, et al., Environ. Technol. 38 (2017) 911–922. doi: 10.1080/09593330.2016.1266393
-
[77]
G. Zhuang, Y. Lin, M. Elvert, et al., Mar. Chem. 160 (2014) 82–90. doi: 10.1016/j.marchem.2014.01.011
-
[78]
H. Liu, J. Chen, M. Chen, et al., Anal. Chim. Acta 1274 (2023) 341496. doi: 10.1016/j.aca.2023.341496
-
[79]
J. Tu, J. Min, Y. Song, et al., Nat. Biomed. Eng. 7 (2023) 1293–1306. doi: 10.1038/s41551-023-01059-5
-
[80]
F. Gao, C. Liu, L. Zhang, et al., Microsyst. Nanoeng. 9 (2023) 1.
-
[81]
J.B. Harkins, B.B. Katz, S.J. Pastor, et al., Anal. Chem. 80 (2008) 2734–2743. doi: 10.1021/ac702214n
-
[82]
K.R. Chalcraft, P. Britz-McKibbin, Anal. Chem. 81 (2009) 307–314. doi: 10.1021/ac8020455
-
[83]
M.M. Rahman, K. Chingin, H. Chen, Chem. Commun. 55 (2019) 9188–9191. doi: 10.1039/c9cc04705k
-
[84]
W. Fang, W. Guo, R. Lu, et al., Nature 626 (2024) 86–91. doi: 10.1038/s41586-023-06917-5
-
[85]
J. Beck, M. Biechele, C. Repik, et al., J. Sep. Sci. 46 (2023) 2200943. doi: 10.1002/jssc.202200943
-
[86]
C. Bruggink, R. Maurer, H. Herrmann, et al., J. Chromatogr. A 1085 (2005) 104–109. doi: 10.1016/j.chroma.2005.03.108
-
[87]
S. Uhlig, M. Kaldhusdal, A. Gjevre, Chromatographia 74 (2011) 133–138. doi: 10.1007/s10337-011-2054-y
-
[88]
S. Wouters, B. Wouters, S. Jespers, et al., J. Chromatogr. A 1355 (2014) 253–260. doi: 10.1016/j.chroma.2014.06.025
-
[89]
C. Bruggink, M. Wuhrer, C.A.M. Koeleman, et al., J. Chromatogr. B 829 (2005) 136–143. doi: 10.1016/j.jchromb.2005.10.006
-
[90]
M. Dong, M. Wu, F. Wang, et al., Anal. Chem. 82 (2010) 2907–2915. doi: 10.1021/ac902907w
-
[91]
B. Quemener, C. Desire, L. Debrauwer, et al., J. Chromatogr. A 984 (2003) 185–194. doi: 10.1016/S0021-9673(02)01729-6
-
[92]
M. Maier, D. Reusch, C. Bruggink, et al., J. Chromatogr. B 1033-1034 (2016) 342–352. doi: 10.1016/j.jchromb.2016.08.001
-
[93]
J. Wang, S.J. Valentine, P. Li, Rapid. Commun. Mass Spectrom. 35 (2021) e9179. doi: 10.1002/rcm.9179
-
[94]
X. Gong, X. Xiong, S. Wang, et al., Anal. Chem. 87 (2015) 9745–9751. doi: 10.1021/acs.analchem.5b01877
-
[95]
W. Chen, Q. Li, J. Luo, et al., Anal. Chem. 96 (2024) 8886–8892. doi: 10.1021/acs.analchem.4c01469
-
[96]
W. Chen, Z. Gao, F. Chu, et al., Anal. Chem. 94 (2022) 15002–15009. doi: 10.1021/acs.analchem.2c02919
-
[97]
W. Chen, K. Yuan, Q. He, et al., Talanta 274 (2024) 125981. doi: 10.1016/j.talanta.2024.125981
-
[98]
M. Wong, H. Tang, S. Man, et al., Rapid. Commun. Mass Spectrom. 27 (2013) 713–721. doi: 10.1002/rcm.6497
-
[99]
Y. Xu, X. Chen, D. Zhang, et al., Food Chem. 461 (2024) 140882. doi: 10.1016/j.foodchem.2024.140882
-
[100]
J. Wang, M. Gao, G. Yan, et al., Anal. Chim. Acta 880 (2015) 77–83. doi: 10.1016/j.aca.2015.04.030
-
[101]
X. Lu, Y. Chen, Y. Zhang, et al., Adv. Mater. 36 (2024) 2307875. doi: 10.1002/adma.202307875
-
[102]
H. Liu, H. Qiu, Chem. Eng. J. 393 (2020) 124691. doi: 10.1016/j.cej.2020.124691
-
[103]
X. Yuan, D.M. Desiderio, J. Mass Spectrom. 37 (2002) 512–524. doi: 10.1002/jms.307
-
[104]
N.S. Tannu, J. Wu, V.K. Rao, et al., Anal. Biochem. 327 (2004) 222–232. doi: 10.1016/j.ab.2004.01.033
-
[105]
E.J. Zaluzec, D.A. Gage, J. Allison, et al., J. Am. Soc. Mass Spectrom. 5 (1994) 230–237. doi: 10.1016/1044-0305(94)85013-5
-
[106]
C. Li, A. DeVor, J. Wang, et al., Rapid. Commun. Mass Spectrom. 36 (2022) e9341. doi: 10.1002/rcm.9341
-
[107]
S. Longobardi, A.M. Gravagnuolo, I. Rea, et al., Anal. Biochem. 449 (2014) 9–16. doi: 10.1016/j.ab.2013.11.021
-
[108]
J. Duan, H. Wang, Q. Cheng, Anal. Chem. 82 (2010) 9211–9220. doi: 10.1021/ac102262m
-
[109]
A. Saeed, M. Najam-ul-Haq, F. Jabeen, et al., Anal. Chem. 85 (2013) 8979–8986. doi: 10.1021/ac4015484
-
[110]
X. Meng, J. Hu, Z. Chao, et al., ACS. Appl. Mater. Interfaces. 10 (2018) 1324–1333. doi: 10.1021/acsami.7b13640
-
[111]
X. Wang, N. Li, D. Xu, et al., Talanta 190 (2018) 23–29. doi: 10.1016/j.talanta.2018.07.066
-
[112]
H. Zhou, S. Xu, M. Ye, et al., J. Proteome Res. 5 (2006) 2431–2437. doi: 10.1021/pr060162f
-
[113]
L. Xu, W. Zhu, R. Sun, et al., Analyst 140 (2015) 3216–3224. doi: 10.1039/C5AN00102A
-
[114]
T.A. Worrall, R.J. Cotter, A.S. Woods, Anal. Chem. 70 (1998) 750–756. doi: 10.1021/ac970969e
-
[115]
B. Tucker, M. Hermann, A. Mainguy, et al., Analyst 145 (2020) 643–650. doi: 10.1039/c9an01782h
-
[116]
Y. Zhou, C. Peng, K.D. Harris, et al., Anal. Chem. 89 (2017) 3362–3369. doi: 10.1021/acs.analchem.6b03934
-
[117]
S. Rankin-Turner, L.M. Heaney, Anal. Sci. Adv. 2 (2021) 193–212. doi: 10.1002/ansa.202000135
-
[118]
H. Wang, J. Liu, R.G. Cooks, et al., Angew. Chem. Int. Ed. 49 (2010) 877–880. doi: 10.1002/anie.200906314
-
[119]
J. Liu, H. Wang, N.E. Manicke, et al., Anal. Chem. 82 (2010) 2463–2471. doi: 10.1021/ac902854g
-
[120]
J. Li, Y. Zheng, W. Mi, et al., Anal. Methods 10 (2018) 2803–2811. doi: 10.1039/c8ay01081a
-
[121]
T. Wang, Y. Zheng, X. Wang, et al., Anal. Chem. 89 (2017) 7988–7995. doi: 10.1021/acs.analchem.7b01296
-
[122]
T. Li, L. Fan, Y. Wang, et al., Anal. Chem. 89 (2017) 1453–1458. doi: 10.1021/acs.analchem.6b02571
-
[123]
M.T. Dulay, R.N. Zare, Rapid. Commun. Mass Spectrom. 31 (2017) 1651–1658. doi: 10.1002/rcm.7952
-
[124]
Y. Zheng, Q. Wang, X. Wang, et al., Anal. Chem. 88 (2016) 7005–7013. doi: 10.1021/acs.analchem.5b04732
-
[125]
N. Riboni, A. Quaranta, H.V. Motwani, et al., Sci. Rep. 9 (2019) 10296. doi: 10.1038/s41598-019-45358-x
-
[126]
Q. Wang, M. Bhattarai, P. Zhao, et al., J. Am. Soc. Mass Spectrom. 31 (2020) 2226–2235. doi: 10.1021/jasms.0c00310
-
[127]
K. Chiu, Q. Wang, H.P. Gunawardena, et al., Int. J. Mass Spectrom. 469 (2021) 116688. doi: 10.1016/j.ijms.2021.116688
-
[128]
H. Chen, S. Yang, M. Li, et al., Angew. Chem. Int. Ed. 49 (2010) 3053–3056. doi: 10.1002/anie.200906886
-
[129]
T. Li, S. Li, J. Shi, et al., Anal. Chim. Acta 1205 (2022) 339760. doi: 10.1016/j.aca.2022.339760
-
[130]
S. Xu, J. Xue, Y. Bai, et al., Anal. Chem. 92 (2020) 15854–15861. doi: 10.1021/acs.analchem.0c03167
-
[131]
M. Zhang, R. Shang, H. Zhang, et al., Anal. Chim. Acta 1313 (2024) 342790. doi: 10.1016/j.aca.2024.342790
-
[132]
Y. Li, Y. Gao, B. Zhan, et al., Chin. Chem. Lett. 33 (2022) 2708–2710. doi: 10.1016/j.cclet.2021.08.119
-
[133]
Z. Ma, Y. Gao, F. Chu, et al., Chin. Chem. Lett. 33 (2022) 4411–4414. doi: 10.1016/j.cclet.2021.12.029
-
[134]
J. Pan, K. Xu, X. Yang, et al., Anal. Chem. 81 (2009) 5008–5015. doi: 10.1021/ac900423x
-
[135]
H. Satoh, T. Yamauchi, M. Yamasaki, et al., J. Vet. Diagn. Invest. 23 (2011) 1202–1207. doi: 10.1177/1040638711425592
-
[136]
Y. Liu, Z. Miao, R. Lakshmanan, et al., Int. J. Mass Spectrom. 325-327 (2012) 161–166. doi: 10.1016/j.ijms.2012.06.006
-
[137]
C.A. Cassou, E.R. Williams, Analyst 139 (2014) 4810–4819. doi: 10.1039/C4AN01085J
-
[138]
A.T. Iavarone, O.A. Udekwu, E.R. Williams, Anal. Chem. 76 (2004) 3944–3950. doi: 10.1021/ac049724+
-
[139]
T.G. Flick, C.A. Cassou, T.M. Chang, et al., Anal. Chem. 84 (2012) 7511–7517. doi: 10.1021/ac301629s
-
[140]
D.J. Clarke, D.J. Campopiano, Analyst 140 (2015) 2679–2686. doi: 10.1039/C4AN02334J
-
[141]
J.C. DeMuth, S.A. McLuckey, Anal. Chem. 87 (2015) 1210–1218. doi: 10.1021/ac503865v
-
[142]
G. Weng, Z. Liu, J. Chen, et al., Anal. Chem. 89 (2017) 10256–10263. doi: 10.1021/acs.analchem.7b01695
-
[143]
A.R. McKay, B.T. Ruotolo, L.L. Ilag, et al., J. Am. Chem. Soc. 128 (2006) 11433–11442. doi: 10.1021/ja061468q
-
[144]
T.G. Flick, S.I. Merenbloom, E.R. Williams, J. Am. Soc. Mass Spectrom. 22 (2011) 1968–1977.
-
[1]
-
-

 DownLoad:
DownLoad:

 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 49
- HTML全文浏览量: 3
 下载:
下载:
