Figure 1.
Summary of the main topics of this review.
Single-atom catalysts partner with defects to open a new chapter in catalysis: Synthesis, performances, and mechanisms
Yuqi Zhu , Suhua Chen , Jianping Zou , Bo Li , Wei Ren , Gaoxia Zhang , Jiajie Wang , Xinyu Liu , Qianhui Li , Huiying Zhang , Fanying Xia

The utilization of fossil fuels has greatly promoted productivity development and economic expansion, marking a major advancement in human history [1-4]. However, this unsustainable and ecologically hazardous energy utilization calls for more advanced and sustainable energy technology innovation and environmental restoration methods [5-8]. Substantial research efforts have yielded remarkable achievements in critical areas including the efficient production of hydrogen energy, carbon dioxide capture/conversion, and the utilization of solar energy [9-14]. Indeed, for most environmental remediation and energy technologies, the high activity, selectivity and durability of catalysts are crucial aspects.
Recently, single-atom catalysts (SACs) have emerged as a cutting-edge direction in heterogeneous catalysis due to their highly dispersed active sites [15-19]. Distinguished from conventional catalysts, SACs exhibit unique ligand microenvironment and electronic structure enable exceptional catalytic performance and reaction specificity [20-22]. Typically, metal single atoms (SAs) and their local coordination environment act as active centers for catalytic reactions and are mainly immobilized on a support through ligand/ionic interactions of adjacent atoms [23,24]. However, SACs preparation is usually accompanied by particle agglomeration owing to the extremely high surface energy [25-29]. Efforts to address this issue have focused on the addition of appropriate amounts of SACs to various supports with high surface areas and desired anchor localization sites. These key aspects can be ensured by strategically introducing defects in the supports, thus ensuring the dispersion/stability of SAs [30-33]. It is worth noting that during the high-temperature synthesis of SACs, interstitial metal atoms can promote the in-situ generation of internal structural defects, thereby alleviating local stress fields [34,35]. These in-situ generation of defects profoundly influence the physicochemical properties of SACs, thus enhancing catalytic performance metrics such as activity and selectivity [36-41]. A comprehensive understanding of defective-SACs interactions is therefore essential for establishing robust structure-property relationships.
Positioning defective-SACs as a central research focus not only enhances catalytic performance but also provides a unique model of defective-SACs interactions for atomic-level catalytic science. This approach promises to advance catalytic materials development from empirical trial-and-error approaches to rational design paradigms, with transformative potential for energy conversion and environmental remediation. Some excellent reviews on the application of defect synergistic SACs in photocatalysis/electrocatalysis as well as biomass conversion have already appeared [42-45]. However, there is still a lack of reviews on the application of defective-SACs in the environmental field. More importantly, the profiling of the different roles played by defects in the constructed defective-SACs has not been designed to be exhaustively described in any of the reported review articles, and an in-depth understanding of the interactions between defects and SACs is crucial for establishing structure-property relationships. At the same time, the time point of defect formation largely determines the "responsibility" it assumes in catalytic structure formation. And this classification basis has not been deeply analyzed in previous reviews. Therefore, it is great need to comprehensively summarize and discuss a series of issues, including the generation of defective-SACs, the influence of defects on SACs, and their outstanding performance in the energy and environmental fields. The article begins with a summary of the synthetic strategies for defective-SACs, which is the first time that the classification is based on the difference in formation time between SACs and defects, with a focus on the important influencing factors in the different synthetic approaches. Immediately thereafter, a comprehensive analysis of the different roles played by defects in the constructed defective-SACs (anchoring sites, electronic structure modulation, site/dynamic lineage formation of new sites, and selective recognition) in order to unravel the mystery of the high activity and stability of defective-SACs. Subsequently, the mechanism behind the dominance of defective-SACs in energy and environmental fields is analyzed in depth from the perspective of the mechanism of each catalytic reaction. The article concludes with the main challenges and prospects of defective-SACs in terms of precise synthesis, accurate characterization and catalytic mechanism studies (Fig. 1).
Given the exceptional performance of defective-SACs in catalytic science, their precise design and preparation have become a key challenge today [46,47]. Till now, researchers have conducted extensive studies and observed that the timing of defect formation plays a critical role in the final catalytic structure [48,49]. In light of this, synthesis strategies can be primarily categorized into two main categories: one-step and stepwise strategy, based on the time order of SAs and defects formation, with particular focus on key influencing factors during the synthesis process.
The one-step pyrolysis method involves the simultaneous high-temperature decomposition of precursors under reducing or inert atmospheres, enabling the synergistic construction of defects and SACs. This is the most direct method for synthesizing defective-SACs. In this process, precursor selection and pyrolysis temperature control are the core factors determining the stability and dispersion of SACs.
The core of precursor selection lies in creating a microenvironment during pyrolysis that inhibits metal aggregation and promotes defect formation. Zhao et al. utilized MgO@CP@Hemin sandwich structures, leveraging the π-π conjugation between coal pitch (CP) and hemin to effectively fix Fe species and suppress their pyrolytic aggregation (Fig. S1a in Supporting information) [50]. During pyrolysis, CP graphitizes and embedded into Fe SACs, releasing lattice strain to form C defects. Based on a similar principle, Zhou et al. pyrolyzed S-encapsulated ZIF-8, enabling precise formation of Pb-N2SV sites and abundant porous structures with C-defect via S volatilization and reaction with gaseous Pb sources, achieving stable dispersion of Pb SAs (Fig. S1b in Supporting information) [51]. Leveraging the advantages of volatile components, Wei et al. designed C-defect engineering of Fe-N4 sites catalysts via a substitution strategy [46]. During the co-pyrolysis of ZIF-8/Fe(acac)3, ZnO NPs generated at 340 ℃ increased with rising temperature but suddenly disappeared at 700 ℃ (Figs. S1c-h in Supporting information). The resulting coordination-unsaturated N atoms effectively captured Fe species and promoted Zn2+/Fe exchanged.
Systematic studies of the effect of pyrolysis temperature on the structure-property characteristics of prepared defective-SACs are equally crucial. Temperature changes significantly alter the defect density and coordination environment of SACs in the final materials. Lv et al. investigated the effect of pyrolysis temperatures (800–1000 ℃) on Co1N-C structures, finding that defect density (Fig. S1i in Supporting information) and amorphous dispersed def-Co-N3 configuration (Fig. S1j in Supporting information) reached a peak at 900 ℃ [52]. However, further temperature increased triggered additional graphitization processes and promoted def-Co-N3 restructuring, leading to a reduction in defect numbers and the formation of Co-N3 structures. This temperature-dependent behavior clearly revealed a delicate balance between the defect maximization region and the structural stability threshold during SAC synthesis. The study of Yuan et al. also confirmed the significant role of temperature in the formation and final state of Co SACs [53].
Beyond regulating defect concentration through pyrolysis temperature, precise control over the location and quantity of defects on the substrate remains a significant challenge [54]. For that, Rong et al. innovatively designed cyanuric acid (CA), 2,4-diaminophenyl-6-phenyl-1,3,5-triazine (DPT), and NiCl2·6H2O coordination precursors at the molecular level, successfully developing vacancy-programmable Ni SACs [32]. They discovered that at 500 ℃, the O and N atoms in CA and DPT coordinated with Ni(Ⅱ) to form an O/N mixed coordination geometry. While at 800 ℃, the Ni-O bonds broke preferentially due to the difference in bond energies between Ni-O and Ni-N, selectively generating O vacancies (OVs). In addition to the aforementioned bond-selective strategy, the unique symbiotic effect between SAs and defects also provides a regulatory pathway. Xu et al. discovered in the Ag-doped CuInS2 system that Ag SAs could significantly reduce the formation energy barrier of neighboring sulfur vacancies (SVs) [55]. However, this symbiotic effect exhibited concentration dependence, and when the Ag loading exceeded 2%, atomic aggregation disrupted the symbiotic state.
Although the one-step pyrolysis synthesis of defective-SACs is convenient to operate, there may be challenges in precisely controlling the dispersion and distribution of SACs during the pyrolysis process. To overcome this bottleneck, a stepwise construction strategy has been developed. This strategy employs a modular design combining "defect pre-positioning and SACs precise anchoring", enabling precise regulation of the structure and composition of SACs [3,56,57]. In this section, we focus more on the generation of defects and how to precisely achieve the dispersion of SACs.
In terms of thermally driven precise anchoring, Yang et al. used a hydrothermal-molten salt synergistic method to prepare uniformly dispersed Fe1/TiO2OVs (Fig. S2a in Supporting information) [58]. Under this strategy, the concentration of TiO2OVs was controlled by adjusting the hydrothermal time and temperature, followed by precise anchoring of Fe SAs onto pre-positioned OVs in the liquid-phase environment provided by molten salt. Breaking through traditional thermodynamic limitations, Liu et al. developed Joule heating technology to rapidly introduce Fe SAs into defect-rich porous carbon spheres (DCS) under millisecond high-temperature shocks [49]. The high-temperature shock enabled the heating and quenching processed to be completed instantaneously, promoting the rapid synthesis of electrocatalysts and opening up possibilities for practical applications.
Shifting to a defect-precise construction strategy, etching technology has become a key tool due to its high-precision control capabilities [59,60]. Dong et al. used Cl2 to etch TiC{100} facets in a fluidized bed reactor to create Ti vacancies (TiVs), and then synthesized Pt-TiVC via auxiliary hydrogen reduction (Fig. S2b in Supporting information) [61]. Among these, etching temperature was the core factor controlling TiVs density. At temperatures reached 800 ℃, excessive etching of Ti atoms within TiC caused outer layer C atoms to lose the constraint of Ti-C bonds, reassembling into graphene ribbons that could not stabilize Pt SAs and cover internal TiVs. In contrast, acid/base solution etching at room-temperature enabled the preparation of defective-SACs by adjusting acid/base concentration, albeit only at extremely low atomic loadings (0.4 mmol%) [62].
Beyond traditional thermochemical methods, photoreduction and plasma technology offer new avenues for precise defect engineering and SAs loading. The photoreduction strategy, with its mild nature, enabled precise control of carrier defect concentration by adjusting ultraviolet light intensity or wavelength/time, as demonstrated by Ren et al. in anchoring Ru SAs on UiO-66 nodes [63]. Similarly, the team of Tao utilized light emitting diode light to drive methanol reduction, precisely anchoring Ru SAs at pre-existing vacancies on Ni(OH)x nanoplates [64]. However, due to the slow rate of photoreduction, migration and aggregation of SAs occurred when the loading exceeded 1.5 wt%. Plasma technology, which utilizes high-energy electrons to generate defects on the surface, can effectively overcome these issues. For example, Zhu et al. treated TiO2 nanowires with H2/N2 plasma to generate OVs, and subsequently achieved high loading (~18.1 wt%) of Sr SAs via a solvothermal method [65]. Indeed, plasma atmosphere selection is crucial. Therefore, Li et al. further revealed that the selection of plasma atmospheres (e.g., O2, Ar, N2, Ar/H2, Ar/NH3) directly influenced defect levels and SAs loading effects through differentiated etching, oxidation, reduction, or doping mechanisms (Fig. S2c in Supporting information) [66]. Experimental results indicated that introducing H2 resulted in more defects than introducing NH3, while Ar plasma was more effective than N2 plasma in enriching defects. And plasma oxidation in an O2 atmosphere was more likely to cause excessive etching and severe structural damage.
The emerging strategy, which synchronizes defect generation with metal anchoring, opens up a new dimension for the efficient preparation of defective-SACs. It is important to note that the one-step strategies mentioned earlier primarily involve the simultaneous construction of defective-SACs via pyrolysis, whereas the emerging strategy outlines approaches for synchronously constructing defective-SACs beyond pyrolysis, offering significantly enhanced efficiency and structural controllability [67]. This section will outline emerging defective-SAC fabrication strategies, with a particular focus on factors affecting SACs dispersion.
Microwave energy fields, with their precise controllability, have emerged as a powerful tool [68]. Fei et al. utilized amine-functionalized graphene oxide (AGO) and trace metal salt precursors to synthesize defective-SACs (M-NG-MW, M = Co, Ni, Cu) [69]. During microwave-assisted thermal reduction, O-containing functional groups decomposed to release CO/CO2, which in situ form C vacancies (CVs) while simultaneously anchoring metal SAs. Interestingly, the combination of microwave technology and plasma can selectively form specific defects. Under microwave-induced plasma treatment, N atoms were selectively eliminated, leading to a structural transition from pentagonal pyrrolidine N defects around Ni sites to a stable pyridine N-dominated Ni-N2 coordination configuration (Fig. S2d in Supporting information) [70].
In the pursuit of high active sites density, breaking through the loading capacity limit of SACs has become a key objective [71,72]. Wang et al. developed a "laser-growing" strategy, which used laser pulses to precisely localize energy in microscopic regions, creating high-density defects in-situ on various substrates at ambient temperature and pressure, thereby achieving a record-breaking SAs loading capacity (42 wt%) [73]. Additionally, physical force field strategies are equally applicable for the preparation of high-loading SACs [74,75]. Tan et al. used mechanical chemical wet ball milling to simultaneously achieve molecular-level disaggregation and dispersion of iron phthalocyanine (FePc) aggregated under shear/compression forces, while creating vacancies on graphene substrates by removing C atoms (Fig. S2e in Supporting information) [76]. The introduction of O-containing groups via ethylene glycol medium further stabilized coordination bonds between dispersed FePc molecules and defect sites.
This chapter outlines one-step pyrolysis, two-step synthesis, and emerging techniques for preparing defective-SACs, including (method comparisons are shown in Table S1 in Supporting information). Controlling energy input (e.g., temperature, solvent concentration, light intensity) is key to tailoring defective-SAC structures. Achieving precise structural control requires focusing on three aspects: (ⅰ) Selecting appropriate precursors to directionally construct defects and anchor SAs; (ⅱ) Balancing energy inputs to avoid overprocessing, which can lead to structural degradation and defect loss, with precise control of pyrolysis temperature and solvent concentration being particularly critical; And (ⅲ) flexibly combining and optimizing method sequences in stepwise strategies, such as using etching to generate defects and light/electrochemical reduction to anchor SAs, thereby customizing synthesis pathways for target catalysts. In this process, defects are not only critical anchor points for SAs, their specific types and the unique coordination configurations they form with SAs will also determine the final catalytic performance. Based on these controllably synthesized materials, the next chapter will delve into the intrinsic mechanisms by which defects exert their effects in catalytic processes.
Defect engineering has become a common approach in customized SACs. However, the diversity and complexity of defects themselves have made their mechanism of action in the SAC system unclear. Typically, defects play a key role in anchoring SAs to achieve high loading and stability of SACs, thereby significantly enhancing catalytic performance. Moreover, defects can also enhance the catalytic activity of SACs by adjusting their electronic structure. More importantly, in the actual reaction, not only do SACs play a primary role, but their adjacent defect sites may also contribute to activity. To uncover the essence of the high performance of defective-SACs, this section will systematically analyze the multifaceted roles of defects within their structures.
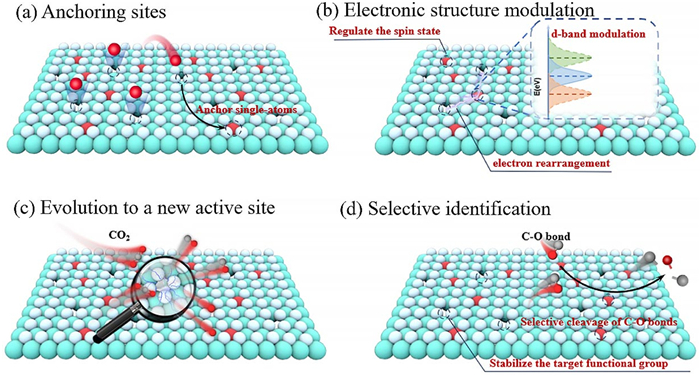
The high surface energy properties of SAs make them highly susceptible to agglomeration [77-79], and anchoring them stably to the carrier surface is the key to preventing agglomeration. Defect structures provide specific binding sites for SAs as natural atomic-level anchors. Due to the complexity of the defect species, the chemical interactions with SAs vary significantly. Therefore, it is necessary to systematically analyze the diverse mechanisms of this atomic-scale anchoring effect to elucidate the specific ways in which different defect types achieve stable anchoring.
Cation vacancies, as a common type of defect, can provide more space for adsorbed SAs to adjust the interfacial electron density and improve the catalytic performance. Ni vacancy-modified Ni(OH)x (VNi-Ni(OH)x) trapped the Pt SAs (Pt-Ni(OH)x) via Pt-O4 coordination (Fig. 2a) [80]. DFT calculations epitomized that an electron delocalization occurred between Pt SAs and bonded O atoms, leading to electron transfer onto Pt SAs. This phenomenon not only greatly improved the electrical conductivity of Pt-Ni(OH)x, but also promoted the activation of the reactants through the interfacial dipole effect, which improved the electrocatalytic performance of VNi-Ni(OH)x (Fig. 2b). Similarly, TiVs constructed in Ti3-xC2TyMXene enabled Pt SAs to occupy vacancy sites via Pt-C bonds (Pt1/Ti3-xC2Ty), inducing a 0.33 |e| charge enriched in Pt sites and significantly reduced CO2 adsorption and activation energies [62].
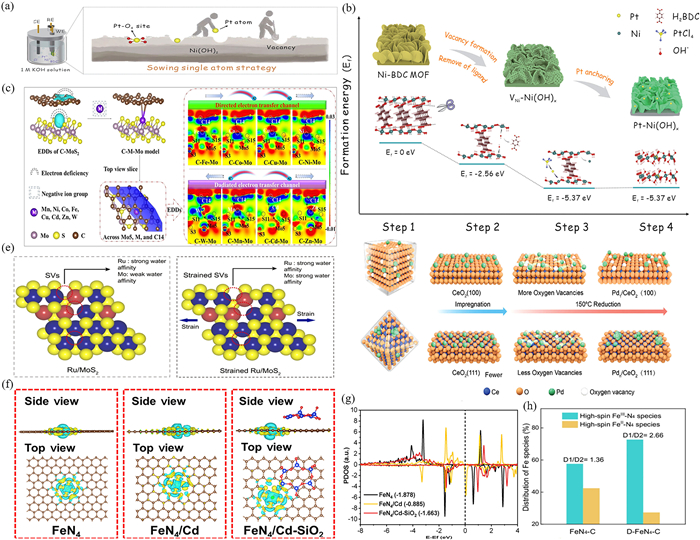
Anionic vacancies mainly act as anchoring sites for SAs via spatial confinement or charge attraction, enhancing SACs metal-metal or metal-support interactions (MSIs) to construct high-active SACs. This was vividly described as the "E-locks" and "E-channels" [81]. The SVs in C-MoS2 formed an "E-lock" with an electronic defect of 0.2 |e| (Fig. 2c), which both trapped and in-situ fixed SAs while enhancing interfacial interactions through confinement effects. When SAs occupied the SVs, they formed C-M-Mo with Mo and C, and E-channels occurred at the interface. Feng et al. intuitively revealed the key role of interfacial effects by comparing the anchoring modes of Ir SAs in the interior of the CoOOH lattice (Ir1/CoOOHlat) with those on the surface (Ir1/CoOOHsur) [82]. The Ir SAs in Ir1/CoOOHsur shared diagonal OVs with CoOOH, forming Ir-OH—Co interfaces, where restricted electronic interactions were more favorable for catalytic processes. Conversely, the formation of Ir-Co bonds in Ir1/CoOOHlat promoted electron adsorption with intermediates.
The defined strategy, as the third defect anchoring strategy, significantly enhances the catalytic activity by modulating the crystal surface-dependent defect distribution and strain effects. Hu et al. compared the differences in Pb species loaded on CeO2(100) and (111) surfaces, revealing the correlation between catalyst stability and surface structure [57]. The abundance of OVs in CeO2(100) crystalline surfaces induced the formation of Pb-O bonds to anchor Pb SAs, whereas the OV-poor (111) crystal face led to Pb clusters formation (Fig. 2d), confirming that high-density OVs were crucial for stabilizing SAs. Jiang et al. innovatively introduced strain-tunable SVs in MoS2 and found that strain amplified the synergistic effect of Ru SAs and SVs [83]. On the one hand, strain enabled Mo to act as a medium to attract and accelerated the transfer of reactants to Ru SAs (Fig. 2e). On the other hand, strain optimized the electronic structure of Ru SAs, thus optimizing the catalytic process.
The activity of a catalyst is essentially determined by the electronic structure of its active sites, so the key to enhancing performance lies in optimizing the electronic properties of these active sites. In defective-SACs systems, SAs are the primary reaction sites, and the introduction of defects is expected to affect their adsorption energy with reactants by adjusting the electronic structure of SAs [84-86]. Notably, this regulation of electronic structure is typically closely related to the anchoring effect of defects on SAs, as detailed in the previous section. Therefore, this section will focus on exploring the effects of defects on charge arrangement, D-band centers, and the spin states of SAs.
Defects significantly affect catalytic performance by reconfiguring the charge distribution of active sites, with the core mechanism being the regulation of the adsorption energy balance of reactants/intermediates. Yang et al. utilized defective graphene (DG) anchored Pt atoms (Pt@DG) to form Pt-C3 coordination structures, resulting in electron transfer from Pt to C atoms (0.079 e–), weakening the adsorption strength of intermediates and optimizing catalytic kinetics [31]. Further studies showed that support-MSIs (SMSIs) also modulated activity through interfacial electron rearrangement. When Pt was anchored to the OVs of CeO2 by Jiang et al., Pt-O-Ce interfacial distortion drove continuous electron transferred from Pt to Ce (Ce4+ → Ce3+) until charge density reached equilibrium, with SMSI intensity positively correlated with interfacial distortion severity [87]. Thereinto, Pt1CeO2{100} stood out in subsequent catalytic processes due to its strongest Pt electron donating ability and high positive charge center formation ability.
In heterogeneous catalysis, the D-band center theory can elucidate the binding properties between reactants and the catalyst surface, while regulating the D-band of SACs is key to optimizing adsorption equilibrium. Taking the NiFe layered double hydroxide (LDH) anchored Ru SAs (Ru1/LDH-MⅢ) as an example, the MⅢ defects contributed to the high oxidation state of Ru and achieved the maximum electron transfer through the construction of the O6/Ni6 double-shell coordination structure [38]. Meanwhile, its D-band electron reduction significantly weakened the Ru(d)-CO(2π*) antibonding orbital occupation, thus promoting the desorption of intermediates. Indeed, the modulation of the D-band by defects is universal. Zhang et al. utilized SiO2-modified CVs to break the symmetry of FeN4, forming a FeN4/Cd-SiO2 configuration with asymmetric charge distribution and surface polarization (Fig. 2f) [88]. This asymmetric electronic structure triggered charge rearrangement, causing the D-band center of Fe to shift upward (Fig. 2g), enhancing the electron transfer capability of FeN4 and optimizing O2 adsorption and intermediates binding energies. Similarly, introducing CVs to construct an asymmetric Fe-N4CV structure yielded the same conclusion [89]. Therefore, the core role of defect engineering lied in precisely optimizing the adsorption/desorption strength of key intermediates by adjusting the position of the D-band center, thus enhancing catalytic performance.
The metal center of SAC is usually the adsorption site for reactants/intermediates in catalytic reactions, in which the spin state of the metal atoms is closely related to the performance of catalytic activity, and defect engineering provides a new way to precisely regulate the spin state. Lai et al. achieved a significant increase in the percentage of high-spin FeⅢ-N4 (doublet 1, D1) by anchoring Fe-N4 on a carbon support ((D-FeN4C) with intrinsic defects (Fig. 2h), suggesting that the defects could be targeted to enrich high-spin active species [90]. Although both FeN4C and D-FeN4C retained high-spin states, the introduction of the defects synchronously increased the number of unpaired electrons and the electron density of the Fe-N4 sites, leading to the catalytic performance jump by synergistically optimizing the spin states and electronic structures.
In multi-reactor catalytic systems, SACs are difficult to achieve optimal performance through a single active site due to the linear limitation of adsorption energy and site competition effect. Instead, defective-SACs show unique advantages owing to their dual-site synergistic mechanism derived from their unsaturated coordination characteristics. Defects can participate in the reaction directly as intrinsic active sites, and can dynamically couple with SACs to generate novel active sites. Therefore, this chapter will focus on exploring the role of defects as intrinsic active sites and their dynamic evolution during reactions, providing new insights for developing efficient multi-step catalytic systems.
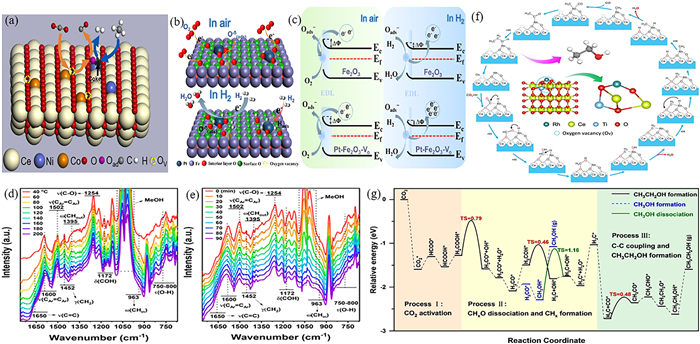
In multi-step catalytic reactions, carbon deposition severely hinders C—H activation, while adsorbed oxygen (Oad) can effectively eliminate carbon deposits. Wu et al. designed a Ni/M-CeO2 system (Ni/M-CeO2 SACs, M = Mg, Co and Zn) system, where Ni SACs facilitated the deep dissociation of CH4 to generate activated carbon species, whereas OV continuously supplied Oad via CO2 to eliminate carbon deposition (Fig. 3a) [91]. Thereinto, Co doping most significantly promoted Ce3+ enrichment, thus regulating Oad density, revealing the progressive metal doping-defect concentration-property relationship. Interestingly, defect-induced regulation the of electronic structure of SACs also achieved a synergistic effect at dual-site. In the constructed Pt on OV-rich Fe2O3 nanosheets (Pt-Fe2O3-VO), VO acted as a strong adsorption site for O–δ species and optimized the interfacial charge distribution by inducing electron depletion layer, which was synergized with the catalytic facilitation of O2(ads) dissociation by Pt SACs [92]. The synergy of the two led to a larger conductivity change and higher corresponding capacity (Fig. 3b) with the reduction of the overall resistance (Fig. 3c) to achieve the simultaneous optimization of response speed and stability.
The dynamic site reconstruction mechanism of defective-SACs provides a new dimension for catalytic activity enhancement. For example, when Co atoms were loaded on 2H MoS2 (Co-SMoS2), Co and SVs exhibited a dynamic equilibrium during hydrogen deoxygenation reactions [93]. During the reaction, Co atoms migrated to MoS2 basal sites to promote SVs formation while creating new active sites, namely the Co-S-Mo interface, significantly lowering the reaction energy barrier to achieve low-temperature efficient catalysis. This dynamic active site evolution is more critical in multi-reactor systems. For example, during the oxygen-dependent oxidation of benzyl (BA) alcohol catalyzed by Co-N-C, O2 and BA competed for adsorption at Co sites [94]. Strong π-π interactions between BA and CoN4 drove nearby CVs to become new adsorption sites.
Accurate modulation of the selectivity of SACs is the cornerstone for achieving atomic-level reaction pathway control. The synergistic integration of SACs and defects enables precise recognition and conversion regulation of specific functional groups, which is not only a key strategy for improving catalytic efficiency, but also a core driver for advancing green chemistry and clean energy development. Herein, we will focus on exploring the synergistic effects between defects and SACs, analyzing how they achieve selective recognition of functional groups, and providing theoretical and practical guidance for designing high-activity, high-selectivity catalysts.
As the second largest biopolymer in lignocellulose, lignin is rich in chemical value for its aromatic structural units, but efficient conversion is still a challenge. Selective cleaving C—O bonds while retaining C=C bonds is key to realize targeted conversion of lignin into high-value chemicals, and the synergistic interaction of SACs with defects provides a new idea for this purpose [95]. For example, Ru SACs loaded on CeO2 (Ru/CeO2-ROV) precisely regulated the hydrogenolysis pathway of the β-aryl ether (β-O-4) bonds (Figs. 3d and e) by the electron-coupling effect between Ru and OVs [96]. In this process, Ru SAs dominated the C—O bonds breaking, while OV optimized the electronic structure of Ru, enhancing substrate adsorption and charge transfer capabilities, thereby enabling selective cleavage of C—O bonds. The strategy was universal, with Ni/CeO2-S selectively attacking the C—O/C—C bond and Pb1/β-FeOOH exclusively hydrogenating the C=C bonds [97,98], fully showcasing the immense potential of defective-SACs to achieve selective control of chemical bonds by regulating electronic structure.
The synergistic interaction between SAs and defects not only enables selective recognition of chemical bonds through the regulation of electronic structure, but also facilitates the formation of Lewis acid-base pairs that directly participate in functional group transformations. The team of Zheng embedding of Rh SAs in Ti doped CeO2 (Rh1/CeTiOx) to form OV-Rh acid-base pairs, in which Rh acted as a nucleophilic site to capture the C atom of CO2, while OV severed as a nucleophilic site to stabilize the O atom (Fig. 3f), synergistically contributing to the breakage of the C—O bond in COOH to produce CO [99]. Meanwhile, the electron-rich region between Rh and Ti drove the electron injected into the CO/CO2 antibonding orbital inhibited excessive CO hydrogenation (CH4), and instead realized the C—C coupling through the stepwise C—O cleavage of the *CHx intermediate. Critical energy barrier analysis showed that the energy required for C—C coupling on Rh1/CeTiOx was significantly lower than that of the CO hydrogenation pathway (Fig. 3g), resulting in the selective generation of ethanol rather than by-products such as methanol.
Overall, defects play an important role for the constructed defective-SACs system, which can be broadly categorized into the following four points (Fig. 4): Overall, defects play a central role in the construction of high-performance defective-SACs systems. Their fundamental function lies in stabilizing and anchoring SAs, thereby ensuring high load capacity and stability, which are prerequisites for efficient catalysis. After anchoring, defects optimize electronic structures through interactions with SAs, regulating the adsorption/desorption behavior of reactants to enhance catalytic activity. Additionally, defects themselves can serve as intrinsic active sites or dynamically collaborate with SAs to form new sites, enhancing adaptability to complex catalytic processes. Furthermore, defects can enhance the selective recognition of specific functional groups by SACs, promoting the directed synthesis of target products, thanks to the electronic regulation of SACs by defects. Therefore, these four functions of defects are interrelated and synergistic, collectively providing a key direction for designing efficient multifunctional catalysts.
Energy problems and environmental pollution are prominent challenges facing the world today. SACs, especially defective-SACs, have developed into advanced catalysts with low cost, high selectivity and robustness due to their unique synergistic effects, and have shown promising prospects in many energy and environmental fields. On this basis, this chapter will comprehensively analyze the kernel linkage between defective-SACs systems and various applications from the perspective of each application mechanism. Particular emphasize the synergistic mechanism between defects and SACs and its broad enhancement of catalytic performance across multiple fields. At the same time, Tables S2 and S3 (Supporting information) summarizes the latest representative advances in defective-SACs, with a focus on structure-activity relationships.
ORR, as a core electrochemical process for energy conversion, directly determines the application scenarios by the choice of its reaction path (4e− to generate H2O or 2e− to generate H2O2). Although precious SACs exhibit excellent intrinsic activity, their practical application is constrained by stability limitations [100]. Introducing defects to construct anchor sites can simultaneously enhance stability and reaction kinetics. Liu et al. demonstrated that defect modulation significantly reduced the adsorption energies (Eads) of Pt on defective graphene, resulting in a stronger anchoring effect compared to samples without defect [101]. This mechanism provided a key pathway for addressing the stability challenges of precious metal SACs.
In the 4e− ORR pathway, O2 adsorption activation and intermediate conversion energy barriers are the core factors governing the reaction efficiency. Liu et al. found that double vacancy defects could optimize the *O2 hydrogenation reaction by activating *O2, specifically by designing Fe-N4 sites with different divalent defects (6, 6a, and 6I) (Fig. S3a in Supporting information) [102]. Compared to the original Fe-N4, these defect-engineered sites exhibited longer O—O bond lengths and shorter Fe-O bond lengths, thereby enhancing the interaction between *O2 and the Fe-N4 sites. From an electronic structure perspective, the hybridization between the Fe 3dz2 (3dyz) and the O2 π* orbital was further enhanced, promoting electron transfer from the Fe-N4 site to O2.
Defective-SACs not only need to regulate the initial O2 adsorption activation but also overcome the high-energy barrier bottleneck of *OH desorption. Therefore, Rong et al. constructed defects around the Co-N4 sites, successfully reducing the energy barrier of the rate-determining step (RDS) [103]. The mechanism lied in edge-defects inducing long-range electron delocalization, increasing the electron density at Co-N4 sites, significantly accelerating *OH desorption, and releasing active sites. This defect also enhanced O2 binding affinity, synergizing with the O2 activation mechanism discovered by Liu et al., which reflected the synergistic effects between different defect functions [102]. Recently, there have been new developments in the regulation of the entire ORR process by defective-SACs. Electronic structure analysis of defect-engineered heteronuclear FeMn-Dual SACs (FeMnDSA/dNC) further revealed the underlying mechanisms [104]. Defect-mediated Fe-Mn interactions enhanced electron transfer from Fe sites to the *OOH intermediate (Fig. S3b in Supporting information), strengthening Fe 3*dz2-O 2p hybridization, which further transformed the RDS in Fe*SA/NC from *OH desorption (0.83 V energy barrier) to *OOH formation (0.40 V) (Fig. S3c in Supporting information). This synergistic interaction between multiple active sites and defects offered a new dimension for ORR catalyst design.
The selectivity difference between the 2e− and 4e− ORR pathways is concentrated in the transformation of the *OOH intermediates, with the former requiring the retention of the O—O bond to generate H2O2, while the latter breaks this bond to form H2O [105,106]. Due to the thermodynamic disadvantage of the 2e− pathway, developing catalysts with both high activity and selectivity is crucial. To this end, Yang et al. constructed Co SACs (Co-B, N—CNTs) rich in OVs based on carbon nanotube, achieving effective and high selective H2O2 production [107]. This was attributed to the fact that OVs not only acted as electron capture centers to promote intermediate activation but also regulated the electronic structure of Co sites to enhance the binding energy of *OOH.
Obviously, regulating the local electronic structure of active sites is an effective strategy for achieving high-selectivity 2e− ORR, which was equally demonstrated by introducing edge-defects [108]. Interestingly, they constructed five types of Co-N4 anchoring configurations on monolayer graphene (Fig. S3d in Supporting information) and made a breakthrough discovery regarding the precise regulation of selectivity by the local geometric configuration of defects. This was because edge-defects drive electrons from Co-N4 to the carbon substrate, causing the D-and center to shift downward and significantly weakening the *OOH bonding energy, with the zigzag configuration (Co-N4/ZZ) exhibiting the most significant effect. Crucially, H2O2 erosion induced oxygen functional group saturation of edge defects, and the dynamic evolution further weakened the *OOH bond, enhancing the selectivity of the 2e− pathway.
Water electrolysis for hydrogen production, a key pathway in the transition to clean energy, is constrained by the high overpotential of the OER [109,110]. While precious metal catalysts (e.g., IrO2/RuO2) can reduce overpotential, they face cost and resource constraints. Therefore, developing non-precious metal alternative catalysts that combine high activity with low cost has become a key research focus. The synergistic effect of defects and SACs offers a new paradigm for addressing this contradiction [111]. Wang et al. anchored ultra-low loading (0.2 wt%) Ru SACs (Ru-Ni-Co-P/NC), which inhibited the inhibition of metal clustering while enhancing the intrinsic activity of OER through defect-limited domain effect and SACs coordination [112]. Defects in OER catalysis exhibit both spatial confinement and electronic regulation functions. Their physical confinement effect suppressed SACs aggregation and increased active site density [112], while induced electronic restructuring optimizes the kinetics of key steps. In terms of deprotonation optimization, the team of Zhao successfully synthesized atomically dispersed Zn with N defects on biomimetic carbon nanotubes (Zn-N-C-2) that effectively promoted the conversion of *OH to *O [113]. The introduced N defects induced electronic reorganization of Zn 4s near the Fermi level, which remarkably reduced the energy barrier for the deprotonation reaction of *OH (Fig. S3e in Supporting information).
Except to electronic regulation, neighboring geometry structure design can also regulate the adsorption configuration of reaction intermediates [114]. Wang et al. introduced neighboring Co2+ vacancies strategy in Ru SACs, inducing *OOH to form hydrogen bonds with surface oxygen [115]. Such interactions fine-tuned the adsorption conformation of *OOH, stabilized intermediates, and reduced the RDS energy barrier for the *O to *OOH reaction in the adsorbate evolution mechanism (AEM) (Fig. S3f in Supporting information). Notably, these Co2+ vacancies imparted narrower band gap to Ru1/VCo-Co(OH)2 via electronic structure modulation, enhancing both electronic conductivity and accelerated OER dynamic.
Traditional OER mechanisms are limited by linear scaling relationships and structural stability bottlenecks, driving the exploration of novel reaction pathways. Hao et al. proposed a proton donor-acceptor mechanism (PDAM) at the Ru-O-Co interface anchored by Co cation vacancies (Ruanc-Co3O4) [116]. As the reaction potential rosed, hydrogen bond vibrations between Ru-O-Co and *OOH strengthen, implying that proton adsorption energy was optimized at the bridging O sites. In contrast, the oxide path mechanism (OPM) proposed by Zhang et al. achieved direct O—O radical coupling (2*O → *OO) through Ru SAs in concert with Co defects, circumventing the risk of lattice oxygen loss (Fig. S3g in Supporting information) [117]. The RDS energy barrier of this pathway was significantly lower than that of traditional OER mechanisms, fundamentally due to Co defects regulating the Ru D-band center, simultaneously promoting O2 desorption kinetics (Fig. S3h in Supporting information). These two innovative mechanisms reconfigured reaction pathways through defect engineering, breaking through the energy barrier limitations of traditional OER.
Hydrogen energy has become the core carrier of clean energy transition due to its high energy density and zero carbon emission characteristics, while the bottleneck of HER technology is the heavy reliance on conventional Pt-based catalysts. Defective-SACs provide an innovative solution to crack the precious metal dosage and activity-stability contradiction. Dong et al. utilized TiVC to anchor Pt atoms, forming Pt-C bonds to stabilize Pt SACs (Fig. S4a in Supporting information) (bond energy −3.6 eV), thereby achieving efficient HER performance [61]. Meanwhile, Wang et al. achieved enhanced electrical conductivity by immobilizing Pt SAs on a monolayer of WO3 (Pt-SA/ML-WO3) with rich O and W vacancies, which resulted from the synergistic effect of O and W vacancies with Pt SAs to form a highly conductive n-type semiconductor [118]. The vacancies increased the DOS near the WO3 Fermi level, shifting it above the conduction band edge and exhibiting typical degenerate semiconductor features. Additionally, Pt-SAs brought in a new energy level, causing Pt-SA/ML-WO3 to display n-type semiconductor features.
Generally, acidic HER consists of two steps: H+ + e− → *H (Volmer) and H+ + *H + e− → H2 (Heyrovsky) or 2*H → H2 (Tafel). Thereinto, the free energy change for the adsorption of *H (ΔG*H) is a key indicator to evaluate the activity, and the ideal catalyst needs to satisfy the ΔG*H tends to zero [119]. The C—Co-MoS2 dual-anchored system constructed by Gong et al. exhibited excellent HER activity, which stemmed from the SMSI-induced C—Co-MoS2 and provided an eclectic microenvironment for H adsorption-desorption via oriented ("E-Channel") of "Mo5 → Co → C14’’ (Fig. S4b in Supporting information) [81]. The study of Park suggested that SMSI could also modulate the d-band center to optimize the adsorption of *H [120]. The Pt-MoAl1-xB system enhanced its SMSI with Pt SAs through the abundant Mo vacancies (MoVs) provided by metal borides nanosheets (MoAl1-xB), which resulted in the lowest D-band center in comparison to other counterparts without defects or SAs. The downward shift of the D-band center led to the decrease of ∆G*H, which further yielded the optimal ∆G*H that accelerated the Heyrovsky process and increased the efficiency of HER in acidic media.
It is worth noting that the introduction of SACs can also reversely regulate defect concentration. Zhu et al. recently found that Pd SACs loading significantly increased the OVs concentration in Pd1.5/WOχ-C, further enhancing the SMSI effect [121]. This bidirectional synergistic effect promoted *H adsorption/desorption kinetics, forming an optimized ΔG*H, while simultaneously lowering the HER reaction energy barrier (Fig. S4c in Supporting information). Although defect engineering has made significant progress in the HER field, existing strategies are largely limited to regulating coordination configurations or geometric environments. Song et al. innovatively constructed an edge plane of porous graphene (Co-N3/EG), simultaneously optimizing coordination configurations and geometric environments [122]. This design enabled *H to adopt a side-adsorbed configuration, weakening the interaction between the H-s orbital and the dxy orbital of Co (Fig. S4d in Supporting information). Electronic density difference and orbital analysis confirmed (Fig. S4e in Supporting information) that the adsorption strength of *H in Co-N3/EG was significantly weaker than in the traditional Co-N3/G system, significantly enhancing the intrinsic activity of HER.
Although alkaline HER can avoid the cost constraints of proton exchange membranes, the high hydrolysis energy barrier of the Volmer step still limits reaction efficiency [123]. For that, Chen and co-workers ingeniously constructed Pt SAs anchored on Ni vacancy-enriched NiSe substrates (PtSA-NiSe-V), where the formed Pt-Se bonds served as electron transfer "bridges" [124]. This unique atomic configuration enabled rapid electron transfer and significantly lowered the reaction barriers for both the ∆G*H2O and ∆G*H+*OH (Fig. S4f in Supporting information). To expand electronic control strategies, Xi et al. designed high-loading tungsten active sites (W-V3S4) anchored on V3S4 nanosheets [125]. This system utilized SVs-induced local electronic restructuring to optimize ΔE H2O while decreasing the ΔG*H+*OH reaction barrier (Figs. S4g-h in Supporting information), significantly accelerating the kinetics of the Volmer step.
CO2RR, as a key technology for carbon neutralization, can selectively convert greenhouse gases into high-value-added carbon-based fuels, offering both environmental and energy benefits. The CO2RR pathway encompasses three core stages: CO2 activation to form intermediates, intermediate C—O bond cleavage/hydrogenation to generate C1 products, and C—C coupling of two intermediates to generate C2+ products. Thereinto, the CO2 activation process achieves ligand activation by regulating the electronic structure of active sites, thereby determining the overall reaction efficiency. Chen et al. revealed the regulatory mechanism of defect engineering on the catalytic performance by constructing Ni SACs with different numbers of pyridinic N vacancies (NVs) (SA-NiNG-NVx) [70]. Studies indicated that NVs reduced the number of coordinating pyridinic N atoms, triggering electronic restructuring between the carrier and Ni SACs, thereby weakening carrier binding. In this scenario, Ni SACs protruded outward, forming CO2 adsorption sites that combined spatial expandability with electronic enrichment properties. Notably, this protrusion effect strengthened with increasing vacancies numbers, but catalytic performance exhibited nonlinear changes. This was because excessive vacancies, while enhancing site exposure, overly weaken SMSIs, and moderated interactions were crucial for maintaining the balance between catalyst stability and activity.
In CO2RR reactions, intermediates adsorption/desorption behavior dominates product selectivity, while defective-SACs offer a new approach to breaking the traditional linear scaling constraints. Zhou et al. systematically constructed 18 asymmetric coordination Pb SACs (Pb-NxSvVz) and revealed a dual weight mechanism [51]: (ⅰ) S atoms acted as electron donors to enhance the electron density at Pb sites, enhancing *COOH intermediates adsorption; And (ⅱ) Vacancy-induced electron depletion promoted *CO desorption (Fig. S5a in Supporting information). This dynamic equilibrium broke the linear constraint on adsorption-desorption, enabling the Pb-N2SV catalysts to occupy the apex in the volcano diagrams, exhibiting the best CO selectivity. Except to intermediates adsorption/desorption, the conversion reaction also dominates product selectivity. Zhang et al. designed Cu-SA-CO and Cu-SA-CO2 catalysts containing OVs, revealing the regulatory mechanism of the *CO hydrogenation pathway by the coordination microenvironment [39]. The study found that in Cu-SA-CO system, OVs surround Cu SACs to form a Ti-Cu-O3 structure, with electronic redistribution promoting *CO hydrogenation. In contrast, Cu-SA-CO2 had a higher energy barrier in the *CO hydrogenation process to produce CH4, primarily favoring the desorption of *CO to CO (Fig. S5b in Supporting information), resulting in a significant reduction in CH4 yield.
The synergistic interaction between defect and SACs not only optimizes the C1 product pathway, but also provides a new paradigm for C—C coupling synthesis of C2+ products by C—C. Shen et al. introduced OV-bridged Cu SACs and Ti SACs to construct a Cu-Ti-OV active unit (Fig. S5c in Supporting information), achieving highly efficient C—C coupling of *CO and *CHO intermediates at the Cu site [126]. The C—C coupling reaction was realized by division of labor: (ⅰ) Cu-Ti-OV converted *CO into *CHO; And (ⅱ) Ti0.91O2 region generated CO2 and diffused to the Cu-Ti-OV sites to realize ectopic coupling, overcoming the efficiency limitations of homotopic coupling. Besides, Xu et al. further elucidated the dynamic role allocation by constructing 2%Ag/CuInS2 (CIS) composites [55]. Ag SACs did not participate in the reaction but induced SVs to reconfigure the catalytic system, resulting in SVs becoming the primary active sites for *CO, while the intrinsic electronic structure of the CIS sites selectively generated *CHO. These two intermediates completed the final C—C coupling on the Cu surface. This "defect-dominated and SACs-assisted" synergistic model opened up a new way to precisely design C2+ products.
Electrocatalytic NRR provides a sustainable pathway for green NH3 synthesis. The NRR process covers three key steps: N2 adsorption and activation → hydrogenation of dinitrogen and N≡N bonds cleavage → NH3 desorption [127]. However, its industrialization is limited by the high bond energy and weak adsorption of N2 molecules, resulting in low Faradaic efficiency (FE) [128]. Defective-SACs overcome this bottleneck by precisely regulating the entire N2 adsorption-activation-desorption. In the report of Liu et al., Y and Sc rare earth SACs were anchored to C defects (Y1/NC, Sc1/NC), revealing that C dual stabilizing-regulating effects of the defects on the SACs [129]. As for stability, C defects prevented SACs from agglomerating through strong anchoring effects. For the regulation, the optimized electronic structure of C defects significantly enhanced N2 adsorption capacity and reduced its free energy. Further, Peng et al. successfully crossed the RDS the NRR from *N2 to *NNH by utilizing the MoVs of MXene to anchor Ru SAs (SA Ru-Mo2CTx) [130]. This was due to the enhanced Ru 3d-C 2p hybridization and upward shift of the D-band center caused by defect introduction, thereby promoting electronic feedback to the N2 antibonding π orbitals.
A non-negligible fact is that the preemption of active sites by HER under acidic conditions severely restricts the NRR selectivity. To this end, Li et al. confined Zr SACs to MXene via OVs (Mo2TiC2O2-ZrSA), achieving highly selective NRR catalysis [131]. The mechanism lied in OVs increasing the Fermi level occupation state, causing the catalyst surface to become positively charged. This property not only suppressed proton binding through electrostatic repulsion to avoid HER competition but also enhanced inert N2 activation, reduced RDS, and accelerated catalytic NRR.
Photocatalytic NRR utilizes solar energy to drive the synthesis of NH3 from N2 and H2O, exhibiting green and sustainable characteristics. However, its efficiency is limited by the difficulty in activating N2 molecules and the recombination of photogenerated carriers [132,133]. Defective-SACs overcome this bottleneck by precisely regulating electron migration pathways. Huang et al. revealed the regulatory mechanism of defect structures on photocatalytic NRR performance by constructing Cu-CN SACs containing T-type/H-type defects [134]. They found that the T-type defects enhanced NRR efficiency through dual effects: (ⅰ) Efficient transfer of photogenerated electrons. T-type defects enhanced the electron gravity of the Cu sites, directing the migration of photogenerated electrons migration and improving the efficiency of photocatalytic NH3 synthesis; And (ⅱ) an increased in the active sites. The enhanced attraction triggered π-conjugated electron cloud distortion and released isolated valence electrons, significantly increasing the number of available activation electrons. Indeed, the electronic interaction between active sites with N2 is also crucial. The defect-engineered Ru1/d-UiO-66 developed by Ren et al. achieved dual enhancement of N2 adsorption/activation (Fig. S5d in Supporting information) [63]. Specifically, the defects increased the specific surface area and transport pathways, while strong electron metal-carrier interactions (EMSI) promoted the injection of Ru d-orbital electrons into the N2 π*-antibonding orbitals, efficiently activating the N≡N bond. Meanwhile, the EMSI induced electron redistribution of the Ru-O bond optimized the intermediate adsorption and further improved the performance.
The high selectivity possessed by defective-SACs provides an innovative solution for multi-path biomass conversion to obtain target products [135-137]. In common hydrogenation mechanisms, Pd SAs and β-FeOOH containing defects (Pd1/β-FeOOH) could selectively hydrogenate C=C bonds in cinnamaldehyde to produce high value-added chemical hydrogenated cinnamaldehyde [98]. The superior performance of Pd1/β-FeOOH stemmed from its ability not only to more easily adsorb cinnamaldehyde, but also to allow the hydrogenated cinnamaldehyde to easily desorb from Pd1/β-FeOOH, facilitating the re-exposure of active sites in subsequent catalytic cycles. And the high selectivity of Pd1/β-FeOOH arose from the confinement of Pb SAs within defects, creating electron-deficient active centers that preferentially adsorbed cinnamaldehyde via the C=C bonds.
Indeed, selectively hydrogenating unsaturated aldehydes with C=O and retaining the C=C bonds is more challenging, as its thermodynamic competitiveness is far lower than that of C=C hydrogenation [138]. To overcome this bottleneck, An et al. constructed Fe-ZIF-800 with a pyrrole-type Fe(Ⅱ)-plN3 structure as active centers, successfully achieving the efficient conversion of furan aldehyde (FF) to furan alcohol (FA) [139]. The pyrrole-type Fe(Ⅱ)-plN3 active center of this catalyst was formed by a defect-induced three-coordination structure with a high-spin 3d6 Fe2+ electronic configuration featuring more unpaired electrons, significantly enhancing interaction with FF. This unique coordination environment promoted the P3 reaction pathway through isopropyl alcohol (IPA) deprotonation (Fig. S5e in Supporting information), enabling FF to stably coordinate with the Fe(Ⅱ) center and directionally activate the C=O bond, while the intact Fe(Ⅱ)-plN4 structure could only react through the energetically unfavorable P1/P2 pathways (Fig. S5f in Supporting information).
Hydrodeoxygenation (HDO) of biomass platform molecules is a key step in the preparation of hydrocarbon fuels. Xia et al. explored the significant role of the Au@NbV-2OV structure (one Au+ occupied the Nb vacancy (NbV) and two OVs) in Au1/Nb2O5 for the efficient of HDO of the lignin model compound methyl catechol [140]. This role lied in the ability of NbV and OC to alter the electronic structure of Au SAs, thereby affecting H2 activation and adsorption. The Au@NbV-2OV not only efficiently cleaved H2 into metal-H (Hδ−) and O—H (H+), but also shifted the d-band center of Au+ sites toward the Fermi level, thus promoting H2 adsorption on the catalyst on the catalyst.
Meanwhile, defective-SACs also play a crucial role in the efficient oxidative synthesis of chemicals such as biomass-derived acids and aldehydes [141]. For example, Tian et al. demonstrated remarkable yields of the high-value product glucaric acid (GLA) by using Pt SAs anchored on defective TiO2 (Pt/def-TiO2) as a photoanode for the selective oxidation of glucose (Fig. S5g in Supporting information) [142]. The high productivity was attributed to the introduction of defects inducing changes in the Pt/def-TiO2 bandgap, separation and transport of supports, and selectivity of intermediate products. Specifically, the introduction of OVs and other disorder defects induced an upward shift of the valence band, allowing additional electron transitions, resulting in a narrower bandgap. For its support separation, def-TiO2 possessed a broad electron conduction region and significant surface band bending, which significantly enhanced the separation and transport of supports. Finally, Ti SAs could regulate the selectivity of GLA by accelerating the oxidation of GLA (Fig. S5h in Supporting information).
Defective-SACs show unique advantages in AOPs for difficult-to-degrade organic pollutants management challenges. Thereinto, the core of sulfate radical-based AOPs (SR-AOPs) lies in the generation of active species through the activation of persulfates (PS, including peroxomonosulfate (PMS) and peroxydisulfate (PDS)) [143,144]. The adsorption and activation of PS in SR-AOPs are the first key factors to advance the subsequent catalytic reaction. Wu et al. designed intrinsic defective-anchored Fe-N4-site catalysts (D-FeN4C) and significantly enhanced the PMS adsorption energy due to shifting the D-band center of the Fe-N4 sites upward through long-range electronic interactions induced by defects [90]. Meanwhile, the structure synchronously promoted the formation of FeN4═O. Specifically, defects induced charge transfer enabled the extension of the S-O bond length (Fig. S6a in Supporting information) while elevating the high-spin FeⅢ-N4 species, lowered the formation energy of FeN4═O and accelerated electron transfer from organic pollutants to Fe-O π* orbitals (Fig. S6b in Supporting information).
Targeted regulation of radical and non-radical reaction pathways in SR-AOPs is not only an important research area for solving various environmental problems, but also the key to enhance the selectivity and efficiency of pollutant degradation [145]. Qin et al. reported that the introduction of NVs in dual SACs (FeNi-NV/CN) increased the contribution of the non-radical electron transfer (ETP) pathway from 50.3% to 71.8% [146]. This phenomenon arose because the NVs promoted an increase in the number of electrons transferred to the PMS by enhancing the electron density of the Fe-Ni sites, thus reconfiguring the interfacial charge distribution and shifting the conventional radical-dominated pathway to the ETP. Complementarily, Liang et al. achieved the radical to 1O2 pathway transition by introducing defects to modulate the coordination number of Co-SACs (Co-Nx, x = 2–4) (Fig. S6c in Supporting information) [147].
Photocatalytic technology has great prospects in pollutant degradation, but it is limited by the low light harvesting efficiency and the rapid recombination of e−/h+ pair of SACs [148]. Defects have been shown to be effective in overcoming these problems when working "side by side" with SACs [149]. As demonstrated in the study by Zhang et al. that the introduction of NVs and Mo SAs enhanced light absorption capacity and support utilization of Mo/NV-TCN (Fig. S6d in Supporting information) [150]. Specifically, the enhanced light absorption resulting from bandgap narrowing was primarily attributed to: (ⅰ) The absence of N atoms in the π-conjugated aromatic ring led to the reconstruction of the band structure and the appearance of new band gap states; And (ⅱ) Mo SAs caused the appearance of Mo 4d orbitals, thereby altering the band structure. In terms of the improved support utilization, on the one hand, NVs could be used as electron capture sites to inhibit the complexation of photogenerated supports. On the other hand, Mo SAs led to the directional transfer of the local charge on the NV-TCN surface, while the NV-induced formation of Mo-2C/2N bonds acted as a bridge for the charge transferred.
To address the challenge of regulating the electron action pathway in the synergistic system of photocatalysis and PMS, Wu et al. designed a core-shell p-n junction catalyst (ZnO@SA-Co-CN) to reveal the directional migration mechanism of photogenerated electrons [151]. They pointed out that such photogenerated electrons triggered the break of O—O bonds in PMS to generate mass ROS. The essential reason for this was thanks to the fact that the presence of OV in ZnO@SA-Co-CN significantly improved light absorption and narrowed the band gap, which facilitated the generation of abundant photogenerated supports. Meanwhile, the OV sites functioned as an electron reservoir to raise the electron density at the top of the VB of ZnO, and the built-in electric field (BIEF) (Fig. S6e in Supporting information) inside the heterojunction served as a pump to drive faster and more efficient electron migration from ZnO to SA-Co-CN (Fig. S6f in Supporting information). Such accelerated electron transfer kinetics amplified the redox cycling at the surface Co SAs sites, thus facilitating PMS activation and ultimately achieving the efficient degradation and mineralization of pollutants in aqueous solutions.
The construction of defective-SACs is undoubtedly a revolutionary breakthrough in chemical synthesis, which bring new possibilities for the modern industry [152,153]. For example, the restriction of Pt SA with three O atoms to CoFe LDH supports (Pt1/LDHV) via cationic vacancies modulation exhibited excellent performance in the anti-Markovian olefin hydrosilylation reaction, which was not comparable to the conventional CoFe-LDH surface-liganded single oxygen atom (Pt1/LDH) [154]. This super-insulating ability was due to the fact that Pt1/LDHV overcame the RDS as the third step (formation of *C3H7Si (OCH3)3) in the olefin hydrosilylation reaction (a four-step process) (Fig. S6g in Supporting information). The introduction of cationic vacancies in Pt1/LDHV significantly enhanced the EMSI, which increased the charge density on the Pt atoms and made it easier for electrons to transfer to the Si-C bond, significantly reducing the RDS energy. More intriguingly, the enhancement of EMSI also promoted the separation of products from the active sites, which led to the recovery of the Pt monatomic sites, further speeding up the reaction rate and improving the efficiency of Pt1/LDHV.
The emissions of VOCs can pose severe threats to both humans and the environment [155]. Fortunately, the defective-SACs exhibit promising prospects for the catalytic combustion of VOCs. Dong et al. achieved excellent catalytic activity and outstanding stability for low-temperature benzene catalytic combustion by stabilizing Pt SAs (Pt1@CeO2) using OVs generated on CeO2 supports as anchors [156]. Owing to the abundant anchoring sites provided by the OVs, the Pt1@CeO2 could achieve a high loading of Pt SAs, which enhanced its catalytic performance of benzene combustion. DFT indicated that the construction of highly-loaded Pt SAs endowed Pt1@CeO2 with a low ring-opening reaction heat (ΔE) of C6H5O2 intermediate (Fig. S6h in Supporting information). This confirmed that the formation of Pt SAs facilitated the ring-opening reaction of C6H5O2, thereby promoting the overall reaction of catalytic combustion of benzene. For another, the firmly anchored Pt SAs into OVs of CeO2 support formed the SMSI, significantly improving the stability of Pt1@CeO2. under water vapour conditions.
The rational design of highly active, selective and durable catalysts is of extraordinary significance for the realization of more advanced and sustainable alternative energy and environmental technologies. Currently, different types of defective-SACs can be constructed through a one-step or stepwise strategy. The introduced defects not only serve as anchor points to ensure the high stability of SACs but also regulate the electronic structure of SACs catalysts, thereby influencing the adsorption/desorption behavior of reactants to enhance catalytic activity. Meanwhile, the unique electronic structure of the defects endows it with a potential active sites function, which can form new sites in the reaction via intrinsic sites/with SACs to better satisfy catalytic reactions involving multiple reactants. It is worth mentioning that the defects can significantly enhance the selective recognition of SACs for specific functional groups, which helps to generate specific products. Ultimately, defective-SACs exhibit excellent performance in energy conversion and environmental fields. Undoubtedly, defective-SACs provide a concrete idea for constructing efficient and ideal catalysts for various catalytic reactions.
However, the following challenges still need to be addressed for future research and practical applications.
(1) Overall, the traditional pyrolysis method, which involves the use of reducing gases/high-temperature treatment processes, still accounts for the majority of methods for synthesizing defective-SACs. The development of more streamlined synthesis methods is particularly important in order to facilitate the practical application of defective-SACs catalysts. Currently, physical radiation methods (plasma treatment, laser-assisted synthesis, Joule heating synthesis and microwave heat shock) show fascination in construction of defective-SACs due to their ultrafast kinetics (within minutes and even seconds), extreme thermodynamics and high efficiency. Besides, it is necessary to explore the large-scale production of defective-SACs at the kilogram level and above, including intermittent and continuous large-scale production, which is expected to meet the goal of daily kilogram level production. And control the defect uniformity during the synthesis process to obtain stable defective-SACs in different batches is a prerequisite for large-scale production.
(2) In general, the introduction of defects causes changes in the active center coordination environment to achieve better catalytic performance. However, overactive defective-SACs catalysts may bring about strong adsorption of reactants/intermediates/surrounding impurities, ultimately resulting in active sites poisoning. On the contrary, when the defective-SACs catalysts are very stable it may not be sufficient to activate the intermediates during catalysis. The rational introduction of defects in SACs can increase the activity of the catalysts, but it may also disrupt the periodic order of the crystal structure, leading to changes in the surface energy and thus affecting the stability of the catalysts. In actual industrial reactions, extreme conditions such as high pressure and acidic environments are often involved, and defective-SACs may become inactive. Therefore, the stability-activity trade-off is important to achieve a wider application of defective-SACs for their engineering applications.
(3) Due to the complexity of supports (defects/heteroatoms/functional groups) and defects (type/number/concentration/coverage) in SACs, the "spark" between them has not been fully deciphered. To fully understand the intrinsic effects of defects on catalytic reactions, it is necessary to reveal the physicochemical dynamics of defective-SACs catalysts during catalytic reactions (including morphology, composition, coordination structure). For example, the development of high-resolution X-ray absorption spectroscopy (XAS) imaging techniques to explore the fine structure of the surface interface of defective-SACs catalysts and the evolution of the sites between the two. Meanwhile, combining advanced characterization techniques such as in situ diffuse reflectance-liquid infrared (DR-LIR), real-time tracking of the adsorption strength of reactants/intermediates on the surface of defective-SACs, and real-time monitoring of the dynamic changes in the microenvironment around the SACs sites. We can explore the role of the defects and the mechanism behind their performance enhancement, and the stability decay mechanisms in depth.
(4) Although defective-SACs are theoretically effective catalysts, they still involve complex synthesis and characterization processes. The traditional approach of conducting a large number of trial-and-error experiments in the laboratory not only results in a waste of time, but also an excessive consumption of resources. In contrast, the use of machine learning (ML) and artificial intelligence (AI)-driven synergy platform to select suitable catalysts in a high-throughput manner to guide catalyst design, which greatly reduces human and material resources is conducive to accelerating catalyst development. However, the application of ML in catalysis is still in its infancy. Future research can apply ML match with high-throughput screening techniques to the design of defective-SACs-predicting their performance, which can be a milestone for the application of defective-SACs in energy conversion and environmental remediation.
(5) Cost-effectiveness and large-scale applications are prerequisites for promoting the practical industrial application of defective-SACs. Combining life cycle assessment (LCA) and economic and technological analysis (TEA) can quantify the potential environmental benefits and risks of industrial application, and balance costs with environmental benefits. Establishing a dedicated LCA database for defective-SACs and promoting policies such as "green catalyst certification standards" can drive their industrial applications. Currently, pilot-scale exploration still needs to be strengthened, so integrating defective-SACs onto carriers and developing reactors suitable for different reaction conditions could be considered to facilitate their large-scale application.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yuqi Zhu: Writing – original draft, Investigation, Conceptualization. Suhua Chen: Supervision, Resources, Project administration, Funding acquisition. Jianping Zou: Supervision, Resources, Project administration. Bo Li: Supervision, Resources, Project administration. Wei Ren: Supervision, Resources, Project administration, Funding acquisition. Gaoxia Zhang: Writing – original draft, Visualization, Conceptualization. Jiajie Wang: Writing – review & editing, Conceptualization. Xinyu Liu: Writing – review & editing, Conceptualization. Qianhui Li: Writing – review & editing. Huiying Zhang: Writing – review & editing. Fanying Xia: Writing – review & editing.
This work was supported by the National Key Research and Development Program of China (No. 2022YFD1700801–3) and Key Laboratory of Jiangxi Province for Persistent Pollutants Prevention Control and Resource Reuse (No. 2023SSY02061). We are grateful for the financial support of the projects and research platform support provided by the laboratory.
Supplementary material associated with this article can be found, in the online version, at doi:
H. Rao, L.C.S. Chmidt, J. Bonin, et al., Nature 548 (2017) 74–77. doi: 10.1038/nature23016
C.W. Li, J. Ciston, M.W. Kanan, Nature 508 (2014) 504–507. doi: 10.1038/nature13249
F.W. Yang, J. Zhang, J.W. Chen, et al., Nano Res. 17 (2024) 5884–5896. doi: 10.1007/s12274-024-6574-9
J. Zhu, L.S. Hu, P.X. Zhao, et al., Chem. Rev. 120 (2020) 851–918. doi: 10.1021/acs.chemrev.9b00248
Y. Jiao, Y. Zheng, M.T. Jaroniec, et al., Chem. Soc. Rev. 44 (2015) 2060–2086. doi: 10.1039/C4CS00470A
C. Tang, S.Z. Qiao, Chem. Soc. Rev. 48 (2019) 3166–3180. doi: 10.1039/c9cs00280d
J.C. Hao, Z.C. Zhuang, J.C. Hao, et al., ACS Nano 16 (2022) 3251–3263. doi: 10.1021/acsnano.1c11145
D.B. Miklos, C. Remy, M. Jekel, et al., Water. Res. 139 (2018) 118–131. doi: 10.1016/j.watres.2018.03.042
Y. Zheng, Y. Jiao, A. Vasileff, et al., Angew. Chem. Int. Ed. 57 (2018) 7568–7579. doi: 10.1002/anie.201710556
C. Hepburn, E. Adlen, J. Beddington, et al., Nature 575 (2019) 87–97. doi: 10.1038/s41586-019-1681-6
X.Y. Shi, H. Xiao, H. Azarabadi, et al., Angew. Chem. Int. Ed. 59 (2020) 6984–7006. doi: 10.1002/anie.201906756
J.Q. Lv, J.F. Xie, A.G.A. Mohamed, et al., Nat. Rev. Chem. 7 (2023) 91–105.
L. Zhou, X.Q. Li, G.W. Ni, et al., Natl. Sci. Rev. 6 (2019) 562–578. doi: 10.1093/nsr/nwz030
J.J. Wang, S.H. Chen, B. Li, et al., Sep. Purif. Technol. 375 (2025) 133785. doi: 10.1016/j.seppur.2025.133785
G.F. Wang, D.L. Huang, M. Cheng, et al., Chem. Eng. J. 488 (2024) 150692. doi: 10.1016/j.cej.2024.150692
L. Jiao, H.L. Jiang, Chem. 5 (2019) 786–804. doi: 10.1016/j.chempr.2018.12.011
X.B. Zheng, P. Li, S.X. Dou, et al., Energ. Environ. Sci. 14 (2021) 2809–2858. doi: 10.1039/d1ee00248a
H.Y. Zhuo, X. Zhang, J.X. Liang, et al., Chem. Rev. 120 (2020) 12315–12341. doi: 10.1021/acs.chemrev.0c00818
B. Li, L. Li, G.X. Zhang, et al., ACS Catal. 15 (2025) 10239–10270. doi: 10.1021/acscatal.5c02018
C.Z. Zhu, S.F. Fu, Q.R. Shi, et al., Angew. Chem. Int. Ed. 56 (2017) 13944–13960. doi: 10.1002/anie.201703864
J.Y. Liu, ACS Catal. 7 (2017) 34–59. doi: 10.1021/acscatal.6b01534
X. Wang, Y.W. Zhang, J. Wu, et al., Chem. Rev. 122 (2022) 1273–1348. doi: 10.1021/acs.chemrev.1c00505
J.B. Xi, H.S. Jung, Y. Xu, et al., Adv. Funct. Mater. 31 (2021) 2008318. doi: 10.1002/adfm.202008318
B. Li, C.Y. Feng, T.M. Wang, et al., Sep. Purif. Technol. 354 (2025) 128955. doi: 10.1016/j.seppur.2024.128955
F. Mo, C.L. Song, Q.X. Zhou, et al., Proc. Natl. Acad. Sci. U. S. A. 120 (2023) e2300281120. doi: 10.1073/pnas.2300281120
Y. Shi, W.M. Huang, J. Li, et al., Nat. Commun. 11 (2020) 4558. doi: 10.1038/s41467-020-18430-8
Y.G. Wu, X.N. Tang, K. Yuan, et al., Energ. Environ. Sci. 16 (2023) 5663–5687. doi: 10.1039/d3ee02474a
H.F. Xiong, A.K. Datye, Y. Wang, Adv. Mater. 33 (2021) 2004319. doi: 10.1002/adma.202004319
L.S. Peng, L. Shang, T.R. Zhang, et al., Adv. Energy Mater. 10 (2020) 2003018. doi: 10.1002/aenm.202003018
X.Y. Liu, Y.X. Zhou, J.K. Lin, et al., Angew. Chem. Int. Ed. 63 (2024) e202406650. doi: 10.1002/anie.202406650
Q. Yang, H.X. Liu, P. Yuan, et al., J. Am. Chem. Soc. 144 (2022) 2171–2178. doi: 10.1021/jacs.1c10814
X. Rong, H.J. Wang, X.L. Lu, et al., Angew. Chem. Int. Ed. 59 (2020) 1961–1965. doi: 10.1002/anie.201912458
J.J. Zhang, X. Yang, G.F. Xu, et al., Adv. Mater. 36 (2024) 2309205. doi: 10.1002/adma.202309205
L. Abdellaoui, Z.W. Chen, Y. Yu, et al., Adv. Funct. Mater. 31 (2021) 2101214. doi: 10.1002/adfm.202101214
S.B. Tian, C. Peng, J.C. Dong, et al., ACS Catal. 11 (2021) 4946–4954. doi: 10.1021/acscatal.1c00455
R. Jiang, L. Li, T. Sheng, et al., J. Am. Chem. Soc. 140 (2018) 11594–11598. doi: 10.1021/jacs.8b07294
L.L. Liao, G.M. Xia, Y.G. Wang, et al., Appl. Catal. B: Environ. Energy 318 (2022) 121826. doi: 10.1016/j.apcatb.2022.121826
J. Jin, X. Han, Y.Y. Fang, et al., Adv. Funct. Mater. 32 (2022) 2109218. doi: 10.1002/adfm.202109218
W.Y. Zhang, C.Y. Deng, W. Wang, et al., Adv. Mater. 36 (2024) 2405825. doi: 10.1002/adma.202405825
J.C. Li, M. Li, N. An, et al., Proc. Natl. Acad. Sci. U. S. A. 118 (2021) e2105628118. doi: 10.1073/pnas.2105628118
Y.X. Lu, T.Y. Liu, C.L. Dong, et al., Adv. Mater. 33 (2021) 2007056. doi: 10.1002/adma.202007056
D.X. Cai, J. Zhang, Z. Kong, et al., ChemCatChem 16 (2024) e202301414. doi: 10.1002/cctc.202301414
N. Goyal, F. Li, Y.B. Hu, J. Mater. Chem. A 12 (2024) 19685–19719. doi: 10.1039/d4ta02110j
Y.Q. Zhang, L. Guo, L. Tao, et al., Small Methods 3 (2019) 1800406. doi: 10.1002/smtd.201800406
D.W. Wang, H. Shan, W. Yin, et al., Fuel 355 (2024) 129439. doi: 10.1016/j.fuel.2023.129439
S.J. Wei, Y.B. Sun, Y.Z. Qiu, et al., Nat. Commun. 14 (2023) 7549. doi: 10.1038/s41467-023-43040-5
S.F. Ji, Y.J. Chen, X.L. Wang, et al., Chem. Rev. 120 (2020) 11900–11955. doi: 10.1021/acs.chemrev.9b00818
R.Y. Pang, H.Y. Xia, X.Y.M. Dong, et al., Adv. Sci. 11 (2024) e2407294. doi: 10.1002/advs.202407294
Y.F. Liu, L.B. Zong, Y.Y. Zhang, et al., Appl. Catal. B: Environ. Energy 361 (2025) 124673. doi: 10.1016/j.apcatb.2024.124673
Y.L. Zhao, H.C. Chen, X.L. Ma, et al., Adv. Mater. 36 (2024) e2308243. doi: 10.1002/adma.202308243
S.H. Zhou, W.B. Wei, X. Cai, et al., Adv. Funct. Mater. 34 (2024) 2311422. doi: 10.1002/adfm.202311422
S. Lv, Y. Zhou, W. Yang, et al., Adv. Funct. Mater. 35 (2025) 2423864. doi: 10.1002/adfm.202423864
S. Yuan, J.W. Zhang, L.Y. Hu, et al., Angew. Chem. Int. Ed. 60 (2021) 21685–21690. doi: 10.1002/anie.202107053
Y.R. Li, S.T. Li, H.W. Huang, Adv. Funct. Mater. 33 (2023) 2304925. doi: 10.1002/adfm.202304925
Y. Xu, P. Wang, M. Zhang, et al., Energ. Environ. Sci. 17 (2024) 5060–5069. doi: 10.1039/d4ee01376j
S.H. Xie, D. Kim, K.L. Ye, et al., J. Rare. Earth. 41 (2023) 941–951. doi: 10.1016/j.jre.2023.01.006
B.T. Hu, K.A. Sun, Z.W. Zhuang, et al., Adv. Mater. 34 (2022) 2107721. doi: 10.1002/adma.202107721
M. Yang, H.B. Li, F.L. Liu, et al., Appl. Catal. B: Environ. Energy 354 (2024) 124071. doi: 10.1016/j.apcatb.2024.124071
G. Yao, Z.Q. Li, Y.H. Zhang, et al., Adv. Funct. Mater. 34 (2024) 2214353. doi: 10.1002/adfm.202214353
J. He, N. Li, Z.G. Li, et al., Adv. Funct. Mater. 31 (2021) 2103597. doi: 10.1002/adfm.202103597
Q.H. Dong, S.G. Ma, J.Y. Zhu, et al., Adv. Funct. Mater. 33 (2023) 2210665. doi: 10.1002/adfm.202210665
D. Zhao, Z. Chen, W.J. Yang, et al., J. Am. Chem. Soc. 141 (2019) 4086–4093. doi: 10.1021/jacs.8b13579
G.M. Ren, J.Y. Zhao, Z.H. Zhao, et al., Angew. Chem. Int. Ed. 63 (2024) e202314408. doi: 10.1002/anie.202314408
Y. Tao, J. Guan, J. Zhang, et al., Angew. Chem. Int. Ed. 63 (2024) e202400625. doi: 10.1002/anie.202400625
H. Zhu, Y.J. Wang, Z.Q. Jiang, et al., Adv. Energy Mater. 14 (2024) 2303987. doi: 10.1002/aenm.202303987
L. Gong, Z. Jiang, Y. Xiong, et al., Int. J. Hydrogen Energ. 48 (2023) 32860–32874. doi: 10.1016/j.ijhydene.2023.04.297
Y.Q. Zhang, J.J. Liu, Y.F. Xu, et al., Chem. Soc. Rev. 53 (2024) 10620–10659. doi: 10.1039/d4cs00217b
Z. Yuan, Y.J. Yu, Q. Xie, et al., Adv. Mater. Interfaces 12 (2025) 2400916. doi: 10.1002/admi.202400916
H.L. Fei, J.C. Dong, C.Z. Wan, et al., Adv. Mater. 30 (2018) 1802146. doi: 10.1002/adma.201802146
C. Jia, S.N. Li, Y. Zhao, et al., Adv. Funct. Mater. 31 (2021) 2107072. doi: 10.1002/adfm.202107072
J.Q. Wang, C. Xi, M. Wang, et al., ACS Catal. 10 (2020) 12575–12581. doi: 10.1021/acscatal.0c03406
R. Kumar, R.K. Singh, D.P. Singh, et al., Coordin. Chem. Rev. 342 (2017) 34–79. doi: 10.1016/j.ccr.2017.03.021
B. Wang, X. Zhu, X.D. Pei, et al., J. Am. Chem. Soc. 145 (2023) 13788–13795. doi: 10.1021/jacs.3c02364
Y.F. Hu, B.L. Li, C.L. Yu, et al., Mater. Today 63 (2023) 288–312. doi: 10.1016/j.mattod.2023.01.019
G.F. Han, F. Li, A. Rykov, et al., Nat. Nanotechnol. 17 (2022) 403–407. doi: 10.1038/s41565-022-01075-7
X.H. Tan, H.P. Li, W. Zhang, et al., Chem. Catal. 2 (2022) 816–835.
S. Wei, A. Li, J. -C. Liu, et al., Nat. Nanotechnol. 13 (2018) 856–861. doi: 10.1038/s41565-018-0197-9
C.T. Campbell, S.C. Parker, D.E. Starr, Science 298 (2002) 811–814. doi: 10.1126/science.1075094
S.B. Simonsen, I. Chorkendorff, S. Dahl, et al., J. Am. Chem. Soc. 132 (2010) 7968–7975. doi: 10.1021/ja910094r
Q. Li, Q. Zhang, W. Xu, et al., Adv. Energy Mater. 13 (2023) 2203955. doi: 10.1002/aenm.202203955
F. Gong, Y. Liu, Y. Zhao, et al., Angew. Chem. Int. Ed. 62 (2023) e202308091. doi: 10.1002/anie.202308091
C. Feng, Z. Zhang, D. Wang, et al., J. Am. Chem. Soc. 144 (2022) 9271–9279. doi: 10.1021/jacs.2c00533
K. Jiang, M. Luo, Z. Liu, et al., Nat. Commun. 12 (2021) 1687. doi: 10.1038/s41467-021-21956-0
C. Wei, S.N. Sun, D. Mandler, et al., Chem. Soc. Rev. 48 (2019) 2518–2534. doi: 10.1039/c8cs00848e
L. Yuan, S.J. Zeng, G.L. Li, et al., Adv. Funct. Mater. 33 (2023) 2306994. doi: 10.1002/adfm.202306994
C.C. Xin, W.Z. Shang, J.W. Hu, et al., Adv. Funct. Mater. 32 (2022) 2108345. doi: 10.1002/adfm.202108345
Z. Jiang, M. Jing, X. Feng, et al., Appl. Catal. B: Environ. Energy 278 (2020) 119304. doi: 10.1016/j.apcatb.2020.119304
Y. Zhang, B. Gong, B. Zhou, et al., Nano Research Energy 4 (2025) e9120180. doi: 10.26599/nre.2025.9120180
H. Tu, H. Zhang, Y. Song, et al., Adv. Sci. 10 (2023) 2305194. doi: 10.1002/advs.202305194
Z. Wu, Z. Xiong, B. Huang, et al., Nat. Commun. 15 (2024) 7775. doi: 10.1038/s41467-024-52074-2
J. Wu, J. Gao, S. Lian, et al., Appl. Catal. B: Environ. Energy 314 (2022) 121516. doi: 10.1016/j.apcatb.2022.121516
S.C. Zhang, X. Chang, L.H. Zhou, et al., ACS Sens. 9 (2024) 2101–2109. doi: 10.1021/acssensors.4c00162
G. Liu, A.W. Robertson, M.M.J. Li, et al., Nat. Chem. 9 (2017) 810–816. doi: 10.1038/nchem.2740
K.T. Huang, H.Q. Fu, W. Shi, et al., J. Catal. 377 (2019) 283–292. doi: 10.1016/j.jcat.2019.06.047
C. Li, X. Zhao, A. Wang, et al., Chem. Rev. 115 (2015) 11559–11624. doi: 10.1021/acs.chemrev.5b00155
K. Zhang, J. Jiang, Z. Liu, et al., ACS Catal. 14 (2024) 16115–16126. doi: 10.1021/acscatal.4c03184
M. Jiang, X.P. Chen, L.L. Wang, et al., Fuel 324 (2022) 124499. doi: 10.1016/j.fuel.2022.124499
Z.J. Li, L.P. Leng, X.W. Lu, et al., Nano Res. 15 (2022) 3114–3121. doi: 10.1007/s12274-021-4028-1
K. Zheng, Y. Li, B. Liu, et al., Angew. Chem. Int. Ed. 61 (2022) e202210991. doi: 10.1002/anie.202210991
X.F. Zhu, X. Tan, K.H. Wu, et al., Angew. Chem. Int. Ed. 60 (2021) 21911–21917. doi: 10.1002/anie.202107790
J. Liu, M.G. Jiao, B.B. Mei, et al., Angew. Chem. Int. Ed. 58 (2019) 1163–1167. doi: 10.1002/anie.201812423
K. Liu, J.W. Fu, Y.Y. Lin, et al., Nat. Commun. 13 (2022) 2075. doi: 10.1038/s41467-022-29797-1
J. Rong, E.R. Gao, N.C. Liu, et al., Energy Storage Mater. 56 (2023) 165–173. doi: 10.1016/j.ensm.2023.01.022
M. Liu, Y. Li, L. Yang, et al., Angew. Chem. Int. Ed. 64 (2025) e202505268. doi: 10.1002/anie.202505268
L.Q. Li, C. Tang, Y. Zheng, et al., Adv. Energy Mater. 10 (2020) 2000789. doi: 10.1002/aenm.202000789
X.X. Yang, Y.C. Zeng, W. Alnoush, et al., Adv. Mater. 34 (2022) 2107954. doi: 10.1002/adma.202107954
L.J. Yang, G. Sun, H.L. Fu, et al., Chem. Eng. J. 472 (2023) 145052. doi: 10.1016/j.cej.2023.145052
Y.H. Tian, M. Li, Z.Z. Wu, et al., Angew. Chem. Int. Ed. 61 (2022) e202213296. doi: 10.1002/anie.202213296
J. Yang, K. An, Z. Yu, et al., ACS Catal. 14 (2024) 17739–17747. doi: 10.1021/acscatal.4c02779
L. Tao, C.Y. Lin, S. Dou, et al., Nano Energy 41 (2017) 417–425. doi: 10.1016/j.nanoen.2017.09.055
J. Yin, J. Jin, M. Lu, et al., J. Am. Chem. Soc. 142 (2020) 18378–18386. doi: 10.1021/jacs.0c05050
D.R. Wang, L. Gu, X.R. Luo, et al., Electroanal. Chem. 924 (2022) 116875. doi: 10.1016/j.jelechem.2022.116875
Z.Y. Zhao, Y.P. Xiong, S. Yu, et al., J. Colloid. Interf. Sci. 650 (2023) 934–942. doi: 10.1016/j.jcis.2023.06.182
H.H. Chen, S.Q. Chen, Z.R. Zhang, et al., ACS Catal. 12 (2022) 13482–13491. doi: 10.1021/acscatal.2c03189
D.D. Wang, J.W. Xue, X.L. Ding, et al., ACS Catal. 12 (2022) 12458–12468. doi: 10.1021/acscatal.2c03476
Y.X. Hao, S.F. Hung, W.J. Zeng, et al., J. Am. Chem. Soc. 145 (2023) 23659–23669. doi: 10.1021/jacs.3c07777
J. Zhang, D.S. Yang, Z. Yang, et al., Appl. Catal. B: Environ. Energy 364 (2025) 124845. doi: 10.1016/j.apcatb.2024.124845
D.L. Wang, H.P. Li, N. Du, et al., Adv. Funct. Mater. 31 (2021) 2009770. doi: 10.1002/adfm.202009770
J.K. Norskov, T. Bligaard, A. Logadottir, et al., J. Electrochem. Soc. 152 (2005) J23–J26. doi: 10.1149/1.1856988
S.J. Park, T.H. Nguyen, D.T. Tran, et al., Energ. Environ. Sci. 16 (2023) 4093–4104. doi: 10.1039/d3ee01314f
K. Zhu, H. Yang, G. Guo, et al., ACS Catal. 15 (2025) 9563–9573. doi: 10.1021/acscatal.5c01725
Q. Song, Z. Gong, J. Liu, et al., Adv. Sci. 12 (2025) 2415665. doi: 10.1002/advs.202415665
T.T. Chao, W.B. Xie, Y.M. Hu, et al., Energ. Environ. Sci. 17 (2024) 1397–1406. doi: 10.1039/d3ee02760k
Z.Y. Chen, X.P. Li, J. Zhao, et al., Angew. Chem. Int. Ed. 62 (2023) e202308686. doi: 10.1002/anie.202308686
M. Xi, H. Zhang, L.F. Yang, et al., Adv. Sci. 12 (2024) 2409855.
Y. Shen, C.J. Ren, L.R. Zheng, et al., Nat. Commun. 14 (2023) 1117. doi: 10.1038/s41467-023-36778-5
W.H. Guo, K.X. Zhang, Z.B. Liang, et al., Chem. Soc. Rev. 48 (2019) 5658–5716. doi: 10.1039/c9cs00159j
Y. Yang, H.L. Wang, X.H. Tan, et al., Adv. Funct. Mater. 34 (2024) 2403535. doi: 10.1002/adfm.202403535
J.Y. Liu, X. Kong, L.R. Zheng, et al., ACS Nano 14 (2020) 1093–1101. doi: 10.1021/acsnano.9b08835
W. Peng, M. Luo, X. Xu, et al., Adv. Energy Mater. 10 (2020) 2001364. doi: 10.1002/aenm.202001364
L. Li, X.Y. Wang, H.R. Guo, et al., Small Methods 3 (2019) 1900337. doi: 10.1002/smtd.201900337
P. Zhang, Y.C. Yu, R. Tuerhong, et al., Inorg. Chem. Front. 12 (2024) 85–117.
L. Li, Y. Yu, S.M. Lin, et al., Catal. Commun. 153 (2021) 106294. doi: 10.1016/j.catcom.2021.106294
P.C. Huang, W. Liu, Z.H. He, et al., Sci. China Chem. 61 (2018) 1187–1196. doi: 10.1007/s11426-018-9273-1
P. Gallezot, Chem. Soc. Rev. 41 (2012) 1538–1558. doi: 10.1039/C1CS15147A
S. De, A.S. Burange, R. Luque, Green. Chem. 24 (2022) 2267–2286. doi: 10.1039/d1gc04285h
Y.S. Wu, H.T. Wang, J.B. Peng, et al., Chem. Eng. J. 454 (2023) 140156. doi: 10.1016/j.cej.2022.140156
J.Y. Fu, J. Lym, W.Q. Zheng, et al., Nat. Catal. 3 (2020) 446–453. doi: 10.1038/s41929-020-0445-x
Z.D. An, P.P. Yang, D.L. Duan, et al., Nat. Commun. 14 (2023) 6666. doi: 10.1038/s41467-023-42337-9
J. Xia, L. Dong, X.H. Liu, et al., ACS Catal. 13 (2023) 6093–6103. doi: 10.1021/acscatal.3c00513
R.X. Ge, Y. Wang, Z.Z. Li, et al., Angew. Chem. Int. Ed. 61 (2022) e202200211. doi: 10.1002/anie.202200211
Z.L. Tian, Y.M. Da, M. Wang, et al., Nat. Commun. 14 (2023) 142. doi: 10.1007/s13131-022-2112-3
D.L. Huang, G.X. Zhang, J. Yi, et al., Chemosphere 263 (2021) 127672. doi: 10.1016/j.chemosphere.2020.127672
S. Giannakis, K.Y.A. Lin, F. Ghanbari, Chem. Eng. J. 406 (2021) 127083. doi: 10.1016/j.cej.2020.127083
Y.Q. Yan, Z.S. Wei, X.G. Duan, et al., Environ. Sci. Technol. 57 (2023) 12153–12179. doi: 10.1021/acs.est.3c05153
J.W. Qin, Q. Wang, B. Han, et al., Appl. Catal. B: Environ. Energy 360 (2025) 124538. doi: 10.1016/j.apcatb.2024.124538
X.Y. Liang, D. Wang, Z.Y. Zhao, et al., Adv. Funct. Mater. 32 (2022) 2203001. doi: 10.1002/adfm.202203001
H.K. Paumo, S. Dalhatou, L.M. Katata-Seru, et al., J. Mol. Liq. 331 (2021) 115458. doi: 10.1016/j.molliq.2021.115458
X.W. Zhang, C.Q. Li, X.L. Wang, et al., Small 18 (2022) 2204793. doi: 10.1002/smll.202204793
C. Zhang, D.Y. Qin, Y. Zhou, et al., Appl. Catal. B: Environ. Energy 303 (2022) 120904. doi: 10.1016/j.apcatb.2021.120904
X.L. Wu, S. Liu, Y. Li, et al., Angew. Chem. Int. Ed. 62 (2023) e202305639. doi: 10.1002/anie.202305639
D.E. Fitzpatrick, C. Battilocchio, S.V. Ley, ACS Cent. Sci. 2 (2016) 131–138. doi: 10.1021/acscentsci.6b00015
B.R. Boswell, C.M.F. Mansson, J.M. Cox, et al., Nat. Chem. 13 (2021) 41–46. doi: 10.1038/s41557-020-00608-8
T.Y. Zhang, X. Yang, J. Jin, et al., Adv. Mater. 36 (2024) 2304144. doi: 10.1002/adma.202304144
C. He, J. Cheng, X. Zhang, et al., Chem. Rev. 119 (2019) 4471–4568. doi: 10.1021/acs.chemrev.8b00408
F. Dong, Y. Meng, W.T. Ling, et al., Appl. Catal. B: Environ. Energy 346 (2024) 123779. doi: 10.1016/j.apcatb.2024.123779

Figure 2 (a) Synthesis of Pt-SAC or the Pt-Ni(OH)x catalyst. (b) Calculated energy diagrams for the generation of Pt-SACs or the Pt-Ni(OH)x catalysts. Reproduced with permission [80]. Copyright 2023, Wiley. (c) EDDs from C-MoS2, structure models of C-M-Mo, top-view slice and the corresponding EDDs in the C-M-Mo. Reproduced with permission [81]. Copyright 2023, Wiley. (d) Schematic representation of the conversion to the corresponding materials on different CeO2 crystal planes (100 and 111). Reproduced with permission [57]. Copyright 2022, Wiley. (e) Comparison of geometries before and after applied strain to Ru/MoS2. Reproduced with permission [83]. Copyright 2021, Springer Nature. (f) Top and side views of the computed differential charge density. (g) The PDOS for Fe d-orbitals in different samples. Reproduced with permission [88]. Copyright 2025, Tsinghua University Press. (h) Different distributions of Fe species from FeN4—C and D-FeN4—C. Reproduced with permission [90]. Copyright 2024, Springer Nature.

Figure 3 (a) Schematic of the carbon removal mechanism on VO. Reproduced with permission [91]. Copyright 2022, Elsevier. (b) The mechanism of H2 sensing for Pt-Fe2O3-VO. (c) Simplified energy band diagrams. Reproduced with permission [92]. Copyright 2024, American Chemical Society. (d) In situ temperature-resolved and (e) in situ-resolved DRIFT spectra (1760–670 cm–1) of D1 on Ru/CeO2-RVO samples. Reproduced with permission [96]. Copyright 2024, American Chemical Society. (f) Schematic of CO2 hydrogenation to ethanol on Rh1/CeTiOx. (g) Free energy diagrams of the CO2 hydrogenation reaction. Reproduced with permission [99]. Copyright 2022, Wiley.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: