Citation:
Jun-Kang Li, Pei-Yao Wang, Jia-Lin Zhang, Shu-Na Zhao, Bo Li, Shuang-Quan Zang. Sulfur-engineered Ni single-atom catalysts for sustainable tandem CO2-to-chemicals conversion across pH-universal windows[J]. Chinese Chemical Letters,
2026, 37(7): 112150.
doi:
10.1016/j.cclet.2025.112150
Sulfur-engineered Ni single-atom catalysts for sustainable tandem CO2-to-chemicals conversion across pH-universal windows
English
Sulfur-engineered Ni single-atom catalysts for sustainable tandem CO2-to-chemicals conversion across pH-universal windows
Henan Key Laboratory of Crystalline Molecular Functional Materials, College of Chemistry and Pingyuan Laboratory, Key Laboratory of Special Functional Molecular Materials (Zhengzhou University), Ministry of Education, Zhengzhou University, Zhengzhou 450001, China
b.
College of Chemistry and Pharmaceutical Engineering, Nanyang Normal University, Nanyang 473061, China
Received Date:
30 September 2025 Accepted Date:
21 November 2025 Revised Date:
18 November 2025 Available Online:
01 July 2026
Abstract:
Tandem CO2 electroreduction integrated with carbonylation reactions offers a promising pathway for transforming greenhouse gases into valuable chemical products. The development of efficient CO2–to–CO electrocatalysts operational across wide potential ranges is crucial to address renewable energy fluctuations and facilitate integrated cascade systems. Herein, a Ni single–atom catalyst (SAC) with sulfur doping in the second shell of Ni–N4 is reported. In situ measurements and theoretical calculations demonstrate that the incorporation S atoms not only modulates the electronic configuration of Ni active sites but also enhances H2O adsorption, enabling rapid CO2 hydrogenation into *COOH intermediates even at high potentials. Consequently, the Ni–N–S/CNS catalyst achieves a Faradaic efficiency for CO2–to–CO (FECO) of 99.3% at –0.7 VRHE and maintains over 90% across pH–universal conditions with ultrawide potential windows: 1400 mV (from –0.3 VRHE to –1.7 VRHE) in alkaline media, 1200 mV (from –0.7 VRHE to –1.9 VRHE) in neutral conditions, and 1000 mV (from –1.3 VRHE to –2.3 VRHE) in acidic environments. Remarkably, this catalyst enables the tandem CO2RR and N–alkylaniline carbonylation process for synthesizing o–aminobenzoates and isatoic anhydrides with high yield. Our findings demonstrate an electrothermo–catalytic tandem strategy for cost–effective CO2 conversion and synthesis of high valuable fine chemicals, thereby broadening the scope of its utilization.
Carbon monoxide (CO), as a crucial one–carbon (C1) building block, plays a vital role in various industrial processes. In particular, carbonylation reactions with CO have been widely utilized for the synthesis of alcohols, carboxylic acids, amides, anhydrides, and other valuable fine chemicals [1-5]. Nevertheless, the inherent toxicity of CO poses significant safety risks in industrial applications, particularly during large–scale storage, transportation, and utilization. Furthermore, direct reliance on this highly toxic, colorless, and odorless gas as a reactant in carbonylation reactions and their industrial scale–up processes remain a critical safety hazard. Therefore, the development of a safe and efficient method for on–demand generation and immediate utilization of CO in carbonylation reactions is highly desirable [6-8].
The excessive use of fossil fuels has caused a continuous rise in atmospheric carbon dioxide (CO2) levels, triggering severe environmental degradation and global energy challenges [9-13]. Employing CO2 as a C1 building block synthesizing high–value chemicals not only contributes to mitigating environmental issues but also facilitates the closure of the carbon cycle. However, due to the high thermodynamic stability of CO2, the conversion of CO2 into desirable products typically requires highly demanding reaction conditions and substantial energy input. Consequently, a tandem strategy – first converting CO2 into CO and then upgrading CO to higher value compounds – represents an effective and rational pathway for CO2 valorization [6,14-17].
The electrocatalytic CO2 reduction reaction (CO2RR), powered by renewable energy under mild and safe conditions, enables selective and efficient CO production, while being widely recognized as one of the most environmentally friendly and effective strategies for CO2 valorization [18-22]. Metal single–atom catalysts (SACs) have recently garnered substantial research interest owing to their optimized atomic utilization efficiency and excellent electrocatalytic CO2RR performance [23-25]. Notably, NiN4 sites immobilized on nitrogen–doped carbon (NC) supports with favorable structural stability have demonstrated widespread application in electrocatalytic CO2–to–CO conversion [26-30]. However, these catalysts exhibit planar–like D4h symmetry and uniform charge distribution, restricting electronic configuration modulation of Ni sites and axial adsorption of *COOH − a critical intermediate in CO formation. Introducing heteroatoms into the second shell, which are bonded to the first shell atoms and distinct from the second coordination sphere in molecular catalysis [31-33] enables modulation of the electron distribution within the NiN4 microenvironment [34-37]. This approach not only maintains the structural stability of NiN4 moiety but also reduces the strong electronegative interactions from proximal N atoms. For example, Tang et al. demonstrated that the S atom in second shell disrupts the uniform charge distribution and modulates the electron configuration of NiN4, thereby lowering the reaction energy barriers of CO2 reduction [38]. In addition, Chen et al. reported that S doping in the second shell of FeN4 can accelerate H2O activation and provide sufficient protons to promote CO2 conversion to the key intermediate *COOH [39]. Furthermore, the introduction of P atoms into the second shell of SnN4 has been shown to facilitate the reduction of Sn4+ to Sn2+, with Sn2+ identified as the true active site for CO2RR to CO [40]. However, although these strategies can achieve high CO selectivity, the majority of electrocatalysts only sustain a Faradaic efficiency for CO (FECO) exceeding 90% within a limited potential window below 1.0 VRHE (versus the reversible hydrogen electrode, RHE). This significantly constrains their applicability under the high CO demand conditions required in industrial–scale tandem electrolysis systems.
Considering the aforementioned factors, second shell coordination engineering was developed for SAC catalysts to convert symmetric NiN4 site into geometrically asymmetric NiN4–S2 configurations. The Ni–N–S/CNS catalyst exhibits an outstanding catalytic activity for CO2RR to CO, characterized by an ultrawide potential window from –0.3 VRHE to –1.7 VRHE in alkaline environments maintaining a FECO above 90% with peak efficiency reaching approximately 100% at –0.7 VRHE. Notably, the catalyst demonstrates durable catalytic performance under varied pH conditions: Sustaining > 90% FECO within operational windows of –0.7 VRHE to –1.9 VRHE (neutral) and –1.3 VRHE to –2.3 VRHE (acidic), highlighting its pH–universal adaptability for CO2RR. In situ ATR–SEIRAS analysis and theoretical calculations demonstrate that S doping induces dual functional modifications reconstructing the electronic structure of Ni active sites while intensifying H2O adsorption energetics. These coupled mechanistic effects enable rapid CO2–to–*COOH transformation kinetics, enabling sustained high activity even at high operational potential. More importantly, the Ni–N–S/CNS catalyst facilitates efficient and stable CO generation, which can be subsequently converted into high–value products, such as o–aminobenzoates and isatoic anhydrides, through alkylaniline carbonylation reactions with high yield. This tandem reaction achieves a safe, green, and efficient sequential conversion of CO2 to CO to value fine chemicals. This work highlights the outstanding CO2RR performance of Ni–N–S/CNS, featuring a pH–universal ultrawide potential window, while establishing a sustainable and cost–effective approach for synthesizing high–value fine chemicals.
The carbon nanosheet (CNS) matrices were synthesized through the pyrolysis of melamine and L–alanine precursors. The resulting material exhibited a sheet–like morphology, as confirmed by aberration–corrected high–angle annular dark field scanning transmission electron microscopy (HAADF–STEM) image (Fig. S1 in Supporting information). Subsequently, a N, S–coordinated Ni complex (Fig. S2 in Supporting information) [41] was immobilized onto the as–synthesized CNS matrices through an adsorption–pyrolysis process at 500 ℃. Notably, the chelating ligands not only effectively suppressed Ni aggregation during thermal treatment but also facilitated electronic configuration modulation of the Ni active sites and/or the CNS matrices through sulfur incorporation. For comparison, a control sample was prepared using Ni(NO3)2 as the Ni precursor under identical conditions, designated as Ni–N–C/CNS, to evaluate the influence of sulfur incorporation on CO2RR performance. Inductively coupled plasma optical emission spectrometry analysis revealed that the Ni contents in Ni–N–S/CNS and Ni–N–C/CNS were 3.12 wt% and 3.74 wt%, respectively.
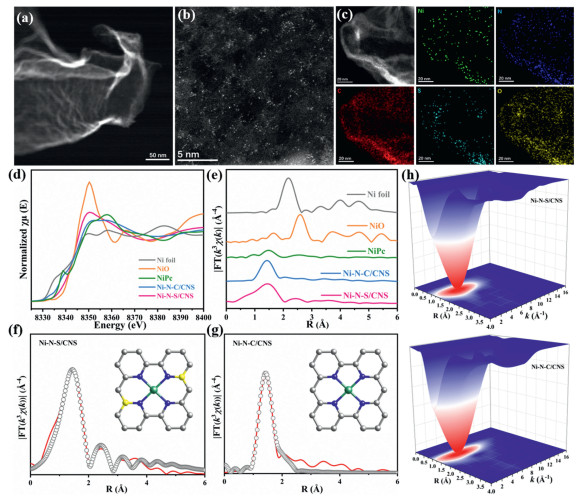
Morphological analysis confirmed that Ni loading did not induce significant alterations to the preformed CNS matrices in Ni–N–S/CNS. Energy–dispersive spectroscopy (EDS) elemental mapping images demonstrated homogeneous dispersion of C, N, S and Ni throughout the entire CNS matrices (Figs. 1a–c). To further distinguish the Ni species at atomic resolution, subangstrom–resolution HAADF–STEM measurements were conducted. As depicted in Fig. 1b, ultra–small bright spots corresponding to single Ni atoms are found on Ni–N–S/CNS, confirming their atomic dispersion rather than nanoparticles or clusters formation. In contrast, Ni–N–C/CNS exhibited a similar structure to Ni–N–S/CNS except the absence of S (Fig. S3 in Supporting information).
Figure 1
Figure 1.
(a) STEM image, (b) HAADF–STEM image, and (c) elemental mapping of Ni–N–S/CNS. (d, e) XANES and k3–weight FT–EXAFS curves of the Ni–N–S/CNS, Ni–N–C/CNS, and references at Ni K–edge. (f, g) The corresponding EXAFS fitting curves of Ni–N–S/CNS and Ni–N–C/CNS at R space, respectively. The inset is the schematic model proposed for catalysts. (h) WT–EXAFS of the Ni–N–S/CNS and Ni–N–C/CNS.
Powder X–ray diffraction (PXRD) results exhibit characteristic peaks identical to those of the graphite structure. The absence of nickel nanoparticles and their associated diffraction peaks in all three samples implies the atomic dispersion of Ni without any agglomeration (Fig. S4 in Supporting information). The Raman spectra of all three samples exhibit two characteristic peaks at 1340 cm–1 (D band) and 1580 cm–1 (G band), corresponding to disordered sp3 carbon and ordered graphitic sp2 carbon, respectively. The ID/IG ratio of Ni–N–S/CNS (1.26) and Ni–N–C/CNS (1.21) was higher than that of CNS matrices (1.17), indicating that the Ni and S species introduced more defects into the CNS matrices (Fig. S5 in Supporting information). Nitrogen adsorption/desorption isotherms were analyzed using the Brunauer–Emmett–Teller (BET) method to determine the specific surface areas and pore characteristics of all three samples (Fig. S6 in Supporting information). These samples showed significant adsorption at low P/P0 (0–0.015) and featured a clear hysteresis loop in the P/P0 range of 0.45–1.0, which indicates their hierarchical microporous/mesoporous structures. The specific surface areas of Ni–N–S/CNS and Ni–N–C/CNS were determined to be 790.41 and 811.48 m2/g, respectively, both of which are higher than that of the CNS matrices (624.67 m2/g). The high surface areas and hierarchically porous structures of Ni–N–S/CNS and Ni–N–C/CNS suggest that the ultrathin nanosheets not only increase the exposure of active sites but also facilitate the charge transfer and mass transport during CO2RR.
Since the electronic properties of active sites are governed by their structural configurations and coordination environments, both X–ray photoelectron spectroscopy (XPS) and absorption fine structure (XAFS) techniques were employed to investigate these features. Ni 2p XPS spectra of Ni–N–S/CNS and Ni–N–C/CNS reveal peaks at ~855.3 eV (Ni 2p3/2) and ~873.0 eV (Ni 2p1/2), which confirm the presence of Ni in the +2 oxidation state (Fig. S7 in Supporting information). As shown in Fig. S8 (Supporting information), the high–resolution N 1s XPS spectra of the as–prepared samples exhibit two distinct peaks at ~398.1 and 401.2 eV, which are attributed to pyridine N and graphite N, respectively. Compared with the NC matrices, an additional peak at ~399.3 eV observed in the other two samples is attributed to the formation of metal–N bonds, indicating the successful anchoring Ni species on N–doped carbon through Ni–N bonds. Increasing Ni–N bonds correlate with decreased pyridine N content, implying pyridine N primarily anchors metal species via metal–N bond formation. Additionally, graphitized N exhibits superior electrical conductivity, which facilitates electron transfer in CO2RR.
To gain deeper insight into the coordination structures of Ni SA in Ni–N–S/CNS and Ni–N–C/CNS samples, XAFS analysis was carried out. Fig. 1d displays the normalized Ni K–edge X–ray absorption near–edge structure (XANES) spectra of Ni–N–S/CNS and Ni–N–C/CNS, in comparison with Ni foil, NiO, and NiPc. The Ni K–edge positions in both samples are close to that of NiO, implying that the Ni species are in the +2 oxidation state. In the Fourier–transformed k3–weighted extended X–ray absorption fine structure (FT–EXAFS) spectra, the absence of a Ni–Ni peak at 2.17 Å in both Ni–N–S/CNS and Ni–N–C/CNS samples suggests the atomic dispersion of Ni atoms on the CNS matrices, which is consistent with the PXRD results. The main peak of both samples is observed at ~1.44 Å, corresponding to the Ni–N first coordination shell. In contrast to the Ni coordination environment in Ni–N–C/CNS, an additional small peak at ~2.42 Å is observed in Ni–N–S/CNS. This distance is much longer than the first–shell Ni–S bond (~1.90 Å) and can be attributed to a second scattering pathway involving Ni–N–S (Fig. 1e) [42-44]. The Wavelet transforms (WT) profiles of the two samples exhibit a maximum intensity at ~(4.2 Å–1, 1.3 Å). This position aligns closely with that of NiPC (4.6 Å–1, 1.4 Å), but is notably distinct from the features of Ni foil (7.3 Å–1, 2.2 Å), thereby providing further evidence for Ni–N coordination (Fig. 1h and Fig. S9 in Supporting information). Furthermore, quantitative FT–EXAFS fitting analysis was conducted to elucidate the coordination configuration of Ni sites in both Ni–N–S/CNS and Ni–N–C/CNS (Figs. 1f and g, Fig. S10 and Table S1 in Supporting information). In Ni–N–S/CNS, the Ni–N coordination number and bond length were identified as 4.1 and 1.97 Å, respectively, whereas those for Ni–N–S were measured at 2.0 and 2.78 Å. These results strongly support the conclusion that Ni is directly coordinated with four N atoms, while S atoms are doped within the second coordination sphere. In contrast, for Ni–N–C/CNS, the fitting results reveal a characteristic NiN4 structural configuration.
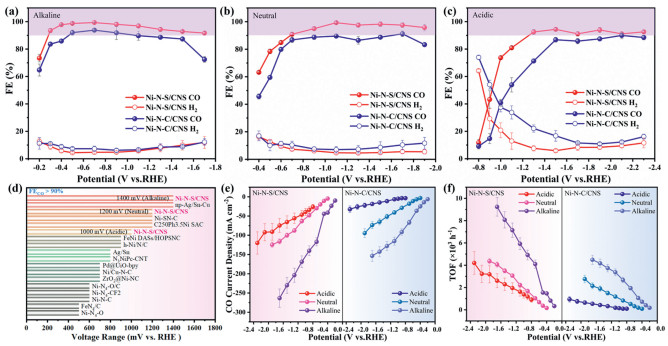
The electrochemical CO2RR catalytic performance of Ni–N–S/CNS and Ni–N–C/CNS was assessed in a standard three–electrode flow cell configuration integrated with a gas diffusion electrode (GDE) under pH–universal electrolytes. As illustrated by the linear sweep voltammetry (LSV) curves (Fig. S11 in Supporting information), Ni–N–S/CNS exhibits lower onset overpotentials and higher cathodic current densities under a flowing CO2 atmosphere relative to those observed in an Ar atmosphere, which indicates the effective CO2RR activity of the samples. The online gas chromatography analysis combined with offline 1H nuclear magnetic resonance spectroscopy confirms that CO and H2 are the sole products formed in pH–universal electrolytes. Ni–N–S/CNS exhibits exceptional FECO across multiple electrolyte environments (Figs. S12 and S13 in Supporting information). In alkaline conditions, the catalyst attains a peak FECO of 99.3% at –0.7 VRHE while maintaining over 90% efficiency throughout the applied potential range of –0.3 VRHE to –1.7 VRHE (Fig. 2a). This robust performance extends to neutral electrolytes, where the system achieves 99.3% FECO at –1.1 VRHE and sustains over 90% efficiency across an extended potential window from –0.7 VRHE to –1.9 VRHE, retaining 95.8% even at –1.9 VRHE (Fig. 2b). Remarkably, the material exhibits exceptional suppression of the hydrogen evolution reaction (HER) under strong acidic conditions, achieving a FECO of 94.4% at –1.5 VRHE with sustained above 90% efficiency across a –1.3 VRHE to –2.3 VRHE operational range, thereby demonstrating its pH–universal catalytic stability (Fig. 2c). Notably, the CO selectivity and high–performance potential window of Ni–N–S/CNS are substantially superior to those reported in previous studies (Fig. 2d and Table S2 in Supporting information) [29,34,45-51]. Comparative analysis reveals Ni–N–C/CNS shows inferior performance, reaching only 93.8% FECO at –0.7 VRHE in alkaline conditions and almost below 90% across the tested potentials in neutral/acidic media (Figs. 2a–c). The CO partial current density (jCO) of Ni–N–S/CNS reaches 263, 124, and 90.8 mA/cm2 at –1.7 VRHE in alkaline, neutral, and acidic electrolytes, respectively, outperforming Ni–N–C/CNS counterparts (Fig. 2e). Crucially, turnover frequency (TOF) analysis, which normalizes for Ni loading, reveals that Ni–N–S/CNS achieves highest values of 9240 h–1 in alkaline, 4370 h–1 in neutral, and 4210 h–1 in acidic electrolytes at the optimal potential, exceeding the corresponding values of Ni–N–C/CNS counterparts (Fig. 2f). These results indicate that sulfur doping effectively modulates the electronic configuration of isolated Ni sites, enhancing their intrinsic catalytic activity. Electrochemical impedance spectroscopy (EIS) was conducted to characterize the charge–transfer properties of the synthesized samples (Fig. S14 in Supporting information). The Nyquist plots revealed that all samples exhibit comparable charge transfer resistance, which suggests similar charge transfer kinetics. Comparative analysis of electrochemical surface areas (ECSA) derived from double–layer capacitance measurements demonstrated that Ni–N–S/CNS and Ni–N–C/CNS exhibit comparable active surface areas, both of which are higher than that of the CNS matrix (Fig. S15 in Supporting information). These collective observations demonstrate that the enhanced CO2RR performance originates not from charger–transfer kinetics and surface area variations but rather correlates directly with the intrinsic catalytic activity of the Ni active sites.
Figure 2
Figure 2.
FECO and FEH2 of Ni–N–S/CNS and Ni–N–C/CNS in (a) alkaline, (b) neutral, and (c) acidic electrolytes. (d) Comparison of potential windows for Ni–N–S/CNS and previously reported electrocatalysts under FECO exceeds 90%. (e) Current density and (f) turnover frequencies (TOF) of CO at different electrolytes.
The long–term stability of Ni–N–S/CNS in a flow cell was investigated at –0.9 VRHE in 1.0 mol/L KOH. The jCO was maintained ≈ 100 mA/cm2 over 28 h, and the FECO remained above 90% throughout the entire process (Fig. S16 in Supporting information). In addition, no significant structural alterations in Ni–N–S/CNS were observed after 28 h CO2RR stability test, as confirmed by XPS spectra, HAADF–STEM imaging and PXRD results (Figs. S17–S21 in Supporting information).
To elucidate the dynamic behavior of reaction intermediates during the CO2RR process, in situ attenuated total reflection surface enhanced infrared absorption spectroscopy (ATR–SEIRAS) was performed (Fig. S22 in Supporting information). The peaks observed at approximately 1402 and 1533 cm–1 are assigned to the symmetric and asymmetric stretching vibrations of the O–C–O bond from the *COOH intermediate, respectively. A relatively weak peak at around 2064 cm–1 can be attributed to *CO intermediates, indicating rapid desorption of *CO species. Particularly, the characteristic band observed at 1650 cm–1 in the Ni–N–S/CNS spectrum is assigned to the H–O–H bending vibrational mode of adsorbed water. This strong co–adsorption promotes hydrogenation of adsorbed CO2, which accelerates *COOH intermediate formation and ultimately improves CO2 reduction efficiency. In contrast, no obvious peaks associated with water adsorption were detected in Ni–N–C/CNS, suggesting that S atoms play an essential role in promoting water molecules adsorption. Similarly, we verified the crucial role of S atoms in promoting water adsorption and subsequent O–H bond cleavage through kinetic isotope effect (KIE) experiments (Fig. S23 in Supporting information).
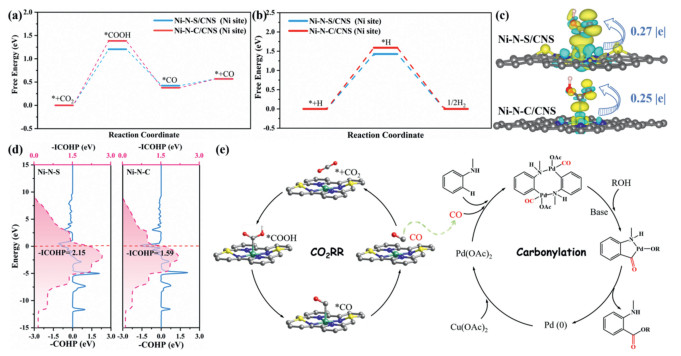
Density functional theory (DFT) calculations were employed to elucidate the mechanism by which S enhances the CO2RR performance of Ni–N–S/CNS. Reaction pathways for CO2–to–CO on Ni–N–S/CNS and Ni–N–C/CNS, as revealed by ATR–SEIRAS results, are illustrated in Fig. 3a. Theoretical calculation results demonstrated that the *COOH formation step is thermodynamically unfavorable, characterized by a higher Gibbs free energy change, suggesting that *COOH formation serves as the rate determining step (RDS). The calculated free–energy barrier for *COOH formation on Ni–N–S/CNS was 1.21 eV, lower than that on Ni–N–C/CNS (1.39 eV), demonstrating significantly reduced energy barriers for Ni–N–S/CNS in CO2RR, consistent with prior experimental observations. Considering the hydrogen evolution reaction (HER) as a competitive process of the CO2RR, the Gibbs free energies associated with HER were also calculated (Fig. 3b). The free energy changes of *COOH formation (ΔG(*COOH)) and *H formation (ΔG(*H)) were compared, as these processes corresponded to the RDS in CO2RR and HER, respectively. On both Ni sites in Ni–N–S and Ni–N–C models, ΔG(*H) values are higher than ΔG(*COOH), indicating that the initiation of CO2RR is thermodynamically more favorable than HER. To further investigate the influence of S on the electronic structure of Ni sites and the formation of *COOH, charge density difference and Bader charge analysis were performed (Fig. 3c). The Bader charge analysis revealed that the Ni site in Ni–N–S model transferred more charges to the *COOH intermediate, thereby lowering the energy barrier of *COOH formation. This finding is consistent with the result obtained from the charge density difference maps. Furthermore, crystal orbital Hamilton populations (COHP) analysis was performed to examine the bonding–antibonding nature of the Ni–*COOH bond, where negative and positive values denote bonding and antibonding contributions, respectively (Fig. 3d). A significantly enhanced occupancy of the Ni–*COOH bonding state was observed at the Ni site in Ni–N–S model compared to that in Ni–N–C model, correlating with stronger *COOH adsorption. Quantitatively, the integrated bonding strength of Ni–*COOH was calculated to be –2.15 eV for the Ni–N–S model, which is more negative than that of the Ni–N–C model (–1.59 eV), revealing an enhancement in the Ni–*COOH bonding strength for the Ni–N–S model. The aforementioned results demonstrate that S incorporation can effectively modulate the electronic structure of Ni sites, notably enhancing electron transfer dynamics between Ni sites and *COOH intermediates, and thereby substantially reducing the energy barrier for the RDS. In addition, the S site in Ni–N–S model exhibits a more negative water adsorption energy (–0.24 eV) compared to the Ni sites (–0.21 eV for the Ni site in Ni–N–S model and –0.20 eV for the Ni site in Ni–N–C model) (Fig. S24 in Supporting information). This indicates that the S site can effectively concentrate water molecules near the Ni site in Ni–N–S model, thereby promoting the protonation of CO2 to generate the *COOH intermediate. In conclusion, the electronic environment of Ni atoms in Ni–N–S/CNS is optimal for stabilizing the key *COOH intermediate during CO2RR to CO. The stronger adsorption of *COOH relative to *H at the Ni sites established a kinetic preference that selectively promotes CO2RR over HER, even under strong acidic conditions.
Figure 3
Figure 3.
Gibbs free–energy diagrams of (a) CO2RR and (b) HER over Ni–N–S/CNS and Ni–N–C/CNS. (c) Charge density difference and Bader charge analysis of *COOH adsorbed on Ni–N–S/CNS and Ni–N–C/CNS. (d) COHP analysis of Ni–*COOH over Ni–N–S/CNS and Ni–N–C/CNS. (e) Proposed mechanism for coupling CO2RR and carbonylation reactions.
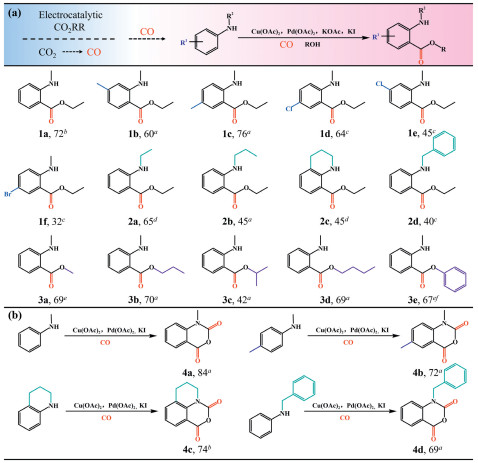
Although electrocatalytic CO2RR can achieve high selectivity toward CO proudction, the current utilization of this reductive product for further transformations remains limited. In this work, we successfully achieved the further conversion of the reductive product CO into high–value fine organic chemicals via a tandem electro–thermo–catalytic system. This approach not only enables the direct conversion of CO2 into high–value–added chemicals but also effectively mitigates the potential safety concerns associated by handing large quantities of toxic CO. As shown in Fig. S25 (Supporting information), the tandem electro–thermo–catalytic system successfully designed by connecting a flow cell with the Schlenk tube. The gaseous products generated via electrocatalytic CO2RR can undergo preliminary purification and then directly used as reactants in the subsequent Pd(Ⅱ)–catalyzed C–H monocarbonylation of N–alkylanilines for the synthesis of o–aminobenzoates (Fig. S26 in Supporting information). The Pd(Ⅱ)–mediated carbonylation process typically proceeds via a pathway initiated by the formation of dimeric palladium intermediate under a CO atmosphere. This is followed by CO insertion into the dimeric palladium intermediate, subsequent alcohol coordination, and reductive elimination, ultimately yielding to the o–aminobenzoates products (Fig. 3e). Within the tandem reaction system, the gaseous products were directed into the Schlenk tube to participate in the Pd(OAc)2–catalyzed oxidative carbonylation of N–methylaniline in DMF at 100 ℃. The reductive product CO generated via electrocatalytic CO2RR was subsequently transformed into o–aminobenzoates – commodity chemicals and highly reactive intermediates. In addition, the negligible amount of hydrogen produced as a by–product during electrocatalytic CO2RR exerted virtually no influence on the Pd(Ⅱ)– catalyzed oxidative carbonylation. After 12 h of reaction, N–methylaniline was successfully transformed into o–aminobenzoate (1a) with a yield of 72%, in agreement with the values reported in the literature (Fig. 4a) [8]. To further assess the effectiveness of tandem reactions, we systematically investigated the catalytic activity of various substrates bearing different functional groups (Fig. 4a). N–Methylanilines substituted with methyl groups on phenyl rings served as reactants under the reaction conditions, enabling the synthesis of corresponding methyl–substituted o–aminobenzoate derivates (1b, 1c) in high yield (76%), which can be attributed to their slightly enhanced nucleophilicity. The m–chloro– and p–bromo–substituted N–methylaniline derivatives underwent efficient conversion to produce corresponding o–aminobenzoate products (1e, 1f) with isolated yield of 45% and 32% respectively, demonstrating carbonylation reactivity's dependence on the substrates' electronic characteristics. In addition, anilines with various alkyl substituents were also explored. N–Ethyl–, N–propyl–, tetrahydroquinoline, as well as N–benzylaniline were employed, yielding the anticipated o–aminobenzoates (2a–2d) in moderate yield ranging from 40% to 65%. Furthermore, various alcohols were examined in the carbonylation reaction. While primary alcohols (methanol, propanol, butanol, and phenol) generated corresponding o–aminobenzoates (3a, 3b, 3d, 3e) with good yields (~70%), the sterically hindered 2–propanol demonstrated significantly lower efficiency (42%).
Figure 4
Figure 4.
(a) aReaction conditions: N–Alkylaniline 1 (0.2 mmol), Pd(OAc)2 (5 mol%), Cu(OAc)2 (1.1 equiv.), KI (20 mol%), KOAc (3 equiv.), and ethanol (5.0 equiv.) under CO (1 atm) in 2.0 mL of DMF at 100 ℃. b1a (1 mmol), DMF (8 mL), 24 h. cIn the absence of KOAc and at 120 ℃. d1.5 equiv. of KOAc. eKOAc (1.5 equiv.). fAlcohol (2.0 equiv.). (b) aReaction conditions: Aniline 1 (0.2 mmol), Pd(OAc)2 (5 mol%), Cu(OAc)2 (2.2 equiv.), KI (0.2 equiv.), CH3CN (2 mL), CO (1 atm), 60 ℃. bPivalic acid (1.0 equiv.) was added.
Furthermore, to highlight the universality of this tandem system, we integrated electrocatalytic CO2RR with Pd(Ⅱ)–catalyzed regioselective carbonylation for the synthesis of isatoic anhydrides, which are important building blocks in organic chemistry (Fig. 4b). Using CH3CN as the solvent in the Pd(Ⅱ)–catalyzed system under 60 ℃ reaction conditions with 2.2 equiv. of Cu(OAc)2 cocatalyst, N–methylaniline successfully converted into the corresponding isatoic anhydride in 84% yield, consistent with literature reports [7]. N–Methylanilines substituted with methyl groups on the phenyl rings were also compatible with this transformation. In addition, the scope was extended to N–alkyl substituents (tetrahydroquinoline, benzyl) on anilines, all of which effectively generated corresponding carbonylation products with moderate to excellent yields. These successful outcomes have clearly demonstrated the potential applications and broad applicability of directing CO2 conversion into value organic chemicals via synergistic coupling of electrocatalytic CO2RR and Pd(Ⅱ)–catalyzed carbonylation.
In summary, we present a facile strategy for engineering the secondary coordination shell to develop a high–performance electrocatalyst featuring atomically dispersed NiN4 sites and dual S doping in the second coordination shell. This Ni–N–S/CNS catalyst exhibited outstanding FECO values, sustaining a plateau above 90% over an ultrawide potential range from –0.3 VRHE to –1.7 VRHE, while achieving near 100% efficiency at –0.7 VRHE under alkaline conditions. Furthermore, the catalyst exhibited sustained efficiency exceeding 90%, with potential windows extending from –0.7 VRHE to –1.9 VRHE under neutral conditions and from –1.3 VRHE to –2.3 VRHE under acidic conditions. Combined in situ ATR–SEIRAS analysis and DFT calculations reveal that S incorporation both alters the electronic structure of Ni catalytic centers and strengthens water molecule adsorption, which collectively promote accelerated CO2–to–*COOH intermediate conversion even under high potentials. More importantly, the Ni–N–S/CNS catalyst ensures efficient and stable CO generation, thereby being successfully applied to tandem alkylaniline carbonylation reactions. This tandem catalytic system achieves high conversion in the synthesis of o–aminobenzoates and isatoic anhydrides, demonstrating significant application potential for the fine chemical production directly from CO2. This study not only highlights the exceptional CO2–to–CO catalytic performance of Ni–N–S/CNS with a pH–universal ultrawide potential window, but also presents a sustainable and cost–economically viable strategy for producing valuable fine chemicals.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Jun-Kang Li: Writing – original draft, Data curation. Pei-Yao Wang: Formal analysis, Data curation. Jia-Lin Zhang: Validation, Investigation. Shu-Na Zhao: Writing – review & editing, Resources. Bo Li: Validation. Shuang-Quan Zang: Supervision, Resources, Investigation.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 92461304, 22375185), and the Natural Science Foundation of Henan Province (No. 252300421181)
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.112150.
[1]
C. Zhu, J. Liu, M.B. Li, J.E. Bäckvall. Chem. Soc. Rev. 49 (2020) 341-353. doi: 10.1039/c9cs00397e
[2]
Q.P. Zhao, W.X. Shi, B. Wang, et al., Angew. Chem. Int. Ed. 64 (2025) e202510693. doi: 10.1002/anie.202510693
H. Cheng, X. Wu, M. Feng, et al., ACS Catal. 11 (2021) 12673-12681. doi: 10.1021/acscatal.1c02319
Figure 1
(a) STEM image, (b) HAADF–STEM image, and (c) elemental mapping of Ni–N–S/CNS. (d, e) XANES and k3–weight FT–EXAFS curves of the Ni–N–S/CNS, Ni–N–C/CNS, and references at Ni K–edge. (f, g) The corresponding EXAFS fitting curves of Ni–N–S/CNS and Ni–N–C/CNS at R space, respectively. The inset is the schematic model proposed for catalysts. (h) WT–EXAFS of the Ni–N–S/CNS and Ni–N–C/CNS.
Figure 2
FECO and FEH2 of Ni–N–S/CNS and Ni–N–C/CNS in (a) alkaline, (b) neutral, and (c) acidic electrolytes. (d) Comparison of potential windows for Ni–N–S/CNS and previously reported electrocatalysts under FECO exceeds 90%. (e) Current density and (f) turnover frequencies (TOF) of CO at different electrolytes.
Figure 3
Gibbs free–energy diagrams of (a) CO2RR and (b) HER over Ni–N–S/CNS and Ni–N–C/CNS. (c) Charge density difference and Bader charge analysis of *COOH adsorbed on Ni–N–S/CNS and Ni–N–C/CNS. (d) COHP analysis of Ni–*COOH over Ni–N–S/CNS and Ni–N–C/CNS. (e) Proposed mechanism for coupling CO2RR and carbonylation reactions.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
