Key Laboratory for Tibet Plateau Phytochemistry of Qinghai Province, Qinghai Engineering Research Center of Modern Tibetan Medicine Development, School of Pharmacy, Qinghai Minzu University, Xining 810007, China
b.
Key Laboratory of Smart Drug Delivery of Ministry of Education, National Key Laboratory of Advanced Drug Formulations for Overcoming Delivery Barriers, School of Pharmacy, Fudan University, Shanghai 201203, China
c.
Shanghai Skin Disease Hospital Tongji University School of Medicine, Shanghai 200443, China
d.
Fudan Zhangjiang Institute, Shanghai 201203, China
e.
Center for Medical Research and Innovation Shanghai Pudong Hospital Fudan University Pudong Medical Center, Shanghai 201399, China
f.
School of Pharmacy, Key Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education, Yunnan University, Kunming 650091, China
g.
Department of Chemistry, State Key Laboratory of Molecular Engineering of Polymers and Chem, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, Shanghai 200438, China
Received Date:
06 September 2025 Accepted Date:
18 November 2025 Revised Date:
17 November 2025 Available Online:
15 July 2026
Abstract:
Bergenin (BG), a bioactive coumarin derivative, suffers from poor solubility and low oral bioavailability, restricting its clinical potential. To overcome these limitations, we developed a lipid prodrug strategy by conjugating BG with bioactive fatty acids of different chain lengths (6C, 12C, 18C) via ester bonds, formulating the conjugates into solid lipid nanoparticles (SLNs), and systematically investigating the structure–activity relationship. Fatty acid conjugation not only enhanced lipophilicity, lipid matrix compatibility, and enzymatic stability but also imparted distinct biological effects that shaped oral absorption. Stearic acids (18C) conferred strong resistance to enzymatic hydrolysis, whereas lauric acid (12C) offered the most favorable balance in improving drug loading, stability, and membrane permeability. Pharmacokinetic studies in rats demonstrated that 12C-BG SLNs achieved the greatest enhancement in oral bioavailability, with an 8.6-fold increase over BG-SLNs and more than 40-fold improvement relative to reported suspensions and phospholipid solid dispersions. Mechanistic studies indicated that absorption was primarily driven by prodrug monomers released during intestinal lipolysis, with a minor contribution from undigested nanoparticles, and moreover the fatty acid chain length strongly influenced cellular permeability and systemic exposure. Collectively, these results underscore the critical role of bioactive fatty acids as conjugating groups in modulating prodrug fate and highlight a promising platform for enhancing the oral delivery of poorly soluble phytochemicals.
As a medicinal plant with a long history of use in traditional medicine systems, Bergenia species (family Saxifragaceae) have demonstrated broad therapeutic potential, particularly in the treatment of respiratory and gastrointestinal disorders [1]. Through systematic phytochemical investigations, bergenin (BG), a pleiotropic coumarin derivative, has been identified as the principal bioactive constituent responsible for these effects [2]. BG exhibits multiple pharmacological functions including expectorant, antitussive, anti-inflammatory, antiarrhythmic, anti-tumor, and metabolic regulatory effects [3,4]. Despite its promising therapeutic activities, the clinical translation of BG is hampered by its low bioavailability, which is mainly ascribed to its extremely low aqueous solubility and poor intestinal permeability [5,6]. Hence, further improving the druggability of BG necessitates the development of advanced formulations that can enhance the oral absorption.
By far, several formulations have been explored to address these limitations associated with poor solubility and permeability, such as solid dispersion formulations, polymer nanocarrier, pharmaceutical crystal engineering and cyclodextrin inclusion complex [7,8]. The most promising one is the lipid-based drug delivery system, as this drug delivery system shows high biocompatible and excels in enhancing the solubility and permeability of hydrophobic molecules simultaneously [9]. During gastrointestinal digestion, lipid components can form mixed micelles (MMs) with endogenous surfactants such as bile salts, which encapsulate lipophilic drugs, thereby enhancing solubility and promoting transmembrane transport across the intestinal mucosa [10]. Compared to other lipid-based carriers, solid lipid nanoparticles (SLNs) offer advantages including low cost, ease of preparation, biocompatibility, and long-term physical stability, Notably, SLNs' unique nanostructure may enable intact particle absorption via holistic pathways, such as transcytosis through intestinal M cells or uptake by Peyer's patches in the gut-associated lymphoid tissue (GALT) [11]. This mechanism bypasses the conventional stepwise drug release-and-absorption process, enhancing the intestinal permeability and bioavailability of poorly soluble drugs, making them particularly attractive for oral drug delivery applications [12].
Despite their advantages, the application of SLNs for BG delivery is constrained by inherent incompatibility between BG and lipid matrices, resulting in suboptimal drug loading (DL) capacities (typically 4%–6%) that ultimately limit oral absorption efficiency [6]. To address these challenges, the lipid–drug conjugate (LDC) strategy has gained increasing attention as a promising alternative approach [13,14]. In this approach, pharmacologically active compounds are covalently conjugated with lipid moieties (e.g., triglycerides, fatty acids, or phospholipids) to form amphiphilic LDCs. This structural modification can enhance drug-lipid matrix compatibility, increase transmembrane permeation, and improve pharmacokinetic profiles as well as the bioavailability of parent drugs [15,16]. By leveraging the lipophilic nature of these prodrugs, this strategy not only overcomes formulation challenges but also synergizes with physiological lipid absorption pathways, thereby amplifying therapeutic efficacy in oral delivery systems.
In this study, the fatty acids of short, medium, and long chain lengths, were utilized to conjugate with BG to obtain three BG-lipid prodrugs, respectively. We then investigated the impact of the lipid chain length on the lipophilicity of the prodrugs, the loading capacities of BG in the SLNs, and finally the oral bioavailability of BG. Additionally, we further explored the impact of the lipid chain length on the gastrointestinal translocation and absorption of intact SLNs through particle labelling with an aggregation-caused quenching (ACQ) probe in the second near-infrared (1000–1700 nm, NIR-Ⅱ) window, which enables an accurate trafficking of nanoparticles in deep tissues by eliminating the interference from free probe signals and reducing tissue scattering/autofluorescence [17,18]. We anticipated that a clear understanding of the in vivo behaviors of SLNs could facilitate the understanding of the structure-activity of various prodrug SLNs and their underlying mechanisms for enhancing the oral absorption of BG.
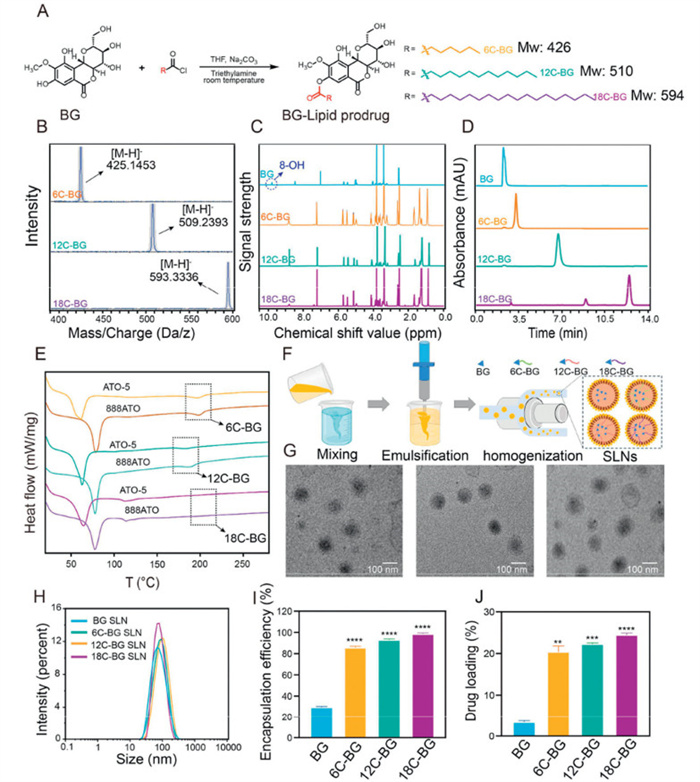
Three BG-lipid prodrugs were obtained by esterifying the 8–hydroxyl group of BG with short-, medium-, and long- chain fatty acids, namely hexanoic acid (6C), lauric acid (12C), and stearic acid (18C), respectively, as schematically illustrated in Fig. 1A. The successful conjugation was confirmed by the high-resolution mass spectra and nuclear magnetic resonance spectra (Figs. 1B and C, Figs. S1–S3 in Supporting information). Structural validation via1H nuclear magnetic resonance (1H-NMR) revealed distinct spectral differences: unmodified BG showed no fatty chain signals, while BG-lipid conjugates exhibited characteristic chemical shift values (δ = 0.8–2.0 ppm) for -CH3 and -CH2 groups, along with the complete disappearance of the 8–hydroxyl singlet (δ = 10.0 ppm), confirming site-specific esterification without altering other functional groups (Fig. 1C). The 13C-NMR analysis confirmed that fatty acid chains are covalently attached to the BG scaffold via ester bonds, directly revealing their structural incorporation. The high-performance liquid chromatography (HPLC) analysis demonstrated that all prodrugs showed high purity (over 93%), evidenced by their minimal impurity peaks (Fig. 1D).
Figure 1
Figure 1.
(A) Scheme of the synthesis pathways of various BG-lipid prodrugs. The mass spectra (B), 1H NMR spectra (C) and HPLC spectra (D) of three prodrugs and BG. (E) The DSC curves of 6C-, 12C-, or 18C-BG that were mixed with ATO-5 and 888ATO, respectively, with the drug/lipid ratio set to 1:3. (F) Schematic illustration of the preparation of various SLNs, respectively. (G) The TEM images of three BG-lipid SLNs. From left to right are 6C-, 12C-, and 18C-BG SLNs, respectively. (H) The particle size distributions of BG, 6C-, 12C- and 18C-BG SLNs measured by dynamic laser scattering, respectively. EE (I) and DL (J) of prodrug and BG in SLNs. **P < 0.01, ***P < 0.001, ****P < 0.0001 (6C-, 12C-, 18C-BG SLN vs. BG SLN). Data expressed as mean ± standard deviation (SD) (n = 3).
To rapidly and effectively assess the compatibility between different prodrug candidates and lipid matrices, we examined the melting behavior of BG, BG-lipid conjugates, two commonly used lipid excipients (ATO-5 and 888ATO) for SLN formulations, and the drug-excipient mixtures using differential scanning calorimetry (DSC) (Fig. 1E, Figs. S4 and S5 in Supporting information). The results showed that unmodified BG displayed a distinct endothermic peak at 227 ℃, corresponding to its crystalline melting point. Upon fatty acid conjugation, the melting points of BG lipid derivatives shifted significantly: 6C-BG, 12C-BG, and 18C-BG exhibited reduced peaks at 197, 190, and 205 ℃, respectively (Fig. S4). These shifts indicate that the introduction of fatty acyl chains disrupted BG's native crystalline lattice, lowering its melting point. Notably, the lipid matrix ATO-5 itself showed a phase transition at 61 ℃, characteristic of its semi-crystalline structure.
After mixing the drug and lipid excipients at varying ratios, different melting behaviors were observed, indicating the different compatibility between various drugs and lipid excipients. It has been estimated that the complete amorphous dispersion of BG-lipid prodrugs in ATO-5 required distinct drug-to-excipient ratios: 1:10 for 6C-BG, 1:3 for 12C-BG, and 1:2 for 18C-BG. In contrast, unmodified BG remained partially crystalline even at a 1:10 ratio, suggesting the bad compatibility between the parent drug and lipid matrix. Notably, chain elongation correlated positively with the lipophilicity of prodrugs, as evidenced by the progressively lower drug-to-excipient ratios needed for complete amorphous dispersion of the prodrug. Comparative studies with another lipid carrier (888ATO) revealed that ATO-5 exhibited markedly stronger compatibility with BG lipids at a 1:3 ratio (Fig. S5). Consequently, ATO-5 was selected as the optimal lipid matrix for subsequent nanoparticle formulation, with a fixed drug-to-excipient ratio of 1:3 to ensure maximal payload integration and stability.
To optimize the formulation of SLNs, we systematically screened surfactants for their ability to stabilize BG-lipid SLNs. Among the candidates tested, Tween 80, polyoxyethylene castor oil, and d-α-tocopherol polyethylene glycol succinate (TPGS), Tween 80 demonstrated superior performance. SLNs formulated with 2% Tween 80 exhibited monodisperse particle size distributions, the highest encapsulation efficiency (EE), and optimal DL (Fig. S6 in Supporting information). In contrast, SLNs prepared with polyoxyethylene castor oil or TPGS showed lower colloidal stability in biorelevant media (fasted state simulated gastric fluid (FaSSGF), PBS, Fasted State Simulated Intestinal Fluid (FaSSIF)). These limitations may stem from suboptimal surfactant-lipid interactions, such as insufficient hydrophobic anchoring of the prodrug or weaker interfacial stabilization, leading to particle aggregation and payload leakage. Based on these findings, ATO-5 (as the lipid matrix), 2% Tween 80 (as the surfactant), and the drug-to-excipient ratio of 1:3 was selected for subsequent experiments to ensure robust nanoparticle performance (Figs. S6–S8 in Supporting information).
BG-lipid loaded SLNs were prepared via high-pressure homogenization (Fig. 1F) [19], with Precirol® ATO-5 and Tween 80 serving as the lipid matrix and emulsifier, respectively. Transmission electron microscopy (TEM) revealed that three BG-lipid SLN formulations all exhibited uniform spherical morphology and were homogeneously dispersed (Fig. 1G). Dynamic light scattering analysis confirmed their narrow particle size distributions, evidenced by their lower polydispersity indices (PDIs) than 0.2 (Fig. 1H and Table S1 in Supporting information). Through controlling the homogenization procedures, we ensured that the average particle sizes of three types of BG-lipid loaded SLNs were nearly same (around 73 nm). We believed that this could effectively eliminate the potential size effect on the subsequent evaluations, as nanoparticle dimensions have been widely demonstrated to profoundly influence drug release kinetics, biodistribution patterns, and cellular internalization efficiency [20]. Furthermore, all SLNs displayed nearly neutral surface as their zeta potentials were all falling within −15~15 mV range, which enabled SLNs to avoid electrostatic interactions with the unstirred mucus layer in the gastrointestinal tract [21].
The EE and DL of BG and BG-lipid SLNs were determined using the classical ultrafiltration centrifugation method. As shown in Figs. 1I and J, the control BG SLNs exhibited a low EE (28%) as well as DL (4%), whereas the BG-lipid SLNs demonstrated significantly higher EEs and DLs (Table S2 in Supporting information). We further evaluated the stability of various BG-lipid SLNs through monitoring of the particle size, PDI, EE, and DL over a period of 7 days. The results demonstrated that the BG-lipid SLNs stored at room temperature (25 ℃) exhibited negligible change in particle sizes and PDIs, indicating their excellent colloidal stability. Moreover, both the EE and DL of various SLNs showed only a slight but not apparent decrease during the 7-day period, suggesting minimal drug leakage (Fig. S9 in Supporting information). These results underscored the high long-term stability of various BG-lipid prodrug SLNs. This is mainly ascribed to the fact that the lipid-drug compatibility progressively improved with the elongation of fatty acid chains, which minimized the drug leakage and thus allowed for higher EEs (86.4%–99.4%) and DLs (21.6%–24.9%). Notably, although existing lipid-based nanocarriers reported in the literature commonly employ much lower drug-to-lipid ratios to encapsulate BG and achieve high EE, their resultant DL capacity remains suboptimal [22]. This fundamental limitation stems from the high lipid content required to ensure carrier stability and impressive EE data, the excessive lipid proportion inevitably compromises the actual drug payload. Our novel prodrug strategy effectively addresses this limitation by co-optimizing both EE and DL capacity.
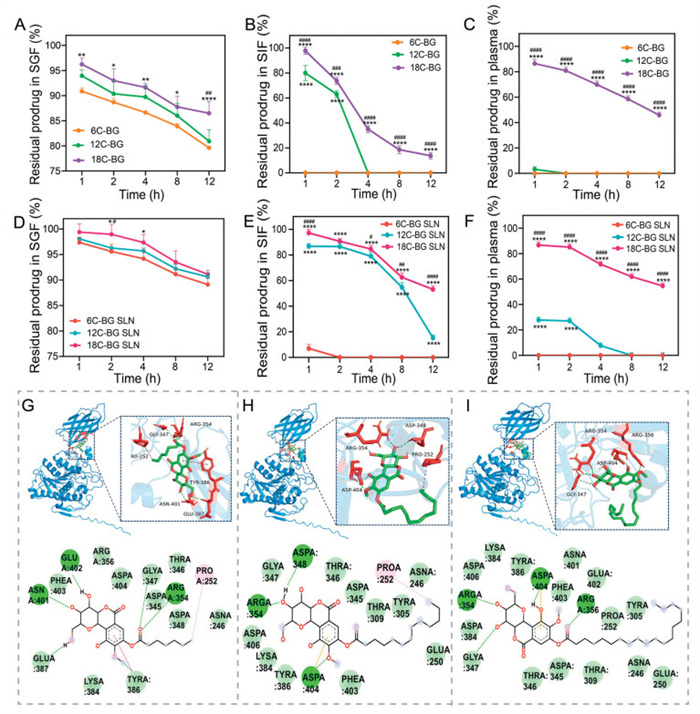
We next evaluated the stability of lipid prodrugs with different chain lengths (6C-, 12C-, and 18C-BG) and their SLNs in three biological media: simulated gastric fluid (SGF), simulated intestinal fluid (SIF), and plasma. As shown in Fig. 2A, all types of BG prodrugs showed gradual degradation in SGF, mainly due to the acid hydrolysis. By contrast, the degradation rates of these prodrugs in SIF and plasma were much faster (Figs. 2B and C), primarily ascribed to the abundant enzymes (such as pancreatic lipases, cholinesterases and carboxylesterases) in these media that could efficiently hydrolyze the ester bonds [23].
Figure 2
Figure 2.
Hydrolysis profiles of various free BG-lipid prodrugs in SGF (A), SIF (B) and plasma (C). Hydrolysis profiles of various BG-lipid prodrugs encapsulated in SLNs in SGF (D), SIF (E) and plasma (F). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (12C-, 18C-BG SLN vs. 6C-BG SLN). #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 (12C-BG SLN vs. 18C-BG SLN). Data expressed as mean ± SD (n = 3). The three-dimensional binding diagram of 6C-BG (G), 12C-BG (H), and 18C-BG (I) with cholinesterase was generated through molecular-protein induced fitting docking.
In addition to the biological media, the conjugated fatty acids also impacted the hydrolysis of prodrugs. The enzymatic hydrolysis rate was found to negatively correlate with the chain length of conjugated fatty acids. For example, 6C-BG underwent near-complete hydrolysis in a very short time (< 1 h) in SIF, while 12C-BG showed medium stability, exhibiting almost fully hydrolyzed within 4 h. By contrast, 18C-BG demonstrated the highest stability, retaining about 18% of the drug after 12 h in SIF. The similar chain length-dependent hydrolysis was also observed in plasma: 6C-BG showed the fastest hydrolysis while 18C-BG exhibited the slowest hydrolysis (Fig. 2C). This is probably due to the fact that the chain length significantly influences the formation efficiency of the enzyme-substrate complex. For prodrugs with short-chain fatty acids, they can more readily access the enzyme's active pocket thanks to their lower hydrophobicity and steric hindrance, leading to higher binding efficiency and consequently easier degradation by the esterases. In contrast, long-chain fatty acids enhance intermolecular hydrophobic interactions of prodrugs and induce high steric hindrance, thus reducing the accessibility of the enzyme [24,25].
Compared with free prodrugs, these BG-lipids that were encapsulated in SLNs showed apparently improved stability in biological media (Figs. 2D–F) as the lipid matrix impeded the interactions between prodrugs and acids/enzymes. For instance, nearly 20% of 12C-BG and 60% of 18C-BG remained in the SLNs after 12-h incubation in SIF (Fig. 2E), respectively, which was not available in free prodrugs. Notably, the SLN-encapsulating 6C-BG was still unstable in SIF and plasma, showing a complete degradation within 2 h (Figs. 2E and F). The combined effect of the poor lipid compatibility and low lipophilicity of 6C-BG probably accounts for the rapid degradation. Under a fixed drug-to-excipient ratio (1:3), the encapsulation of 6C-BG in SLNs may be inadequate, leaving a fraction of free prodrugs unbound or weakly associated with the lipid matrix. Furthermore, the limited lipophilicity of 6C-BG, due to its short hydrocarbon chain, probably exacerbated the drug leakage by reducing hydrophobic interactions with the lipid carrier. In contrast, 12C-BG and 18C-BG have medium/long-chain fatty acids and progressively enhanced lipid solubility, thus they are more stably incorporated within SLNs. This could effectively minimize premature drug release, and confer SLN-encapsulating 12C-BG and 18C-BG insusceptibility to hydrolysis by enzymes in SIF and plasma, respectively (Figs. 2E and F).
To further explore the impact of the lipid chain length on the enzymatic degradation of prodrugs, we investigated the binding efficacies of short-, medium-, and long-chain BG-lipids with cholinesterase through molecular docking simulations. The results showed that the binding affinities of 6C-BG, 12C-BG, and 18C-BG with cholinesterase were 6.7, 5.8, and 5.2 kcal/mol, respectively, confirming that the binding efficacy of BG-lipid prodrugs with esterases decreases with the elongation of the chain length. Unlike the short-chain 6C-BG, which tightly anchors within the catalytic pocket through hydrogen bonding, van der Waals forces, and π-π stacking (Fig. 2G), the medium-chain 12C-BG exhibits significantly reduced π-π contacts (indicated by red dashed lines in Fig. 2H) due to steric hindrance from its aliphatic chain, retaining only partial hydrogen bonding. The long-chain 18C-BG undergoes severe steric clash, forcing molecular reorientation that restricts interactions to weak hydrophobic engagements with peripheral residues (Fig. 2I).
Subsequently, the in vitro lipolysis of various SLN formulations were investigated based on a representative pH-stat lipolysis model. The results demonstrated that all BG-lipid SLNs showed similar lipolysis trends, with small variations in lipolysis rates (Fig. S10 in Supporting information). The lipolysis rates of various SLNs were observed to slightly depend on the conjugated lipid chain lengths. Compared with the counterparts, 18C-BG SLNs displayed the slowest lipolysis. For instance, within the initial 10-min lipolysis period, 93%–94% of 6C-BG and 12C-BG SLNs were degraded, while only 86% of the 18C-BG SLNs were degraded. Notably, the BG SLNs and 6C-BG SLNs showed similar lipolysis rate, probably owing to the rapid enzymatic degradation of 6C-BG prodrugs. By contrast, the enhanced lipid compatibility and enzymatic stability of 18C-BG may impede the enzymatic action of lipases through steric hindrance, thus leading to a slightly slow lipolysis.
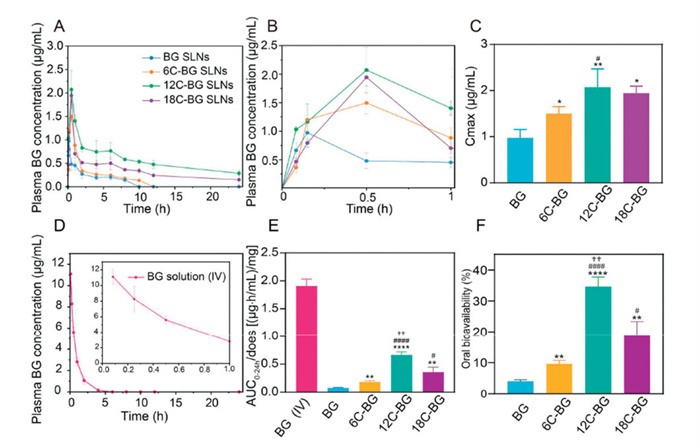
The oral absorption of various BG-lipid SLNs was comparatively analyzed in rats by monitoring the plasma concentrations of BG, with the BG SLNs as the control. All animal experiments were conducted according to the protocol approved by the Animal Care and Use Ethics Committee of School of Pharmacy, Fudan University (approval No. 2021–03-YJ-HHS-29). As shown in Figs. 3A and B, the pharmacokinetic curves revealed that all BG-lipid SLNs showed an enhanced systemic exposure of BG than the control group (BG SLNs), evidencing that the lipid conjugation favors oral absorption of BG. Notably, the plasma BG concentrations of all BG-lipid prodrug SLNs were higher than that of BG SLNs at 0.5–24 h following oral administration. In all SLN formulations, BG SLNs showed the fastest oral absorption, reaching its maximum concentration at 0.25 h (Fig. S11 in Supporting information). By contrast, the time to maximum concentration (Tmax) of BG-prodrug SLNs increased to 0.42–0.5 h (6C-BG: 0.42 ± 0.12 h; 12C-BG: 0.5 ± 0.0 h; 18C-BG: 0.5 ± 0.0 h), which was mainly due to the fact that additional time is needed to release the parent drug from various prodrugs.
Figure 3
Figure 3.
(A) Plotting the plasma BG concentrations versus time after oral administration of BG SLN or various BG-lipid prodrug SLNs to rats (at an equivalent BG dose of 20 mg/kg) within 24 h. (B) The enlargement of pharmacokinetic profiles of BG SLN and various BG-lipid prodrug SLNs within the initial 1 h. (C) Comparison of Cmax of four SLNs. (D) Plotting the plasma BG concentrations vs. time after intravenous injection of BG (5 mg/kg) to rats within 24 h. (E) AUC0–24h/dose of four SLNs and intravenous injection of BG. (F) Oral bioavailability of four groups of SLNs. *P < 0.05, **P < 0.01, ****P < 0.0001 (6C-, 12C-, 18C-BG SLN vs. BG SLN). #P < 0.05, ####P < 0.0001 (12C-, 18C-BG SLN vs. 6C-BG SLN). ††P < 0.01 (12C-BG SLN vs. 18C-BG SLN). Data expressed as mean ± SD (n = 3).
The fatty acid conjugation also increased the maximum plasma concentrations (Cmax) of BG. Among these formulations, 12C-BG SLNs showed the highest Cmax, around 2.1 folds of that in the BG SLNs group (Fig. 3C). And the Cmax of BG in 6C-BG SLNs and 18C-BG SLNs are significantly higher than that in the control group, as well. These results indicate that the conjugation with fatty acids significantly enhance the oral absorption of BG, with lauric acid showing the best performance. This was further supported by the fact that BG was detectable in plasma within 24 h post-administration in the groups of 12C-BG SLNs and 18C-BG SLNs while was undetectable after 10 h in the control group.
To calculate the absolute oral bioavailability of various SLNs, we plotted the pharmacokinetic profile of BG solution post intravenous administration simultaneously, which showed that although the plasma BG concentration was much higher than that in oral group within the initial 1 h, it underwent a rapid decline and became undetectable at 6 h post administration (Fig. 3D). To eliminate the dose effect, the dose-normalized area under the curve (AUC) in all groups were calculated and comparatively analyzed (Fig. 3E). The results confirmed that all BG-lipid prodrug SLNs significantly enhanced the oral absorption of BG, compared with the control (BG SLNs). The enhancement extents in 6C-BG SLNs, 12C-BG SLNs, and 18C-BG SLNs were measured to be 239.87% (P < 0.01), 864.05% (P < 0.0001), and 467.97% (P < 0.001), respectively (Table S3 in Supporting information). Based on the normalized AUC value of intravenous BG solution, the absolute oral bioavailability of BG in the BG-SLN group was calculated to be 4.008%. By contrast, the oral bioavailability in lipid prodrug formulations were much higher, with the 12C-BG SLNs being the highest. Specifically, the absolute oral bioavailability of 6C-BG SLNs, 12C-BG SLNs, and 18C-BG SLNs are 9.608% ± 1.127%, 34.610% ± 3.086%, and 18.769% ± 4.606%, respectively (Fig. 3F).
We also attempted to compare the performance of various BG-lipid SLNs with the reported oral formulations based on AUC0–24h/dose values. It is of note that 12C-BG SLNs exhibited a 43.2- to 118-fold increase in bioavailability compared to crude BG suspensions [26,27], a 44-fold improvement over BG–phospholipid complex solid dispersion systems [28], and a threefold enhancement over cationic liposomes and nanostructured lipid carriers [29]. The outperformed absorption enhancement of 12C-BG SLNs over BG SLNs in this study and other reported BG formulations underscores the significance of our prodrug strategy for enhancing the druggability and efficacy of BG.
We assumed that multiple synergy mechanisms contributed to the absorption enhancement of BG by our BG-lipid prodrug nanoplatform. Firstly, the covalent conjugation of BG with fatty acids increases its lipophilicity, which in turn improves its EE within SLNs and facilitates the epithelial uptake. Secondly, the released BG prodrugs during the hydrolysis of SLNs in the gastrointestinal tract show high membrane permeability and are easily absorbed into the circulation. Thirdly, the released fatty acids after prodrug hydrolysis, particularly lauric acid, may act as a permeation-enhancer. These multiple actions may enhance the paracellular permeability and promote the absorption of both the prodrugs, parent drugs, and moreover the undigested SLNs. Additionally, the solid lipid core of the SLNs slows lauric acid release, preserving a locally effective concentration of drug and excipient, thereby sustaining favorable absorption conditions.
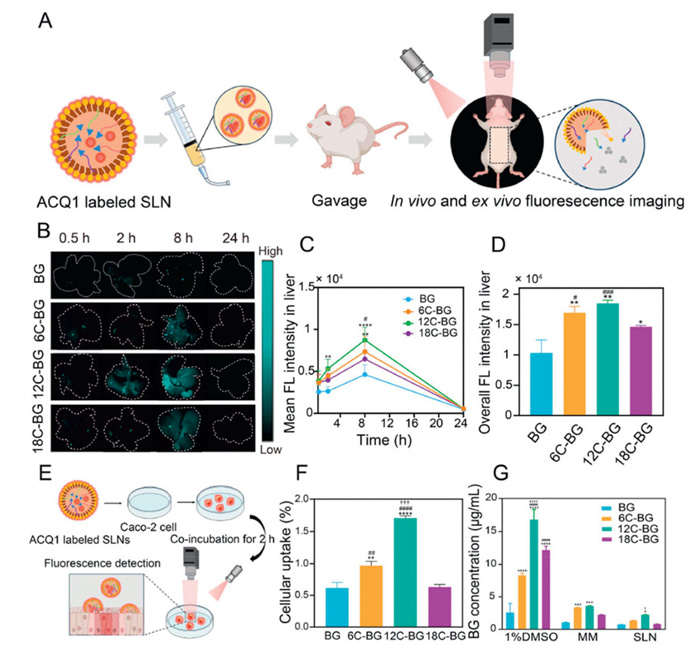
To verify these mechanisms, we further investigated and quantitively analyzed the gastrointestinal residence, distribution, and transport kinetics of various SLNs in vivo and ex vivo, respectively, through labeling by our recently developed NIR-Ⅱ ACQ probe (ACQ-1) (Fig. 4A and Figs. S14A–D in Supporting information). These ACQ probes emit intense fluorescence when encapsulated in the SLNs, while immediately become non-emissive upon release into aqueous biological medium during the degradation of SLNs (Fig. 4A). Hence, the fluorescence of ACQ probes strongly indicates the integrity of SLN particles. In vivo imaging results revealed that the abdominal fluorescence of rats after oral administration of various SLNs gradually declined and nearly disappeared after 12 h (Fig. S12 in Supporting information), indicating the degradation, absorption, and excretion of SLNs within the gastrointestinal tract.
Figure 4
Figure 4.
(A) Schematic of in vivo and ex vivo imaging using ACQ1-labeled particles. Representative ex vivo images (B) and mean (C) and total (D) fluorescence intensity of livers versus time after oral administration of BG and BG-lipid SLNs to rats. (E) Schematic diagram of intestinal epithelial cell uptake of BG-lipid SLNs labeled with ACQ1. (F) Comparison of fluorescence intensity with initial fluorescence intensity after 2-h incubation with Caco-2 cells quantitative changes in liver fluorescence intensity after oral administration of BG-lipid SLNs in rats. (G) Cell uptake of BG-lipid, BG-lipid MMs, BG-lipid SLNs after incubation with Caco-2 cells for 2 h. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (6C-, 12C-, 18C-BG SLN vs. BG SLN). #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 (12C-, 18C-BG SLN vs. 6C-BG SLN). †P < 0.05, ††P < 0.01, †††P < 0.001, ††††P < 0.0001 (12C-BG SLN vs. 18C-BG SLN). For all panels except (F): #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 (12C-, 6C-BG SLN vs. 18C-BG SLN). Data expressed as mean ± SD (n = 3).
To investigate the potential interspecies differences affecting particle distributions and transportations, we conducted the in vivo imaging studies in mice simultaneously, which showed similar results to that in rats (Fig. S13A in Supporting information). In both species, the nanoparticles were initially localized in the stomach and progressively migrated to the small intestine and colon over time. Notably, these four types of SLNs exerted similar retention profiles in the abdominal regions (Fig. S13C in Supporting information), indicating that the variations in the physicochemical properties of the lipid prodrugs showed negligible impact on the gastrointestinal distribution or transit of SLNs.
To confirmed the gastrointestinal transport and retention of various SLNs, rats were sacrificed at predetermined time points post oral administration of various fluorescently labelled SLNs, respectively, and the whole gastrointestinal segments were collected for ex vivo fluorescence imaging (Fig. S13B in Supporting information). At 0.5 h, strong fluorescence signals of SLNs were detected in the stomach and jejunoileum, the main site for the degradation and absorption of SLNs, indicating the rapid transit of SLNs from the stomach to the small intestine. Within 2 h, a significant proportion of SLNs had migrated to the cecum, while the fluorescence in the stomach and small intestine markedly declined (Fig. S14 in Supporting information). Then, the fluorescence signals of SLNs continued to decrease, and only weak signals remained in the colon and rectum at 8 h. Finally, the fluorescence of SLNs became nearly undetectable after 24 h, indicating complete degradation, absorption and excretion of the nanoparticles. Similar to the in vivo quantitative results, no significant differences were observed in the transport or retention profiles within the gastrointestinal tract among the four SLN formulations (Fig. S13D in Supporting information), further confirming that the fatty acid conjugation did not influence the overall gastrointestinal transition of SLNs.
The absorption of various SLNs were investigated by ex vivo imaging of livers and other main organs, which showed that the fluorescence signals of various SLNs were only detectable in the liver, while negligible in the heart, lungs, spleen, and kidneys (Fig. 4B and Fig. S15 in Supporting information). This is probably ascribed to the high uptake of particles by Kupffer cells in the liver tissues post oral absorption of SLNs. All groups showed a similar fluorescence trend in the liver: a weak signal at 0.5 h, increased signal at 2 h, peaked signal at 8 h, and disappeared signal at 24 h (Fig. 4C). Among these SLNs, 12C-BG SLNs showed the strongest liver signal, suggesting their highest hepatic accumulation and oral absorption (Fig. 4D).
We further investigated the cellular uptake of various SLNs using Caco-2 monolayers that simulated intestinal epithelium (Fig. 4E). After incubating various SLNs (BG SLNs, 6C-BG SLNs, 12C-BG SLNs, and 18C-BG SLNs) with the monolayers for two hours, the cellular uptake in four groups were all around 0.5%–1.5% of initial particle amounts (Fig. 4F). Among these SLNs, 12C-BG SLNs showed the highest cellular uptake, which aligned with the pharmacokinetic results.
Considering the role of the released free prodrugs and the formed MMs during the lipolysis of SLNs in the enhanced cellular uptake of BG, we incubated free BG and various BG-lipid prodrug solutions, MMs, and SLNs with Caco-2 monolayers to comparatively analyze their cellular uptake efficiencies. The results showed that the monomer solution showed the highest uptake, significantly exceeding that of the MM and SLN groups (Fig. 4G). This implies that the epithelial absorption of various prodrug monomers probably makes the main contribution to the enhanced oral bioavailability of BG. And the superior cellular uptake of BG-lipid prodrug monomers over the BG monomer are primarily due to their increased lipophilicity, which facilitate their interactions with cell membranes. Notably, 12C-BG consistently showed the highest cellular uptake when encapsulated in MMs and SLNs, respectively.
This difference is probably attributed to the properties of the conjugated medium-chain fatty acids (12C), which have been confirmed to promote cellular absorption of various drugs. It has been widely reported that lauric acid can also increase the membrane perturbation and integrity by insertion into the cell membrane, thus enhancing the transcellular flux of drug payloads and nanoparticles [30-34]. Furthermore, like other medium-chain fatty acids, lauric acid shows high potency in activating phospholipase C, triggering the production of inositol trisphosphate and diacylglycerol, thereby rapidly and reversibly opening tight junctions and facilitating the paracellular permeability of drug molecules with various molecular weights and moreover nanoparticles [35]. Besides, our P-gp inhibition assay also suggested that the conjugated lauric acid may act as a P-gp inhibitor (Fig. S16 in Supporting information), which aligned with the previously reported result [36]. Consequently, after the oral administration of 12C-BG SLNs, most nanoparticles underwent lipolysis, releasing the free prodrug monomers. With the further degradation of 12C-BG, the released lauric acids worked as a good permeability enhancer, increasing the cell intestinal permeability of prodrugs, parent drugs and undigested nanoparticles through multiple mechanisms. This finally led to the superior oral bioavailability of BG in the 12C-BG SLNs over other formulations.
In this study, we successfully established a lipid prodrug strategy to enhance the oral bioavailability of BG, through conjugating the parent drug molecules with fatty acids of varying chain lengths (6C, 12C, 18C), respectively. All lipid prodrugs exhibited enhanced lipophilicity and lipid matrix compatibility compared to the parent drug, leading to markedly higher DL in the conventional lipid-based delivery systems. Notably, the fatty acid chain length had a significant impact on the stability of prodrugs against enzymes. With the elongation of fatty acids, the enzymatic degradation of prodrugs was significantly depressed, mainly due to steric hindrance. Strikingly, the medium chain-length fatty acid (lauric acid) conjugation showed the greatest enhancement in the oral bioavailability of BG, around 8.6 folds of the native BG SLNs and more than 40-fold increase compared with the reported suspensions and phospholipid solid dispersions. Mechanistic investigations based on particle trafficking revealed that the conjugated fatty acids showed little impact on the lipolysis and gastrointestinal translocation of SLNs.
While fluorescence imaging revealed the hepatic uptake of nanoparticles, especially the 12C-BG SLNs, the cellular experiment results suggested that the uptake of prodrug monomers released during lipolysis primarily contributed to the epithelial uptake of BG. Besides, all the SLN-encapsulating prodrugs underwent rapid degradation into parent drug after oral administration as we failed to detect all the prodrugs (below the quantification limit of 1 µg/mL) in vivo. This further indicated that the oral absorption of intact SLNs were minimal, thus making a limited contribution to the total absorption of BG. Notably, the cell experiments also strongly evidenced that these fatty acids significantly influenced the cell monolayer permeability of both free prodrugs and particle-encapsulated prodrugs. Thanks to the excellent permeability enhancement capability of lauric acid, 12C-BG showed the highest cell uptake either in free form or encapsulated in nanoparticles, which aligned with the distribution of SLNs in the liver. Collectively, these results indicate that BG-lipid SLNs are absorbed primarily as prodrug monomers released during intestinal lipolysis, with a minor fraction taken up as intact nanoparticles. This study highlights the pivotal role of fatty acid chain length in modulating prodrug stability, cellular uptake, and systemic exposure. We believe that the combination of lipid prodrug design with SLN technology not only overcomes the inherent limitations of BG but also offers a versatile platform for improving the oral delivery of other poorly soluble phytochemicals.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by Natural Science Foundation of Qinghai Province (No. 2024-ZJ-911) and National Natural Science Foundation of China (No. 82104082).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.112134.
Figure 1
(A) Scheme of the synthesis pathways of various BG-lipid prodrugs. The mass spectra (B), 1H NMR spectra (C) and HPLC spectra (D) of three prodrugs and BG. (E) The DSC curves of 6C-, 12C-, or 18C-BG that were mixed with ATO-5 and 888ATO, respectively, with the drug/lipid ratio set to 1:3. (F) Schematic illustration of the preparation of various SLNs, respectively. (G) The TEM images of three BG-lipid SLNs. From left to right are 6C-, 12C-, and 18C-BG SLNs, respectively. (H) The particle size distributions of BG, 6C-, 12C- and 18C-BG SLNs measured by dynamic laser scattering, respectively. EE (I) and DL (J) of prodrug and BG in SLNs. **P < 0.01, ***P < 0.001, ****P < 0.0001 (6C-, 12C-, 18C-BG SLN vs. BG SLN). Data expressed as mean ± standard deviation (SD) (n = 3).
Figure 2
Hydrolysis profiles of various free BG-lipid prodrugs in SGF (A), SIF (B) and plasma (C). Hydrolysis profiles of various BG-lipid prodrugs encapsulated in SLNs in SGF (D), SIF (E) and plasma (F). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (12C-, 18C-BG SLN vs. 6C-BG SLN). #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 (12C-BG SLN vs. 18C-BG SLN). Data expressed as mean ± SD (n = 3). The three-dimensional binding diagram of 6C-BG (G), 12C-BG (H), and 18C-BG (I) with cholinesterase was generated through molecular-protein induced fitting docking.
Figure 3
(A) Plotting the plasma BG concentrations versus time after oral administration of BG SLN or various BG-lipid prodrug SLNs to rats (at an equivalent BG dose of 20 mg/kg) within 24 h. (B) The enlargement of pharmacokinetic profiles of BG SLN and various BG-lipid prodrug SLNs within the initial 1 h. (C) Comparison of Cmax of four SLNs. (D) Plotting the plasma BG concentrations vs. time after intravenous injection of BG (5 mg/kg) to rats within 24 h. (E) AUC0–24h/dose of four SLNs and intravenous injection of BG. (F) Oral bioavailability of four groups of SLNs. *P < 0.05, **P < 0.01, ****P < 0.0001 (6C-, 12C-, 18C-BG SLN vs. BG SLN). #P < 0.05, ####P < 0.0001 (12C-, 18C-BG SLN vs. 6C-BG SLN). ††P < 0.01 (12C-BG SLN vs. 18C-BG SLN). Data expressed as mean ± SD (n = 3).
Figure 4
(A) Schematic of in vivo and ex vivo imaging using ACQ1-labeled particles. Representative ex vivo images (B) and mean (C) and total (D) fluorescence intensity of livers versus time after oral administration of BG and BG-lipid SLNs to rats. (E) Schematic diagram of intestinal epithelial cell uptake of BG-lipid SLNs labeled with ACQ1. (F) Comparison of fluorescence intensity with initial fluorescence intensity after 2-h incubation with Caco-2 cells quantitative changes in liver fluorescence intensity after oral administration of BG-lipid SLNs in rats. (G) Cell uptake of BG-lipid, BG-lipid MMs, BG-lipid SLNs after incubation with Caco-2 cells for 2 h. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (6C-, 12C-, 18C-BG SLN vs. BG SLN). #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 (12C-, 18C-BG SLN vs. 6C-BG SLN). †P < 0.05, ††P < 0.01, †††P < 0.001, ††††P < 0.0001 (12C-BG SLN vs. 18C-BG SLN). For all panels except (F): #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 (12C-, 6C-BG SLN vs. 18C-BG SLN). Data expressed as mean ± SD (n = 3).

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
