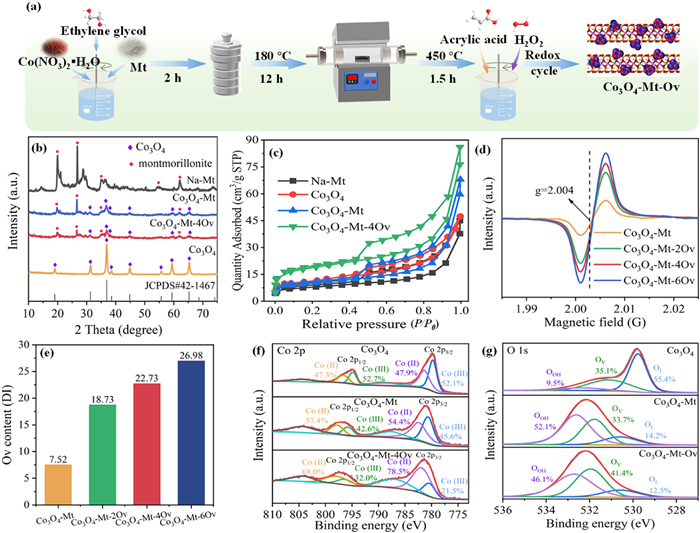
Figure 1.
(a) Schematic illustration of the synthesis of the Co3O4−MT-OV. (b) XRD, (c) BET, (d) EPR, (e) OV contents, (f) Co 2p HR-XPS and (g) O 1s HR-XPS of the catalysts.
Surface oxygen vacancy trigger peroxymonosulfate activation with cobalt modified montmorillonite for ofloxacin degradation: The synergistic effect of 1O2 and Co(Ⅳ)=O
Xiao-qiang Cao , Xingyao Liu , Yaqi Wang , Haining Wang , Bo Wei , Yanan Shang , Yizhen Zhang , Yujiao Kan , Yang Zhang , Xing Xu , Longlong Zhang

The Fenton-like process has drawn considerable attention as a prospective technology for water decontamination [1,2]. Specifically, extensive research has been conducted on the advanced oxidation processes (AOPs) relying on peroxymonosulfate (PMS) due to their various oxidation pathways that can generate highly reactive radicals (e.g., •OH (1.9−2.7 V vs. NHE) and SO4•− (2.5−3.1 V/NHE)) or highly selective non-radical species including singlet oxygen (1O2, 2.2 V vs. NHE), high-valent metal, and metals-PMS complex [3,4]. It is notable that non-radical species (e.g., 1O2 and Co(Ⅳ)=O) have gained extensive attention in the selective oxidation of organic contaminants owing to their inherent merits like potent electrophilicity and selectivity, a long half-life, and strong resilience to environmental interference [5,6]. Nevertheless, the ordinary production procedures of the non-radical species through PMS activation by metallic nanoparticles or complexes presented unsatisfactory results with low selectivity [7].
Defect engineering has emerged as a highly promising strategy for optimizing heterogeneous catalytic PMS activation, demonstrating significant potential to selectively modulate the PMS activation pathway toward non-radical species generation [8,9]. Vacancy defects can adjust the electronic structure of metal oxides, leading to the formation of unsaturated coordination sites [10]. Additionally, these defects optimize the adsorption energy of reactants on the catalyst surface. As a result, the reaction energy barrier between the catalyst and PMS is reduced, enhancing catalytic activation and accelerating the degradation of pollutants [11]. Recent researches have extensively explored various types of vacancy defects, such as oxygen vacancy (OV), nitrogen vacancy (NV) and sulfur vacancy (SV), due to their substantial potential in water treatment applications [12–14]. Among them, OV is among the most extensively investigated in PMS-activated systems [15]. In metal oxides, oxygen atoms within the crystal structure can be released under external conditions, such as elevated temperatures, leading to the formation of OV. For example, Wu et al. synthesized a series of Fe-Co LDH catalysts with varying levels of OV by modulating the calcination temperature and demonstrated the significant role of OV in PMS activation [16]. Besides, it has been reported that OV had the potential to improve site exposure and facilitate mass diffusion in cobalt-related catalysts, as well as enhance the adsorption of PMS on catalyst surfaces [17]. The localization of sufficient electrons around the OV supports a single-electron-transfer non-radical process, which can induce the formation of 1O2 via direct electron transfer [18,19]. Whereas, some recent studies have revealed that 1O2, derived from the transformation of superoxide radicals (O2•−), may serve as the predominant reactive species responsible for pollutant degradation in OV-enriched metal oxide/PMS systems [20]. Meanwhile, the role of OV in OV-rich catalysts for PMS activation remains controversial, as OV typically coexist with reducing metal active sites. These metal sites can also directly activate PMS through electron transfer processes, complicating the attribution of catalytic activity [21]. Therefore, further studies are required to reveal the role of OV on PMS activation and elucidate the underlying mechanism.
Among transition metals, cobalt (Co) is recognized as the most efficient activator for PMS activation [17]. Recent studies have extensively documented Co2+-mediated PMS systems, with particular attention given to the non-radical species generation pathway [22]. However, some critical challenges remain, such as secondary pollution from Co2+ leaching and inefficient catalyst recovery [23], both of which hinder its practical application. While stable cobalt oxides can mitigate metal leaching and address recovery issues, they often suffer from rapid deactivation due to nanoparticle aggregation and sluggish Co(Ⅲ)/Co(Ⅱ) redox cycling [24]. Encapsulating cobalt oxides (e.g., Co3O4, CoOx) onto the metal support presents a viable strategy to mitigate Co2+ leaching, suppress nanoparticle aggregation and enhance mass transfer kinetics. Natural montmorillonite (Mt) has emerged as a superior alternative to traditional template materials (e.g., carbonaceous materials, zeolites, and mesoporous matrices) for immobilizing metal oxide catalysts, owing to its unique combination of abundant availability, cost-effectiveness, layered architecture, and cation exchange capacity [25]. The two-dimensional lamellar structure of Mt, composed of outer tetrahedral silicon layers and an inner octahedral sheet, not only facilitates the efficient flow of aqueous matrices but also enhances the oxidative degradation of pollutants [26]. Crucially, native cations (e.g., K+, Na+, and Ca2+) adsorbed on basal surfaces or within interlayer spaces can be easily exchanged, enabling the strategic incorporation of active Co(Ⅱ) to tailor catalytic properties [27]. Furthermore, the interlayer domain of Mt functions as a nanoconfined "microchemical reactor", significantly improving mass transfer kinetics and prolonging the residence time for contaminant degradation [28]. Additionally, the dynamic expansion and contraction of these nanoconfined spaces provide a high surface area for the tunable dispersion of active species and create abundant adsorption sites for organic molecules, further optimizing catalytic performance [29].
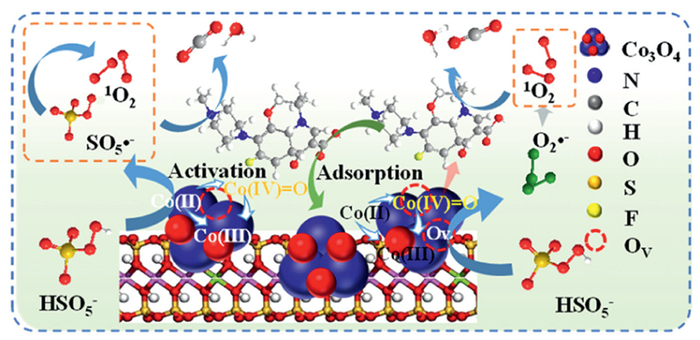
In this study, a series of nano-Co3O4-encapsulated montmorillonite catalysts with varying levels of OV was synthesized for PMS activation. Ofloxacin (OFL), a frequently identified fluoroquinolone antibiotic in surface water and wastewater, was designated as the target pollutant to evaluate their catalytic efficiencies. The relationship between OV levels and OFL degradation was established. Results showed that OV in nano-Co3O4-encapsulated montmorillonite catalysts can selectively induce the activation of PMS to produce 1O2 and Co(Ⅳ)=O. The underlying mechanism was further revealed through comprehensive experiment and theoretical calculation. The findings offer significant insights into the OV-mediated heterogeneous Fenton-like reaction for the selective oxidation of emerging organic contaminants in water and wastewater.
The typical synthesis steps of Co3O4−Mt-OV are shown in Fig. 1a. Typically, Co3O4−Mt was firstly synthesized via a combination of solvent-thermal and high-temperature calcination techniques (Text S1 in Supporting information). After that, Co3O4−Mt-xOV with varied contents of OV was obtained chemical etching reduction and subsequent oxidation (Text S1), where x was the reduction/oxidation cycle times. As shown in Fig. 1b, the X-ray diffraction (XRD) pattern of Na-Mt exhibited characteristic diffraction peaks of montmorillonite (JCPDS No. 29–1498) at 2θ = 7.08°, 19.76°, 29.40°, 36.04°, and 61.82°, corresponding to the (001), (100), and (440) crystal planes, respectively. For pristine Co3O4, the observed peaks at 19.0°, 31.3°, 36.9°, 38.5°, 44.9°, 55.7°, 59.4°, and 65.2° were assigned to the (111), (220), (311), (222), (400), (422), (511), and (440) planes of spinel Co3O4 (JCPDS No. 42–1467) [30]. Notably, the absence of impurity phases confirmed the high crystallinity and phase purity of the synthesized Co3O4. The Co3O4−Mt composite demonstrated a biphasic structure containing both montmorillonite and Co3O4 components, confirming successful immobilization of Co3O4 nanoparticles on the montmorillonite carrier. Whereas, the characteristic diffraction peak intensity of Co3O4 and Mt in Co3O4−MT was reduced in comparation with that of pure Co3O4, which meant that Co3O4 particles were well dispersed and crystallinity was reduced in the presence of montmorillonite carrier. The XRD patterns of Co3O4−Mt-4OV and Co3O4−Mt were basically the same, which indicated that the crystal structure of the catalyst was not affected after chemical etching of OV. However, Nevertheless, the slightly diminished peak intensity observed in Co3O4−Mt-4OV may be attributed to partial lattice distortion induced by increased OV concentration [31].
The isotherm adsorption curves of all samples showed the characteristic of typical type Ⅳ curve (Fig. 1c), implying the mesoporous properties of the catalysts. The pore size distribution further verified the abundance of micropores and mesopores in these catalysts (Fig. S1 in Supporting information). The surface area of Co3O4−Mt-4OV was detected to be 61.646 m2/g, which was about twice that of Co3O4−Mt (33.264 m2/g). Hence, the generation of OV can increase the specific surface area of Co3O4−Mt, favoring the utilization of active sites in the catalytic reactions. The existence and content of OV in different samples were further measured by electron paramagnetic resonance (EPR). From Fig. 1d, the asymmetric EPR signals at g = 2.004 were clearly observed in Co3O4−Mt-4OV catalysts, which was attributed to the unpaired electrons on the capture surface OV [32,33]. The intensity of OV signals significantly enhanced by increasing the number of REDOX cycles in the preparation process. To be specific, the OV content of Co3O4−Mt-6OV reached 3.59 times than that of Co3O4−Mt (Fig. 1e). The full-scan spectrum of X-ray photoelectron spectroscopy (XPS) showed that Co, Al, Si and O were the dominant components in Co3O4−Mt-4OV (Fig. S2 in Supporting information). The C 1s core level peak of 284.8 eV is used as energy reference for charge correction. The high-resolution XPS spectrum were further obtained to determine the valence distributions of Co in the different samples. Two components of Co 2p3/2 and Co 2p1/2 spin–orbit doublets were observed in the high-resolution Co 2p XPS spectra (Fig. 1f). The Co 2p3/2 XPS spectra suggested the presence of Co(Ⅲ), Co(Ⅱ) and satellite species in Co3O4−Mt-4OV with the peaks centered at around 794.8, 781.4 and 788.5 eV, respectively [34]. Based on the area intensity, the Co3O4−Mt-4OV possessed the highest ratios of Co(Ⅱ)/Co(Ⅲ) compared to Co3O4 and Co3O4−Mt, which was hope to significantly facilitate the activation process. Such changing trend was also observed in the previous study, which might be attributed to the stepwise formation of OV [35]. The O 1s XPS spectra of Co3O4 can be deconvolved into three main peaks corresponding to lattice oxygen (OL), oxygen vacancy (OV) and hydroxyl oxygen (OOH) with peak centers of 529.7, 531.1 and 532.3 eV, respectively (Fig. 1g) [36]. The O 1s peaks of Co3O4−MT and Co3O4−MT-4OV were slightly shifted towards higher binding energy, which might be due to the formation of chemical bonds between montmorillonite and Co3O4 nanoparticles [37]. The relative abundance of OV in Co3O4−MT-4OV (41.4%) was higher than that in Co3O4−MT (33.7%), which aligned with the EPR results. As Fig. S2 shows, the positive binding energy shifts of Al 2p and Si 2p spectrum of Co3O4−Mt and Co3O4−MT-4OV were observed, further suggesting that a Co-O-Al(Si) bond might be formed between Co3O4 and montmorillonite [38].
From the scanning electron microscope (SEM) images, a massive scale structure and smooth surface was observed in the montmorillonite (Fig. 2a). The prepared Co3O4 nanoparticles are in a spherical shape of about 20 nm, but a large number of nanoparticles accumulate into large clusters and are easily agglomerated (Fig. 2b). Compared with Na-Mt, the surface of the Co3O4−Mt-4OV sample became very rough and loose (Fig. 2c). In addition, the EDS element mapping showed that Co, Mg, O, Si, and Al elements were evenly dispersed on the surface of Co3O4−Mt-4OV (Fig. 2d). The transmission electron microscope (TEM) images of Co3O4−Mt-4OV further proved that Co3O4 nanoparticles with a diameter of 5–10 nm were uniformly fixed on the interlayer and surface of montmorillonite (Figs. 2e-g), which might be due to the confinement effect preventing the agglomeration of Co3O4 nanoparticles effectively. This is because nano-confinement effect and interaction in confined space can significantly affect the physical and chemical properties of Co3O4, leading to a smaller size and higher dispersion [39]. Characteristic lattice fringes of 0.23, 0.24, 0.28, and 0.46 nm were observed via HRTEM, corresponding to the (222), (311), (220), and (111) crystal planes of Co3O4 nanoparticles (Figs. 2h and i), respectively. These observations agreed with the XRD results. Additionally, evident grain boundaries between Co3O4 nanoparticles were observed, suggesting the presence of abundant defects due to the formation of numerous grain boundaries. This is consistent with the observation of significant lattice distortion in the catalyst, as indicated by the red circle in Fig. 2i, which implied the existence of OV [40].

The adsorption properties of the modified montmorillonite catalysts for OFL were evaluated. Merely 5.1% OFL can be adsorbed by Na-Mt after 60 min (Fig. S3a in Supporting information), since OFL as a macromolecular organic matter is hard to enter the montmorillonite layer. The adsorption of OFL with Co3O4 was 25.1%, while it was markedly increased to 69.1% with Co3O4−MT (Fig. S3a). With the increase of OV content, the adsorption efficiency of Co3O4−Mt-2OV, Co3O4−Mt-4OV and Co3O4−Mt-6OV for OFL gradually increased to 73.0%, 76.0% and 80.3% after 60 min, respectively, indicating that OV might play an important role in the adsorption process. The adsorption of OFL by the catalysts conformed to the quasi-secondary kinetic curve (Fig. S3b in Supporting information), suggesting that the process was primarily governed by chemisorption [41]. Previous studies have demonstrated that the enhancement of the catalyst's adsorption capacity can facilitate the catalytic degradation of pollutants [42]. Given the significant adsorption capacity of these catalysts for pollutants, in this study, the catalysts were pre-mixed with the pollutants for 30 min prior to the catalytic experiments. This pre-treatment ensured that the adsorption capacity of the catalysts was nearly saturated, thereby minimizing the influence of adsorption on the outcomes of the catalytic degradation experiments. PMS alone can only degrade 5.5% of OFL within 60 min, and the removal efficiency slightly increased to 13.1% with the activation of PMS by Na-Mt (Fig. 3a). Under the same conditions, 83.9% of OFL was degraded in the Co3O4/PMS system with the reaction rates (kobs) of 0.041 min-1 (Fig. 3b). The kobs value of OFL degradation significantly enlarged to 0.161 min-1 by the Co3O4−Mt/PMS system, which might be attributed to the optimal dispersion of Co3O4 on montmorillonite surface abundant in hydroxyl groups. Besides, the interlayer structure of montmorillonite offers a localized environment for the accumulation of PMS and pollutants, facilitating the degradation of OFL by the nanoconfinement effect [43]. Notably, after chemical etching of OV, the catalytic performance of Co3O4−Mt-2OV, Co3O4−Mt-4OV and Co3O4−Mt-6OV catalysts was further improved, and kobs of OFL degradation were significantly increased to 0.441, 0.487 and 0.553 min-1, respectively (Fig. 3b), suggesting that the augmentation of OV content could markedly enhance the catalytic reaction. As shown in Fig. 3c, the kobs of OFL degradation was linearly correlated (R2 = 0.98) with the OV content in the Co3O4−Mt-xOV, which evidenced the crucial role of OV for PMS activation. Due to their comparable catalytic performance between Co3O4−Mt-4OV and Co3O4−Mt-6OV, Co3O4−Mt-4OV requiring a simpler preparation process was selected for further study. The catalytic activity of Co3O4−Mt-4OV was compared with the reported relating catalysts of PMS. The modified kobs (m-kobs) was calculated by multiplying the observed rate constant by the pollutant concentration and then dividing by the catalyst dosage. As shown in Table S2 and Fig. S4 (Supporting information), the Co3O4−Mt-4OV is one of the most efficient PMS heterogeneous activators, with an m-kobs of 97.88 × 10–3 min-1, which is 1.1–36 times greater than those of reported Co-based and Ov-mediated heterogeneous catalysts.
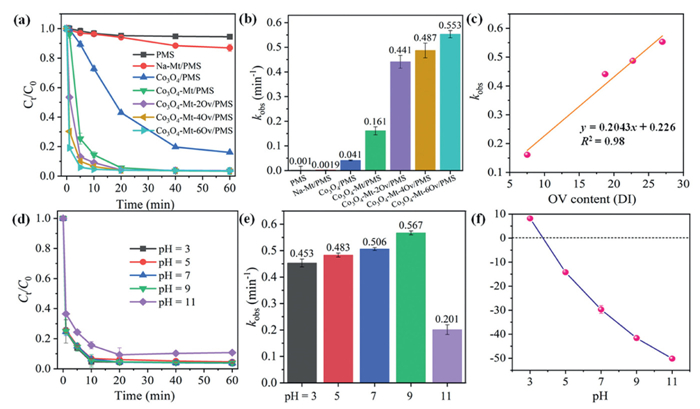
As shown in Fig. 3d, the removal of OFL was not obviously changed within the pH range of 3.0–9.0, maintaining over 95% of OFL degradation. In a strong alkaline environment (pH 11.0), the degradation efficiency of OFL was reduced to 89.2% with the degradation rate decreased to 0.201 min-1 (Fig. 3e). The pKa1 and pKa2 of PMS are 0 and 9.442, respectively [44]. Thus, PMS was dominantly existed in the form of SO52− that was a less oxidized dianion and generated through deprotonation. Besides, the zeta potential test revealed that the surface of Co3O4−Mt-4OV was negatively charged under strongly alkaline solution (Fig. 3f). Thus, the electrostatic repulsion between SO52− and Co3O4−Mt-4OV surface could prevent their catalytic reaction. The negative charge of OFL in alkaline conditions was not conducive to enter the montmorillonite layer. Additionally, PMS became more unstable under alkaline conditions, leading to increased unnecessary consumption (Fig. S5 in Supporting information). The above reasons together resulted to the inhibition of OFL degradation at pH 11.0.
To identify the role of reactive oxygen species (ROS) in the Co3O4−Mt-4OV/PMS system, quenching experiments were performed using different scavengers. Among them, MeOH can effectively quench •OH (k•OH = (1.2–2.8) × 109 L mol-1 s-1) and SO4•− (kSO4•− = (1.6–7.7) × 107 L mol-1 s-1), while tert–butyl alcohol (TBA) can be only used to quench •OH (k•OH = 109 L mol-1 s-1) [45]. As Fig. 4a shows, the degradation of OFL was hardly inhibited with MeOH and TBA, indicating that the contributions of •OH and SO4•− were insignificant. EPR test was performed using DMPO as electron spin trapping agents to verify the generation of •OH and SO4•− in the Co3O4−Mt-4OV/PMS system. From Fig. 4d, the peaks for DMPO-•OH and DMPO-SO4•− adducts were not detected, demonstrating the absence of •OH and SO4•− in the activation of PMS with Co3O4−Mt-4OV. Further, p-BQ and FFA were employed as typical scavengers for O2•− (kO2•− = 9.0 × 108–1.0 × 109 L mol-1 s-1) and 1O2 (k1O2 = 1.2 × 108 L mol-1 s-1), respectively [46]. The degradation of OFL was obviously inhibited by p-BQ (Figs. 4a and b), while PMS consumption was barely affected (Fig. S6 in Supporting information). This suggested that the inhibition of OFL degradation by p-BQ was indeed caused by the scavenging for O2•−, and hence, O2•− might contribute to the removal of OFL. The DMPO-trapped EPR spectra of O2•− directly indicated the formation of O2•− (Fig. 4e). However, unlike SO4•−, •OH, and 1O2, O2•− typically acts as a reductant rather than an oxidant in aqueous systems due to its high solvation energy in water [47]. Previous studies have also demonstrated that O2•− exhibited limited reactivity in direct oxidation of organic contaminants except for the quinones due to its low redox potential (−0.33 V vs. NHE) [48], but can function as a precursor for 1O2 formation [49]. Hence, it is challenging for O2•− to directly contribute to the oxidation of OFL, but might emerge indirectly as an intermediate product of 1O2. Based on the above analysis, the degradation of OFL in the Co3O4−Mt-4OV/PMS system is more likely to occur predominantly via non-radical pathways. As expected, the addition of FFA significantly suppressed OFL degradation (Figs. 4a and b) while exhibiting negligible effects on PMS consumption (Fig. S6), suggesting that 1O2 likely served as the predominant reactive species in this system. Moreover, replacing the solvent from H2O to D2O, which can increase the lifetime of 1O2 by approximately tenfold [50], led to enhanced degradation efficiency and reaction rate constants for OFL (Fig. 4c). This highlighted the essential role of 1O2 in facilitating the degradation of OFL. Notably, a typical 1:1:1 triple-state signal of TEM-1O2 was detected in PMS solution, and the peak intensity significantly increased once Co3O4−Mt-4OV was introduced (Fig. 4f), strongly evidencing the generation of 1O2 during the catalytic reaction.
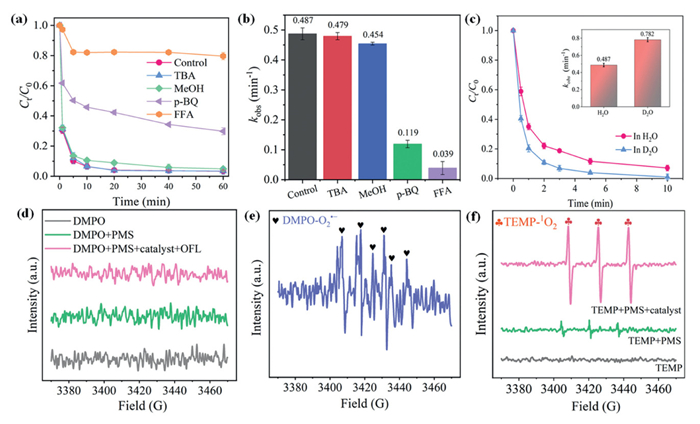
Experimental evidence from N2 purging confirmed that the primary source of 1O2 in the Co3O4−Mt-4OV/PMS system stemmed from catalyst-mediated PMS activation rather than dissolved oxygen, as OFL degradation remained unaffected under anaerobic conditions (Fig. S7 in Supporting information). Subsequent investigation focused on elucidating the formation mechanism and evolutionary pathway of 1O2 within this catalytic system. Although the above results have identified O2•− as potential precursors for 1O2 formation, their direct conversion faces thermodynamic limitations [51]. Meanwhile, the classical Haber-Weiss reaction (O2•− → •OH→ 1O2) can also be ruled out, since •OH was not detected in the system (Fig. 4d). Recent studies have found that some metal active sites serve as critical mediators in the transformation process from O2•− to 1O2. For example, Yi et al. demonstrated the conversion of O2•− to 1O2 in a molybdenum-assisted Fenton system, where Mo6+ is reduced by O2•−, yielding 1O2 and Mo4+ (Mo6+ + O2•− → 1O2 + Mo4+) [51]. Similarly, Liu et al. revealed that high-valent cobalt species (e.g., Co(Ⅳ)/Co(Ⅲ)) can oxidize O2•−, producing 1O2 alongside Co(Ⅲ)/Co(Ⅱ) (≡Co(Ⅳ)/Co(Ⅲ) + O2•− → 1O2 + Co(Ⅲ)/Co(Ⅱ)) [52]. Building on these findings, the formation of high-valent cobalt species and their potential catalytic role in the O2•−/1O2 conversion was further explored.
Notably, the addition of DMSO, a high-valent cobalt-oxo species (Co(Ⅳ)=O) scavenger, inhibited approximately 70% of OFL degradation (Fig. S8 in Supporting information), suggesting that Co(Ⅳ)=O played a significant role. The high conversion of PMSO to PMSO2 (above 87%, Fig. S9 in Supporting information) further evidenced the generation of Co(Ⅳ)=O, since PMSO2 is a known product of oxygen transfer between Co(Ⅳ)=O and PMSO. To further verify the role of Co(Ⅳ)=O, we examined the degradation of a 1O2 probe (1,3-diphenylisobenzofuran, DPBF) with and without the presence of DMSO. Notably, DPBF degradation decreased to 35% with DMSO, compared to 97% in the control (Fig. S10 in Supporting information), confirming that Co(Ⅳ) contributed to 1O2 generation. We subsequently examined the TEMP-1O2 signal in the presence of DMSO. Significantly, introducing DMSO resulted in a significant suppression of the TEMP-1O2 signal (Fig. S11 in Supporting information). Hence, considering that the interlayer confinement effect can drive quantum state transitions through valence electron redistribution, the generated Co(Ⅳ)=O species facilitates O2•− in surpassing the reaction energy barrier and subsequently converting to 1O2 [53,54].
The content of OV before and after catalyst use was determined by EPR method. The peak intensity of OV was observed to decrease following the reaction (Fig. 5a), potentially due to the occupation of OV sites by adsorbed oxygen, leading to the formation of relatively weakly bonded surface-adsorbed oxygen and lattice oxygen [55]. During the reaction process, pollutants or degradation intermediates can also cover the OV on the catalyst surface, indicating that OV was consumed in the activation of PMS. Similar results were obtained by O 1s high-resolution XPS spectra before and after the use of Co3O4−Mt-4OV catalyst (Fig. S12c in Supporting information). This further implied that the OV on the surface of Co3O4−Mt-4OV catalyst played an important role in PMS activation.
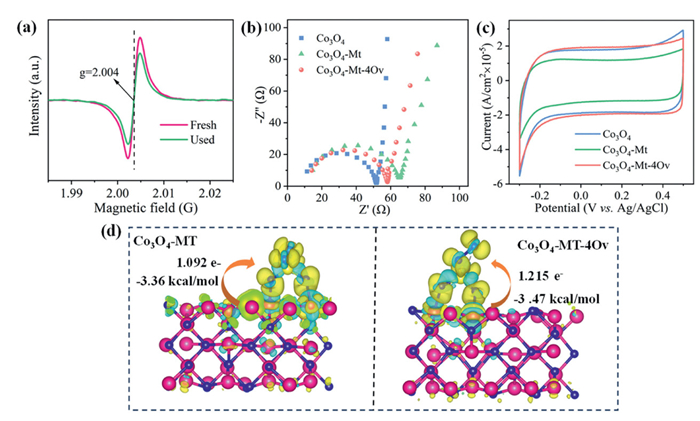
Electrochemical impedance spectroscopy (EIS) analysis was conducted to evaluate the charge transfer capabilities of the catalysts. As shown in Fig. 5b, Co3O4 displayed the smallest semicircle diameter, suggesting the lowest charge transfer resistance among the tested samples. The introduction of montmorillonite (Mt) increased the charge transfer resistance in Co3O4−Mt. However, Co3O4−MT-4OV, with its abundant oxygen vacancies, exhibited a significantly reduced charge transfer resistance, indicating a more efficient electron transfer between the catalyst and PMS. Further supporting this observation, cyclic voltammetry (CV) tests revealed that Co3O4−MT-4OV demonstrated higher current density and superior reduction capability compared to Co3O4−Mt (Fig. 5c), highlighting the beneficial role of OV in enhancing electron transfer performance.
To further unveil the significance of OV in Co3O4−Mt-4OV, density functional theory (DFT) calculation was conducted to compare the adsorption energy and the charge transfer of Co3O4−Mt and Co3O4−Mt-4OV in PMS activation. Results showed that Co3O4−Mt-4OV had a higher adsorption energy (−3.47 kcal/mol) than that of Co3O4−Mt (−3.36 kcal/mol) (Fig. 5d), indicating that the introduce of OV can enhance the adsorption of PMS, favoring the following catalytic reaction. The Bader analysis results indicated that the charge transfer to PMS was 1.092 e- for the Co3O4−Mt configuration and 1.215 e- for the Co3O4−Mt-4OV configuration (Fig. 5d). This increased charge transfer can substantially enhance the cleavage of O—H bonds, thus promoting the formation of SO5•− and O2•− at the Co(Ⅱ) site [57]. Collectively, these findings indicated that the Ov in Co3O4−Mt-4OV played a multifaceted and critical role in activating PMS. OVs acted as electron-rich sites that enhance PMS adsorption via electrostatic interactions or charge transfer, particularly the terminal oxygen (Ot) in PMS. Additionally, OVs synergized with neighboring metal sites (e.g., Co3+/Co2+) to form dual active centers, promoting the electron transfer and stabilizing intermediates.
To characterize the evolution of cobalt species in Co3O4−Mt-4OV during catalytic reactions, XPS analysis was performed to track the chemical state variations before and after the reaction. The high-resolution XPS spectra of Co 2p and O 1s are shown in Fig S12 (Supporting information), with characteristic peaks provided in Table S1 (Supporting information). The XPS analysis of Co 2p regions revealed significant cobalt speciation evolution during the catalytic process. In the Co 2p1/2 spectra, the Co(Ⅲ) component at 798.16 eV exhibited an obvious increase from 32.0% to 38.9% after reaction, while the Co(Ⅱ) content correspondingly decreased from 68.0% to 61.0% (Fig. S12b in Supporting information). Parallel trends were observed in the Co 2p3/2 region, where the Co(Ⅲ)/Co(Ⅱ) ratio displayed analogous progression. These redox dynamics strongly suggested that Co(Ⅱ) served as the catalytic initiator, driving the interconversion cycle among Co(Ⅳ)/Co(Ⅲ)/Co(Ⅱ) species. Notably, the absence of detectable Co(Ⅳ) species is attributed to the metastable nature of excited Co(Ⅳ), which undergoes ultrafast non-radiative decay to the Co(Ⅲ) ground state [56]. Compared with the fresh catalyst, the content of OV in Co3O4−Mt-4OV after use slightly decreased (Fig. S12c in Supporting information). It is noteworthy that the relative intensities of lattice oxygen and adsorbed oxygen increased from 12.5% and 46.1% to 15.6% and 50.1%, respectively. This phenomenon can be attributed to the consumption of oxygen vacancies, which facilitated the formation of relatively weakly bound surface-adsorbed oxygen (e.g., ≡Co(Ⅱ)−OH) and lattice oxygen [58]. Interestingly, previous studies reported that ≡Co(Ⅱ)−OH complexes could induce the formation of ≡Co(Ⅲ)-O-O-SO3⁻ intermediates upon reaction with HSO5⁻ of PMS, which subsequently evolved into Co(Ⅳ)=O species [13,59]. Hence, it can be inferred that ≡Co(Ⅱ) mediated the ultimate formation of Co(Ⅳ)=O through a two-electron transfer pathway. Accompanied by the continuous Co(Ⅱ)/Co(Ⅲ)/Co(Ⅳ) cycling, 1O2 was generated through the co-transformation of Co(Ⅳ)=O and O2•−. Beyond that, ≡Co(Ⅲ)-mediated electron donation to HSO₅⁻ facilitated its deprotonation to generate SO5•− (HSO5− → SO5•− + H+ + e-), which subsequently underwent disproportionation reaction for the generation of 1O2 (SO5•−+ SO5•−→ 2SO4− + 1O2) [60].
Based on the above results, the possible catalytic mechanism was proposed (Fig. 6). Initially, the curled and stacked structure of Co3O4−Mt-4OV undergoes layer expansion in OFL solution, forming semi-confined microcavities that function as nanoreactors. Under dynamic aqueous conditions, both OFL molecules and PMS anions become concentrated within these expanded interlayer spaces through adsorption and confinement effects. Within the constrained environment, surface-bound ≡Co(Ⅱ) species rapidly couple with PMS through coordination, where HSO5− activation via ≡Co(Ⅱ) triggers the generation of SO4•−, alongside oxidized ≡Co(Ⅲ) species. The cleavage of PMS O—O bonds initiates chain reactions that yield O2•− through dual electron transfer pathways. The transient Co(Ⅲ)-OOSO3 intermediate undergoes radical-mediated O—O bond reorganization, evolving into Co(Ⅳ)=O that ultimately cycle back to ≡Co(Ⅵ)/Co(Ⅲ) redox pairs. The generated Co(Ⅳ)=O can contribute to the degradation of OFL through both direct and indirect oxidation process. In indirect pathway, Co(Ⅳ)=O subsequently mediate the conversion of accumulated O2•− into 1O2 through electron transfer processes. As a powerful oxidizing species, Co(Ⅳ)=O can also directly contribute to the oxidation of OFL. Hence, the generated 1O2 and Co(Ⅳ)=O predominantly driven the degradation of OFL.
Various inorganic anions (e.g., Cl−, H2PO4−, HCO3−/CO32− and SO42−) existed in actual wastewater, which might affect the degradation of organic pollutants by AOPs to varying degrees [61,62]. It can be seen from Fig. S13 (Supporting information) and Fig. 7a that the presence of Cl−, H2PO4− and SO42− at low (1 mmol/L) and high concentration (50 mmol/L) showed negligible influence on the system, and the OFL removal efficiency still reached > 96% within 60 min. Upon the addition of 50 mmol/L HCO3− to the system, the degradation of OFL was slightly inhibited, resulting in a reduction of the degradation efficiency to 90.0%. HCO3−/CO32− can bind to the Co(Ⅱ) site on the catalyst surface to inactivate it, thus preventing the adsorption and activation for PMS. Moreover, as a representative organic matter, humic acid (HA) showed little effect on OFL degradation (Fig. S14 in Supporting information). In general, the Co3O4−MT-4OV/PMS system showed a strong anti-interference ability to complex water matrices, which further evidenced the significant contribution of non-radical oxidation for OFL.
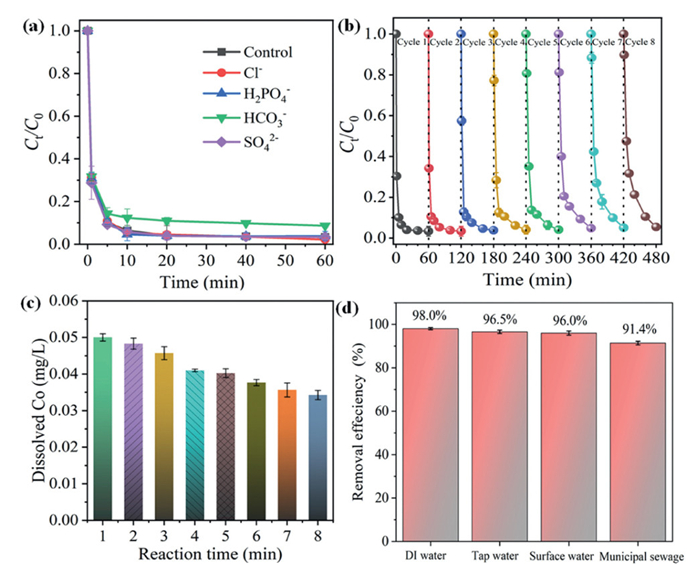
The reusability and stability of catalysts are crucial for the long-term economic viability of wastewater treatment processes. Consequently, to assess both the reusability of Co3O4−MT-4OV composites in degrading OFL within PMS systems and the stability of the catalysts, eight consecutive cycles of stability experiments were conducted under identical operating conditions. As illustrated in Fig. 7b, the Co3O4−MT-4OV catalyst exhibited consistent high catalytic performance over eight cycles, with the removal efficiency of OFL consistently exceeding 94.6%. The degradation rate demonstrated a marginal decrease as the number of cycles increased, which might be caused by the loss of metal active sites. The intermediate products generated during the reaction can induce surface and interlayer passivation of montmorillonite. This passivation would hinder the entry of PMS and OFL into the interlayer of montmorillonite, thereby diminishing the catalytic efficiency. The Co ion leaching after the reaction was further detected. Results showed that the Co ion concentration after 60-min reaction in the first cycle was only 0.05 mg/L, and gradually decreased as the number of cycles increased (Fig. 7c), indicating the high chemical stability of Co3O4−MT-4OV during PMS activation. Such a trace amount of Co2+ in the solution led to a limited degradation of OFL with PMS (Fig. S15 in Supporting information). Hence, the contribution of the leached Co2+ in the Co3O4−MT-4OV/PMS system was insignificant, considering that the percolation is a gradual, cumulative process. The superior performance and stability of the Co3O4−MT-4OV catalyst can be attributed to the nano-confinement effect of montmorillonite, which enhanced the adsorption of PMS and OFL and subsequently improved the utilization of catalytic sites within montmorillonite. Moreover, earlier research has indicated that carbon-based catalysts are typically prone to carbon corrosion issues [63]. In comparison, metal oxides and OV active sites exhibit little sensitivity to such challenges. The application of nano-confinement techniques can improve the environmental resilience of catalysts, leading to enhanced stability and reusability. As a result, the Co3O4−MT-4OV catalyst showcased superior performance in terms of stability and reusability. Moreover, the Co3O4−MT-4OV/PMS system was almost not affected when operated in the actual waters (tap water, surface water, and municipal sewage) (Fig. 7d). The performance of degrading various organic contaminants with the Co3O4−Mt-4OV/PMS process was further evaluated. Results showed that all these six contaminants (including sulfamethoxazole (SMX), sulfadiazine (SDZ), carbamazepine (CBZ), ciprofloxacin (CIP), enrofloxacin (ENF) and tetracycline (TTC)) can be completely degraded within 10 min (Fig. S16 in Supporting information), suggesting that Co3O4−Mt-4OV/PMS process was effective for various organic contaminants degradation. These findings suggested that the Co3O4−MT-4OV/PMS system held significant potential for practical applications.
To better elucidate the degradation process of OFL, the main intermediate products of OFL degradation were detected using HPLC-MS-MS. Nine transformation products (TPs), namely TP-112, TP-150, TP-158, TP-200, TP-205, TP-279, TP-318, TP-338 and TP-378 were identified, and the key information of the intermediates was shown in Fig. S17 and Table S3 (Supporting information). Based on the results, three degradation pathways of OFL were proposed, as shown in Fig. S18 (Supporting information). In Pathway Ⅰ, the piperazine ring of OFL underwent hydroxylation to form TP-378, followed by ROS attacking the quinolone ring to generate TP-338. Further ROS attack led to piperazine ring cleavage, ultimately forming TP-158. In Pathway Ⅱ, the decarboxylation of OFL occurred to produce TP-318, after which ROS attack the quinolone ring, breaking the carbon-carbon double bond and inducing hydrolysis. The piperazine ring decomposed, and the oxazine ring opened to form TP-200, which further underwent defluorination to yield TP-150. In Pathway Ⅲ, the piperazinyl substituent of OFL was first attacked by ROS to form TP-279, followed by oxazine ring opening, addition reactions, and decarboxylation to generate TP-205. Finally, under further ROS reactions, TP-112 was produced.
The Co3O4−Mt-4Ov/PMS process can effectively degrade OFL and ultimately mineralize it into CO2 and H2O. However, some intermediates retain an intact fluoroquinolone core, which may pose ecological hazards. Therefore, toxicity predictions were conducted using the ecological structure activity relationship (ECOSAR) program to analyze the acute and chronic toxicity of the degradation intermediates. As shown in Fig. S19 (Supporting information), the toxicity of three intermediates (TP-378, TP-158, and TP-112) was lower than that of OFL, while the toxicity of other byproducts became higher, indicating that some intermediates carry greater ecological risks. In particular, TP-318, TP-150, and TP-205 exhibited higher toxicity, likely due to decarboxylation at the quinolone substituent position or the presence of highly toxic aniline structures. Although some intermediates are more toxic, the system achieves a high mineralization efficiency of 90.3% (Fig. S20 in Supporting information). Therefore, by optimizing reaction conditions to ensure complete degradation, the potential toxicity risks posed by intermediate byproducts could be largely reduced. Moreover, an analysis of the degradation pathways revealed that all three intermediates in Pathway Ⅱ exhibit high toxicity. This observation suggested that future research should focus on regulating degradation pathways and stages to prevent the formation of toxic products.
In this study, different levels of oxygen vacancy (OV) were successfully constructed on the Co3O4-encapsulated montmorillonite (namely Co3O4−Mt-xOV, x = 2, 4, 6) through a two-step process involving chemical etching reduction and oxidation. Results showed that the performance of activated PMS was markedly improved through the introduction of OV on Co3O4−Mt. The degradation rate of ofloxacin (OFL) in the Co3O4−Mt-xOV/PMS process was positively correlated with the content of OV, suggesting the significant role of OV for PMS activation. Comprehensive experimental and theoretical analysis unveiled that OV enabled the modification of the electronic structure in Co3O4 and enhanced the adsorption capacity for PMS and OFL. Moreover, OV could modulate the catalytic decomposition of PMS, thereby predominantly generating 1O2 and Co(Ⅳ)=O, which behaved as the primary reactive species for OFL oxidation. This non-radical-mediated process enabled the system to selectively remove OFL from the contaminated water without interference from the background matrix of the water body. The Co3O4−Mt-4OV also possessed outstanding chemical stability and reusability, making it a highly viable candidate for practical applications. The knowledge obtained in this study will advance the understanding of vacancy defect engineering in PMS activation, and guide the design of efficient catalysts. This research holds significant importance for the rational design and synthesis of environmentally friendly natural mineral materials rich in OV, as well as for the advancement of highly efficient environmental catalytic materials.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Xiao-qiang Cao: Writing – review & editing, Writing – original draft, Visualization, Supervision, Funding acquisition. Xingyao Liu: Methodology, Investigation, Data curation. Yaqi Wang: Visualization, Methodology, Investigation. Haining Wang: Software, Investigation. Bo Wei: Software, Methodology. Yanan Shang: Validation, Supervision. Yizhen Zhang: Project administration, Data curation. Yujiao Kan: Project administration, Conceptualization. Yang Zhang: Methodology, Investigation. Xing Xu: Supervision, Resources. Longlong Zhang: Writing – review & editing, Writing – original draft, Supervision, Project administration, Methodology, Funding acquisition.
The research work was supported by National Natural Science Foundation of China (Nos. 22476116, 52074176, 52400090), Natural Science Foundation of Shandong Province (Nos. ZR2024ME156, ZR2024QB138), and Qingdao Natural Science Foundation (No. 24-4-4-zrjj-70-jch).
Supplementary material associated with this article can be found, in the online version, at doi:
J. Yang, S. Wang, X. Luo, et al., Chin. Chem. Lett. 36 (2025) 110996. doi: 10.1016/j.cclet.2025.110996
C. Zhang, N. Ding, Y. Pan, et al., Chin. Chem. Lett. 35 (2024) 109579. doi: 10.1016/j.cclet.2024.109579
Y. Lin, Y. Wang, Z. Wang, et al., Nat Commun. 15 (2024) 10032. doi: 10.1038/s41467-024-54225-x
Z. Weng, Y. Lin, S. Guo, et al., Angew. Chem. Int. Ed. 62 (2023) e202310934. doi: 10.1002/anie.202310934
Q. Fang, H. Yang, S. Ye, et al., Water Res. 245 (2023) 120614. doi: 10.1016/j.watres.2023.120614
Y. Wang, Y. Lin, S. He, et al., J. Hazard. Mater. 461 (2024) 132538. doi: 10.1016/j.jhazmat.2023.132538
J. Zhen, J. Sun, X. Xu, et al., Angew. Chem. Int. Ed. 63 (2024) e202402669. doi: 10.1002/anie.202402669
T. Zhang, S. Wu, N. Li, et al., J. Hazard. Mater. 449 (2023) 130971. doi: 10.1016/j.jhazmat.2023.130971
Z. Fang, J. Qi, W. Chen, et al., Appl. Catal. B 338 (2023) 123084. doi: 10.1016/j.apcatb.2023.123084
L. Kong, G. Fang, Z. Fang, et al., Chem. Eng. J. 416 (2021) 128996. doi: 10.1016/j.cej.2021.128996
F. Wang, Y. Xiang, Y. Zhang, X, et al., Chin. Chem. Lett. 37 (2026) 111315. doi: 10.1016/j.cclet.2025.111315
S. Chen, J. Li, W. Zhou, et al., Coord. Chem. Rev. 507 (2024) 215749. doi: 10.1016/j.ccr.2024.215749
J. Jiang, Z. Zhao, J. Gao, et al., Environ. Sci. Technol. 56 (2022) 5611–5619. doi: 10.1021/acs.est.2c01913
Q. Wang, J. Lu, M. Yu, et al., Environ. Pollut. 333 (2023) 121990. doi: 10.1016/j.envpol.2023.121990
S. Xue, C. Cheng, J. Kang, et al., Chin. Chem. Lett. 36 (2025) 110776. doi: 10.1016/j.cclet.2024.110776
P. Li, Y. Lin, S. Zhao, et al., Appl. Catal. B 298 (2021) 120596. doi: 10.1016/j.apcatb.2021.120596
J. Lim, Y. Yang, M.R. Hoffmann, Environ. Sci. Technol. 53 (2019) 6972–6980. doi: 10.1021/acs.est.9b01449
J. Wang, X. Duan, J. Gao, et al., Water Res. 185 (2020) 116244. doi: 10.1016/j.watres.2020.116244
L. Yang, Y. Jiao, X. Xu, et al., ACS Sustainable Chem. Eng. 10 (2022) 1899–1909. doi: 10.1021/acssuschemeng.1c07605
J. Hu, X. Zeng, G. Wang, et al., Chem. Eng. J. 400 (2020) 125869. doi: 10.1016/j.cej.2020.125869
X. Dong, X. Duan, Z. Sun, et al., Appl. Catal. B 261 (2020) 118214. doi: 10.1016/j.apcatb.2019.118214
Q. Liu, H. Qie, Z. Sun, et al., Chin. Chem. Lett. 34 (2023) 108397. doi: 10.1016/j.cclet.2023.108397
R. Yao, J. Pinals, R. Dorakhan, et al., ACS Catal. 12 (2022) 12227–12245. doi: 10.1021/acscatal.2c02525
J. Liang, L. Fu, K. Gao, X. Duan, Appl. Catal. B 315 (2022) 121542. doi: 10.1016/j.apcatb.2022.121542
P. Xu, H. Qin, Q. Tian, et al., Chem. Eng. J. 504 (2025) 158836. doi: 10.1016/j.cej.2024.158836
W. Gao, X. Cao, L. Hou, et al., Chin. Chem. Lett. 37 (2026) 111095. doi: 10.1016/j.cclet.2025.111095
N. Chen, M. Huang, C. Liu, et al., Water Res. 165 (2019) 114997. doi: 10.1016/j.watres.2019.114997
X. Li, I.C. Bourg, Environ. Sci. Technol. 58 (2024) 1109–1118. doi: 10.1021/acs.est.3c08253
X. Liu, C. Tournassat, S. Grangeon, et al., Nat. Rev. Earth Environ. 3 (2022) 461–476. doi: 10.1038/s43017-022-00301-z
X. Liu, W. Wang, C. Du, Y. Su, Appl. Clay Sci. 228 (2022) 106625. doi: 10.1016/j.clay.2022.106625
X. Zhang, X. Yang, S. Chen, et al., Chem. Eng. J. 452 (2023) 139192. doi: 10.1016/j.cej.2022.139192
X. Chen, X. Peng, L. Jiang, et al., Chem. Eng. J. 427 (2022) 130945.
B. Lei, W. Cui, P. Chen, et al., ACS Catal. 12 (2022) 9670–9678. doi: 10.1021/acscatal.2c02390
Y. Li, M. Wang, J. Tian, et al., Appl. Surf. Sci. 669 (2024) 160588. doi: 10.1016/j.apsusc.2024.160588
S. Yuan, X. Liu, W. Liao, et al., Geochim. Cosmochim. Acta 223 (2018) 422–436. doi: 10.3390/icem18-05277
Z. Wang, R. Lin, Y. Huo, et al., Adv. Funct. Mater. 32 (2022) 2109503. doi: 10.1002/adfm.202109503
X. Xue, R. Chen, H. Chen, et al., Nano Lett. 18 (2018) 7372–7377. doi: 10.1021/acs.nanolett.8b03655
J.H. Jhang, J.A. Boscoboinik, E.I. Altman, J. Chem. Phys. 152 (2020) 084705. doi: 10.1063/1.5142621
Z. Zhou, F. Yu, J. Ma, Environ. Chem. Lett. 20 (2022) 563–595. doi: 10.1007/s10311-021-01355-z
Y. Wang, M. Liu, Y. Zang, et al., J. Environ. Manage. 321 (2022) 115971. doi: 10.1016/j.jenvman.2022.115971
L. Cao, Y. Chai, Y. Zhang, et al., J. Environ. Manage. 375 (2025) 124217. doi: 10.1016/j.jenvman.2025.124217
T. Jiang, B. Wang, B. Gao, et al., J. Hazard. Mater. 442 (2023) 130075.
W. Li, Y. Nan, Z. Zhang, Q. You, Z. Jin, Chem. Eng. J. 398 (2020) 125449.
Y. Li, X. Zhao, Y. Yan, et al., Chem. Eng. J. 376 (2019) 121302.
F. Ghanbari, M. Moradi, Chem. Eng. J. 310 (2017) 41–62.
Z. Hao, W. Hou, C. Fang, et al., J. Hazard. Mater. 439 (2022) 129618.
Y. Guo, J. Long, J. Huang, et al., Water Res. 215 (2022) 118275.
Z. Luo, Y. Yan, R. Spinney, et al., Water Res. 261 (2024) 122023.
L. Zhang, X. Zhou, L. Yang, et al., Appl. Catal. B 349 (2024) 123897.
W. Chen, Y. Jiang, M. Zhao, et al., Anal. Chem. 95 (2023) 5340–5345. doi: 10.1021/acs.analchem.2c05459
Q. Yi, J. Ji, B. Shen, et al., Environ. Sci. Technol. 53 (2019) 9725–9733. doi: 10.1021/acs.est.9b01676
T. Liu, S. Xiao, N. Li, et al., Nat. Commun. 14 (2023) 2881.
Y. Zou, J. Li, J. Tan, et al., Chem. Eng. J. 471 (2023) 144531.
P. Xu, H. Qin, Q. Tian, et al., Chem. Eng. J. 504 (2025) 158836.
X. Hu, J. Wang, J. Wang, et al., Appl. Catal. B 318 (2022) 121879.
Y. Zong, H. Zhang, X. Zhang, et al., Appl. Catal. B 300 (2022) 120722.
M. Ta, T. Zhang, T. Wang, et al., J. Mater. Chem. A 12 (2024) 17529–17543. doi: 10.1039/d4ta02383h
Z. Su, W. Yang, C. Wang, et al., Environ. Sci. Technol. 54 (2020) 12684–12692. doi: 10.1021/acs.est.0c03981
Y. Zeng, F. Wang, D. He, et al., Chem. Eng. J. 459 (2023) 141642.
C. Ruan, L. Zhang, Z. Song, et al., ChemistrySelect 10 (2025) e202405300.
J. Wang, S. Wang, Chem. Eng. J. 411 (2021) 128392.
M. Zhao, Y. Liu, M. Feng, et al., Chem. Eng. J. 489 (2024) 151371.
H. Luo, H. Fu, H. Yin, Q. Lin, J. Hazard. Mater. 426 (2022) 128044.

Figure 1 (a) Schematic illustration of the synthesis of the Co3O4−MT-OV. (b) XRD, (c) BET, (d) EPR, (e) OV contents, (f) Co 2p HR-XPS and (g) O 1s HR-XPS of the catalysts.

Figure 2 SEM images of (a) Na-Mt, (b) Co3O4, (c) Co3O4−Mt-4OV, and (d) element mapping of Co3O4−Mt-4OV. TEM images of (e) Na-Mt, (f) Co3O4, (g) Co3O4−Mt-4OV, and HRTEM images of (h) Co3O4, (i) Co3O4−Mt-4OV.

Figure 3 (a) Kinetics of OFL degradation in different systems, and (b) the corresponding degradation rate (kobs). (c) The linear relationship between OV content of Co3O4−Mt-xOV and kobs of OFL. (d) Effects of solution pH on the degradation of OFL in the Co3O4−Mt-4OV/PMS system, and (e) the corresponding kobs values. (f) Zeta potential of Co3O4−Mt-4OV changed with varying solution pH. Experiment conditions: [OFL]0 = 20 mg/L and T = 20 ℃, if not specified catalyst dosage = 0.2 g/L, [PMS]0 = 1.0 mmol/L and pH0 = 7.0.

Figure 4 (a) OFL degradation in the Co3O4−Mt-4OV/PMS process with and without the existence of different quenchers, and (b) the corresponding kobs values. (c) Degradation of OFL in the D2O solution (insert: the corresponding kobs values). The EPR spectra of (d) DMPO-SO4•− and DMPO-•OH, (e) DMPO—O2•− and (f) TEMP-1O2. Experiment conditions: catalyst dosage = 0.2 g/L, [PMS]0 = 1.0 mmol/L, [OFL]0 = 20 mg/L, pH0 = 7.0 and T = 20 ℃, if applied [MeOH]0 = [TBA]0 = 100 mmol/L, [FFA]0 = [p-BQ]0 = 10 mmol/L.

Figure 5 (a) EPR spectra of Co3O4−Mt-4OV catalyst before and after use. (b) Electrochemical impedance spectroscopy and (c) cyclic voltammetry spectra of Co3O4, Co3O4−Mt and Co3O4−Mt-4OV. (d) The calculated charge density difference of PMS adsorbed on (a) Co3O4−Mt and (b) Co3O4−Mt-4OV.

Figure 7 (a) Effects of common anions in water on OFL degradation in the Co3O4−Mt-4OV/PMS process. (b) Performance of OFL degradation during the cycling tests of Co3O4−Mt-4OV, and (c) the leaching of cobalt ions during the cycling tests. (d) Degradation of OFL in the Co3O4−Mt-4Ov/PMS process in real waters. Experiment conditions: Catalyst dosage = 0.2 g/L, [PMS]0 = 1.0 mmol/L, [OFL]0 = 20 mg/L, pH0 = 7.0 and T = 20 ℃, if applied [Cl−]0 = [H2PO4−]0 = [HCO3−]0 = [SO42−]0 = 50 mmol/L.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: