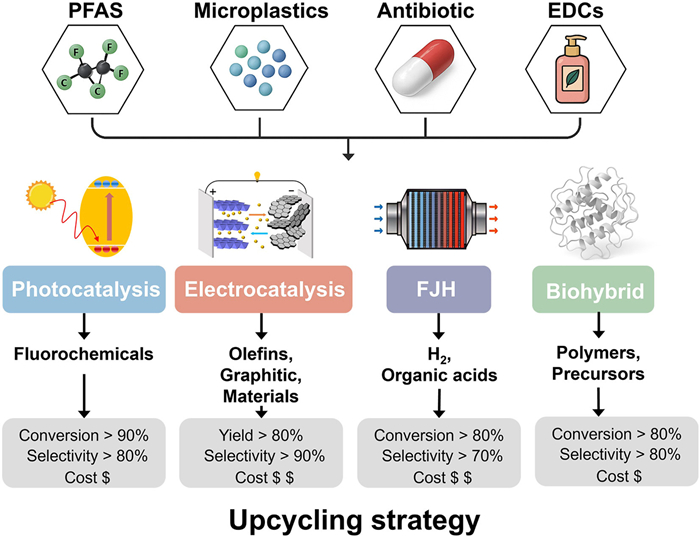
Figure 1.
Emerging pollutant-specific upcycling strategies and performance indicators across catalytic platforms including photocatalysis, electrocatalysis, flash Joule heating (FJH), and biohybrid systems.
Reuse emerging pollutants towards the circular economy
Shuai Yue , Zhiyong Zhao , Mengxue Yang , Yixiao Liu , Pengfei Wang , Chenxin Xie , Sihui Zhan

The accelerating global consumption of synthetic chemicals and materials has triggered a corresponding surge in the release of emerging pollutants—substances that are not yet comprehensively regulated but increasingly recognized for their environmental persistence, bioaccumulation, and adverse effects on human and ecological health [1–3]. These pollutants encompass a broad and expanding category, including per- and polyfluoroalkyl substances (PFAS), microplastics, antibiotics, and endocrine-disrupting chemicals (EDCs), which are now ubiquitously detected across water, soil, air, biota, and even human tissues [4,5].
PFAS, often dubbed “forever chemicals” for their exceptional stability, have been detected in over 98% of tested human blood samples globally and are widespread in drinking water, wildlife, and food systems [6,7]. Microplastics, plastic fragments smaller than 5 mm, have permeated the deepest ocean sediments, Arctic snow, and airborne particulate matter, raising concerns about their role as vectors for toxic chemicals and pathogens [8,9]. Antibiotics, frequently discharged via municipal and agricultural pathways, not only persist in aquatic environments but also contribute to the escalating crisis of antimicrobial resistance (AMR) [10,11]. EDCs, such as bisphenol A (BPA), phthalates, and nonylphenols, can disrupt hormonal pathways even at trace levels and have been linked to reproductive, developmental, and immunological disorders [12,13].
Despite advances in wastewater treatment technologies, most existing methods, including adsorption, incineration, and advanced oxidation processes (AOPs), are designed for containment or destruction [14–17]. These approaches are often energy-intensive, produce secondary pollution, and fail to recover the elemental and molecular value embedded in pollutants [18–20]. Moreover, the chemical diversity and structural complexity of emerging pollutants complicate the development of one-size-fits-all solutions, highlighting the urgent need for transformative remediation paradigms.
In this context, pollutant upcycling, a process that selectively converts pollutants into value-added products, has emerged as a promising alternative aligned with circular economy and net-zero carbon goals [21]. Rather than pursuing complete mineralization to CO2 and H2O, upcycling strategies leverage catalytic transformations to reclaim valuable elements such as carbon, nitrogen, fluorine, and hydrogen, enabling the production of fuels, platform chemicals, and functional materials from waste (Fig. 1) [22,23].
Recent breakthroughs across pollutant categories exemplify the potential of this approach. For PFAS, Miyake’s group demonstrated visible-light-driven organic photocatalysts capable of defluorinating perfluorooctanoic acid (PFOA) and fluorinated polymers under mild conditions [24], while Kang’s twisted carbazole photocatalysts enabled the room-temperature breakdown of polytetrafluoroethylene (PTFE) into carbonaceous and fluorine-rich products [25]. Gao’s team further advanced this field using ultrasound-driven contact-electrocatalysis to activate PTFE and enhance PFAS defluorination via intrinsic interfacial electric fields [26].
In the domain of microplastics, Ma et al. developed an orthogonal transformation framework guided by solid-state NMR fingerprinting, enabling selective depolymerization of mixed plastic waste into high-value chemicals through tandem catalytic steps [27]. Tang’s group engineered Zn/b-ZnO catalysts capable of converting polyolefins into lubricant-range oils with high turnover frequencies and energy efficiency [28], while Guo’s PdMn single-atom nanofibers achieved industrial-level current densities for electrocatalytic PET reforming into glycolic acid and hydrogen [29].
Efforts in antibiotic remediation have also progressed from mere degradation to integrated valorization. Hu’s Fe-N,O-doped graphene catalyst harnesses intrinsic surface electric fields to degrade fluoroquinolones without external energy input [30], whereas Xu’s SA-TCPP homojunction system enhances H2O2 generation for simultaneous antibiotic and resistance gene destruction via self-Fenton photocatalysis [31]. Complementarily, Lai’s Fe-BNQDs@g-C3N4 catalyst offers a PAA-activated AOP platform with superior selectivity and efficiency for sulfonamide degradation [32].
For EDCs, Zhao’s TiO2 photoanodes enable selective recognition and breakdown of phthalates in complex effluents by targeting functional motifs [33]. Wang’s Fc-modified Bi-MOFs unlock differential oxidant activation for bisphenol degradation [34], while Li’s WS2-enhanced PMS system facilitates high-efficiency BPA removal through radical and non-radical pathways, supported by DFT-guided mechanistic elucidation [35].
Crucially, these advances are not confined to lab-scale demonstrations. Emerging techno-economic assessments and life cycle analyses suggest that pollutant upcycling can reduce carbon footprints by up to 80% compared to conventional treatments, while offering significant economic returns by converting a fraction of pollutant mass into marketable products [36]. Such strategies directly support the United Nations Sustainable Development Goals (SDGs), particularly SDG 6 (clean water), SDG 12 (responsible consumption and production), and SDG 13 (climate action) [37].
Yet, critical gaps remain. Many existing reviews narrowly focus on individual pollutant classes or degradation pathways without integrating insights across materials chemistry, catalysis, process engineering, and environmental policy. Furthermore, there is a paucity of analyses that address geographic disparities in research efforts or synthesize progress toward circular economy frameworks.
In this review, we present a comprehensive analysis of the state-of-the-art in pollutant upcycling across four major contaminant classes—PFAS, microplastics, antibiotics, and EDCs. We highlight catalytic mechanisms, global research contributions, material innovations, and valorization pathways. Through critical evaluation of recent breakthroughs and future directions, we aim to establish a multidisciplinary roadmap for converting pollutant liabilities into chemical assets, thus redefining the future of environmental remediation.
Perfluoroalkyl and polyfluoroalkyl substances (PFAS) are a class of synthetic organofluorine compounds in which hydrogen atoms along the carbon backbone are partially or fully substituted by fluorine [38,39]. Owing to the exceptional strength of the carbon–fluorine (C–F) bond, PFAS exhibit extraordinary chemical and thermal stability [40]. The presence of polar functional groups further enhances their surface activity and aqueous solubility, contributing to their widespread use across industrial and commercial sectors over the past eight decades [41]. Common subclasses include perfluoroalkyl sulfonic acids (PFSAs), perfluoroalkyl carboxylic acids (PFCAs), and various precursors such as fluorotelomer alcohols (FTOHs), perfluoroalkyl sulfonamides (FASAs), and sulfonamide derivatives, which can be transformed into PFSAs and PFCAs through environmental processes [42]. PFSAs and PFCAs are typically ionic and highly soluble, enabling long-range waterborne transport, whereas their neutral precursors tend to volatilize and undergo atmospheric oxidation, ultimately yielding more persistent ionic PFAS [43].
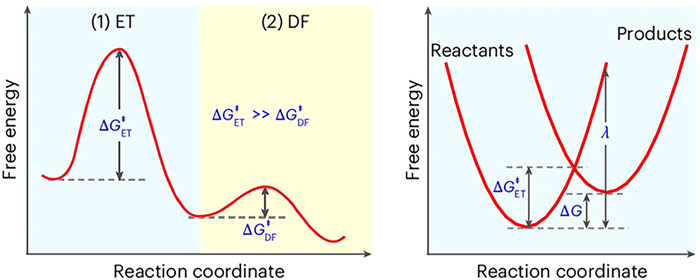
Traditional PFAS treatment technologies have prioritized complete mineralization to non-toxic end products. However, in light of intensifying global carbon constraints and the urgent need for sustainable materials management, growing interest is being directed toward valorization strategies that convert PFAS into chemically tractable or even valuable intermediates. A mechanistic understanding of PFAS degradation is crucial to advancing these pathways. Zhang et al. [44] recently proposed a two-step degradation mechanism dominated by an electron transfer (ET)-limited defluorination (DF) process (Fig. 2). In this framework, the free energy barrier associated with ET (ΔGET‡) determines the reaction rate, whereas the subsequent C–F bond cleavage proceeds with markedly lower activation energy. By selectively modulating ΔGET‡, targeted defluorination at the α- or ε-positions of CF2 units becomes feasible under mild conditions, allowing the fluorinated backbone to be retained for downstream derivatization. This offers the potential to generate functionalized intermediates for use in surfactants, specialty polymers, or electronic materials, thereby supporting a shift from PFAS destruction to chemical valorization and fluorine circularity.
The environmental persistence of PFAS is most evident in wastewater streams, where they pose significant ecological and human health risks. Efficient removal not only mitigates contamination but also opens avenues for fluorine recovery. Kang et al. [25] demonstrated photocatalytic defluorination of a variety of PFAS, including PTFE, PFOS, and PFOA, using a highly twisted carbazole-core superphotoreductant (Fig. S1 in Supporting information). Operating at moderate temperatures (40–60 ℃), the process enables defluorination with concomitant formation of valuable byproducts such as carbonates, formates, oxalates, and trifluoroacetates, and recovers fluorine as inorganic salts. This method provides a promising alternative to conventional treatment routes, offering dual benefits of pollutant removal and resource recovery, and advancing the concept of sustainable wastewater management.
Despite these advances, scalable, energy-efficient, and high-throughput strategies for PFAS conversion remain elusive—particularly for legacy compounds such as PFOA and PFOS. Many existing technologies are hampered by high energy requirements and the risk of generating toxic fluorinated byproducts. Addressing these limitations, James et al. [45] developed a rapid and scalable electrothermal mineralization approach based on flash Joule heating (FJH), which enables efficient mineralization of PFAS-laden granular activated carbon (PFAS-GAC) (Fig. 3a). In this method, PFAS-GAC mixed with sodium or calcium salts is subjected to sub-second electrical pulses, resulting in instantaneous heating (>3000 ℃) and conversion of >90% of organofluorine and >99% of PFOS into benign fluoride salts (Fig. 3c). For example, at 130 V and 1.00 s residence time, 96% of the fluorine in PFOA was mineralized to inorganic fluoride in the presence of NaOH (Fig. 3d). These findings position FJH as a tunable, fast, and scalable platform for PFAS mineralization.
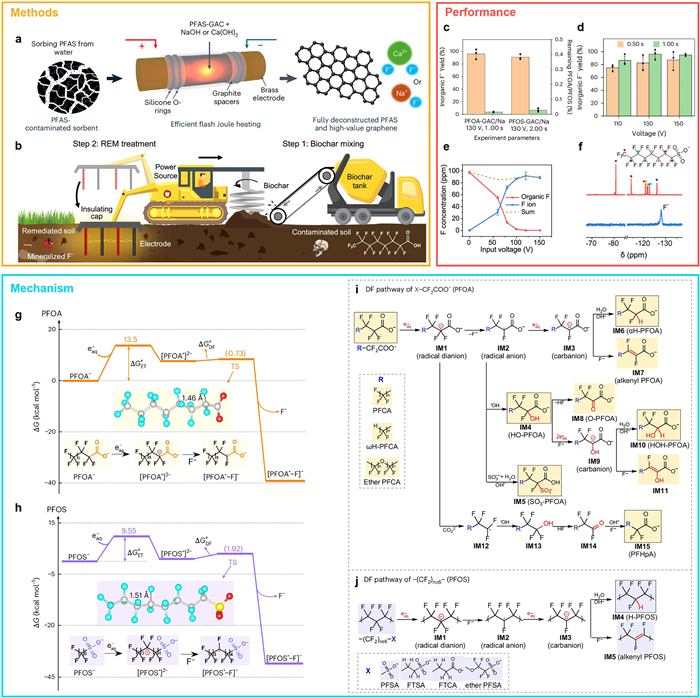
This electrothermal strategy was further extended to PFAS-contaminated soils through rapid electrothermal mineralization (REM) (Fig. 3b) [45]. By incorporating biochar as a conductive additive, the system raised soil temperatures above 1000 ℃ within seconds via current pulsing. The inherent calcium content in soil reacted with released fluoride to form CaF2, yielding a stable and environmentally benign residue (Fig. 3e). 19F NMR spectra confirmed complete PFAS removal, with only hydrated fluoride peaks (128 ppm) detectable after treatment. The extent of C–F bond cleavage scaled positively with applied voltage, underscoring the method's tunability and economic promise for field applications (Fig. 3f) [46].
Mechanistic elucidation of the degradation pathway further reinforces the ET-limited nature of PFAS breakdown [44]. For both PFOA and PFOS, ΔGET‡ values (13.5 and 9.55 kcal/mol, respectively) significantly exceed those of subsequent defluorination steps (ΔGDF‡ < 2 kcal/mol at preferred α- and ε-CF2 positions; Figs. 3g and h), identifying ET as the rate-determining step. These computational insights are supported by experimental Eyring analyses. High-resolution mass spectrometry revealed a series of defluorinated intermediates, such as αH-PFOA, OH-PFOA, and SO3-PFOA, arising from stepwise electron transfer and radical transformations (Figs. 3i and j). The accumulation or disappearance of these species correlates with their respective ΔG‡ values, with high-barrier intermediates like αH-PFOA (13.2 kcal/mol) persisting, while low-barrier intermediates (<3 kcal/mol) rapidly transform. These detailed energy profiles offer a comprehensive mechanistic map for guiding selective PFAS degradation and resource recovery efforts.
Beyond electrothermal strategies, mechanochemical defluorination has emerged as a promising low-energy pathway for PFAS mineralization and fluorine recovery. Gouverneur and colleagues recently developed an innovative phosphate-mediated mechanochemical approach capable of efficiently degrading perfluorinated polymers such as polytetrafluoroethylene (PTFE) under ambient conditions (Figs. 4a and b) [47]. In this method, PTFE is combined with tripotassium phosphate (K3PO4, at 1.25 equiv. per fluorine atom) and subjected to ball milling at 35 Hz for 3 h. The mechanical force drives the defluorination reaction, converting fluorine atoms into water-soluble potassium fluoride (KF, yield up to 84%) and potassium fluorophosphate (K2PO3F, yield ~15%) (Fig. 4c). Notably, the carbon skeleton of PTFE is not fully destroyed but undergoes transformation into graphitized carbon, suggesting both substantial structural reorganization and potential for material reuse.
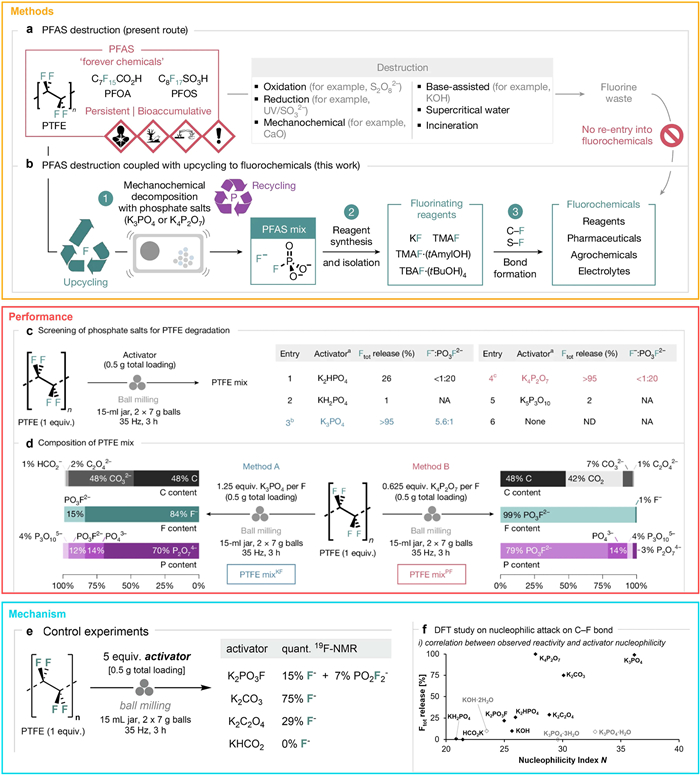
To confirm the completeness of the reaction and the purity of the products, the authors employed solid-state nuclear magnetic resonance (NMR) and X-ray diffraction (XRD) analyses. Solid-state NMR provided insights into the local chemical environments of fluorine and phosphorus, while XRD revealed the crystalline phases of the inorganic products. The combined data confirmed the complete degradation of PTFE, with no residual polymer detectable post-reaction. Moreover, phosphate recovery was high, with up to 96% of the reagent reclaimed for reuse—highlighting the circular potential of this process and its compatibility with sustainable fluorine chemistry (Fig. 4d).
To further elucidate the reactivity trends among phosphate species, density functional theory (DFT) calculations were performed. The results revealed a strong correlation between phosphate nucleophilicity and PFAS degradation efficiency (Figs. 4e and f). Among the tested candidates, K3PO4 exhibited the lowest SN2 reaction barrier (ΔG‡ = 31.8 kcal/mol), consistent with its superior defluorination performance. In comparison, K4P2O7 showed a slightly higher barrier (ΔG‡ = 33.2 kcal/mol), despite producing high-purity K2PO3F. These findings not only provide mechanistic insights into phosphate-driven PFAS degradation but also offer a theoretical framework for the rational selection of phosphate reagents in future mechanochemical defluorination strategies.
Microplastics are plastic particles, fragments, or fibers smaller than 5 mm in size. They were first defined by Thompson et al. [48] in 2004. When the particle size is below 1 μm, they are referred to as nanoplastics. Since Carpenter et al. [49] first detected microplastics in the Sargasso Sea in 1972, their presence has been widely identified across ecosystems, including the atmosphere, soil, water, urban sewage systems, and even in food, drinking water, and biological samples, including those from humans [50]. The potential risks of microplastics to the environment and human health have drawn increasing concern. Microplastics in the environment can be categorized into primary and secondary sources. Primary microplastics are industrially manufactured particles or fibers, such as plastic microbeads in personal care products and microfibers from textiles. These are directly released into the environment through product use and fabric washing [51]. Secondary microplastics, on the other hand, form when larger plastic waste in the environment breaks down due to physical, chemical, and biological processes [52]. These are the major contributors to microplastic pollution. Larger plastics undergo weathering and degradation, eventually fragmenting into micro- and nano-sized particles. Microplastics come in various shapes and types. Based on shape, they are classified as fibers, fragments, particles, and foams. Chemically, they include materials like polyethylene (PE), polyvinyl chloride (PVC), polypropylene (PP), polystyrene (PS), and polyamide (PA) [53]. These microplastics can originate from both petrochemical-based and bio-based sources. They are also divided into biodegradable and non-biodegradable types. Other important microplastic sources include tire wear particles and silicone rubber particles.
Given the large carbon emissions and microplastic pollution resulting from poor waste management, there is increasing interest in developing strategies to convert carbon-rich waste into valuable chemicals. Catalytic technologies that convert waste plastics into high-value chemicals not only help reduce CO2 emissions but also save crude oil and generate economic benefits [54,36,55]. Over the years, three main strategies for managing plastic waste have emerged: (1) Traditional landfill and incineration; (2) harmless degradation of plastics into biodegradable organic molecules; and (3) chemical recycling through catalytic processes to enable circular reuse. Landfilling and incineration are environmentally harmful and fail to offer economic benefits. Although the degradation of waste plastics into green molecules and CO2 is a potential solution, it faces limitations in large-scale application. In contrast, chemical recycling, through catalytic technologies, can address plastic waste while reducing greenhouse gas emissions [56]. However, challenges such as high energy consumption and inefficient product distribution remain, hindering widespread adoption. Therefore, there is a growing emphasis on innovation in catalytic technologies, particularly through photocatalysis and electrocatalysis [57–59]. These methods, combined with tailored catalysts, can offer low energy input, enhanced product distribution, and contribute to a zero-plastic-waste environment and carbon neutrality (Fig. 5).
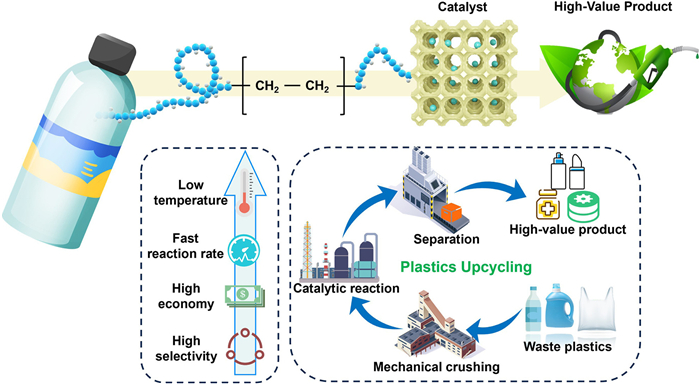
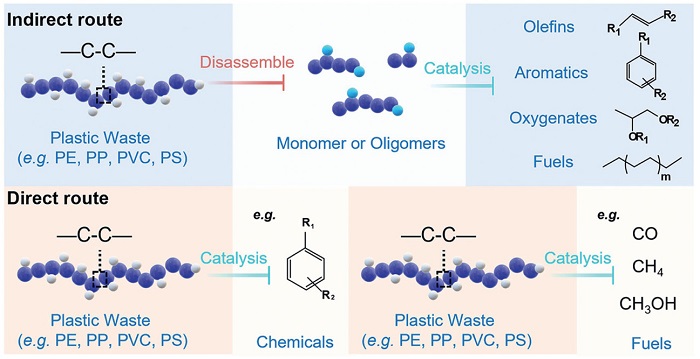
Catalytic plastic valorization strategies can be divided into two primary pathways: indirect and direct upcycling (Fig. 6). Indirect approaches involve initial chemical disassembly, such as hydrolysis, alcoholysis, or ammonolysis, of polymers like PE, PP, PVC, and PS into monomers or oligomers, followed by catalytic conversion into target products such as olefins, aromatics, oxygenates, or fuels [60]. This pathway enables precise control of intermediate structure and functionality, facilitating selective downstream transformation. In contrast, direct upcycling bypasses intermediate formation, utilizing catalytic processes such as hydrogenolysis, oxidative cleavage, and aromatization to convert polymer backbones directly into small-molecule products [61]. This approach often relies on in situ activation of C–C or C–H bonds via reactive oxygen species (ROS) or radical pathways, enabling simplified processing and enhanced energy efficiency. Together, these two strategies provide a comprehensive framework for transforming diverse plastic wastes into economically and environmentally valuable chemicals.
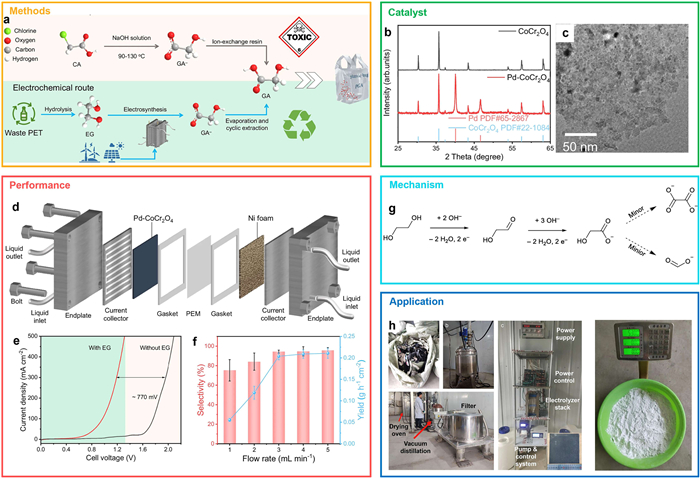
One promising example of indirect upcycling involves the electrochemical recycling of polyethylene terephthalate (PET) into biodegradable polyglycolic acid (PGA) [62]. This process integrates alkaline hydrolysis of PET into ethylene glycol (EG), electrooxidation of EG to glycolic acid (GA), followed by acidification, purification, and polymerization to yield PGA (Fig. 7a). Using renewable electricity and avoiding toxic reagents, this method offers both environmental and economic advantages. In a recent study, Chen et al. engineered a Pd–CoCr2O4 catalyst optimized for local pH control and hydroxyl radical coverage on the electrode surface (Figs. 7b and c). In semi-industrial continuous-flow electrolysis (324 cm2 × 5 cells), the system achieved a GA production rate of 0.32 kg/h with 93.0% selectivity at a current density of 280 mA/cm2, and a final PGA yield of 87% from 20 kg of waste PET (Figs. 7d-f). In situ infrared spectroscopy revealed that EG molecules were first adsorbed on the catalyst surface, then deprotonated and oxidized to aldehyde intermediates, which subsequently formed glycolate. Further oxidation produced minor amounts of formic acid, oxalic acid, and citric acid (Fig. 7g). Notably, the process enabled >99% recovery of both NaOH and EG, along with 91% recovery of GA (purity >99%), achieving near-zero emissions and low-cost operation (Fig. 7h).
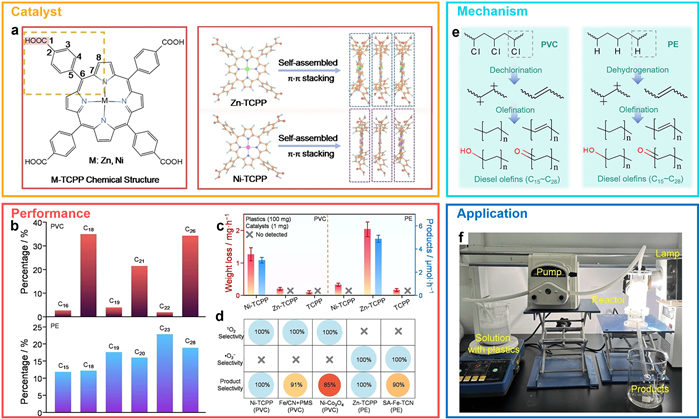
As a demonstration of direct upcycling, our group developed a general photocatalytic strategy to selectively convert polyolefin wastes into diesel-range olefins (C15-C28) by controlling ROS species [63]. Tetrakis(4-carboxyphenyl)porphyrin (TCPP) catalysts incorporating different central metal atoms were used to modulate reactivity (Fig. 8a). For PVC, Ni–TCPP exhibited the highest depolymerization rate (1.28 mg/h) and diesel olefin yield (3.06 μmol/h), while Zn–TCPP was more effective for PE, achieving a depolymerization rate of 2.05 mg/h and a diesel olefin yield of 4.91 μmol/h (Figs. 8b-d). In contrast, metal-free TCPP showed limited activity. ROS-selective systems confirmed that singlet oxygen (1O2) favored PVC activation via electrophilic attack on C–Cl bonds, while superoxide anions (•O2–) promoted dehydrogenation and C–H bond cleavage in PE (Fig. 8e). Both ROS types facilitated formation of carbocation intermediates, leading to chain scission and the generation of olefinic C=C bonds. Reactor-level evaluation demonstrated excellent performance: For PVC, Ni–TCPP achieved a weight loss rate of 22.0 mg/h, diesel olefin yield of 43.9 μmol, and a carbon conversion rate of 53.7%, underscoring its potential for practical implementation in solar-driven plastic reforming systems (Fig. 8f).
Antibiotics are natural or synthetic substances that inhibit or kill microorganisms. Originally, antibiotics were secondary metabolites produced by microorganisms such as bacteria, fungi, and actinomycetes, or by higher animals and plants. Over time, both environmental and synthetic antibiotics have expanded, now covering over 20 categories and >300 types, including sulfonamides, tetracyclines, quinolones, β-lactams, macrolides, and others. These antibiotics are widely used in medicine and agriculture [64]. Between 2000 and 2010, global antibiotic consumption rose by 36%, with a substantial increase in both human and agricultural use [65]. China, as a major producer and consumer, accounts for over 30% of the global market, with annual antibiotic production around 200,000 tons from 2013 to 2020 [66].
In response to concerns over antibiotic pollution, numerous treatment methods have been developed, including adsorption, photocatalysis, biodegradation, and electrochemical treatments [67–70]. However, many of these methods are unsustainable due to their high energy consumption or long processing times. More importantly, these treatments typically degrade the antibiotics without recovering energy or resources. Antibiotic wastewater, with organic matter concentrations around 400–500 mg/L chemical oxygen demand (COD), holds considerable potential chemical energy—more than double that of typical activated sludge systems [71].
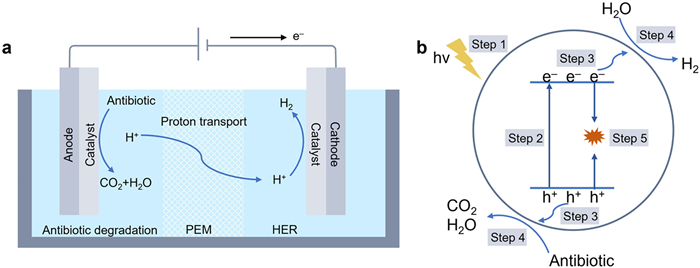
To enable both pollutant removal and energy recovery, recent advances have focused on electrocatalytic and photocatalytic technologies (Fig. 9). In electrocatalysis, antibiotics can replace oxygen as anodic substrates, thereby circumventing the oxygen evolution reaction (OER). At the anode, antibiotics are oxidized into CO2 and H2O, while at the cathode, protons are reduced to generate hydrogen [72]. This dual-function system enables simultaneous antibiotic degradation and green hydrogen production. In photocatalytic systems, light absorption by semiconductors generates electron-hole pairs [73,74]. The photogenerated holes (h+) drive the oxidation of antibiotic molecules into CO2 and H2O, while the electrons (e–) reduce water or protons to produce H2 gas [75]. This offers a sustainable route for environmental remediation coupled with clean energy generation.
Deng et al. [76] exemplified this approach by designing an electrochemical system for sulfamethoxazole (SMX) degradation and hydrogen evolution. Using Fe2O3/NiFe2O4-based electrodes (Fig. 10a, anode: Fe2O3/NiFe2O4–400@NF; cathode: Fe2O3/NiFe2O4–300@NF), over 90% of SMX was degraded within 3 h, and nearly complete mineralization was achieved within 4 h (Figs. 10b and c). Hydrogen production remained stable, with negligible changes in cell voltage despite the presence of SMX—likely due to the SMX oxidation replacing the OER, offsetting the increased electrolyte resistance (Figs. 10d and e). These findings confirm the viability of integrating pollutant removal and H2 production with minimal energy penalty.
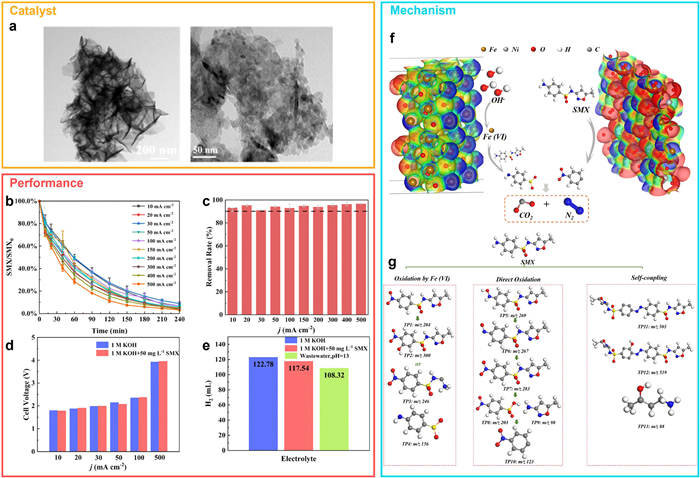
Density functional theory (DFT) calculations revealed that Fe3+ sites in Fe2O3 possess higher electrophilicity than those in NiFe2O4, making them more favorable for OH– adsorption and subsequent Fe(Ⅵ) generation. The presence of Ni promotes charge transfer toward Fe, modulating electrostatic potential and enhancing oxidative reactivity (Fig. 10f). Approximately 20% of SMX degradation was attributed to Fe(Ⅵ), while catalytic sites at NiFe2O4 nanosheet corners further enhanced performance. Liquid chromatography-mass spectrometry (LC-MS) identified degradation intermediates and elucidated three transformation pathways. The isoxazole ring, the main electrophilic center, was found to be the initial target of Fe(Ⅵ), while the amino group underwent direct oxidation via electron donation (Fig. 10g).
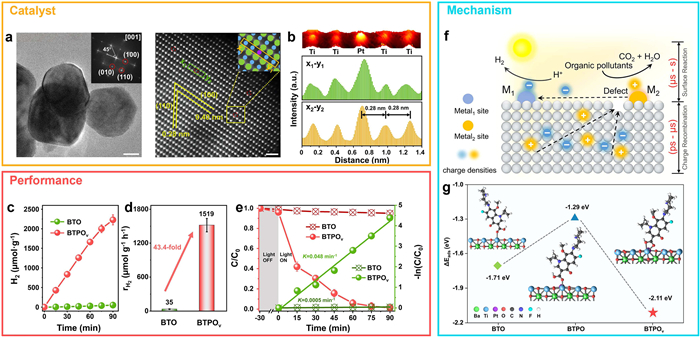
Photocatalytic strategies have also demonstrated dual-function potential. Our group developed a Pt-doped BaTiO3 catalyst with engineered oxygen vacancies (BTPOv) (Figs. 11a and b), achieving a hydrogen production rate of 1519 μmol g–1 h–1 during moxifloxacin (MOX) degradation—representing a 43-fold improvement over pristine BaTiO3 (Figs. 11c-e) [77]. DFT calculations indicated that the Ti3+ sites adjacent to oxygen vacancies facilitate electron transfer to Pt for H* adsorption and reduction, while photogenerated holes remain localized to Ti3+ sites for MOX oxidation (Fig. 11f). The adsorption energy of MOX on BTPOv (−2.11 eV) was significantly higher than on BTO (−1.71 eV) or BTPO (−1.29 eV), underscoring the enhanced catalytic activity of unsaturated Ti coordination environments (Fig. 11g). These results confirm the synergistic role of structural defects and metal doping in promoting bifunctional photocatalytic performance.
Endocrine-disrupting chemicals (EDCs) are widely present in diverse sectors, including transportation, construction, agriculture, electronics, food, and personal care [78]. Among the most common EDCs is di(2-ethylhexyl) phthalate (DEHP), a plasticizer extensively used in the production of flexible polyvinylchloride (PVC) products such as electrical wires, films, flooring, and synthetic leather. Structurally related compounds—diisobutyl phthalate (DIBP), dibutyl phthalate (DBP), and butylbenzyl phthalate (BBP)—share similar applications. However, conventional municipal wastewater treatment plants often fail to completely remove these compounds, resulting in their residual presence in treated effluents and biosolids [79]. Moreover, landfill leachate and land-applied sludge further contribute to their environmental dissemination.
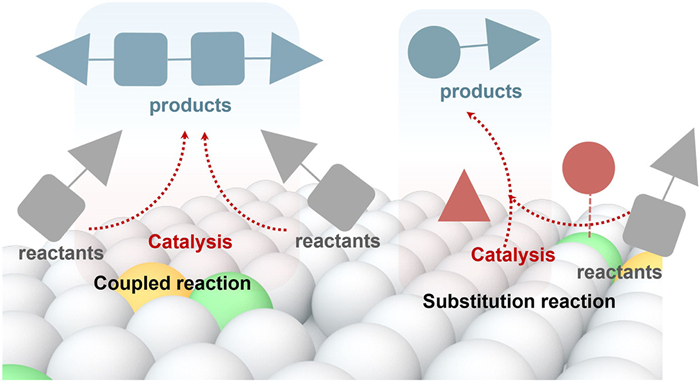
Phenolic compounds, a significant subclass of EDCs, are widely distributed in industrial effluents, pharmaceuticals, and plastic products, posing persistent threats to both ecosystems and human health [80]. Recent advances have highlighted two complementary strategies for the transformation of phenolic EDCs: Enzyme-catalyzed oxidative coupling polymerization and photocatalytic selective dehalogenation (Fig. 12). The former leverages enzymatic oxidation to convert phenolic pollutants into high-molecular-weight, low-mobility polymeric products, thereby mitigating their environmental persistence while generating materials with potential value. The latter strategy targets halogenated phenols, enabling site-selective dehalogenation via photocatalysis, and facilitates the conversion of toxic species into environmentally benign and functionally useful intermediates. Together, these approaches represent a shift toward integrated detoxification and resource valorization in the treatment of phenolic EDCs.
Lu et al. [81] developed a biocatalytic oxidative polymerization platform by encapsulating two enzymes, catalase (CAT) and horseradish peroxidase (HRP), within zeolitic imidazolate framework-8 (ZIF-8) nanostructures to form CAT/HRP@ZIF-8 hybrid nanomotors (termed enzyme@MOFtors) (Fig. 13a). These enzyme-powered nanomotors were integrated with an ultrafiltration (UF) membrane system (MOFtor-UF) to remove a range of phenolic contaminants, including 17β-estradiol, bisphenol A (BPA), acetaminophen, and various substituted phenols (Fig. 13b). Removal efficiency strongly correlated with pollutant hydrophobicity, as quantified by XLogP values. Compounds with XLogP > 2, such as BPA and DCP, exhibited removal rates exceeding 90%, owing to enhanced interfacial interactions with the hydrophobic ZIF-8 shell (Fig. 13c).
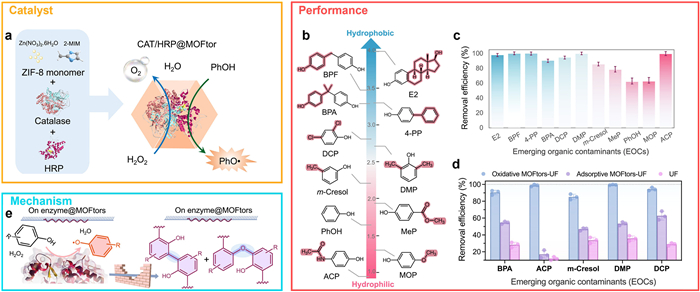
Control experiments revealed that traditional ultrafiltration alone was ineffective for small-molecule phenolics. However, the combination of HRP catalysis and hydrophobic interactions in the enzyme@MOFtor system substantially improved removal efficiency. For example, BPA and DMP removal increased from ~50% (via adsorption alone) to over 90% when enzymatic oxidation was incorporated (Fig. 13d). In contrast, low-hydrophobicity compounds like acetaminophen (XLogP = 0.5) exhibited only modest enhancement through adsorption, underscoring the critical role of HRP-mediated oxidation in their removal. High-resolution mass spectrometry (HRMS) analysis of the washed surface residues revealed oligomeric polyphenol chains (4–11 units), consistent with poly(styrene oxide) (PPO)-type structures. These were further confirmed via 1H and 13C NMR spectroscopy, supporting a biocatalytic polymerization mechanism involving HRP-generated phenoxy radicals. The increase in molecular weight and hydrophobicity of the polymer products facilitated their capture and removal via ultrafiltration. Notably, both the polymeric byproducts and active enzymes could be recovered by dissolving the ZIF-8 shell, and the enzymatic activity was largely retained post-release, demonstrating system recyclability (Fig. 13e).
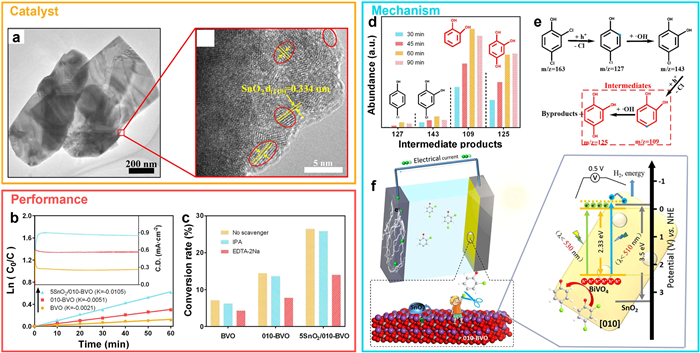
In a complementary approach, Xie et al. [82] developed a selective photocatalytic platform for chlorinated EDCs. A SnO2/BiVO4 (010)-facet-exposed photoanode was synthesized via hydrothermal methods, significantly enhancing charge separation and adsorption capacity (Fig. 14a). This system enabled efficient conversion of 2,4-dichlorophenol (2,4-DCP) into non-toxic value-added intermediates such as catechol and pyrogallol (Figs. 14b and c). LC-MS analysis identified a series of intermediates, including 4-chlorophenol, 4-chlorobenzene-1,2-diol, and 1,2,4-benzenetriol (Figs. 14d and e). Mechanistic insights suggested that ortho-chlorine removal occurs first via H+-mediated oxidation, followed by hydroxylation and subsequent para-dechlorination (Fig. 14f). The main products, 1,2-benzenediol and 1,2,4-benzenetriol, are valuable precursors for fertilizers, metalworking fluids, and photographic developers. Together, these two strategies, biocatalytic polymerization and selective photocatalytic substitution, represent promising pathways for the detoxification and valorization of phenolic EDCs. They exemplify a paradigm shift from pollutant degradation to molecular transformation, opening avenues for circular resource recovery while mitigating environmental hazards.
Despite encouraging laboratory-scale progress, the real-world deployment of pollutant upcycling technologies still faces a number of technical and systemic challenges [83]. Several proof-of-concept strategies have failed to achieve scalable performance due to fundamental limitations in catalyst stability, feedstock complexity, and process integration.
For instance, many photocatalytic systems exhibit rapid deactivation under long-term illumination or in the presence of complex wastewater matrices containing natural organic matter and inorganic ions, which scavenge reactive species and block active sites [84]. Similarly, electrocatalytic systems may suffer from passivation due to fouling, electrode corrosion, or the accumulation of intermediates that disrupt electron transfer, especially when scaled beyond laboratory volumes [85].
In the case of PFAS degradation, several early photochemical and plasma-based treatments demonstrated only partial defluorination or generated toxic byproducts such as short-chain fluorinated fragments or perfluoroalkyl ethers [86]. Moreover, some electrothermal methods, although effective, have been criticized for their high energy intensity and limited selectivity, raising concerns over their net sustainability benefits [87].
Polymer upcycling technologies also face limitations in terms of feedstock heterogeneity and product separation. For example, catalytic systems optimized for specific plastics (e.g., PET or PE) often perform poorly with mixed plastic waste, which remains the norm in real-world waste streams [88]. Additionally, downstream separation of complex product mixtures, especially when targeting high-purity chemicals or fuels, can be prohibitively expensive without energy-efficient purification technologies [89].
These challenges highlight the need for robust techno-economic evaluation, life-cycle analysis, and real-time process monitoring when transitioning from lab-scale feasibility to industrial application. Transparent reporting of limitations and failed scale-up attempts will be crucial in refining future research directions and building public and investor confidence in circular economy innovations.
While these technical hurdles underscore the complexity of pollutant upcycling, they also reveal opportunities to innovate around the concept of resource recovery, particularly when pollutants are reconceptualized not merely as contaminants to destroy, but as unconventional feedstocks rich in elemental and molecular value [90].
The recovery of valuable resources from pollutants like endocrine disruptors, antibiotics, microplastics, and perfluorinated compounds (PFCs) is becoming increasingly feasible through advanced treatment technologies [91–93]. These pollutants, often seen as environmental hazards, can be transformed into valuable chemicals that can drive economic growth. Endocrine disruptors and antibiotics, for example, can be converted into high-value pharmaceutical intermediates or specialty chemicals, while microplastics and PFCs can be upcycled into carbon-based materials or fluorochemicals with industrial applications.
Endocrine disruptors can be effectively recovered through electrochemical oxidation or photocatalytic transformation, processes that not only detoxify these pollutants but also generate pharmaceutical precursors, such as hydroquinone derivatives [94]. Similarly, antibiotics in wastewater can be degraded via photocatalysis to produce hydrogen or organic acids like formic acid, utilizing their molecular structures as sacrificial reagents. These approaches align with the waste-to-value concept, where waste streams are viewed as potential resources for chemical production rather than as waste to be discarded [95]. Microplastics, which pose significant environmental risks, are increasingly being explored as carbon precursors for high-performance materials like carbon nanotubes and graphene [96]. Pyrolysis and catalytic conversion processes can effectively transform microplastic waste into valuable nanostructures, which have a projected market value exceeding $100 billion annually, given their wide-ranging applications in electronics, energy storage, and advanced composites [97]. Perfluorinated compounds, known for their persistence in the environment, can be decomposed through plasma or reductive defluorination techniques. These processes release fluorine atoms, which can be captured and used to synthesize fluoropolymers and refrigerants [98]. The market for fluorine-based products is valued at $20 billion annually, presenting significant economic potential.
The global implementation of these recovery strategies could lead to substantial economic gains. For example, converting just 5% of antibiotic waste streams could generate $2 billion annually in hydrogen fuel revenue. Additionally, recovering endocrine disruptors as specialty chemicals could reduce pharmaceutical manufacturing costs by 10%. Microplastic-derived nanomaterials could drive sustainable innovation across industries, while the upcycling of PFCs would reduce reliance on mined fluorine resources. While emerging technologies for pollutant resource recovery face challenges related to scalability, efficiency, and energy costs, advancements in catalytic design, separation technologies, and system integration offer promising solutions. By turning pollutants into valuable resources, we have the potential to redefine wastewater treatment and foster a global transition to circular economies.
To assess the economic viability of pollutant upcycling, we evaluate a representative case of microplastic treatment in a domestic wastewater resource-recovery plant serving a city of 500,000 people in China. The facility treats approximately 100,000 m3 of wastewater per day, recovering up to 5 tons of microplastics daily [99]. These microplastics are catalytically photoreformed into diesel-range olefins and green hydrogen via a solar-assisted process. The system includes a microplastic storage unit, an integrated photoreforming reactor, and a product separation module (Fig. 15a).
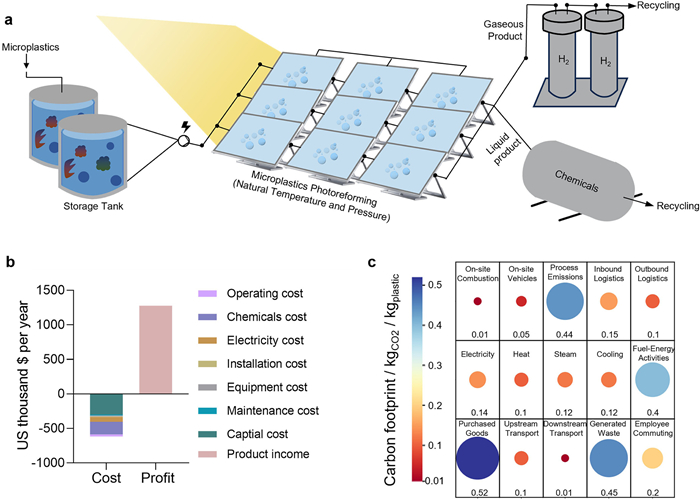
A detailed techno-economic analysis (TEA) reveals that the facility could generate annual economic returns of approximately $661,167 USD. As shown in Fig. 15b and Table S1 (Supporting information), the primary revenue streams include the sale of value-added olefins and hydrogen, while key cost components involve capital expenditure (CAPEX) for photoreactor infrastructure, catalyst replacement, electricity, maintenance, and labor. A breakdown of these cost categories is presented in Fig. S2 (Supporting information), which indicates that catalyst and infrastructure account for over 60% of total annualized costs.
To enhance generalizability, we introduce a generalized economic evaluation framework in Table S2 (Supporting information), encompassing four key pillars: (1) CAPEX (reactors, pre-treatment, and separation units), (2) OPEX (energy, catalyst, labor), (3) product revenue (based on market value of olefins and H2), and (4) carbon credits (linked to CO2 mitigation). Each component is provided with a typical range based on available data from literature and industry reports, allowing for cross-platform comparison.
In terms of environmental performance, life-cycle carbon analysis indicates that this approach emits approximately 2.91 kg CO2 per kg of microplastic treated, which is 14.43 kg lower than traditional landfill disposal. At full capacity, the system can prevent ~26,537 tons of CO2 emissions annually (Fig. 15c), contributing directly to carbon neutrality targets. Importantly, a sensitivity analysis (Fig. S3 in Supporting information) demonstrates that the system’s profitability is most affected by fluctuations in olefin prices and catalyst replacement frequency, underscoring the need for robust market conditions and durable catalysts.
To enhance the scalability analysis, we further evaluated the transition from laboratory-scale prototypes to full-scale industrial deployment. Photoreforming and electrocatalytic technologies, in particular, have demonstrated strong potential for integration into real-world infrastructures such as municipal wastewater treatment facilities and decentralized resource recovery hubs. For instance, a continuous-flow electrocatalytic system for pharmaceutical wastewater treatment was successfully operated at a pilot scale with a throughput of 300 L/h, maintaining high removal efficiency under real effluent conditions. Likewise, a photocatalytic module for PFAS-contaminated leachate was incorporated into a landfill treatment line, achieving over 80% defluorination with sustained performance over a 60-day period. These demonstrations highlight the practical viability of upcycling technologies in large-volume and compositionally variable water matrices. Modular reactor configurations, automated process controls, and solar integration further support scalability, while long-term operation under fluctuating conditions validates economic feasibility for future industrial implementation.
Policy support remains essential for scaling such technologies. Early-stage public investment and subsidies for CAPEX-intensive components can help lower adoption barriers. Policymakers should prioritize upcycling routes in circular economy roadmaps, particularly those integrating membrane separation, thermocatalysis, and advanced adsorption for pre-treatment. Public education and market incentives will also play vital roles in promoting microplastic valorization. Despite current challenges such as energy demand and reactor scalability, the growing policy momentum toward sustainable water and resource management is expected to accelerate the deployment of such circular solutions.
In the long term, resource-oriented wastewater treatment facilities could redefine urban infrastructure by simultaneously addressing environmental, economic, and energy challenges, transforming pollution burdens into commodity value streams.
The successful deployment of pollutant upcycling technologies at scale depends not only on scientific innovation but also on supportive policy frameworks. Governments worldwide are beginning to recognize the need for regulatory mechanisms that shift from linear waste disposal to circular resource utilization [100]. For instance, the European Union’s Chemicals Strategy for Sustainability and Circular Economy Action Plan explicitly call for the reuse and recovery of hazardous substances, including PFAS and microplastics, under extended producer responsibility (EPR) schemes [101]. Similarly, the U.S. EPA’s 2021 PFAS Strategic Roadmap outlines commitments to support technology development for safe and cost-effective PFAS destruction and reuse [102].
Policy interventions are particularly critical at the early stages of technology commercialization. These include subsidies for pilot-scale upcycling projects, preferential procurement for recycled materials, and tax incentives for industries adopting green remediation technologies. For example, China’s 14th Five-Year Plan prioritizes resource-efficient wastewater treatment and has earmarked funding for innovations in pollutant valorization and circular economy infrastructure [103]. International cooperation, such as the OECD’s work on persistent pollutants and the UNEP’s Global Partnership on Plastic Pollution, can facilitate knowledge transfer and harmonized standards for safe and efficient resource recovery from emerging pollutants [104].
Importantly, regulations must evolve to account for the dual value of these contaminants: as environmental threats and as reservoirs of valuable elements (e.g., fluorine, carbon, nitrogen). Life-cycle-based policy instruments that internalize the environmental costs and economic benefits of upcycling will be key to incentivizing industry participation. In this context, integrating pollutant upcycling into national climate strategies, ESG frameworks, and sustainability metrics will enable broader systemic transitions. As scientific evidence increasingly supports the viability of pollutant-to-resource pathways, policy alignment will determine their real-world impact.
While pollutant upcycling has made remarkable strides in laboratory research, its widespread implementation hinges on overcoming several critical scientific, engineering, and policy barriers. A key technical priority is improving catalyst durability and recyclability under realistic operating conditions, especially when exposed to complex, heterogeneous pollutant matrices. Developing robust, long-lived catalysts that retain high selectivity and activity over extended cycles is essential for economic feasibility. Simultaneously, reactor engineering must evolve to support continuous-flow, scalable systems with efficient light capture, heat management, and mass transfer, especially for solar-powered and electrocatalytic processes.
Another research priority is advancing separation and purification technologies for product recovery. Many upcycling routes yield complex mixtures; thus, efficient downstream separation of target chemicals (e.g., fluorochemicals, olefins, H2) with minimal energy input is critical to close the economic loop. Integrated catalyst–reactor–separation design will be necessary to streamline conversion and valorization in a single platform. In parallel, data-driven process control (e.g., real-time monitoring, AI-optimized reaction parameters) can enhance system adaptability to fluctuating waste inputs.
Looking ahead, tailored upcycling strategies must be developed for each pollutant class. For PFAS, the focus lies in developing energy-efficient, selective defluorination pathways that yield functionalized intermediates instead of complete mineralization, alongside scalable thermal or mechanochemical platforms for fluorine recovery. For microplastics, research should emphasize photocatalytic or piezoelectric systems that combine high conversion rates with solar compatibility, and that can be modularly integrated into wastewater treatment infrastructure. Antibiotic upcycling calls for bifunctional electrocatalytic systems that simultaneously degrade contaminants and recover energy as hydrogen fuel. In contrast, endocrine disruptors require gentle, selective conversion routes, such as enzyme-mediated polymerization or photocatalytic dehalogenation, to minimize harmful byproducts while enabling recovery of high-value molecular building blocks. Cross-cutting innovations in multifunctional catalysts, selective bond activation, and hybrid reactor–separation platforms will be vital to meet the diverse challenges posed by these pollutants.
On the policy front, implementing Extended Producer Responsibility (EPR) schemes for PFAS-containing materials, plastics, and pharmaceuticals can shift the financial burden of end-of-life management toward manufacturers. Subsidies and tax incentives for recovered materials and green hydrogen production can also accelerate the commercial viability of upcycling technologies. Furthermore, mandatory pollutant disclosure and regulatory frameworks for upcycled products are needed to build consumer trust and industrial demand.
Finally, successful deployment requires system-level integration of upcycling technologies into existing wastewater treatment infrastructure, landfill leachate management, and resource recovery hubs, particularly in rapidly urbanizing or water-stressed regions. Transdisciplinary collaborations—bridging chemistry, environmental engineering, economics, and public policy—will be crucial to translate lab-scale breakthroughs into field-ready solutions. By uniting technological innovation with supportive policy instruments and lifecycle-informed system design, pollutant upcycling can evolve from a niche concept to a central pillar of the circular economy—transforming environmental liabilities into economic and societal value.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Shuai Yue: Writing – original draft. Zhiyong Zhao: Writing – review & editing. Mengxue Yang: Writing – review & editing. Yixiao Liu: Writing – review & editing. Pengfei Wang: Writing – review & editing. Chenxin Xie: Writing – review & editing. Sihui Zhan: Writing – review & editing, Funding acquisition.
This work was supported by the Natural Science Foundation of China (Nos. 22225604, U24A20518, and 22422605), the Tianjin Commission of Science and Technology as key critical technologies R&D projects (No. 23YFZCSN00010), the Frontiers Science Center for New Organic Matter (No. 63181206), Young Scientific and Technological Talents (Level Two) in Tianjin (No. QN20230206), the Fundamental Research Funds for the Central Universities, Nankai University (Nos. 63231195 and 63253207), and Haihe Laboratory of Sustainable Chemical Transformations. The first author is funded by the Shanghai Tongji Gao Tingyao Environmental Science & Technology Development Foundation.
Supplementary material associated with this article can be
found, in the online version, at doi:
W.W. Li, H.Q. Yu, B.E. Rittmann, Nature 528 (2015) 29–31. doi: 10.1038/528029a
R. Rosal, A. Rodriguez, J. Antonio Perdigon-Melon, et al., Water Res. 44 (2010) 578–588. doi: 10.1016/j.watres.2009.07.004
J.L. Schnoor, Environ. Sci. Technol. 37 (2003) 375A-375A. doi: 10.1021/es032604j
T. Liu, Y. Zhang, Z. Shan, et al., Nat. Water 1 (2023) 1059–1067. doi: 10.1038/s44221-023-00162-w
R. Ni, K.N. Opoku, X. Li, et al., Chin. Chem. Lett. 36 (2025) 110813. doi: 10.1016/j.cclet.2024.110813
M.G. Evich, M.J.B. Davis, J.P. McCord, et al., Science 375 (2022) 512.
S. Glass, H.A. Santiago-Cruz, W. Chen, et al., Nat. Water 3 (2025) 644–654. doi: 10.1038/s44221-025-00433-8
S. Zhao, K.F. Kvale, L. Zhu, et al., Nature 641 (2025) 51–61. doi: 10.1038/s41586-025-08818-1
S. Yue, P.F. Wang, B.N. Yu, et al., Adv. Energy Mater. 13 (2023) 2302008. doi: 10.1002/aenm.202302008
W.J. Xue, X.Y. Shi, J.M. Guo, et al., Water Res. 253 (2024) 121309. doi: 10.1016/j.watres.2024.121309
K. Tian, L.M. Hu, L.T. Li, et al., Chin. Chem. Lett. 33 (2022) 4461–4477. doi: 10.1016/j.cclet.2021.12.042
D. Chen, K. Kannan, H.L. Tan, et al., Environ. Sci. Technol. 50 (2016) 5438–5453. doi: 10.1021/acs.est.5b05387
Y. Xiao, D.M. Han, M. Currell, X.F. Song, Y.H. Zhang, Water Res. 245 (2023) 120645. doi: 10.1016/j.watres.2023.120645
Z.F. Yin, C.J. Cui, H. Chen, et al., Small 16 (2020) 1902301. doi: 10.1002/smll.201902301
S. Yue, Z.Y. Zhao, T. Zhang, et al., Environ. Sci. Technol. 58 (2024) 22865–22879. doi: 10.1021/acs.est.4c06688
Z. Zhao, G. Yang, P. Wang, et al., Nat. Commun. 16 (2025) 5861. doi: 10.1038/s41467-025-60964-2
K. Zhu, X.Y. Liang, Y.W. Chen, et al., Coord. Chem. Rev. 519 (2024) 216110. doi: 10.1016/j.ccr.2024.216110
Z.Y. Zhao, T. Zhang, S. Yue, et al., ChemPhysChem 25 (2024) 202300726. doi: 10.1002/cphc.202300726
M.X. Yang, Z.Y. Zhao, T.Y. Zhi, et al., Carbon Neutral 4 (2025) e70017.
X. Li, W.R. Bu, K. Zhu, et al., Sep. Purif. Technol. 337 (2024) 126382. doi: 10.1016/j.seppur.2024.126382
R.C. Cao, M.Q. Zhang, Y.C. Jiao, et al., Nat. Sustain. 6 (2023) 1685–1692. doi: 10.1038/s41893-023-01234-1
Z.Y. Zhao, S. Yue, G.H. Yang, P.F. Wang, S.H. Zhan, Trans. Tianjin Univ. 30 (2024) 1–26. doi: 10.1007/s12209-024-00383-4
K. Zhu, W.L. Qin, Y.W. Chen, et al., Nano Today 58 (2024) 102462. doi: 10.1016/j.nantod.2024.102462
X. Liu, A. Sau, A.R. Green, et al., Nature 637 (2025) 601–607. doi: 10.1038/s41586-024-08327-7
H. Zhang, J.X. Chen, J.P. Qu, Y.B. Kang, Nature 635 (2024) 610–617. doi: 10.1038/s41586-024-08179-1
Y.F. Wang, J. Zhang, W.K. Zhang, et al., Angew. Chem. Int. Ed. 63 (2024) e202402440. doi: 10.1002/anie.202402440
M.Q. Zhang, Y.D. Zhou, R.C. Cao, et al., Nature 643 (2025) 395–403. doi: 10.1038/s41586-025-09088-7
J. Zhao, B.N. Liu, L.Q. Xiong, et al., Nat. Commun. 16 (2025) 1726. doi: 10.1038/s41467-024-55584-1
S.L. Liu, K. Ma, H.F. Teng, et al., Adv. Mater. 37 (2025) 202411148.
Y. Wang, P. Zhang, W. Liao, et al., Environ. Sci. Teachnol. 59 (2025) 13516–13527. doi: 10.1021/acs.est.5c03405
X. Yu, Z.P. Wang, S. Kim, et al., Appl. Catal. B: Environ. 371 (2025) 125209. doi: 10.1016/j.apcatb.2025.125209
S. Li, Y.L. Yang, J.F. Niu, et al., Environ. Sci. Technol. 58 (2024) 21871–21881. doi: 10.1021/acs.est.4c08224
J.Z. Cai, B.L. Niu, H.Y. Zhao, G.H. Zhao, Environ. Sci. Technol. 55 (2021) 2618–2627. doi: 10.1021/acs.est.0c07106
Y. Gao, F. Wang, J. Tang, et al., Chem. Eng. J. 495 (2024) 153651. doi: 10.1016/j.cej.2024.153651
Y.D. Chen, Y. Shao, O.Y. Li, et al., Chem. Eng. J. 442 (2022) 135961. doi: 10.1016/j.cej.2022.135961
C. Jehanno, J.W. Alty, M. Roosen, et al., Nature 603 (2022) 803–814. doi: 10.1038/s41586-021-04350-0
S. Guevara, I.P. Julián, J. Sustain. Res. 1 (2019) e190019.
A.R. Santiago, S. Yin, J. Elbert, et al., J. Am. Chem. Soc. 145 (2023) 9508–9519. doi: 10.1021/jacs.2c10963
M.B. Gu, L.J. Duan, Z.Z. Zhang, et al., Chem. Eng. J. 505 (2025) 159124. doi: 10.1016/j.cej.2024.159124
K.X. Fu, J.J. Huang, F. Luo, et al., Environ. Sci. Technol. 58 (2024) 16669–16689.
A. Podder, A. Sadmani, D. Reinhart, N.B. Chang, R. Goel, J. Hazard. Mater. 419 (2021) 126361. doi: 10.1016/j.jhazmat.2021.126361
D.H. Ma, C.I. Olivares, Environ. Sci. Technol. 59 (2025) 10734–10749. doi: 10.1021/acs.est.5c00906
Y.L. Liu, M. Sun, Water Res. 207 (2021) 117781. doi: 10.1016/j.watres.2021.117781
S.D. Tan, R.Y. Wang, K.M. Wang, et al., Nat. Water 3 (2025) 734–745. doi: 10.1038/s44221-025-00449-0
P. Scotland, K.M. Wyss, Y. Cheng, et al., Nat. Water 3 (2025) 486–496. doi: 10.1038/s44221-025-00404-z
Y. Cheng, B. Deng, P. Scotland, et al., Nat. Commun. 15 (2024) 6117. doi: 10.1038/s41467-024-49809-6
L. Yang, Z. Chen, C.A. Goult, T. Schlatzer, R.S. Paton, V. Gouverneur, Nature 640 (2025) 100–106. doi: 10.1038/s41586-025-08698-5
E.J. Carpenter, S.J. Anderson, H.P. Miklas, B.B. Peck, G.R. Harvey, Science 178 (1972) 749. doi: 10.1126/science.178.4062.749
J.L. Chen, J. Wu, P.C. Sherrell, et al., Adv. Sci. 9 (2022) 2103764. doi: 10.1002/advs.202103764
Y. Chae, Y.J. An, Environ. Pollut. 240 (2018) 387–395. doi: 10.1016/j.envpol.2018.05.008
E. Hernandez, B. Nowack, D.M. Mitrano, Environ. Sci. Technol. 51 (2017) 7036–7046. doi: 10.1021/acs.est.7b01750
X.W. Wu, X.L. Zhao, R.Z. Chen, et al., Water Res. 221 (2022) 118825. doi: 10.1016/j.watres.2022.118825
K. Ragaert, L. Delva, K. Van Geem, Waste Manage 69 (2017) 24–58. doi: 10.1016/j.wasman.2017.07.044
S. Yue, Y. Liu, Z. Zhao, et al., Proc. Natl. Acad. Sci. U. S. A. 122 (2025) e2508636122. doi: 10.1073/pnas.2508636122
S. Yue, Z.Y. Zhao, Y.X. Liu, et al., Adv. Funct. Mater. 35 (2025) 09766. doi: 10.1002/adfm.202509766
X. Chen, Y. Wang, L. Zhang, ChemSusChem 14 (2021) 4137–4151. doi: 10.1002/cssc.202100868
C. Xing, C. Mao, S. Wang, et al., Nat. Catal. 8 (2025) 556–568. doi: 10.1038/s41929-025-01349-y
T. Zhang, P.F. Wang, S. Yue, et al., Sci. China Chem. 67 (2024) 1161–1174. doi: 10.1007/s11426-023-1914-0
W. Utetiwabo, L. Yang, M.K. Tufail, et al., Chin. Chem. Lett. 31 (2020) 1474–1489. doi: 10.1016/j.cclet.2020.01.003
M.Q. Zhang, M. Wang, B. Sun, et al., Chem 8 (2022) 2912–2923. doi: 10.1016/j.chempr.2022.08.004
W. Zhang, S. Kim, M.L. Sarazen, et al., Angew. Chem. Int. Ed. 64 (2025) 202500559. doi: 10.1002/anie.202500559
Y.X. Wang, F.L. Liu, J. Chen, et al., Nat. Commun. 16 (2025) 4440. doi: 10.1038/s41467-025-59667-5
S. Yue, Z.Y. Zhao, T. Zhang, et al., Angew. Chem. Int. Ed. 63 (2024) 202406795. doi: 10.1002/anie.202406795
Y. Mao, B. Yu, P. Wang, et al., Nat. Commun. 15 (2024) 6364. doi: 10.1038/s41467-024-50238-8
E.Y. Klein, I. Impalli, S. Poleon, et al., Proc. Natl. Acad. Sci. U. S. A. 121 (2024) e2411919121. doi: 10.1073/pnas.2411919121
F. Li, S. Yue, Z. Zhao, et al., Mater. Today Sustain. 27 (2024) 100904.
W. Tian, J. Lin, H. Zhang, et al., J. Hazard. Mater. 423 (2022) 127083. doi: 10.1016/j.jhazmat.2021.127083
Y. Bao, K. Xiao, S. Yue, et al., Surf. Inter. 40 (2023) 103107.
F. Li, P. Wang, T. Zhang, et al., Angew. Chem. Int. Ed. 62 (2023) 202313298. doi: 10.1002/anie.202313298
J. Tang, Z. Li, X. Xiao, et al., Water Res. 268 (2025) 122683. doi: 10.1016/j.watres.2024.122683
B.L. Phoon, C.C. Ong, M.S.M. Saheed, et al., J. Hazard. Mater. 400 (2020) 122961. doi: 10.1016/j.jhazmat.2020.122961
X. Wang, X. Li, S. Li, et al., Chem. Eng. J. 516 (2025) 163941. doi: 10.1016/j.cej.2025.163941
S. Yue, Z. Zhao, T. Zhang, et al., Mater. Today Energy 40 (2024) 101482. doi: 10.1016/j.mtener.2023.101482
S. Yue, L. Chen, M. Zhang, et al., Nano-Micro Lett. 14 (2022) 15. doi: 10.1007/s40820-021-00749-6
Y. Zhi, C. Gu, H. Ji, et al., Chin. Chem. Lett. 36 (2025) 110234. doi: 10.1016/j.cclet.2024.110234
Q. Zhang, Y. Tong, Z. Wang, et al., J. Mater. Chem. A 11 (2023) 6129–6143. doi: 10.1039/d2ta08850a
T. Zhang, Z. Zhao, D. Zhang, et al., Proc. Natl. Acad. Sci. U. S. A. 120 (2023) e2302873120. doi: 10.1073/pnas.2302873120
C. Duh-Leong, M.V. Maffini, C.D. Kassotis, et al., Nat. Rev. Endocrinol. 19 (2023) 600–614. doi: 10.1038/s41574-023-00872-x
J.A. Citulski, K. Farahbakhsh, Environ. Sci. Technol. 44 (2010) 8367–8376. doi: 10.1021/es102403y
T. Ruan, P. Li, H. Wang, et al., Chem. Rev. 123 (2023) 10584–10640. doi: 10.1021/acs.chemrev.3c00056
S. Xu, Z. Feng, L. Bao, et al., Matter 8 (2025) 102216. doi: 10.1016/j.matt.2025.102216
J. Yang, N. Sun, Z. Zhang, et al., ACS Appl. Mater. Interfaces 12 (2020) 28264–28272. doi: 10.1021/acsami.0c06892
C. Luo, S. Liu, T. Wu, et al., Green Chem. 27 (2025) 7445–7471. doi: 10.1039/d5gc01305d
S. Yue, W. Hu, J. Wang, et al., Chem. Eng. J. 435 (2022) 135005. doi: 10.1016/j.cej.2022.135005
F.M. Li, L. Huang, S. Zaman, et al., Adv. Mater. 34 (2022) 2200840. doi: 10.1002/adma.202200840
B. Saawarn, B. Mahanty, S. Hait, et al., Environ. Res. 214 (2022) 114004. doi: 10.1016/j.envres.2022.114004
Q. Dong, S. Hu, L. Hu, et al., Nat. Chem. Eng. 1 (2024) 680–690. doi: 10.1038/s44286-024-00134-1
Y. Shi, X. Diao, N. Ji, et al., ACS Catal. 15 (2024) 841–868.
L.R. Lynd, G.T. Beckham, A.M. Guss, et al., Environ. Energy Sci. 15 (2022) 938–990. doi: 10.1039/d1ee02540f
X. Zhao, B. Boruah, K.F. Chin, et al., Adv. Mater. 34 (2022) 2100843. doi: 10.1002/adma.202100843
Q. Shi, Y.Q. Zhu, X. Liu, et al., J. Am. Chem. Soc. 147 (2025) 14004–14014. doi: 10.1021/jacs.5c04034
S. Yue, G. Xiao, Q. Shi, et al., Adv. Funct. Mater. 35 (2025) 2503631. doi: 10.1002/adfm.202503631
W. Yu, N. Fang, R. Wang, et al., Adv. Funct. Mater. 34 (2024) 202314894.
V. UshaVipinachandran, S. Rajendran, K.H.B. Haroon, et al., ACS Appl. Nano Mater. 3 (2020) 11659–11687. doi: 10.1021/acsanm.0c02974
F. Haque, C.H. Fan, Y.Y. Lee, J. Clean. Prod. 415 (2023) 137873. doi: 10.1016/j.jclepro.2023.137873
J. Kang, L. Zhou, X.G. Duan, et al., Matter 1 (2019) 745–758. doi: 10.1016/j.matt.2019.06.004
Y. Dong, B.Q. Jia, F.Y. Fu, et al., Angew. Chem. Int. Ed. 55 (2016) 13504–13508. doi: 10.1002/anie.201607455
D.J. Sheldon, M.R. Crimmin, Chem. Soc. Rev. 51 (2022) 4977–4995. doi: 10.1039/d1cs01072g
L.S. Zhang, J.Y. Liu, Y.S. Xie, et al., J. Clean. Prod. 291 (2021) 125968. doi: 10.1016/j.jclepro.2021.125968
T. Domenech, B. Bahn-Walkowiak, Ecol. Econ. 155 (2019) 7–19. doi: 10.1016/j.ecolecon.2017.11.001
T. Mohr, I. Schliebner, M. Neumann, et al., Environ. Sci. Eur. 36 (2024) 99. doi: 10.1186/s12302-024-00932-7
A. Aponte, A. Engel, D. Bauer, et al., Brookhaven National Laboratory Report, Upton, NY (United States), 2023.
L. Ji, Y.A. Sun, J.W. Liu, et al., Sci. Total Environ. 881 (2023) 163435. doi: 10.1016/j.scitotenv.2023.163435
D.C. Muir, P.H. Howard, Environ. Sci. Technol. 40 (2006) 7157–7166. doi: 10.1021/es061677a

Figure 1 Emerging pollutant-specific upcycling strategies and performance indicators across catalytic platforms including photocatalysis, electrocatalysis, flash Joule heating (FJH), and biohybrid systems.

Figure 2 Schematic illustration of the rate-limiting electron transfer step in PFAS conversion. The activation free energy for the electron transfer step (ΔGET‡) is consistently higher than that for defluorination (ΔGDF‡), supporting an ET-limited mechanism. Energy diagram based on Marcus theory, depicting the relationship among ΔGET‡, reaction free energy (ΔG), and reorganization energy (λ). Copied with permission [44]. Copyright 2025, Springer Nature.

Figure 3 Electrothermal defluorination of PFAS-contaminated materials via flash Joule heating and rapid electrothermal mineralization. (a) Schematic of the experimental setup for flash Joule heating (FJH). (b) Conceptual illustration of the rapid electrothermal mineralization (REM) process for bulk soil remediation. (c) Summary of mineralization efficiency and degradation of PFOA- and PFOS-loaded granular activated carbon (GAC), with >90% of inorganic fluoride recovered. (d) Yield of inorganic fluoride from FJH treatment of PFOA-GAC using 1.2 molar equivalents of sodium per mole of fluorine; data are presented as mean ± SD from three independent experiments (n = 3). Copied with permission [45]. Copyright 2025, Springer Nature. (e) Voltage-dependent variation in organic fluorine content (red line) and mineralized fluoride (blue line) in PFOS-contaminated soil. (f) 19F NMR spectra of PFOS after REM treatment, showing exclusive formation of hydrated fluoride. Copied with permission [46]. Copyright 2024, Springer Nature. (g, h) Calculated potential energy surfaces showing that the activation barrier for the electron transfer (ΔGET‡) exceeds that of defluorination (ΔGDF‡) for both PFOA and PFOS, confirming ET as the rate-limiting step. (i, j) Proposed defluorination pathways for PFOA and PFOS, respectively. Copied with permission [44]. Copyright 2025, Springer Nature.

Figure 4 Catalytic transformation of PFAS into valuable fluorochemicals via phosphate activation and mechanistic insights. (a) Representative strategies for PFAS degradation. (b) This work: PFAS mineralization coupled with fluorochemical upcycling. (c) Screening of phosphate salts as activators for PTFE, with total fluoride yields (F– and PO3F2–) quantified via 19F NMR spectroscopy in 10% D2O/H2O with NaOTf as internal standard. (d) Elemental quantification of PTFE mixtures by 13C, 19F, and 31P NMR. (e) Control experiments using reaction by-products as activators, with fluoride products analyzed by 19F NMR. (f) DFT calculations revealing the relationship between reactivity and oxyanion nucleophilicity or SN2 activation barriers; hydrated salts shown in grey. Copied with permission [47]. Copyright 2025, Springer Nature.

Figure 5 Catalytic upcycling of plastic waste into high-value products through chemical processes.

Figure 7 Integrated electrosynthetic strategy for GA production from EG and PET waste: Catalyst design, flow cell optimization, and scalable implementation. (a) Schematic of glycolic acid (GA) production from ethylene glycol (EG). (b) XRD patterns of CoCr2O4 and Pd-CoCr2O4 catalysts. (c) High-resolution transmission electron microscopy (HRTEM) image of Pd-CoCr2O4 revealing nanostructured features. (d) Flow-cell schematic illustrating the continuous electrosynthesis of GA and co-generation of H2. (e) Linear sweep voltammetry (LSV) profiles of the flow-cell with and without EG at 10 mL/min. (f) GA selectivity and yield as a function of flow rate at 1.25 V. (g) Proposed reaction pathway for EG oxidation to GA on Pd-CoCr2O4 in 1 mol/L KOH. (h) Scale-up process photographs: PET waste pre-treatment, stirred reactor, electrolyzer stack with control system, and product separation setup (filtration, distillation, drying). Copied with permission [62]. Copyright 2025, Springer Nature.

Figure 8 Mechanistic insights into polyolefin photoreforming enabled by supramolecular assembly and ROS mediation. (a) Schematic illustration of the self-assembly of Zn-TCPP and Ni-TCPP via π–π stacking interactions. (b) Carbon atom distribution of gaseous products from polyolefin photoreforming as determined by GC–MS analysis. (c) Weight loss rate and product formation rate during polyolefin photoreforming. (d) Correlation between reactive oxygen species (ROS) type and product selectivity in systems dominated by individual ROS. (e) Proposed depolymerization pathways for PVC and PE under photocatalytic conditions. (f) Photograph of the experimental setup used for plastic photoreforming. Copied with permission [63]. Copyright 2024, Wiley.

Figure 9 Integrated electrocatalytic and photocatalytic pathways for simultaneous antibiotic degradation and hydrogen generation. (a) Schematic diagram of an electrochemical cell for antibiotic degradation at the anode and hydrogen evolution at the cathode, separated by a proton exchange membrane (PEM). (b) Proposed mechanism for photocatalytic antibiotic degradation coupled with water splitting, highlighting key charge transfer and transformation steps.

Figure 10 Electrocatalytic degradation of sulfamethoxazole coupled with hydrogen evolution using Fe2O3–NiFe2O4 heterostructures. (a) Ttransmission electron microscopy (TEM) images showing nanoparticles and nanosheets of Fe2O3–NiFe2O4. (b) Degradation profiles of sulfamethoxazole (SMX) at varying current densities. (c) SMX removal rate within 4 h under different current conditions. (d) Cell voltage response and (e) hydrogen yield in various electrolytes. (f) Electrostatic potential distributions of Fe2O3 and NiFe2O4. (g) Proposed degradation pathways of SMX during the electrocatalytic process. Copied with permission [76]. Copyright 2023, RSC.

Figure 11 Defect-engineered BTPOv photocatalysts for enhanced antibiotic degradation via photo-heating effect. (a) TEM image, fast Fourier transform (FFT) pattern, and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of vacancy-rich BTPOv. (b) Surface profile and elemental line scan along the selected x–y region. (c) Time-dependent degradation of phenol (PHE) over BTO and BTPOv. (d) PHE degradation rates on BTO and BTPOv photocatalysts. (e) Degradation efficiency and pseudo-first-order kinetics of moxifloxacin (MOX) removal using BTO and BTPOv under the photo-thermal process. (f) Schematic illustration of the photoinduced charge transfer and heat-assisted catalysis mechanism. (g) Calculated adsorption energies of MOX on BTO, BTPO, and BTPOv surfaces. Copied with permission [77]. Copyright 2023, National Academy of Sciences.

Figure 12 Dual catalytic strategies for phenolic EDCs conversion: coupled polymerization and selective substitution pathways.

Figure 13 Bioengineered nanomotors for selective removal of emerging organic contaminants via enzymatic cascade and adsorptive filtration. (a) One-step synthesis of CAT/HRP enzyme-integrated ZIF-8 nanomotors and their cascade reaction mechanism. (b, c) Correlation between contaminant hydrophobicity (XLogP) and removal efficiency across a range of emerging organic contaminants (EOCs). (d) Comparative performance of catalytic oxidation, adsorption, and membrane filtration processes in EOC removal. (e) Proposed coupling and polymerization mechanisms facilitated by the biocatalytic nanomotors. Copied with permission [81]. Copyright 2025, Elsevier.

Figure 14 Engineered SnO2/BiVO4 heterostructure for photoelectrochemical and selective conversion of 2,4-dichlorophenol via high-energy electron transfer pathways. (a) TEM and HRTEM images of the 5SnO2/010-BVO heterostructure. (b) Photoelectrochemical performance for 2,4-DCP conversion at 0.5 V vs. Ag/AgCl. (c) Radical-trapping analysis to identify reactive species. (d) Time-resolved LC–MS spectra showing intermediate product formation. (e) Proposed reaction pathway for 2,4-DCP degradation. (f) Schematic illustration of photogenerated high-energy electron transfer driving controlled molecular transformation. Copied with permission [82]. Copyright 2020, American Chemical Society.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:


 下载:
下载: