Figure 1.
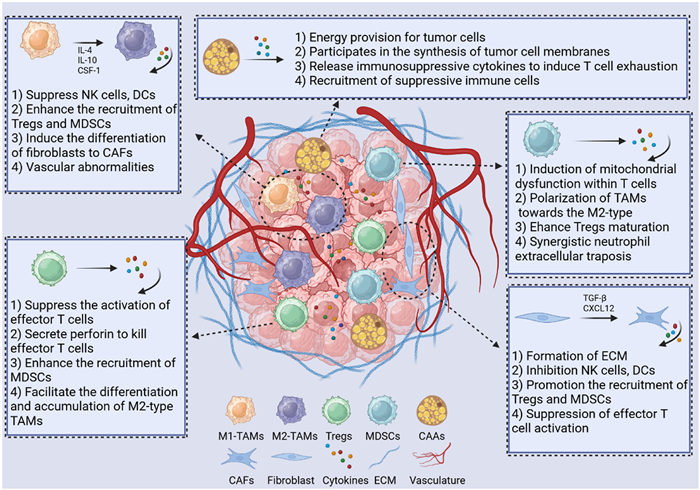
Illustrative schematic depicting diverse tumor stromal cell types and their respective roles within the ITME.
Nanodelivery strategies modulating tumor stromal cells for reverting the immunosuppressive tumor microenvironment
Jiayi Sun , Luyao Huang , Wenfeng Jia , Yitong Liu , Li Xiang , Xing Yang , Fan Tong , Xiaobo Wang , Huile Gao , Yi Zhang

As the worldwide incidence of cancer escalates, a range of novel therapeutic approaches is being developed. Among these, cancer immunotherapy functions by stimulating the host's immune system to eradicate malignant tumors [1,2]. In comparison to conventional treatment methods, it offers multiple benefits, including the enhancement of long-lasting immune memory and a reduction in related toxicity. Its development has transpired over a century, transitioning from empirical methodologies to more precise regulation [3]. In 1891, William Coley administered Coley’s toxins directly into the lesions of patients with sarcoma, resulting in observable tumor regression. This event represents the initial evidence supporting the notion that immune activation can effectively counteract tumors [4]. In 1957, Burnet proposed the concept of “immune surveillance”, positing that the immune system possesses the capability to identify and eradicate nascent tumors [5]. Subsequently, in 1975, Köhler and Milstein advanced the field by developing hybridoma technology, which facilitated the generation of monoclonal antibodies [6]. Between 1995 and 2000, James Allison and Tasuku Honjo elucidated the pivotal functions of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death receptor 1 (PD-1) in the immune suppression, thereby establishing a novel domain of immune checkpoint inhibitors (ICIs) [7]. These seminal researches provided a foundational for contemporary cancer immunotherapy. As the 21st century commenced, tumor immunotherapy began to gain more prominence, culminating in the approval of sipuleucel-T, a therapeutic cancer vaccine for prostate cancer, by the Food and Drug Administration in April 2010, marking it as the first therapeutic cancer vaccine to achieve such approval [8]. In 2018, the contributions of ICIs were recognized with the Nobel Prize in Physiology or Medicine [9]. The CTLA-4 antibody ipilimumab, along with anti-PD-1/programmed cell death ligand 1 (PD-L1) antibodies such as pembrolizumab and nivolumab, exhibited remarkable clinical efficacy [10–12]. Furthermore, cell therapies, including chimeric antigen receptor T-cell immunotherapy (CAR-T) and CAR-natural killer (NK), have demonstrated significant benefits in the treatment of specific cancer types [13].
Despite significant advancements in tumor immunotherapy, the response rate for cold tumors remains below 20% [14,15]. Unlike hot tumors, cold tumors are characterized by a lack of immune cell infiltration, resulting in an immune desert state [16,17]. A primary challenge is the presence of the immunosuppressive tumor microenvironment (ITME). The formation of ITME can be attributed to three key mechanisms: (1) Physical barriers: The aberrant activation of cancer-associated fibroblasts (CAFs) leads to the synthesis of a dense extracellular matrix (ECM), which directly obstructs the migration of T cells. Additionally, CAFs anchor T cells within the matrix through the secretion of chemokines, such as C-X-C motif chemokine ligand 12, thereby inhibiting their access to the tumor microenvironment (TME) [18]. (2) Immunosuppressive networks: The accumulation of immunosuppressive cell types, results in the secretion of substantial quantities of immunosuppressive molecules such as transforming growth factor beta (TGF-β), interleukin (IL)-10, and arginase (Arg). These molecules directly inhibit T cell functionality and promote T cell exhaustion [19]. (3) Metabolic dysregulation: Tumor cells and associated stromal cells produce elevated levels of lactate via the Warburg effect, leading to the acidification of the microenvironment [20]. Additionally, tumor cells and myeloid-derived suppressor cells (MDSCs) exhibit high expression of indoleamine 2,3-dioxygenase (IDO), which catalyzes the degradation of tryptophan, while MDSCs express Arg 1, resulting in the depletion of arginine. These metabolic alterations contribute to T cell inactivation or functional impairment [20,21].
The establishment of the ITME is fundamentally linked to the biological activities of tumor cells, immune cells, and tumor stromal cells. In the initial phases of immunotherapy research, a significant emphasis was focused on modulating or killing tumor cells [22,23], T cells [24], or NK cells [25], which are considered the direct contributors to the TME. However, as investigations into cancer immunotherapy have progressed, it has become evident that this strategy is fraught with limitations, including high mutation rates of targets, readily acquire resistance, and the difficulty of overcoming physical barriers. Consequently, there has been a shift towards the development of immunotherapies that modulate tumor stromal cells [26–28]. These tumor stromal cells encompass CAFs, tumor-associated macrophages (TAMs), MDSCs, regulatory T cells (Tregs), cancer-associated adipocytes (CAAs), and pericytes, etc., all of which play critical roles in the formation of the ITME [29–31]. The strategies for modulating tumor stromal cells can be broadly categorized into five principal types to reverse the immunosuppressive state: (1) Breaking the physical barrier, (2) reducing the recruitment of immunosuppressive cell populations, (3) directly eliminating immunosuppressive cells, (4) improving the metabolic environment within the ITME, and (5) synergistically augmenting existing immunotherapeutic strategies [32–35].
The emergence of nanotechnology has significantly transformed the development of drug delivery systems (DDS) [36–38]. The integration of nano-DDS with modulated tumor stromal cell immunotherapy has the potential to enhance delivery efficiency, including improved tumor permeability, retention, and bioavailability, while simultaneously minimizing adverse effects [39,40]. Furthermore, these nano-DDS facilitate precise targeting and the incorporation of multiple functions [41–43]. nano-DDS can exploit characteristics of the ITME as internal stimuli, such as mildly acidic pH [44], elevated glutathione (GSH) levels [45,46], hypoxia [47], and specific enzymatic activities [48], to initiate drug release. Furthermore, nano-DDS can achieve precise spatiotemporal control by responding to external stimuli, including laser irradiation [49], ultrasound [50], magnetic fields [51], and temperature variations [52]. More significantly, by integrating multiple responsive stimuli, multi-responsive nano-DDS can be engineered to mitigate off-target effects, address heterogeneity within lesions, and facilitate sequential therapeutic responses [53–55].
Biomimetic nano-DDS not only reduce systemic clearance but also leverage homing mechanisms to traverse tissue barriers effectively [56,57]. Charge-reversal nanoparticles minimize clearance while enhancing tumor penetration [58], whereas shape-transforming and size-modulating nanoparticles maintain efficient tumor penetration and simultaneously increase tumor retention [39,40,59]. Self-propelled nanomotors overcome mobility constraints inherent to the TME [60], and DNA-based nanorobots offer precise targeting capabilities, high programmability, and deep tissue penetration [61]. Collectively, these ongoing innovations in nano-DDS have substantially advanced the progress of cancer immunotherapy.
In this review, we not only elucidated the benefits of modulating tumor stromal cells and employing nanodrug delivery systems but also established a precise structural framework for designing compatible nano-DDS by thoroughly detailing the immunosuppressive mechanisms linked to each distinct type of tumor stromal cell. We provided a comprehensive summary of tailored nano-DDS designed to utilize these mechanisms, encompassing approaches such as clearance, reprogramming, and inhibition of immunosuppressive cell recruitment. Moreover, we highlighted the integrated paradigm of utilizing nano-DDS to simultaneously modulate multiple cellular populations. Clinically, this review underscored the significance of these strategies as core solutions to overcoming drug resistance in “cold tumors”, effectively alleviating immunosuppression, enhancing the efficacy of existing immune therapeutics such as PD-1/PD-L1 inhibitors, and thereby expanding the patient population that may benefit from immunotherapy. Moreover, this review exemplified the profound interdisciplinary integration of materials science, nanotechnology, immunology, and clinical medicine. Beyond summarizing recent progress, we candidly proposed current challenges, including delivery efficiency, scalability of production, and tumor heterogeneity, identifying critical technical obstacles that must be surmounted by researchers and engineers. This review aims to accelerate the translation of these innovations from laboratory research to clinical application.
Tumor immunity is generally categorized into three distinct phases: elimination, equilibrium, and escape [62]. The development of a tumor immunosuppressive microenvironment predominantly occurs during escape and the later stages of equilibrium. The initial phase of tumor immunity involves the immune system's recognition and eradication of tumor cells that express new antigens (elimination phase). Subsequently, under persistent immune pressure, tumor cells with low immunogenicity are preferentially selected for survival, entering the equilibrium phase. In this phase, these cells are subject to immune surveillance while continuing to evolve at a gradual pace. Ultimately, tumor cells may undergo mutations, such as the loss of antigens and the upregulation of PD-L1 expression, and actively recruit or polarize surrounding tumor stromal cells, thereby collaboratively establishing a robust ITME (escape phase) [63–65].
Key characteristics of this ITME include the presence of CAFs that form a dense ECM, creating physical barriers and releasing inhibitory cytokines [66]; the formation of abnormal blood vessels that contribute to hypoxia and nutrient deprivation [67,68]; and significant infiltration by immunosuppressive cells [69]. Collectively, these elements impede the infiltration of immune cells, suppress their functionality, and induce a state of exhaustion, thereby facilitating evasion of immune detection. Modulating tumor stromal cells presents a promising strategy to dismantle physical barriers, enhance immune cell infiltration, reverse inhibitory signals, and remodel the ITME, ultimately improving the effectiveness of immunotherapy. Next, we will elucidate the roles of various tumor stromal cells in the establishment of the ITME, which is essential for the development of compatible therapeutic strategies (Fig. 1).
Macrophages, a category of phagocytic cells, function as the initial defense line against pathogenic damage to tissues. TAMs represent a specific subtype of macrophages that infiltrate the ITME, constituting over 50% of tumor stromal cells and being the predominant immune cell type in the majority of solid tumors [70]. Some TAMs are derived from circulating Ly6C+C-C-Motif Receptor 2 (CCR2)+ monocytes, which migrate from the bloodstream to tumor tissues in response to specific chemokine signals [71,72]. Others originate from locally resident macrophages within the tumor that are derived from CX3C chemokine receptor 1+Kit+ myeloid progenitor cells [73]. Upon receiving signals from the tumor, these macrophages undergo activation and are subsequently reprogrammed into TAMs. During elimination phase, the majority of TAMs exhibit the M1 phenotype, which is associated with anti-tumor activity. However, as immune editing progresses, M1 phenotype TAMs polarize into the pro-tumor M2 phenotype, influenced by cytokines secreted by the tumor, including IL-4, IL-10, and macrophage colony-stimulating factor 1 (CSF-1) [74,75]. Recent research has indicated that elevated expression of LSM1 (a part of the cytoplasmic protein complex Lsm1–7-Pat1) in tumors is positively correlated with increased infiltration of M2-type TAMs, thereby identifying it as a key accomplice in tumor immune evasion [76].
The primary mechanisms through which TAMs facilitate the development of the ITME are delineated as follows: (1) Direct suppression of adaptive immunity: TAMs exhibit elevated levels of PD-L1, which interacts with PD-1 on T cells, resulting in the downregulation of co-stimulatory molecules such as CD80/CD86 [77,78]. This interaction leads to T cell dysfunction or inactivation. Additionally, TAMs impair T cell CD3ζ chain functionality by secreting Arg 1, degrade tryptophan via IDO to generate kynurenine, which activates the aryl hydrocarbon receptor pathway, thereby inhibiting T cell proliferation [79–82]. Furthermore, they secrete cytokines such as TGF-β and IL-10, which suppress the activation of NK cells and the maturation of dendritic cells (DCs) [83–85]. (2) Recruitment of suppressive immune cells: TAMs are capable of secreting chemokines such as C—C motif ligand (CCL) 22 and IL-10 to attract Tregs [86,87]. They also release IL-10 and TGF-β to promote the proliferation of MDSCs [88–90]. Through the positive feedback loop of CCL2, TAMs continuously recruit monocytes, which subsequently differentiate into M2-type TAMs [91]. (3) Remodeling of the physical barriers: TAMs contribute to aberrant angiogenesis by secreting cytokines such as vascular endothelial growth factor (VEGF), fibroblast growth factor, and platelet-derived growth factor (PDGF), which culminates in the establishment of a hypoxic microenvironment. This process is associated with the upregulation of PD-L1 and Arg 1 expression on TAMs [92]. Additionally, TAMs uptake tumor-derived lactate, which activates G protein-coupled receptor 132 signaling, promoting the secretion of VEGF and IL-10 [93,94]. TAMs can also stimulate fibroblasts to differentiate into CAFs, thereby creating a dense physical barrier that obstructs T cell infiltration [94,95]. (4) Epigenetic stabilization of pro-tumor phenotypes: TAMs inhibit anti-tumor genes by upregulating histone deacetylases and stabilize M2 phenotype epigenetic memory through DNA methylation modifications [96–98].
Tregs represent a specialized subtype of immunosuppressive T cells, distinguished by the expression of CD4+ and CD25+ markers, as well as the upregulation of the pivotal regulatory transcription factor forkhead box protein P3 (FoxP3). The primary role of Tregs is to uphold immune homeostasis by mitigating excessive immune responses. The classification and nomenclature of Tregs have been standardized, encompassing two principal populations: (1) natural Tregs (nTregs), which are generated in the thymus and differentiate into nTregs upon exposure to self-antigens, constituting approximately 5%–10% of the total Treg population; and (2) induced Tregs, which are derived from FoxP3+ cells through stimulation with TGF-β in conjunction with IL-2 or IL-10, representing over 90% of Tregs infiltrating tumors [99,100].
The intricate mechanisms through which Tregs facilitate ITME primarily encompass the following components: (1) Direct contact-mediated immune suppression: Tregs inhibit the activation of effector T cells by engaging with antigen-presenting cells (APCs) via the high expression of CTLA-4, which subsequently suppresses CD28 signaling. Furthermore, Tregs can directly release perforin, forming pores in the membranes of effector T cells, and deliver granzyme B, thereby inducing apoptosis in these cells. (2) Secretion of immunosuppressive cytokine: Tregs produce cytokines such as TGF-β, IL-10, IL-35, and adenosine, which collectively inhibit the proliferation and differentiation of effector T cells. (3) Induction of metabolic exhaustion: Tregs, characterized by the expression of high-affinity IL-2 receptors (CD25), effectively compete for IL-2 uptake, consequently suppressing the activation and survival of effector T cells. Additionally, Tregs promote the expression of IDO, which depletes tryptophan in the TME, further obstructing T cell proliferation. (4) Recruitment or polarization of immunosuppressive cells: Tregs can enhance the expansion of MDSCs through the secretion of immunosuppressive cytokines, and they can also upregulate IDO expression in APCs via CTLA-4, leading to the polarization of macrophages towards the M2 phenotype [101–103].
MDSCs represent a subtype of myeloid cells that are typically generated and released by the bone marrow in an immature form in response to local inflammatory stimuli, such as infection or tissue injury. This response aims to mitigate excessive inflammation and safeguard the host from autoimmune reactions. Tumors exploit this physiological mechanism by enhancing the production and recruitment of MDSCs, thereby establishing the ITME that can inhibit host immune responses, facilitate the formation of new blood vessels, and remodel surrounding tissues with components that support tumor stroma [104,105]. MDSCs are generally categorized into two distinct subpopulations: polymorphonuclear MDSCs (PMN-MDSCs), which constitute approximately 80%–90% of the total MDSCs population and exhibit a phenotype akin to neutrophils (CD11b+ Gr1high/Ly6G+ Ly6Clow), characterized by elevated expression of Arg [106]; and monocytic-like MDSCs (M-MDSCs), which account for 10%–20% of MDSCs and display a phenotype similar to monocytes (CD11b+ Ly6G− Ly6Chigh), with high levels of inducible nitric oxide synthase (iNOS) [107]. The abnormal expansion of these cells is mediated through the signal transducer and activator of transcription 3/5 (STAT3/STAT5) signaling pathway within the bone marrow, thereby contributing to the establishment of an immunosuppressive network [21].
The intricate mechanisms through which MDSCs facilitate the establishment of an ITME can be delineated into several key components: (1) Metabolic reprogramming: PMN-MDSCs exhibit elevated expression of Arg 1, which catalyzes the degradation of arginine, a critical amino acid for T cell functionality, thereby contributing to T cell dysfunction [108]. M-MDSCs synthesize nitric oxide via iNOS, which obstructs the mitochondrial respiratory chain in T cells, ultimately leading to T cell apoptosis [109]. Additionally, the depletion of cysteine and the activity of nicotinamide adenine dinucleotide phosphate oxidase result in the accumulation of reactive oxygen species (ROS), which inflict damage on T cell chemokine receptors, culminating in T cell inactivation or death [110]. (2) Cellular cooperation: The pronounced expression of PD-L1 and galectin-9 facilitates direct interactions with T cell receptors PD-1 and TIM-3, thereby collaborating with Tregs and TAMs to promote T cell exhaustion [111,112]. Furthermore, MDSCs secrete TGF-β and IL-10, which drive the differentiation of naïve CD4+ T cells into Tregs and skew TAMs towards the M2 phenotype, ultimately establishing an immunosuppressive axis involving MDSCs, Tregs, and TAMs [21]. (3) Synergistic neutrophil extracellular traposis (NETosis): Within the ITME, the processes of NETosis and PMN-MDSCs contribute to immunosuppression and tumor progression through a mutually reinforcing cycle. Upon activation by tumor-derived factors such as IL-8 and granulocyte-CSF, PMN-MDSCs can either directly activate neutrophils or undergo NETosis themselves, driven by elevated levels of ROS and granzyme (myeloperoxidase/neutrophil elastase). The damage-associated molecular patterns and pro-inflammatory mediators released from neutrophil extracellular traps (NETs) serve to recruit, amplify, and activate PMN-MDSCs [113–115]. This synergistic interplay engenders a self-perpetuating cycle that exacerbates immunosuppression, fosters tumor progression, and induces tissue damage, thereby emerging as a pivotal factor in tumor immune evasion.
Fibroblasts play a crucial role in maintaining the structure and functionality of normal tissues by synthesizing the ECM and facilitating tissue repair processes [116]. Within the ITME, CAFs represent a highly activated stromal cells that arise from either cellular transformation or phenotypic activation. These CAFs predominantly originate from the aberrant activation of previously quiescent fibroblasts at the tumor site, the differentiation of recruited mesenchymal stem cells following their migration to the tumor, the transformation of epithelial or endothelial cells, and the differentiation of adipocytes or the tumors. CAFs contribute to ECM formation by secreting components such as collagen I, collagen III, fibronectin, and hyaluronic acid, with collagen cross-linking significantly impeding T cell migration by as much as 90% [117]. Furthermore, CAFs secrete various immunosuppressive cytokine, including TGF-β, IL-6, CXCL12, CSF-1, and IL-34, which collectively suppress T cell and NK cell activity, hinder DCs maturation, promote the expansion of Tregs and MDSCs, and facilitate the polarization of TAMs towards the M2 phenotype. Additionally, CAFs can deplete arginine and tryptophan in the ITME via the action of Arg 1 and IDO, which inhibits T cell proliferation and diminishes T cell cytotoxicity through lactate secretion [66,118]. Notably, myosin light chain 9 expressed in CAFs has been shown to modulate the immune microenvironment in colorectal cancer and to promote tumor progression in an autocrine fashion. CAFs also express a variety of immune checkpoint molecules, including upregulated PD-L1/2, CD73/NT5E, and fibroblast activation protein (FAP), all of which effectively inhibit T cell activity [119–121].
CAAs represent atypical adipocytes that undergo differentiation in response to stimuli from tumor cells within the ITME. These cells are predominantly found in adipose tissue infiltrative malignancies, including breast, ovarian, and colorectal cancers. The defining features of CAAs encompass a transformation from large unilocular lipid droplets to smaller multilocular lipid droplets, akin to brown adipose tissue, a decrease in fatty acid synthesis, an increase in lipolysis and fatty acid oxidation, and the secretion of pro-inflammatory mediators, adipokines, and matrix metalloproteinases (MMPs) [122]. This results in a diminished capacity for normal energy storage, with their role shifting towards nutrient provision for tumors and the suppression of antitumor immunity. CAAs primarily arise from the white adipose tissue that surrounds tumors and undergo phenotypic alterations induced by tumor-derived factors [123]. Specifically, VEGF and PDGF secreted by tumors recruit preadipocytes to the invasive tumor front, where they differentiate into CAAs under hypoxic conditions. Adipose stem cells are influenced by an imbalance in leptin and adiponectin levels, leading to aberrant differentiation into a pro-cancer phenotype via peroxisome proliferator-activated receptor gamma signaling [124–126].
CAAs contribute to the establishment of an ITME through various mechanisms, including the elevated expression of fatty acid binding protein 4 (FABP4), which facilitates the transport of free fatty acids (FFAs) to tumor cells, thereby supplying them with β-oxidation substrates that enhance their proliferation and metastatic potential [127,128]. Additionally, CAAs upregulate lipophagy-related genes, such as autophagy-related gene 5, through hypoxia-inducible factor 1-alpha (HIF-1α), enabling the breakdown of their lipid droplets to release cholesterol for tumor membrane synthesis. They secrete IL-6, TGF-β, and leptin, which promote the expansion of Tregs, and exhibit high levels of PD-L1, which directly induces T cell exhaustion [122,129]. Furthermore, CAAs attract MDSCs via chemokines such as CCL2 and CCL5, thereby inhibiting the cytotoxic activities of NK cells and T cells. They also release IL-8 and monocyte chemoattractant protein-1, facilitating the polarization of TAMs towards a M2 phenotype [130–132].
Moreover, Wang et al. elucidated a novel mechanism whereby FFAs are released from CAAs following lipolysis stimulated by tumor-derived secretions. These FFAs are subsequently transferred into tumor cells, enhancing FFA β-oxidation and thereby facilitating tumor progression [133]. CAAs produce lactate and ketone bodies, further contributing to ITME acidification and impairing the functionality of effector immune cells. They downregulate adiponectin, a factor that typically exerts tumor-suppressive effects, thereby alleviating its inhibitory influence on the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway, resulting in an excess of pro-inflammatory mediators. Additionally, CAAs secrete exosomes containing non-coding RNA, such as miR-144 and miR-126, which convey pro-tumor signals to immune cells. They also release MMPs that degrade the ECM, thereby facilitating tumor invasion. Similarly, CAAs can induce the enzyme IDO and secrete Arg, leading to the depletion of tryptophan and arginine, which inhibits T cell proliferation and function. As a pivotal contributor to the tumor progression and metastasis, CAAs foster the development of the ITME through multiple mechanisms, including nutrient provision, cytokines secretion, matrix remodeling, and direct immune cell suppression. Consequently, modulating CAAs with agents such as FABP4 inhibitors, diacylglycerol acyltransferase 1 inhibitors, and leptin receptor antagonists may represent a novel strategy for tumor immunotherapy [133–136].
In tumor immunotherapy, conventional DDS encounter considerable obstacles, including the rapid clearance of drugs, inadequate accumulation at tumor sites, poor penetration to dense barriers, and the off-target effects. In response to these challenges, nano-DDS have emerged as a promising strategy that can significantly improve drug delivery efficiency and enable precise targeting [137–139]. These systems offer several advantages, including responsiveness to the ITME, ease of functional modification for the integration of multiple functions, biodegradability, and the potential to overcome drug resistance [140–142]. As researchers gain a more comprehensive understanding of the ITME and the role of tumor stromal cells, numerous innovative nano-DDS have been developed to specifically modulate tumor stromal cells (Table 1) [143–167].
 DownLoad:
CSV
DownLoad:
CSV
| Nano-DDS | Modulatory or targeting cells | Therapeutic mechanisms | Ref. |
| M2NP | TAMs | Elimination of M2-type TAMs | [147] |
| V(Hb)@DOX | TAMs | Elimination of M2-type TAMs | [148] |
| M@SINPs | TAMs | Transition of M2-type TAMs to the M1 phenotype | [149] |
| BisCCL2/5i | TAMs | Reducing TAMs recruitment | [150] |
| ME@C | TAMs | Targeting M2-type TAMs and activating CD8+ T cells | [151] |
| tLyp1-hNPs | Tregs | Targeting Tregs and inhibiting the phosphorylation of STAT3 and STAT5 | [152] |
| NPsiCTLA-4 | Tregs | Targeting Tregs and diminished the ratio of CD4+ FoxP3+ Tregs | [153] |
| SNPs | Tregs | Targeting Tregs while simultaneously blocking CTLA-4 and PD-1 | [154] |
| Anti–CTLA-4×SIRPα heterodimer | Tregs | Exposing Tregs to macrophage-mediated phagocytosis | [155] |
| Nb-Fc1B | Tregs | Selectively elimination of CCR8+ ti-Tregs | [156] |
| BDM | MDSCs | Enhancing targeting and mitigating the myelotoxic effects of drugs | [157] |
| I-4-P@PssP | MDSCs | Inhibiting MDSCs recruitment | [158] |
| dLy-6C-LAMP | MDSCs | Utilizing the intrinsic homing properties of M-MDSCs to traverse the BBB | [159] |
| Immunecare-DISC | MDSCs | Eliminating MDSCs and activating Toll-like receptor 3 | [160] |
| IEVs-PFD/138 | CAFs | Suppressing CAFs proliferation, migration, and secretion of profibrotic factors | [161] |
| JQ1&PFD@CTL | CAFs | Metabolic reprogramming and ECM degradation | [162] |
| oP@ALG | CAFs | Diminishing ECM deposition | [163] |
| PEG-SAB-Lip | CAFs | Induction of CAF quiescence and ECM degradation | [164] |
| DT-GM1 | TAMs, GSCs | Eliminating or repolarizing M2-type TAMs and eradicating GSCs | [165] |
| NICER | CSCs, tumors | Activation of CD8+ T cells and suppression of YTHDF1 expression | [166] |
| HPA/AS/CQ@Man-EM | TAMs, tumors | Repolarizing M2-type TAMs and elevating ROS levels and reducing VEGF expression | [167] |
In the ITME, the M2-type TAMs represent the predominant category. They possess the capability to shield tumors from the inhibitory influences of adaptive immune responses by means of their expression or secretion of proteolytic enzymes, immunosuppressive agents, and growth factors. Consequently, M2-type TAMs are identified as a primary target for therapeutic interventions aimed at modulating ITME. Common mechanisms for modulating TAMs to reshape the ITME include: eliminating M2-type TAMs, inhibiting TAMs recruitment, reprogramming TAMs towards the M1 phenotype, and mitigating metabolic suppression.
Qian et al. introduced a dual-targeting peptide-lipid nanoparticle (M2NP) specifically designed to target M2-like TAMs by binding to the scavenger receptor (Fig. 2A) [147]. Initially, a bispecific construct was generated by conjugating the α-peptide, a GSG linker, and M2pep, resulting in the compound designated as α-M2pep. Subsequently, leveraging the amphiphilic properties of the α-peptide, α-M2pep was effectively incorporated with phospholipids and a near-infrared dye core to assemble a core–shell fluorescent lipid nanoparticle targeting M2-type TAMs, herein referred to as M2NPs. Moreover, they modified an anti-CSF-1R siRNA with cholesterol (chol-siCD115) to mimic the endogenous delivery patterns of cholesterol by high-density lipoprotein to enhance its in vivo trafficking. M2NPs facilitates the delivery of siRNA aimed at silencing the CSF-1 receptor, leading to the selective eradication of M2-type TAMs within tumors. This intervention significantly diminishes immunosuppressive cytokines, enhances the infiltration of CD8+ T cells, and prolongs survival in melanoma models. Similarly, Wang et al. developed a biomimetic red blood cell nano-DDS (V(Hb)@DOX) that targets the M2-type TAMs marker receptor CD163 to deliver doxorubicin (DOX) (Fig. 2B). This approach effectively eliminates M2-type TAMs, markedly reduces levels of IL-10 and TGF-β, and inhibits metastasis in breast cancer as well as recurrence in colon cancer [148].
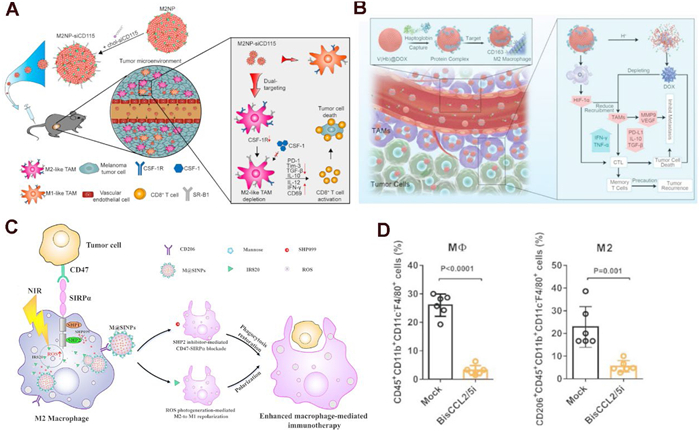
Compared to elimination strategies, polarization strategies aim to reconfigure the immune microenvironment, fostering long-lasting immunological memory and synergizing with ICIs to elicit a more robust and enduring anti-tumor response. Gong et al. created a nano-DDS utilizing albumin nanoparticles (M@SINPs) for the concurrent delivery of the photosensitizer IR820 and the src homology 2 domain-containing protein tyrosine phosphatase (SHP2) inhibitor SHP099 (Fig. 2C) [149]. This system is capable of generating ROS within TAMs upon laser irradiation, thereby facilitating the transition of M2-type TAMs to the M1 phenotype. Furthermore, the inhibition of SHP2 disrupts the CD47-signal regulatory protein alpha signaling pathway, thereby restoring the phagocytic activity of M1-type macrophages.
Wang et al. developed a method to encapsulate the mRNA of the BisCCL2/5i antibody within a clinically approved lipid nanoparticle platform, which selectively binds to and neutralizes CCL2 and CCL5. This intervention significantly reduces TAMs recruitment and effectively mitigates immune suppression (Fig. 2D) [150]. When BisCCL2/5i mRNA was combined with PD-1 ligand inhibitors, it resulted in prolonged survival in murine models of primary liver cancer, colorectal cancer, and pancreatic cancer. Nevertheless, these therapeutic strategies frequently neglect the potential role of M2-type TAMs as APCs. Cui et al. demonstrated that M2-type TAMs possess elevated lysosomal cysteine protease activity compared to other APCs, and they discovered that inhibiting this enzymatic activity could substantially enhance antigen cross-presentation and activate CD8+T cells [168]. Building on this finding, Qiao et al. employed mesoporous silica nanoparticles loaded with the cysteine protease inhibitor E64, which were coated with galactose ligand-modified cancer cell membranes derived from surgical tumors, for postoperative cancer immunotherapy [151]. This innovative nano-DDS specifically targets M2-type TAMs and facilitates the release of E64 within lysosomes, thereby enhancing antigen cross-presentation and directly activating CD8+ T cells, which in turn inhibits the proliferation of B16-OVA melanoma. Following synergistic interaction with ICIs, this method also effectively curtailed postoperative tumor recurrence in 4T1 tumors, offering novel insights into TAMs-modulated immunotherapy.
While Tregs are crucial for sustaining immune homeostasis and mitigating autoimmunity, their immunosuppressive effects within the ITME are recognized as a significant mechanism facilitating tumor immune evasion. Consequently, Tregs are regarded as a critical target for nano-DDS designed to reverse immunotherapeutic strategies. Current immunotherapeutic strategies modulating Tregs include various approaches: the direct elimination of Tregs, and inhibiting Tregs recruitment.
Ou et al. have developed a hybrid nanoparticle conjugated with the tLyp1 peptide, specifically designed to target Tregs (Fig. 3A) [152]. This nanoparticle enhances the downregulation of Tregs-mediated suppression by imatinib through the inhibition of phosphorylation of STAT3 and STAT5. Furthermore, CTLA-4 is another prevalent target for Tregs modulation. Li et al. synthesized nanoparticles (NPsiCTLA-4) capable of encapsulating CTLA-4 small interfering RNA utilizing biocompatible and biodegradable PEG-PLA copolymers and BHEM-Chol (Fig. 3B) [153]. The NPsiCTLA-4 significantly diminished the ratio of CD4+ FoxP3+ Tregs and effectively inhibited tumor growth in murine models of melanoma, thereby extending survival time. Zhang et al. developed hybrid spherical nucleotide nanoparticles (SNPs) loaded with a CTLA-4 siRNA aptamer (cSNP) and PD-1 siRNA (pSNP) (Fig. 3C) [154]. These hybrid SNPs demonstrated a synergistic immune-stimulating effect by targeting Tregs while simultaneously blocking CTLA-4 and PD-1, which elicited a robust anti-tumor immune response that ultimately suppressed the growth of melanoma tumors and colorectal adenocarcinomas in mice.
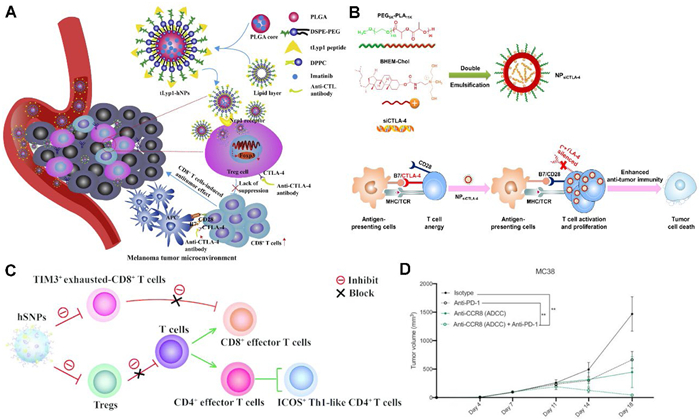
Notably, some researchers have engineered a nanodrug that precisely targets Tregs by exploiting the overexpression of CD47 (the “don't eat me” signal) and CTLA-4. Zhang et al. designed a molecular clamp structure comprising an anti-CTLA-4×signal regulatory protein α (SIRPα) heterodimer [155]. This molecular clamp effectively binds to tumor Tregs via the CTLA-4 antibody, while SIRPα inhibits the CD47, thereby exposing Tregs to macrophage-mediated phagocytosis. The Fc region on the connecting arm activates Fc receptors on macrophages, initiating phagocytosis and ultimately leading to the clearance of Tregs, thereby simultaneously dismantling their “don't eat me” defense and activating the “eat me” signal in macrophages. This approach enhances the suppressive tumor immune microenvironment, inhibits tumor growth at only low doses, and fosters long-term immune memory.
The presence of the aforementioned targets in not only Tregs but also in other critical immune cell types, such as effector T cells, may result in significant adverse effects. In addition to the previously discussed molecular clamp strategy, researchers have identified a subset of highly activated Tregs (ti-Tregs), which exhibit the most potent immunosuppressive properties in murine tumor models (including lung cancer, colon cancer, and melanoma) as well as in human tumor samples (notably non-small cell lung cancer and melanoma). These ti-Tregs are characterized by the elevated expression of the CCR8 receptor protein, which is upregulated via the NF-κB signaling pathway. In light of this, Damme et al. conducted a screening for nanobodies that target various epitopes of the CCR8 extracellular domain, utilizing immunized llamas [156]. They subsequently developed two bispecific tetravalent Nb-Fc therapeutic nanoproducts, designated Nb-Fc1A and Nb-Fc1B. Nb-Fc1A is designed solely to inhibit CCR8 signaling, whereas Nb-Fc1B additionally facilitates NK cell-mediated cytotoxicity. The findings demonstrate that Nb-Fc1B can selectively eliminate CCR8+ ti-Tregs within the ITME with a clearance efficiency exceeding 90%, without adversely affecting peripheral Tregs or CD4+FoxP3⁻ T cells. Furthermore, when combined with anti-PD-1 therapy, Nb-Fc1B was capable of completely eradicating tumors and establishing long-term immune memory in the MC38 model (Fig. 3D).
MDSCs play a crucial role in the immune suppression associated with tumors. These cells are capable of inhibiting the activity of various immune cells, including CD8+ T cells, NK cells, and B cells. Furthermore, MDSCs can recruit Tregs and induce the polarization of TAMs towards the M2 phenotype. They also contribute to tumor angiogenesis, facilitate the process of epithelial-mesenchymal transition, secrete MMPs, and can differentiate into osteoclasts, all of which enhance the invasion and metastasis of tumor cells. Consequently, a variety of immunotherapeutic strategies modulating MDSCs have been developed. These strategies can be broadly classified into four categories: (1) Inhibiting the functional activity of MDSCs; (2) directly eliminating MDSCs; (3) preventing the recruitment of MDSCs; and (4) inducing the differentiation of MDSCs into mature myeloid cells.
In recent years, black phosphorus (BP) has garnered significant attention due to its distinctive electronic structure and broad absorption peak within the visible light spectrum, which confer strong singlet oxygen generation capacity and superior photothermal properties. Lan et al. synthesized BP nanosheets with 200 nm, subsequently loading them with the chemotherapeutic agent gemcitabine (GEM). The nanosheets were then coated with MDSCs membranes, resulting in a composite denoted as BP@decitabine (Dacogen)@MDSCs (BDM) (Fig. 4A) [157]. Membrane proteins derived from MDSCs confer upon BDM the capability to enhance targeting and mitigate the myelotoxic effects of decitabine (Dacogen). Concurrently, the photothermal and photodynamic properties of BP facilitate hyperthermia and ROS-induced mitochondrial damage, while also enhancing antitumor immunity through immunogenic cell death. Furthermore, the localized administration of decitabine (Dacogen) promotes tumor cell demethylation and induces cell cycle arrest. The results demonstrated a substantial reversal of ITME alongside a notable decrease in tumor progression (Fig. 4B). This integrative multi-modal therapeutic strategy, combining photothermal therapy, photodynamic therapy, chemotherapy, and immunotherapy, offers a promising avenue for effective cancer treatment.
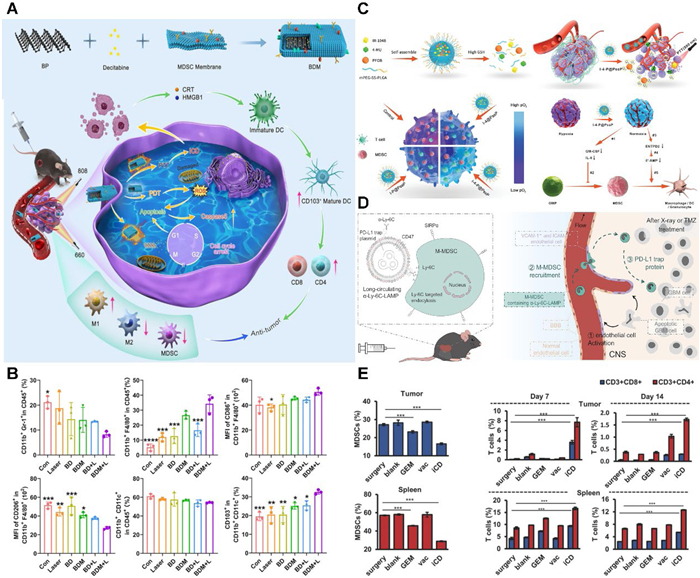
Hypoxia is known to induce HIF-1α, which subsequently upregulates ENTPD2, facilitating MDSCs maturation, while also enhancing the production of GM-CSF and IL-6 to promote the differentiation of bone marrow granulocyte-macrophage progenitor cells into MDSCs. This underscores tumor hypoxia as a critical factor driving MDSCs recruitment. In response, Yang et al. developed a GSH-responsive amphiphilic polymer nanoparticle (I-4-P@PssP), which comprises a tumor GSH-responsive mPEG-ss-poly(lactic-co-glycolic acid carrier, the photothermal drug IR-1048, the oxygen-carrying contrast agent perfluorooctyl bromide (PFOB), and 4-methylumbelliferone (4-MU), which inhibits hyaluronic acid synthesis (Fig. 4C) [158]. I-4-P@PssP enhances the hypoxic ITME through direct oxygen delivery from PFOB and sustained oxygen delivery resulting from 4-MU-induced vascular normalization, thereby significantly inhibiting MDSCs recruitment. Following combined treatment with photothermal therapy, the primary tumor volume was reduced by 90%, and metastatic nodules in the lungs and liver were decreased by 85%, resulting in a 2.3-fold increase in survival.
Moreover, Yu et al. have innovatively developed a dLy-6C-LAMP nano-DDS that actively recruits and enriches M-MDSCs post-radiotherapy and chemotherapy (Fig. 4D) [159]. This system effectively targets peripheral M-MDSCs, leveraging their inherent homing capabilities to deliver therapeutic genes directly into glioblastoma (GBM). It facilitates the delivery of a secretory PD-L1 Trap protein that broadly inhibits the PD-1/PD-L1 pathway, thereby effectively activating anti-tumor immunity, significantly reducing recurrence, and prolonging survival. The approach of utilizing endogenous cells as carriers to traverse biological barriers for targeted delivery offers novel insights for the treatment of other brain diseases that are protected by the blood-brain barrier (BBB).
Lim et al. have engineered a three-dimensional scaffold, termed immuneCare-DISC, aimed at enhancing anti-tumor immunity through dual mechanisms [160]. Firstly, GEM is utilized as an agent to eliminate MDSCs, thereby mitigating immune suppression within the ITME. Secondly, the system employs whole tumor lysate antigens in conjunction with nano-adjuvants that carry Toll-like receptor 3 agonists, functioning as a cancer vaccine to bolster immune stimulation. Additionally, a nano-gel containing poly(I:C) has been introduced as a nano-adjuvant to elicit a robust cellular immune response. The findings demonstrated that immuneCare-DISC effectively eliminates both tumor and splenic MDSCs via GEM, alleviates T cell suppression, activates DCs maturation through the vaccine, promotes the secretion of inflammatory cytokines such as interferon-γ and tumor necrosis factor-α, and significantly increases the populations of CD4+ and CD8+ T cells in both the tumor and spleen (Fig. 4E). In the 4T1 model, 25% of the mice exhibited no tumor recurrence, 100% survived beyond 30 days, and 60% showed no metastatic nodules.
Compared to other tumor stromal cells, CAFs have emerged as crucial therapeutic targets for modulating the ITME due to their spatial heterogeneity, metabolic reprogramming capacity, and dynamic plasticity [169]. Strategies modulating CAFs typically reverse the ITME through three primary mechanisms: disrupting physical barriers, inhibiting the secretion of immunosuppressive factors, and regulating metabolic reprogramming.
Bone marrow mesenchymal stem cells (BMSCs) possess inherent antifibrotic properties are readily taken up by CAFs. Zhou et al. developed engineered extracellular vehicles (EVs) derived from BMSCs, which were surface-modified with an integrin α5-targeting peptide to enhance their specific enrichment in CAFs [161]. They were co-loaded with miR-138–5p (a miRNA inhibiting CAFs activation) and pirfenidone (PFD, an antifibrotic small-molecule drug), forming IEVs-PFD/138 (Fig. 5A). Results demonstrated that integrin α5 peptide modification doubled the enrichment efficiency of miR-138–5p in CAFs, significantly suppressing CAFs proliferation, migration, and secretion of profibrotic factors (TGF-β, IL-6, CXCL12), while reducing tumor stiffness and solid stress. Furthermore, the released PFD directly inhibited TGF-β expression, augmenting the antifibrotic effect. IEVs-PFD/138 combined with GEM significantly enhanced therapeutic efficacy across three mouse models (subcutaneous, orthotopic, patient-derived tumor xenograft), providing strong evidence for clinical translation.
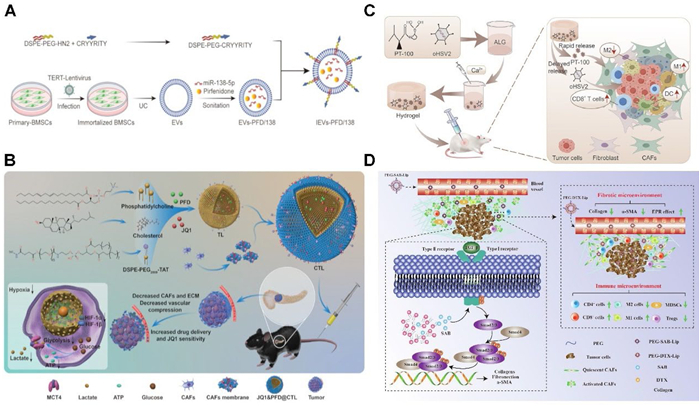
Research has revealed that in pancreatic cancer, CAFs within the ITME induce the formation of a dense stromal barrier (occupying up to 50% of the tumor volume), which impedes drug penetration and promotes chemoresistance. Additionally, CAFs have been shown to upregulate bromodomain-containing protein 4 expression, thereby diminishing the efficacy of the BET inhibitor JQ1. Consequently, targeting CAFs within the pancreatic ITME holds potential not only to enhance the sensitivity of cancer cells to JQ1 but also to improve drug perfusion and oxygen delivery, leading to reduced glycolysis and constrained energy supply. Building on this premise, Zhang et al. developed a nano-DDS named CTL comprising liposomes modified with TAT cell-penetrating peptides and coated with CAFs-derived cell membranes, designed to co-deliver PFD and JQ1 (Fig. 5B) [162]. The JQ1&PFD@CTL formulation exhibited robust CAFs-targeting capabilities both in vitro and in vivo. Through mechanisms involving metabolic reprogramming and ECM degradation, treatment with JQ1&PFD@CTL achieved a pronounced antitumor effect, with an inhibition rate of 79.79%.
Oncolytic viruses (OVs) elicit immune responses through their selective infection of tumor cells; however, following intratumoral administration, they frequently disseminate into healthy tissues, such as the liver, resulting in toxicity. Furthermore, the ECM produced by CAFs generates elevated stromal pressure, which impedes both OVs penetration and immune cell infiltration. Addressing this, Hou et al. developed a calcium ion-responsive sodium alginate (ALG) hydrogel encapsulating a rapidly released small-molecule (the FAP inhibitor PT-100, targeting CAFs) and a sustained-release component (the oncolytic HSV-2 virus, oH2), forming oP@ALG (Fig. 5C) [163]. Their findings demonstrated that intratumoral injection of oP@ALG induced calcium ion-mediated formation of a stable porous matrix, enhancing intratumoral retention of oH2 by 18-fold, while PT-100 effectively suppressed CAFs proliferation. In a subcutaneous tumor model, treatment with oP@ALG significantly diminished ECM deposition and inhibited tumor progression, achieving a tumor inhibition rate of 73.5%.
Bioactive compounds derived from traditional Chinese herbal medicine are increasingly recognized for their role in cancer immunomodulation. Salvianolic acid B (SAB) exhibits multiple therapeutic properties, including anticancer, anti-inflammatory, antioxidant, and anti-fibrotic effects. Building on these attributes, Chen et al. developed a PEGylated liposomal formulation encapsulating SAB (PEG-SAB-Lip) aimed at modulating and inactivating CAFs (Fig. 5D) [164]. This strategy seeks to enhance nano-DDS efficiency, remodel the ITME, and improve the efficacy of chemotherapy and immunotherapy. Their findings demonstrated that PEG-SAB-Lip effectively inhibited TGF-β secretion and downregulated Smad 2/3 phosphorylation, leading to reduced α-smooth muscle actin expression and induction of CAF quiescence. Consequently, ECM deposition was diminished, facilitating improved nanodrug penetration. Furthermore, the treatment significantly increased intratumoral infiltration of CD8+ and CD4+ T cells, M1-type TAMs, as well as pro-inflammatory cytokines interferon-γ and tumor necrosis factor-α. Concurrently, there was a marked reduction in immunosuppressive populations, including MDSCs, Tregs, and the cytokines IL-6 and IL-10, collectively indicating effective remodeling of the ITME.
The ITME constitutes a complex, integrated system in which tumor cells, stromal cells, immune cells, and their diverse signaling molecules interact intricately rather than functioning in isolation. Modulation of a single subtype of tumor stromal cells can result in compensatory mechanisms by other tumor stromal cell populations, which may upregulate alternative factors or signaling pathways. This compensatory response can diminish the efficacy of the therapeutic intervention and potentially contribute to the development of treatment resistance. Consequently, by harnessing the synergistic mechanisms of multiple regulation, developing multifunctional nano-DDS capable of simultaneously modulating various cells can induce antitumor immune responses and effectively overcome treatment resistance to a degree that markedly exceeds the outcomes attained through single-modulation therapeutic strategies. For instance, s(DGL)n@Apt effectively addresses physical drug resistance and immune suppression associated with the dense stromal environment of pancreatic cancer by concurrently modulating ECM and MDSCs [170]. Similarly, IR780-peptide nanocomposites selectively target and modulate CAFs via a photothermal mechanism, thereby disrupting the cancer cell niche and preventing tumor recurrence and drug resistance [171]. Additionally, Wan et al. have developed a strategy that simultaneously delivers a STING agonist and a CCR2 antagonist, which activates the STING signaling pathway while inhibiting the CCL2/CCL7–CCR2 axis. This dual modulation regulates DCs, TAMs, and MDSCs, ultimately overcoming the immune resistance of pancreatic ductal adenocarcinoma [172]. The design of effective multicellular synergistic modulatory nano-DDS is guided by three fundamental principles: (1) Synergistic modulating, wherein the selection of multiple modulatory sites is deliberate and based on their biological synergism or sequential interactions, enabling bidirectional amplification of therapeutic efficacy. (2) Spatiotemporally controlled release, which necessitates that the nano-DDS can programmably and precisely release multiple therapeutic agents in accordance with specific functional demands. For instance, incorporating responsive segments with varying response speed allows for the initial release of ECM-degrading agents, followed by drugs that modulate tumor stromal cells or stimulate immune responses, thereby enhancing overall treatment outcomes. (3) Safety considerations, emphasizing the use of biocompatible and biodegradable materials to ensure that the nanocarrier can be safely metabolized and eliminated post-therapy, thus minimizing risks of inflammation or immune reactions associated with prolonged retention.
Yang et al. engineered a dual-targeting nano-DDS (DT-GM1) composed of self-assembled monosialotetrahexosylganglioside (GM1) and DSPE-PEG, designed as a carrier for the DOX (Fig. 6A) [165]. In both in vitro and in vivo models of GBM, DT-GM1 demonstrated the capacity to traverse the BBB passively, attributed to its lipid-based characteristics. Furthermore, it exhibited precise targeting of M2-type TAMs and glioblastoma stem cells (GSCs) via the M2pep peptide and PTPRZ1 antibody, respectively. This strategy effectively eliminates M2-type TAMs and GSCs, thereby inhibiting the PTN-PTPRZ1 signaling pathway. Concurrently, it disrupts the tumor-promoting feedback loop between these cell populations and facilitates the polarization of TAMs from the M2 to the M1 phenotype. Such remodeling of the ITME presents a new direction for targeted immunotherapeutic interventions against GBM.
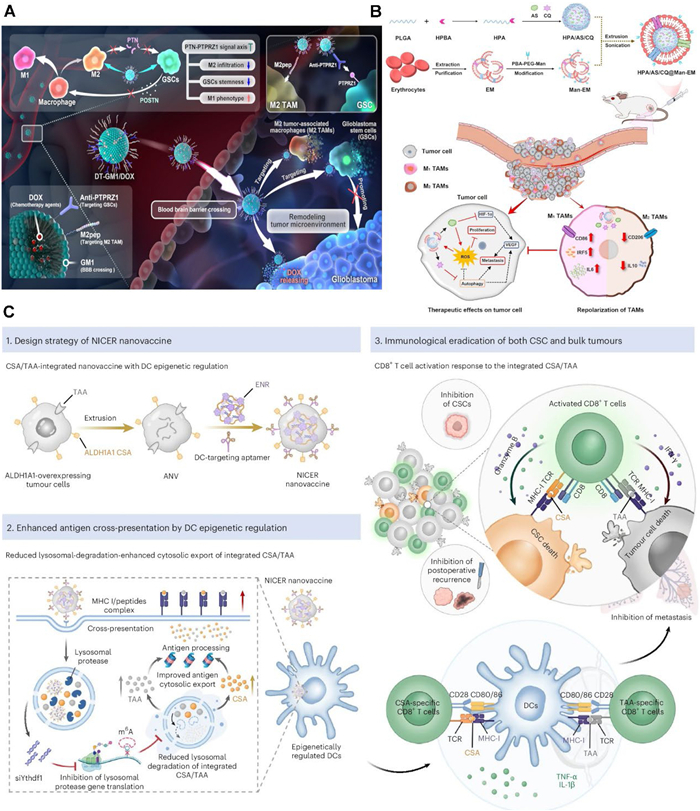
The treatment of colorectal cancer is often constrained by insufficient consideration of the ITME, particularly the M2-type TAMs. The antimalarial agents artesunate (AS) and chloroquine (CQ) exhibit broad-spectrum anticancer properties. AS exerts its antitumor effects by inducing ROS, apoptosis, and ferroptosis, whereas CQ inhibits autophagy and facilitates the repolarizing of M2-type TAMs into pro-inflammatory M1-type TAMs. Building on these mechanisms, Peng et al. engineered a dual-modulated nanodelivery system (HPA/AS/CQ@Man-EM), which encapsulated both AS and CQ and effectively modulated tumor cells as well as TAMs (Fig. 6B) [167]. Results in vitro and in vivo revealed that HPA/AS/CQ@Man-EM impedes tumor cell proliferation and angiogenesis by elevating ROS levels and reducing VEGF expression. Concurrently, it promotes the repolarization of M2-type TAMs to the M1 phenotype, thereby remodeling the ITME. This work provides a promising prospect in the development of clinical regimens for CRC treatment.
A recent investigation highlights that postoperative recurrence and metastasis of solid tumors constitute the principal factors contributing to treatment failure, predominantly due to the persistence of residual cancer stem cells (CSCs). To mitigate this challenge, You et al. introduced a dual-targeting nanovaccine approach termed NICER (Nanovaccine with Integrated CSA/TAA and Epigenetic Regulation) (Fig. 6C) [166]. This strategy concurrently targets CSC-specific antigens (CSA) and tumor-associated antigens (TAAs), thereby eliciting activation of both CSCs-specific CD8+ T cells and conventional tumor cell-specific CD8+ T cells. Furthermore, upon uptake by DCs, the siRNA targeting YTH N6-methyladenosine binding protein 1 (YTHDF1) effectively silences YTHDF1 expression, which normally suppresses lysosomal protease activity in DCs, thus enhancing antigen cross-presentation efficiency. The results demonstrated a 90% reduction in tumor-infiltrating ALDH1A1high CSCs and an over 80% decrease in tumor volume in the 4T1 model. Moreover, in spontaneous metastasis tumor and postoperative recurrence models, the NICER treatment group exhibited pronounced anti-metastatic and anti-recurrence efficacy.
Tumor stromal cells are widely recognized for their pivotal role in modulating the ITME and contributing significantly to resistance against anti-tumor immunotherapies. The application of nano-DDS to modulate these stromal cells represents a strategically important approach in tumor immunotherapy. Nano-DDS not only surmount immunosuppressive barriers and provide precise spatiotemporal control but also facilitate the integration of multi-targeted and multi-modulated therapeutic strategies, thereby improving the efficacy of tumor immunotherapy. Recent advancements in nanoengineering-based immunotherapeutic strategies aimed at stromal cells and ITME reprogramming have yielded promising outcomes, presenting numerous potential avenues for clinical translation.
However, nanodelivery systems designed to modulate tumor stromal cells continue to encounter multifaceted challenges. Conventional nanocarriers predominantly depend on the enhanced permeability and retention effect to facilitate tumor accumulation, but clinical evidence indicates that merely 0.7% of the administered dose successfully localizes to the tumor site. Investigations in pancreatic cancer models reveal that only approximately 21% of endothelial cells engage in nanocarrier transport through caveolae-mediated transcytosis, leading to heterogeneous drug distribution within the ITME. Furthermore, batch-to-batch variability in nanomedicine production constitutes a significant challenge to clinical translation. For instance, slight discrepancies during the synthesis of mannose-modified nanoparticles (MnCNPs) can result in substantial alterations in nanoparticle properties, thereby impacting their therapeutic efficacy and safety profiles. Achieving consistent physicochemical properties of MnCNPs, particularly regarding particle size, surface functionalization, and drug encapsulation efficiency, during large-scale manufacturing remains a critical challenge. Additionally, targeting ligands, which are essential for specificity, are susceptible to degradation or detachment during storage and transportation, raising concerns about the stability and shelf life of these formulations.
Beyond the intrinsic limitations of the nanomedicines themselves, the heterogeneity of the tumor stroma cells and the ITME further compromises the therapeutic effectiveness of the nano-DDS. Considerable variability exists in the phenotypic characteristics of stromal cells across different tumor types. For instance, CD206+ M2-type TAMs are frequently observed in certain solid tumors, whereas in gliomas, CD163+ M2-type TAMs constitute the predominant infiltrating immune cell population. This phenotypic heterogeneity poses challenges for the development of nanotechnology-based therapeutic systems with broad-spectrum efficacy. Furthermore, the modulation of various stromal cell populations encounters several significant challenges. Approaches aimed at targeting TAMs and Tregs often suffer from inadequate specificity. For instance, many receptors employed to identify M2-polarized TAMs, such as CD206 and scavenger receptors, are also expressed at low levels on other immune cells, including DCs, as well as on certain non-immune cells. Similarly, principal targets of Tregs, such as CTLA-4 and CD25, are also present on effector T cells, particularly activated effector T cells. Consequently, interventions directed against these molecules risk inadvertently impairing antitumor immune responses by affecting cells that are essential for immune activation, thereby diminishing therapeutic efficacy. Moreover, Tregs are crucial for maintaining peripheral immune tolerance and preventing autoimmune pathologies. Their depletion or functional inhibition in non-target tissues can provoke severe autoimmune adverse effects, including colitis, dermatitis, and hypophysitis, representing a major barrier to the clinical translation of such therapies. The metabolic suppression mechanisms employed by MDSCs are complex and redundant; thus, inhibition of a single metabolic pathway may induce compensatory upregulation of alternative pathways. Additionally, given the persistent supply of MDSCs, selective elimination of these cells either in peripheral tissues or within the tumor microenvironment yields limited therapeutic benefit. Excessive disruption of CAFs can paradoxically promote tumor metastasis and compromise the integrity of the tumor vasculature. Although strategies involving the coordinated modulation of multiple cell types may mitigate some of these limitations, their inherent complexity and the potential for unpredictable systemic toxicities pose significant challenges. In addition, interpatient variability further complicates the accurate prediction of therapeutic responses. The limitations inherent in preclinical models, such as discrepancies between animal models and human disease pathology, as well as insufficient replication of the ITME, have contributed to the unsuccessful clinical translation of numerous promising nanomedicines.
Currently, nano-immunotherapy modulating tumor stromal cells is at a pivotal juncture, transitioning from fundamental research toward clinical implementation. The existing challenges represent both obstacles and opportunities for innovation within the field. An optimal future nano-DDS should integrate efficient delivery mechanisms, ITME remodeling, and real-time monitoring capabilities. The incorporation of computational science-assisted design holds significant potential to accelerate the development timeline. Overcoming heterogeneity barriers necessitates precise classification based on patient-specific stromal characteristics. The application of artificial intelligence and big data analytics, in conjunction with multi-omics profiling and radiomic features, to develop personalized delivery strategies that inform clinical decision-making constitutes a promising direction for future research. Furthermore, from an industrial perspective, the advancement of innovative manufacturing processes aimed at cost reduction is imperative to enhance the accessibility of these advanced therapies to a wider patient population. Achieving successful clinical translation demands multidisciplinary collaboration and the development of novel methodologies. Additionally, synergistic cooperation across diverse industries and scientific domains is essential to realize the full therapeutic potential of this emerging technology for cancer patients.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jiayi Sun: Data curation. Luyao Huang: Writing – original draft. Wenfeng Jia: Writing – review & editing. Yitong Liu: Visualization. Li Xiang: Formal analysis. Xing Yang: Investigation. Fan Tong: Writing – review & editing. Xiaobo Wang: Methodology. Huile Gao: Conceptualization. Yi Zhang: Funding acquisition.
This work was supported by the National Key Research and Development Program of China (No. 2023YFC3504402) and Sichuan Science and Technology Program (Nos. 2025ZNSFSC1806, 2024NSFSC0701).
J.C. Del Paggio, Nat. Rev. Clin. Oncol. 15 (2018) 268–270. doi: 10.1038/nrclinonc.2018.27
P.N. Kelly, AAAS Sel. Symp. 359 (2018) 1344–1345. doi: 10.1126/science.359.6382.1344
M. Zhang, C. Liu, J. Tu, et al., Mol. Cancer 24 (2025) 136. doi: 10.1007/978-981-97-8812-5_14
H.C. Nauts, W.E. Swift, B.L. Coley, Cancer Res. 6 (1946) 205–216.
M. Burnet, Br. Med. J. 1 (1957) 779. doi: 10.1136/bmj.1.5022.779
G. Köhler, C. Milstein, Nature 256 (1975) 495–497. doi: 10.1038/256495a0
A. van Elsas, A.A. Hurwitz, J.P. Allison, J. Exp. Med. 190 (1999) 355–366. doi: 10.1084/jem.190.3.355
M.A. Cheever, C.S. Higano, Clin. Cancer. Res. 17 (2011) 3520–3526. doi: 10.1158/1078-0432.CCR-10-3126
P.W. Huang, J.W. Chang, Biomed. J. 42 (2019) 299–306. doi: 10.1016/j.bj.2019.09.002
T. Yau, Y.K. Kang, T.Y. Kim, et al., JAMA Oncol. 6 (2020) e204564. doi: 10.1001/jamaoncol.2020.4564
J.M. Llovet, F. Castet, M. Heikenwalder, et al., Nat. Rev. Clin. Oncol. 19 (2022) 151–172. doi: 10.1038/s41571-021-00573-2
A.X. Zhu, R.S. Finn, J. Edeline, et al., Lancet Oncol. 19 (2018) 940–952. doi: 10.1016/S1470-2045(18)30351-6
L. Peng, G. Sferruzza, L. Yang, L. Zhou, S. Chen, Cell. Mol. Immunol. 21 (2024) 1089–1108. doi: 10.1038/s41423-024-01207-0
Q. Sun, Y. Zhu, D. Zhao, et al., T Technol. Cancer. Res. Treat. 24 (2025) 15330338251356387. doi: 10.1177/15330338251356387
J. Yu, M. Li, B. Ren, et al., Front. Pharmacol. 14 (2023) 1261575. doi: 10.3389/fphar.2023.1261575
Q. Chen, T. Sun, C. Jiang, Nano-micro Lett. 13 (2021) 92. doi: 10.1007/s40820-021-00622-6
Z. Wang, K. Chen, Brain-X 1 (2023) e33. doi: 10.1002/brx2.33
H. Jia, X. Chen, L. Zhang, M. Chen, J. Hematol. Oncol. 18 (2025) 36. doi: 10.1186/s13045-025-01688-0
Y. Xu, J. Xiong, X. Sun, H. Gao, Acta Pharm. Sin. B 12 (2022) 4327–4347. doi: 10.1016/j.apsb.2022.11.001
M. Certo, C.H. Tsai, V. Pucino, P.C. Ho, C. Mauro, Nat. Rev. Immunol. 21 (2021) 151–161. doi: 10.1038/s41577-020-0406-2
S. He, L. Zheng, C. Qi, Mol. Cancer 24 (2025) 5. doi: 10.1109/cisat66811.2025.11181725
H. Cabral, H. Kinoh, K. Kataoka, Acc. Chem. Res. 53 (2020) 2765–2776. doi: 10.1021/acs.accounts.0c00518
H. Gao, Curr. Drug Metab. 17 (2016) 731–736. doi: 10.2174/1389200217666160630203600
D. Schmid, C.G. Park, C.A. Hartl, et al., Nat. Commun. 8 (2017) 1747. doi: 10.1038/s41467-017-01830-8
N. Kim, H.H. Lee, H.J. Lee, et al., Arch. Pharm. Res. 42 (2019) 591–606. doi: 10.1007/s12272-019-01143-y
Y. Cheng, X.J. Han, X.T. Lai, X.W. Wei, Phytomedicine 135 (2024) 156093. doi: 10.1016/j.phymed.2024.156093
Z.Z. Lu, L. Ma, L. Mei, et al., Int. J. Pharm. 628 (2022) 122303. doi: 10.1016/j.ijpharm.2022.122303
F. Tong, H.L. Hu, Y.Y. Xu, et al., Acta Pharm. Sin. B 13 (2023) 3471–3488. doi: 10.1016/j.apsb.2022.11.003
R. Malla, L. Pappu, K.C. Amajala, M.A. Kamal, Curr. Med. Chem. 29 (2022) 6197–6216. doi: 10.2174/0929867329666220324143215
H. Deng, G. Wang, S.Y. Zhao, et al., Front. Pharmacol. 14 (2023) 1228962. doi: 10.3389/fphar.2023.1228962
Y.H. Wei, R.W. Li, Y.S. Wang, et al., Int. J. Nanomed. 19 (2024) 10129–10144. doi: 10.2147/IJN.S466315
Y.K. Li, X.D. Shen, H.T. Ding, et al., Acta Pharm. Sin. B 14 (2024) 3680–3696. doi: 10.1016/j.apsb.2024.03.010
D.D. Wan, Y.L. Yang, Y.Y. Liu, et al., J. Control. Release 317 (2020) 43–56. doi: 10.1016/j.jconrel.2019.11.011
S.Q. He, L.L. Wang, D.X. Wu, et al., Acta Pharm. Sin. B 14 (2024) 765–780. doi: 10.1016/j.apsb.2023.10.006
K.C. Kao, S. Vilbois, C.H. Tsai, P.C. Ho, Nat. Cell. Biol. 24 (2022) 1574–1583. doi: 10.1038/s41556-022-01002-x
M.S. Goldberg, Nat. Rev. Cancer 19 (2019) 587–602. doi: 10.1038/s41568-019-0186-9
Y. Luo, X. He, Q. Du, et al., Exploration 4 (2024) 20230134. doi: 10.1002/EXP.20230134
Y. Jin, Y. Huang, H. Ren, et al., Biomaterials 305 (2024) 122463. doi: 10.1016/j.biomaterials.2023.122463
W. Jia, Y. Wang, R. Liu, X. Yu, H. Gao, Adv. Funct. Mater. 31 (2021) 2009765. doi: 10.1002/adfm.202009765
W. Yu, R. Liu, Y. Zhou, H. Gao, ACS Cent. Sci. 6 (2020) 100–116. doi: 10.1021/acscentsci.9b01139
W. Jia, B. Gong, J. Chen, et al., Adv. Funct. Mater. 34 (2024) 2408581. doi: 10.1002/adfm.202408581
L.L. Wang, S.S. He, R. Liu, et al., Acta Pharm. Sin. B 14 (2024) 2263–2280. doi: 10.1016/j.apsb.2023.12.001
C. Yang, H. Ming, B.W. Li, et al., J. Control. Release 376 (2024) 659–677. doi: 10.1016/j.jconrel.2024.10.043
X. He, J. Xie, J. Zhang, et al., Adv. Sci. 9 (2022) 2104286. doi: 10.1002/advs.202104286
Q. Yang, G. Wu, Y. Yang, et al., Adv. Funct. Mater. 34 (2024) 2402194. doi: 10.1002/adfm.202402194
Y. Wang, F. Yang, J. Wang, et al., Chin. Chem. Lett. (2025), doi: 10.1016/j.cclet.2025.111771.
Z. Li, P. Liu, W. Chen, et al., J. Nanobiotechnology 21 (2023) 221. doi: 10.1186/s12951-023-01939-7
W. Jia, R. Liu, Y. Wang, et al., Acta Pharm. Sin. B 12 (2022) 3354–3366. doi: 10.1016/j.apsb.2022.03.010
C. Hu, T. Lei, Y. Wang, et al., Biomaterials 255 (2020) 120159. doi: 10.1016/j.biomaterials.2020.120159
Z. Chen, L. Sang, Z. Qixi, et al., Mater. Today Bio 32 (2025) 101661. doi: 10.1016/j.mtbio.2025.101661
Y. Zhang, J. Li, P. Habibovic, Bioact. Mater. 15 (2022) 372–381.
Z. Zhang, Y. Wang, M.M. Rizk, et al., Biomater. Adv. 134 (2022) 112716. doi: 10.1016/j.msec.2022.112716
R. Cai, M. Wang, M. Pan, et al., Bioact. Mater. 53 (2025) 737–753.
N. Peng, X. Ding, Z. Wang, et al., Carbohydr. Polym. 204 (2019) 32–41. doi: 10.1016/j.carbpol.2018.09.084
Y. Liu, X. Ran, G. Zhou, Y. Liu, W. Tan, ACS Nano 19 (2025) 18164–18175. doi: 10.1021/acsnano.4c16628
L. Tang, M. Xie, J. Li, et al., Chin. Chem. Lett. 34 (2023) 107801. doi: 10.1016/j.cclet.2022.107801
S.Q. Guan, C.H. Liu, Y.P. Wang, et al., Chin. Chem. Lett. (2025), doi: 10.1016/j.cclet.2025.111692.
X. Chen, L. Liu, C. Jiang, Acta Pharm. Sin. B 6 (2016) 261–267. doi: 10.1016/j.apsb.2016.05.011
L. Tang, Y. Yin, Z. Zhang, et al., Chem. Eng. J. 493 (2024) 152590. doi: 10.1016/j.cej.2024.152590
W. Yu, R. Lin, X. He, et al., Acta Pharm. Sin. B 11 (2021) 2924–2936. doi: 10.1016/j.apsb.2021.04.006
H.R. Singh, E. Kopperger, F.C. Simmel, Trends Mol. Med. 24 (2018) 591– 593. doi: 10.1016/j.molmed.2018.05.001
G.P. Dunn, A.T. Bruce, H. Ikeda, L.J. Old, R.D. Schreiber, Nat. Immunol. 3 (2002) 991–998. doi: 10.1038/ni1102-991
R.D. Schreiber, L.J. Old, M.J. Smyth, Science 331 (2011) 1565–1570. doi: 10.1126/science.1203486
S. Ruan, Y. Huang, M. He, H. Gao, Adv. Sci. 9 (2022) 2200027. doi: 10.1002/advs.202200027
M.M. Gubin, M.D. Vesely, Clin. Cancer Res. 28 (2022) 3917–3928. doi: 10.1158/1078-0432.ccr-21-1804
X. Xiang, Y.R. Niu, Z.H. Wang, et al., Cytokine. Growth. Factor. Rev. 67 (2022) 35–48. doi: 10.1016/j.cytogfr.2022.07.006
J. Hu, X. Li, L. Yang, H. Li, Biomed. Pharmacother. 151 (2022) 113068. doi: 10.1016/j.biopha.2022.113068
Z. Lamplugh, Y. Fan, Front. Immunol. 12 (2021) 811485. doi: 10.3389/fimmu.2021.811485
J.Y. Mun, S.H. Leem, J.H. Lee, H.S. Kim, Front. Immunol. 13 (2022) 864739. doi: 10.3389/fimmu.2022.864739
I. Vitale, G. Manic, L.M. Coussens, G. Kroemer, L. Galluzzi, Cell Metab 30 (2019) 36–50. doi: 10.1016/j.cmet.2019.06.001
K. Wu, K. Lin, X. Li, et al., Front. Immunol. 11 (2020) 1731. doi: 10.3389/fimmu.2020.01731
R.A. Franklin, M.O. Li, Trends. Cancer 2 (2016) 20–34. doi: 10.1016/j.trecan.2015.11.004
P. Pathria, T.L. Louis, J.A. Varner, Trends. Immunol. 40 (2019) 310–327. doi: 10.1016/j.it.2019.02.003
B. Toledo, L. Zhu Chen, M. Paniagua-Sancho, et al., J. Hematol. Oncol. 17 (2024) 44. doi: 10.1186/s13045-024-01559-0
Y. Singh, V.K. Pawar, J.G. Meher, et al., J. Control. Release 254 (2017) 92–106. doi: 10.1016/j.jconrel.2017.03.395
Y.D.T. Tzeng, J.H. Hsiao, P.Y. Chu, et al., Pharmacol. Res. 198 (2023) 107008. doi: 10.1016/j.phrs.2023.107008
S. Singhal, J. Stadanlick, M.J. Annunziata, et al., Sci. Transl. Med. 11 (2019) eaat1500. doi: 10.1126/scitranslmed.aat1500
S.M. Morrissey, F. Zhang, C. Ding, et al., Cell. Metab. 33 (2021) 2040–2058 e2010. doi: 10.1016/j.cmet.2021.09.002
V.R. Moulton, A.R. Gillooly, M.A. Perl, A. Markopoulou, G.C. Tsokos, PLoS One 10 (2015) e0131073. doi: 10.1371/journal.pone.0131073
V.R. Moulton, G.C. Tsokos, J. Biol. Chem. 285 (2010) 12490–12496. doi: 10.1074/jbc.M109.091660
M. Czystowska-Kuzmicz, A. Sosnowska, D. Nowis, et al., Nat. Commun. 10 (2019) 3000. doi: 10.1038/s41467-019-10979-3
H. Oweira, I. Lahdou, S. Mehrle, et al., J. Clin. Med. 11 (2022) 4794. doi: 10.3390/jcm11164794
S. Viel, A. Marçais, F.S.F. Guimaraes, et al., Sci. Signal. 9 (2016) ra19.
V. Zaiatz-Bittencourt, D.K. Finlay, C.M. Gardiner, J. Immunol. 200 (2018) 3934–3941. doi: 10.4049/jimmunol.1701461
S. Song, Y. Wang, J. Wang, et al., Eur. J. Cancer 48 (2012) 2252–2259. doi: 10.1016/j.ejca.2011.12.009
D. Wang, L. Yang, D. Yue, et al., Cancer Lett. 452 (2019) 244–253. doi: 10.1016/j.canlet.2019.03.040
Q. Zhu, X. Wu, Y. Wu, X. Wang, Oncol. Rep. 36 (2016) 3472–3478. doi: 10.3892/or.2016.5136
J. Chen, Y. Ye, P. Liu, et al., Hum. Immunol. 78 (2017) 113–119. doi: 10.4103/2211-5056.214233
H. Chen, Y. Chen, H. Liu, et al., Front. Immunol. 9 (2018) 1179. doi: 10.3389/fimmu.2018.01179
B. Bierie, H.L. Moses, Cytokine Growth Factor Rev. 21 (2010) 49–59. doi: 10.1016/j.cytogfr.2009.11.008
L. Gao, F.Q. Wang, H.M. Li, et al., Oncotarget 7 (2016) 87037. doi: 10.18632/oncotarget.13523
Y. Yang, S. Li, K.K. To, et al., J. Exp. Clin. Cancer Res. 44 (2025) 145. doi: 10.1142/9789819814770_0007
Y. Liu, C. Jiang, C. Xu, L. Gu, Cancer Cell Int. 23 (2023) 253. doi: 10.1186/s12935-023-03103-5
P. Chen, H. Zuo, H. Xiong, et al., Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 580–585. doi: 10.1073/pnas.1614035114
G. Gunaydin, Front. Oncol. 11 (2021) 668349. doi: 10.3389/fonc.2021.668349
Y. Xu, H. Zhang, D. Nie, Front. Immunol. 16 (2025) 1521550. doi: 10.3389/fimmu.2025.1521550
W. Zhang, Q. Han, Y. Ding, et al., Cell. Mol. Life Sci. 80 (2023) 14. doi: 10.1117/12.2677145
I. Vogel, S.P. Bapat, Cancer Cell 43 (2025) 1002–1004. doi: 10.1016/j.ccell.2025.04.004
Y. Yan, L. Huang, Y. Liu, et al., J. Hematol. Oncol. 15 (2022) 104. doi: 10.1186/s13045-022-01322-3
S. Yenyuwadee, K. Aliazis, Q. Wang, et al., Semin. Cancer Biol. 86 (2022) 187–201. doi: 10.1016/j.semcancer.2022.08.004
J.Waibl Polania, E.C. Lerner, D.S. Wilkinson, A. Hoyt-Miggelbrink, P.E. Fecci, Front. Immunol. 12 (2021) 777073. doi: 10.3389/fimmu.2021.777073
C. Li, P. Jiang, S. Wei, X. Xu, J. Wang, Mol. Cancer 19 (2020) 116. doi: 10.1186/s12943-020-01234-1
J. Yang, H. Bae, Exp. Mol. Med. 55 (2023) 1996–2004. doi: 10.1038/s12276-023-01080-3
V. Bronte, P. Serafini, E. Apolloni, P. Zanovello, J. Immunother. 24 (2001) 431–446. doi: 10.1097/00002371-200111000-00001
K. Nakamura, M.J. Smyth, Cell. Mol. Immunol. 17 (2020) 1–12. doi: 10.1038/s41423-019-0306-1
V. Damuzzo, L. Pinton, G. Desantis, et al., Cytometry B: Clin. Cytom. 88 (2015) 77–91. doi: 10.1002/cyto.b.21206
D.I. Gabrilovich, Cancer Immunol. Res. 5 (2017) 3–8. doi: 10.1158/2326-6066.CIR-16-0297
M.E. Heuvers, F. Muskens, K. Bezemer, et al., Lung Cancer 81 (2013) 468–474. doi: 10.1016/j.lungcan.2013.06.005
C. Wu, M.E. Muroski, J. Miska, et al., Nanomed. Nanotechnol. Biol. Med. 16 (2019) 126–137. doi: 10.1016/j.nano.2018.11.015
M.K. Srivastava, P. Sinha, V.K. Clements, P. Rodriguez, S. Ostrand-Rosenberg, Cancer Res. 70 (2010) 68–77.
Y. Yamauchi, S. Safi, C. Blattner, et al., Am. J. Respir. Crit. Care Med. 198 (2018) 777–787. doi: 10.1164/rccm.201708-1707oc
Y. Zhang, M. Zhang, X. Li, Z. Tang, L. He, K. Lv, Mol. Immunol. 83 (2017) 62–71. doi: 10.1016/j.molimm.2017.01.013
S. Zilio, P. Serafini, Vaccines 4 (2016) 31. doi: 10.3390/vaccines4030031
X. Cao, Q. Lan, H. Xu, et al., Int. Immunopharmacol. 143 (2024) 113500. doi: 10.1016/j.intimp.2024.113500
Á. Teijeira, S. Garasa, M. Gato, et al., Immunity 52 (2020) 856–871 e858. doi: 10.1016/j.immuni.2020.03.001
F.S. Younesi, A.E. Miller, T.H. Barker, F.M. Rossi, B. Hinz, Nat. Rev. Mol. Cell Biol. 25 (2024) 617–638. doi: 10.1038/s41580-024-00716-0
R. Kalluri, Nat. Rev. Cancer 16 (2016) 582–598. doi: 10.1038/nrc.2016.73
Z. Zhang, L. Chen, Cancer Nanotechnol. 16 (2025) 2. doi: 10.1186/s12645-024-00302-1
M.A. Huber, R.D. Schubert, R.U. Peter, et al., J. Invest. Dermatol. 120 (2003) 182–188. doi: 10.1046/j.1523-1747.2003.12035.x
J. Zhou, X.H. Wang, Y.X. Zhao, et al., J. Cancer 9 (2018) 4635. doi: 10.7150/jca.28583
A.P. Minz, D. Mohapatra, M. Dutta, et al., Cancer Immunol. Immunother. 72 (2023) 4261–4278. doi: 10.1007/s00262-023-03562-9
B. Dirat, L. Bochet, M. Dabek, et al., Cancer Res. 71 (2011) 2455–2465. doi: 10.1158/0008-5472.CAN-10-3323
M. Lo Iacono, C. Modica, G. Porcelli, et al., Biomolecules 12 (2022) 702. doi: 10.3390/biom12050702
C. Bouche, D.F. Quail, Cancer Res. 83 (2023) 1170–1172. doi: 10.1158/0008-5472.can-23-0505
R. Munteanu, A. Onaciu, C. Moldovan, et al., Pharmaceutics 12 (2020) 402. doi: 10.3390/pharmaceutics12050402
C. Wu, S. Dong, R. Huang, X. Chen, Cancers 15 (2023) 726. doi: 10.3390/cancers15030726
L. Yu, W. Wei, J. Lv, Y. Lu, Z. Wang, C. Cai, Cancer Lett. 604 (2024) 217271. doi: 10.1016/j.canlet.2024.217271
J. Zeng, E.R. Sauter, B. Li, Trends. Mol. Med. 26 (2020) 437–440. doi: 10.1016/j.molmed.2020.03.004
B. Wu, X. Sun, H.B. Gupta, et al., OncoImmunology 7 (2018) e1500107. doi: 10.1080/2162402X.2018.1500107
A.M. Santander, O. Lopez-Ocejo, O. Casas, et al., Cancers 7 (2015) 143–178. doi: 10.3390/cancers7010143
V. D’Esposito, D. Liguoro, M.R. Ambrosio, et al., Oncotarget 7 (2016) 24495. doi: 10.18632/oncotarget.8336
Z. Guo, Z. Zhu, X. Lin, et al., Biomark Res. 12 (2024) 166. doi: 10.54254/2753-7048/70/20241048
Y.Y. Wang, C. Attané, D. Milhas, et al., JCI Insight 2 (2017) e87489.
C. Attané, D. Milhas, A.J. Hoy, C. Muller, Curr. Med. Chem. 27 (2020) 3984–4001. doi: 10.2174/0929867325666180426165001
K.M. Bussard, L. Mutkus, K. Stumpf, C. Gomez-Manzano, F.C. Marini, Breast Cancer Res. 18 (2016) 84. doi: 10.1186/s13058-016-0740-2
Q. Wu, B. Li, Z. Li, et al., J. Hematol. Oncol. 12 (2019) 95. doi: 10.1186/s13045-019-0778-6
L.L. Zhu, X.Z. Yu, T. Cao, et al., Acta Pharm. Sin. B 13 (2023) 2464–2482. doi: 10.1016/j.apsb.2023.03.004
M. Zhao, J. Li, J.W. Liu, et al., J. Control. Release 335 (2021) 320–332. doi: 10.1016/j.jconrel.2021.05.036
Y.T. Zhang, M. Jiang, G.S. Du, et al., Acta Pharm. Sin. B 13 (2023) 3518–3534. doi: 10.1016/j.apsb.2022.03.017
W.J. Meng, L. Huang, J.M. Guo, et al., Pharmaceutics 16 (2024) 1549. doi: 10.3390/pharmaceutics16121549
Z.X. Zhu, S.Q. Cao, H.W. Li, et al., J. Control. Release 381 (2025) 113603. doi: 10.1016/j.jconrel.2025.113603
Y.Z. Lin, X. Wang, S. He, et al., Acta Pharm. Sin. B 14 (2024) 854–868. doi: 10.1016/j.apsb.2023.08.014
Y.X. Zhang, J. Zhou, Y.Y. Wang, et al., J. Control. Release 381 (2025) 113606. doi: 10.1016/j.jconrel.2025.113606
Z. Zeng, Y. Liu, Q.L. Wen, et al., Drug Deliv. 28 (2021) 943–956. doi: 10.1080/10717544.2021.1921076
Q.W. Yu, Y. Qiu, J.P. Li, et al., J. Control. Release 321 (2020) 564–575. doi: 10.1016/j.jconrel.2020.02.040
C.Y. Xia, M. Li, G.Y. Ran, et al., J. Control. Release 335 (2021) 557–574. doi: 10.1016/j.jconrel.2021.05.034
Y. Qian, S. Qiao, Y. Dai, et al., ACS Nano 11 (2017) 9536–9549. doi: 10.1021/acsnano.7b05465
Y. Wang, J. Yu, Z. Luo, et al., Adv. Mater. 33 (2021) 2103497. doi: 10.1002/adma.202103497
Y. Gong, W. Gao, J. Zhang, et al., J. Nanobiotechnol. 22 (2024) 341. doi: 10.1186/s12951-024-02622-1
Y. Wang, K. Tiruthani, S. Li, et al., Adv. Mater. 33 (2021) 2007603. doi: 10.1002/adma.202007603
G. Qiao, S. Li, X. Pan, et al., Sci. Adv. 10 (2024) eadk7955. doi: 10.1126/sciadv.adk7955
W. Ou, R.K. Thapa, L. Jiang, et al., J. Control. Release 281 (2018) 84–96. doi: 10.1016/j.jconrel.2018.05.018
S.Y. Li, Y. Liu, C.F. Xu, et al., J. Control. Release 231 (2016) 17–28. doi: 10.1016/j.jconrel.2016.01.044
J. Zhang, D. Liu, J. Liu, et al., Biomater. Sci. 8 (2020) 4757–4766. doi: 10.1039/d0bm00908c
A. Zhang, Z. Ren, K.F. Tseng, et al., Sci. Transl. Med. 13 (2021) eabg8693. doi: 10.1126/scitranslmed.abg8693
H. Van Damme, B. Dombrecht, M. Kiss, et al., J. Immunother. Cancer 9 (2021) e001749. doi: 10.1136/jitc-2020-001749
Z. Lan, W.J. Liu, W.W. Yin, et al., J. Nanobiotechnology 22 (2024) 174. doi: 10.1186/s12951-024-02417-4
Z. Yang, H. Zuo, Y. Hou, et al., Small 20 (2024) 2406860. doi: 10.1002/smll.202406860
T. Yu, K. Wang, J. Wang, et al., J. Control. Release 369 (2024) 199–214. doi: 10.1016/j.jconrel.2024.03.043
H. Phuengkham, C. Song, S.H. Um, Y.T. Lim, Adv. Mater. 30 (2018) 1706719. doi: 10.1002/adma.201706719
P. Zhou, X. Du, W. Jia, K. Feng, Y. Zhang, STTT 9 (2024) 151.
Y. Zhang, R. Yu, C. Zhao, et al., Adv. Sci. 11 (2024) 2305279. doi: 10.1002/advs.202305279
X. Hou, M. Liu, H. Wu, et al., Chin. Chem. Lett. 36 (2025) 110106. doi: 10.1016/j.cclet.2024.110106
Y. Chen, M. Hu, S. Wang, et al., Int. J. Pharm. 623 (2022) 121953. doi: 10.1016/j.ijpharm.2022.121953
M. Yang, B. Wang, Y. Yin, et al., J. Control. Release 353 (2023) 63–76. doi: 10.1016/j.jconrel.2022.11.025
Q. You, G. Wu, H. Li, et al., Nat. Nanotechnol. 20 (2025) 1298–1311. doi: 10.1038/s41565-025-01952-x
J. Peng, J. Zhou, R. Sun, et al., Int. J. Biol. Macromol. 244 (2023) 125163. doi: 10.1016/j.ijbiomac.2023.125163
C. Cui, K. Chakraborty, X.A. Tang, et al., Nat. Nanotechnol. 16 (2021) 1394–1402. doi: 10.1038/s41565-021-00988-z
G. Zhao, F. Guo, W. Yan, et al., Mater. Today Bio 32 (2025) 101882. doi: 10.1016/j.mtbio.2025.101882
H. Chen, Q. Guo, Y. Chu, et al., Biomaterials 287 (2022) 121599. doi: 10.1016/j.biomaterials.2022.121599
Y.N. Tan, Y.P. Li, J.D. Huang, et al., Cancer Lett. 522 (2021) 238–254. doi: 10.1016/j.canlet.2021.09.031
Z. Wan, H. Huang, R.E. West Ⅲ, et al., Mater. Today 62 (2023) 33–50. doi: 10.1016/j.mattod.2022.11.008

Figure 1 Illustrative schematic depicting diverse tumor stromal cell types and their respective roles within the ITME.

Figure 2 The examples of nano-DDS for modulating TAMs. (A) Design of the M2NP for M2-like TAM-specific molecular-targeted immunotherapy. Copied with permission [147]. Copyright 2017, American Chemical Society. (B) Schematic illustration of engineered endogenous TAM-targeted biomimetic nano-RBC to reprogram ITME for enhanced cancer chemo-immunotherapy. Copied with permission [148]. Copyright 2021, Wiley‐VCH GmbH. (C) Schematic illustration of M@SINPs-mediated M2-to-M1 repolarization and phagocytosis restoration of TAMs for improved cancer immunotherapy. Copied with permission [149]. Copyright 2025, BioMed Central Ltd. (D) The percentage of macrophages and M2-type macrophages in the total immune cells following treatment (n = 6). Copied with permission [150], Copyright 2021, Wiley‐VCH GmbH.

Figure 3 The examples of nano-DDS for modulating Tregs. (A) Schematic illustration of tLyp1-hNPs. Copied with permission [152]. Copyright 2018, Published by Elsevier B.V. (B) The Structural Characteristics and Functional Mechanism of NPsiCTLA-4. Copied with permission [153]. Copyright 2016, Elsevier B.V. (C) Illustration of the hSNP enhanced synergistic anti-tumor effect mechanism of CTLA-4 and PD-1 blockades. Copied with permission [154]. Copyright 2020, The Royal Society of Chemistry. (D) s.c. MC38 tumor growth in mice treated with isotype (black, closed circle), anti-PD-1 (black, circle), anti-CCR8 (green, closed circle) or the combination of anti-PD-1 and anti-CCR8 (ADCC) (green, circle) (n = 10). Copied with permission [156]. Copyright 2020, The Royal Society of Chemistry.

Figure 4 The examples of nano-DDS for modulating MDSCs. (A) Schematic overview illustrating the preparation and the anti-tumor therapy of BDM. (B) The percentage of tumor-infiltrating CD11b+ Gr-1+ MDSCs, CD11b+ F4/80+ Macrophages. The MFI of tumor-infiltrating CD86+ CD11b+ F4/80+ M1-like Macrophages and CD206+ CD11b+ F4/80+ M2-like Macrophages, the percentage of tumor-infiltrating CD11b+ CD11c+ DCs and CD103+ CD11b+ CD11c+ matured DCs. Copied with permission [157]. Copyright 2025, BioMed Central Ltd. (C) Schematic illustration of synthesis and working principle of I-4-P@PssP NPs. Copied with permission [158]. Copyright 2024, Wiley‐VCH GmbH. (D) αLy-6C-LAMP targeting peripheral, especially splenic, M-MDSCs and eliciting antitumor immune responses. Copied with permission [159]. Copyright 2025, Elsevier B.V. (E) FACS analysis demonstrating infiltrating MDSCs (CD11b+Gr1+) on day 7 and CD8+ and CD4+ T cells on days 7 and 14 in recurring tumors and the spleen. Copied with permission [160]. Copyright 2018, WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim.

Figure 5 The examples of nano-DDS for modulating CAFs. (A) Schematic representation of engineered BMSCs-derived EVs for the co-delivery of miR-138–5p mimic and PFD to pancreatic CAFs. Copied with permission [161]. Copyright 2025, Springer Nature Limited. (B) The synthesis of JQ1&PFD@CTL, along with its role in inhibiting CAFs and the formation of the stromal barrier, enhancing drug delivery, improving the hypoxic tumor environment, inhibiting tumor glycolysis, and simultaneously increasing the sensitivity of JQ1. Copied with permission [162]. Copyright 2023, The Authors. Advanced Science published by Wiley‐VCH GmbH. (C) Schematic illustration of the preparation process of oP@ALG and the mechanism of oP@ALG after intratumoral injection. Copied with permission [163]. Copyright 2025, Elsevier B.V. (D) Illustrative schematic depicting the antitumor mechanisms of PEG-SAB-Lip and PEG-DTX-Lip. Copied with permission [164]. Copyright 2025, Elsevier B.V.

Figure 6 The examples of nano-DDS for multicellular cooperative modulating. (A) Schematic illustration of DT-GM1/DOX composition and targeting of M2 TAMs and GSCs across BBB to block PTN-PTPRZ1 signaling axis synergistically with DOX against GBM. Copied with permission [165]. Copyright 2025, Elsevier B.V. (B) Schematic illustration on the design of Man-EM camouflaged HPA NPs encapsulating both AS and CQ (HPA/AS/CQ@Man-EM) for CRC treatment. Copied with permission [167]. Copyright 2025, Elsevier B.V. (C) Schematic of CSA/TAA-integrated NICER nanovaccine, and the immune response against both CSCs and bulk tumors with improved antigen cross-presentation of DCs in an epigenetic manner by limiting lysosomal degradation and enhancing cytosolic export of the internalized antigens. TCR, T cell receptor. Copied with permission [166]. Copyright 2025, Springer Nature Limited.
Table 1. The table to summarize relevant content includes different modulatory cells and therapeutic mechanisms for various cells.
| Nano-DDS | Modulatory or targeting cells | Therapeutic mechanisms | Ref. |
| M2NP | TAMs | Elimination of M2-type TAMs | [147] |
| V(Hb)@DOX | TAMs | Elimination of M2-type TAMs | [148] |
| M@SINPs | TAMs | Transition of M2-type TAMs to the M1 phenotype | [149] |
| BisCCL2/5i | TAMs | Reducing TAMs recruitment | [150] |
| ME@C | TAMs | Targeting M2-type TAMs and activating CD8+ T cells | [151] |
| tLyp1-hNPs | Tregs | Targeting Tregs and inhibiting the phosphorylation of STAT3 and STAT5 | [152] |
| NPsiCTLA-4 | Tregs | Targeting Tregs and diminished the ratio of CD4+ FoxP3+ Tregs | [153] |
| SNPs | Tregs | Targeting Tregs while simultaneously blocking CTLA-4 and PD-1 | [154] |
| Anti–CTLA-4×SIRPα heterodimer | Tregs | Exposing Tregs to macrophage-mediated phagocytosis | [155] |
| Nb-Fc1B | Tregs | Selectively elimination of CCR8+ ti-Tregs | [156] |
| BDM | MDSCs | Enhancing targeting and mitigating the myelotoxic effects of drugs | [157] |
| I-4-P@PssP | MDSCs | Inhibiting MDSCs recruitment | [158] |
| dLy-6C-LAMP | MDSCs | Utilizing the intrinsic homing properties of M-MDSCs to traverse the BBB | [159] |
| Immunecare-DISC | MDSCs | Eliminating MDSCs and activating Toll-like receptor 3 | [160] |
| IEVs-PFD/138 | CAFs | Suppressing CAFs proliferation, migration, and secretion of profibrotic factors | [161] |
| JQ1&PFD@CTL | CAFs | Metabolic reprogramming and ECM degradation | [162] |
| oP@ALG | CAFs | Diminishing ECM deposition | [163] |
| PEG-SAB-Lip | CAFs | Induction of CAF quiescence and ECM degradation | [164] |
| DT-GM1 | TAMs, GSCs | Eliminating or repolarizing M2-type TAMs and eradicating GSCs | [165] |
| NICER | CSCs, tumors | Activation of CD8+ T cells and suppression of YTHDF1 expression | [166] |
| HPA/AS/CQ@Man-EM | TAMs, tumors | Repolarizing M2-type TAMs and elevating ROS levels and reducing VEGF expression | [167] |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: