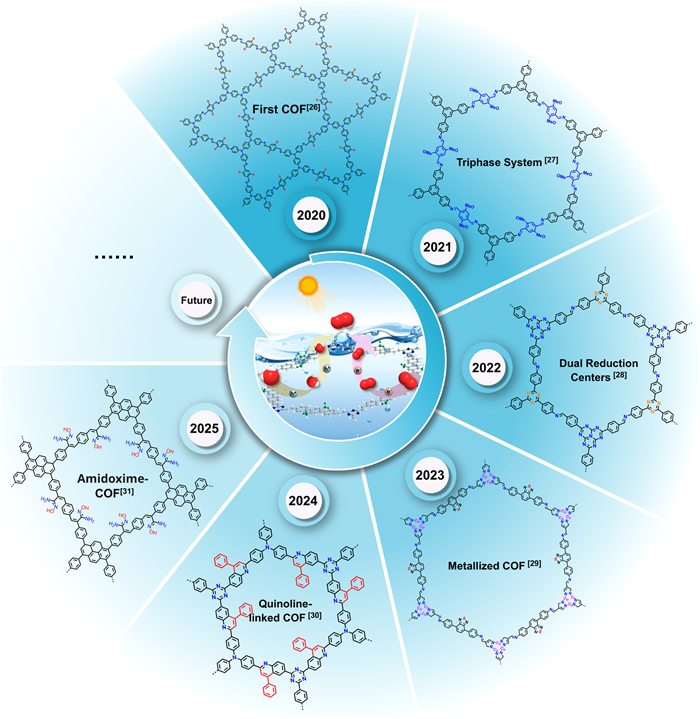
Figure 1.
Selected examples of COFs used for H2O2 photosynthesis.

Hydrogen peroxide (H2O2), an environmentally friendly oxidant and energy-dense liquid fuel, serves as one of the 100 most critical industrial chemicals. It has extensive applications in chemical synthesis, environmental remediation, and biomedical engineering. The global market reached US$10.9 billion in 2023, with annual demand exceeding 4.4 million tons [1–4]. Currently, the industrial synthesis of H2O2 predominantly relies on the anthraquinone oxidation process, accounting for over 95% of global output. Besides, the direct Nreaction of hydrogen and oxygen catalyzed by noble metal catalysts has emerged as a supplementary approach. However, these conventional methods face critical limitations, including high operational costs, substantial energy consumption, and environmental pollution from byproduct emissions [5–7]. Furthermore, the high concentration of commercially produced H2O2 poses safety risks during storage and transport, and typically requires dilution for practical applications (e.g., < 0.1 wt% for water treatment) [8,9]. Therefore, the development of environmentally friendly and economical technologies for the on-site synthesis of directly utilizable H2O2 is of great importance.
Molecular oxygen (O2) is the most environmentally benign and abundant available oxidant on the planet. Photocatalytic O2 reduction reactions (ORR) provide a sustainable and appealing alternative technology for directly synthesizing clean and ready-to-use H2O2 from pure water and air [10–13]. In principle, the two-electron (2e−) ORR process for H2O2 photosynthesis includes either the direct one-step 2e− (Eq. 1, all potentials are reported with reference to the normal hydrogen electrode (NHE)) or the consecutive two-step 2e− (Eqs. 2 and 3) route [14]. The generated H2O2 can further gain an e− and be reduced to a hydroxyl radical (·OH) and a hydroxide ion (OH−), i.e., 3e− ORR process (Eq. 4) [15]. Besides, the 1e− ORR produces superoxide radicals (·O2−) that easily react with photogenerated holes (h+) to produce singlet oxygen (1O2) (Eq. 5), and the 4e− ORR pathway always competes with the 2e− (Eq. 6) [16,17]. Despite extensive research on inorganic semiconductors and organic polymers, most systems still suffer from low activity, poor selectivity, and insufficient mechanistic understanding [18–22]. Addressing these limitations requires atomic-scale insights into the intrinsic relationship between catalyst active sites and O2 adsorption/reduction behaviors, which is crucial for rationally engineering high-performance photocatalytic systems.
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
|
|
(6) |
Covalent organic frameworks (COFs), a kind of crystalline organic porous materials constructed by integrating organic units through covalent bonds, have emerged as thriving candidates in H2O2 photosynthesis due to their high crystallinity, tunable electronic band structures, and well-defined active sites [23–25]. The vast building units, rich bonding linkages, and diverse topological networks of COFs endow them with structures fully designable. This unique feature allows precise regulation of electronic properties via customized configurations, thereby optimizing catalytic activity and selectivity for H2O2 generation. Furthermore, the structures of COFs are precisely identifiable at the atomic level, providing unparalleled opportunities to elucidate the nature of the active sites and the structure-property relationships. Since the pioneering work by Voort et al. in 2020 [26], research on COFs-based H2O2 photosynthesis has grown exponentially (Fig. S1 in Supporting information). In particular, significant breakthroughs have been achieved in microenvironment engineering of COFs for regulating the activity and pathways of H2O2 photosynthesis (Fig. 1) [26–31]. Over five years of rapid development, COFs have emerged as the leading candidates for the photosynthesis of clean and ready-to-use H2O2 via photocatalytic O2 activation [32]. Although some reviews on COFs for H2O2 photosynthesis have been published, a critical review summarizing the relationship between the microenvironment of COFs and the efficiency of H2O2 photosynthesis remains conspicuously absent.
In this review, we provide a critical overview of the progress and challenges in microenvironment engineering of COFs for H2O2 photosynthesis by ORR. To begin with, the principles of photocatalytic O2-to-H2O2 conversion and outlining key strategies for microenvironment engineering of COFs are introduced. Subsequently, the research progress of microenvironment engineering of COFs in H2O2 photosynthesis is critically scrutinized and summarized. In this part, the significant role of precise microenvironment engineering of COFs in regulating O2 adsorption/activation behaviors, e− transport and separation, and proton utilization will be emphasized. Finally, the challenges and potential advancements in this area are projected. This review aims to provide valuable theoretical guidance for developing next-generation COFs towards boosted H2O2 photosynthesis.
The photosynthesis of H2O2 primarily occurs via the ORR, a complex multistep process involving electron or energy transfer pathways that generate various reactive oxygen species (ROSs) (Fig. 2a). The formation of undesired ROSs reduces O2 utilization efficiency, diminishing both the activity and selectivity of H2O2 photosynthesis. Furthermore, some highly oxidative ROSs (e.g., ·OH) can accelerate catalyst degradation, which compromises the structural stability and catalytic longevity of photocatalysts.
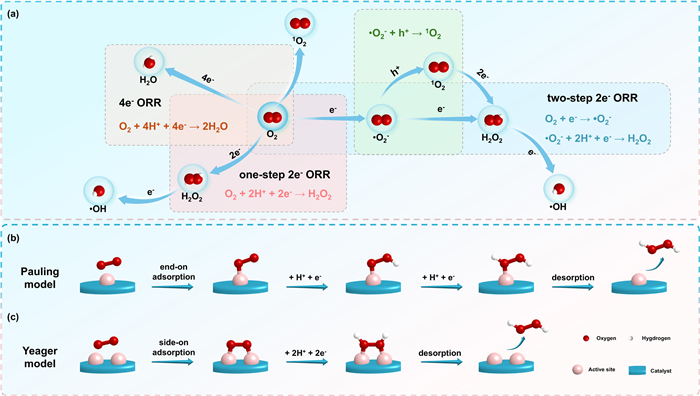
The adsorption and activation behaviors of O2 play a decisive role in the pathway of ORR. Various experimental and theoretical studies have identified two initial O2 adsorption models in H2O2 photocatalysis: the end-on adsorption (Pauling model) and the side-on adsorption (Yeager model) (Figs. 2b and c) [33,34]. These distinct O2 adsorption configurations dictate the ORR reaction pathways. In the “end-on” configuration, the O2 molecule adsorbs at single active sites (Fig. 2b), and then acquires an electron in its π* antibonding orbital during the formation of the intermediate ·O2− (Fig. S2 in Supporting information, observed by in-situ diffuse reflection infrared Fourier transform spectroscopy (DRIFTS) at ~1100 cm−1) [35,36]. The ·O2− can easily react with photogenerated h+ to produce 1O2 (Fig. S2 in Supporting information), introducing competing side reactions. In contrast, when two active sites on the catalyst are close together, each oxygen atom can coordinate with active atoms through Yeager-type adsorption (Fig. 2c). This pathway often results in the formation of 1,4-peroxide (*OO*, in-situ DRIFTS at ~820 cm−1) [37], which subsequently combines with e− and protons to directly from H2O2 via a one-step 2e- ORR pathway. Thus, tailoring the active sites in catalysts can effectively enhance the selectivity of H2O2 production. Photocatalysts with well-defined active sites exhibit superior performance by preferentially stabilizing the desired adsorption configuration and reaction pathway.
The microenvironment of COFs refers to the localized chemical and physical environment within their highly ordered porous structures, collectively defined by organic building units, functional groups, pore surface chemistry, and host-guest interactions. This microenvironment exhibits the following key characteristics: (1) Precise pore geometry (size, shape, and topology); (2) chemical functionality on pore walls; (3) nanoconfinement effects arising from framework rigidity/flexibility and stacking modes; (4) local polarity and electronic tuning via guest binding. Taking the classic two-dimensional (2D) COFs as an example, different symmetric units are used as nodes or linkers, resulting in a wide variety of layer structures and tunable pore geometries. A vast library of monomers and covalent bond-forming reactions provides an ideal platform for fine-tuning the microenvironment engineering of COFs. Based on the artificial operable modulations, this section will systematically introduce factors that affect the microenvironment engineering of COFs, including structural regulation, crystallinity, dimensions, interlayer stacking, and topology (Fig. 3).
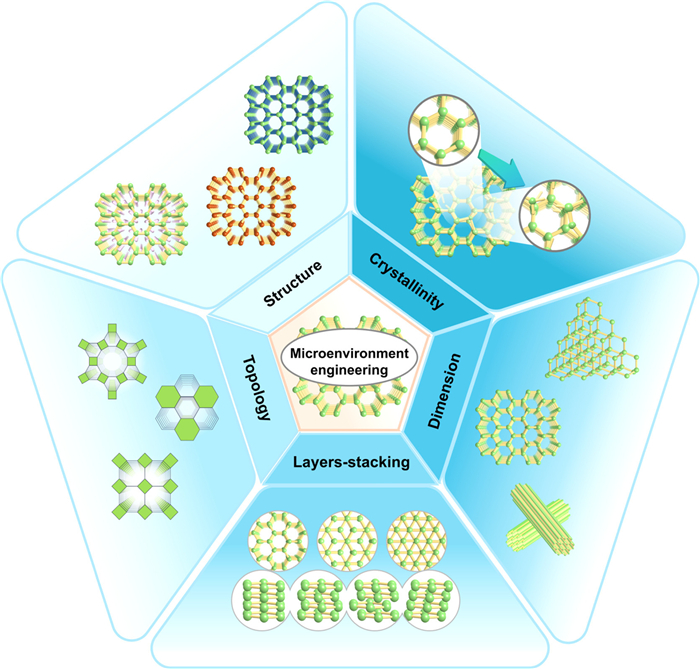
In in-situ synthesis, although the original ligands may lack significant catalytic activity, catalytic sites can be strategically engineered and uniformly distributed within frameworks of COFs through rational structural design. Moreover, post-synthetic modifications such as halogenation, pendant groups appending, or metal coordination enable fine-tuning the chemical environment of the active sites without compromising structural integrity. This modular approach allows optimization of photocatalytic performance while retaining the framework’s crystallinity and porosity.
The linking bonds in COFs, such as imine and triazine bonds, can serve as acidic or basic active sites for certain catalytic reactions [38]. Strategic bond engineering further enables the optimization of charge transport dynamics and active sites functionality. Transformations from imine to quinoline, azole, or amide linkages have also been achieved [39–41], with N, O, or S atoms in these linkages or other sites in the framework acting as typical electron-rich species, potentially contributing to photocatalytic behavior. These modified linkages synergistically modulate local electronic environments and provide accessible coordination sites, significantly enhancing photocatalytic performance in targeted reactions.
Crystallinity, defined as the long-range periodic arrangement of atoms or molecules in a material, plays a pivotal role in photocatalytic applications of COFs [42]. The highly ordered structure of crystalline COFs enables efficient separation and transport of photogenerated e−-h+ pairs, reducing recombination and thereby enhancing photocatalytic efficiency [43]. Enhanced crystallinity can improve catalytic performance by (ⅰ) maximizing active sites density and uniformity, (ⅱ) optimizing the reactant adsorption kinetics, and (ⅲ) ensuring structural stability under irradiation.
As the most extensively studied family of COFs, 2D COFs feature covalent connections that extend within planar layers, facilitating rapid and directional molecule transport between pores or layers. Their structural dimensions are highly tunable via synthetic strategies, typically ranging from nanoscale to microscale. For instance, powder-form 2D COFs often exhibit crystal sizes of tens to hundreds of nanometers, while 2D COF single crystals up to 0.2 mm in size, with in-plane lattice parameters usually falling in the range of 2–5 nm depending on building block design. Additionally, the 2D plane provides a large surface area, making it easier to introduce functional groups through post-modification, significantly enhancing design versatility. However, the non-covalent interactions (π-π stacking or dipole interactions) in stacked 2D frameworks can somewhat limit the accessibility of active sites [44]. By comparison, three-dimensional (3D) COFs possess more complex pore structures, with their 3D morphology allowing for the stable positioning of COF particles in space and forming a highly cross-linked network. This 3D configuration not only significantly increases the specific surface area but also indirectly enhances the number of active sites, effectively addressing the issue of active sites occlusion due to interlayer stacking in 2D COFs [45]. One-dimensional (1D) COFs are created by covalently linking building blocks in 1D, resulting in porous nanoribbons that offer advantages such as easier dispersion and greater exposure to edge sites [46].
The energy band structure of COFs can be influenced by the interlayer stacking modes [47,48]. Stacking often results in the emergence of several local energy minima within a narrow energy window, which in the best-case scenario can lead to polymorphic stacking modes, such as eclipsed (AA stacking), staggered (AB stacking or ABC stacking), and disordered stacking [49]. In 2D COFs, interlayer π-π interactions not only enhance structural stability and facilitate vertical (z-direction) e− transfer [50] but also create nanoconfined environments that modulate reactants adsorption and diffusion. However, this stacking mode hinders the in-plane e− transport within the COFs, a limitation that becomes more pronounced in AA stacking mode COFs. Notably, an appropriate degree of interlayer shift may expose potential active sites.
Another interlayer factor that affects the microenvironment engineering of COFs is interlayer distance. Strong π-π interactions between layers often lead to tight stacking, which can restrict mass transport of reactants and reduce accessibility to catalytic sites, thereby limiting catalytic efficiency. Ligand molecules with large steric effects, such as pyrene, triazine, and carbazole, can create spatial barriers between layers due to their larger molecular sizes, increasing the interlayer distance and optimizing the e−-h+ pairs separation efficiency [51,52].
The reticular chemistry of COFs can be determined by the pre-designed precursors and topological diagrams. Topological diagrams enable the design of COFs with different skeletons and porosities, which is the source of COFs’ diversity. The successful realization of the COFs’ complex and diverse topologies relies on the varied structural characteristics of building blocks, which include different sizes, docking sites, reactive groups, chiral centers and functional groups [53]. The geometry of the building blocks directly dictates the topology of COFs, while the functions of these blocks determine the types of chemical bonds formed and the properties and distribution of active sites. Since COFs with different topologies possess unique pore sizes and distributions, profoundly influencing their electronic transport pathways and light absorption characteristics [54]. These changes, in turn, directly affect the nature of the catalytic sites within COFs, especially those selective active sites whose catalytic behavior can be significantly altered by modifications in topology. Therefore, it is crucial to consider how the topological structure regulates active sites and influences photocatalytic performance when designing COF-based photocatalysts. This concept has gradually gained attention, but it has yet to receive sufficient emphasis [55].
In general, the rational design of porous materials as catalysts is achieved by incorporating catalytic active sites into their framework. The intrinsic properties of COFs are primarily determined by the chemical structure of their skeleton. Introducing functional groups with specific roles (e.g., hydroxyl, carboxyl, halogen, metal atoms/clusters) into COFs can significantly regulate their electronic structure and surface chemistry, further enhancing their photocatalytic performances. These functional groups can be integrated into the COF frameworks through two synthetic approaches: in situ synthesis and post-synthetic modification.
Unsaturated bonds are frequently utilized to modify COF skeletons due to their high e− density and conjugation effects, which enhance their electronic transport properties. Additionally, the presence of unsaturated bonds significantly influences the geometric structure of molecules (double bonds typically impart a planar structure to the molecule, while triple bonds often result in a linear arrangement), which optimizes the electronic transport pathway and facilitates efficient charge carrier separation [56]. Zhu et al. demonstrated that diacetylene units significantly enhanced the COFs’ O2 adsorption capacity and adjusted the ORR to be streamlined into a one-step 2e− process [57]. Chen et al. introduced vinyl groups as pendant groups into the COF skeletons. Compared to PDA-COF with imine N atoms as O2 adsorption sites, the vinyl groups in the DVA-COF became new adsorption sites for O2 and exhibited stronger adsorption affinity. Furthermore, the introduction of vinyl groups changed the O2 adsorption sites configuration from Pauling-type to Yeager-type, promoting a one-step 2e− ORR pathway and thus improving the photocatalytic efficiency of H2O2 production in DVA-COF [34].
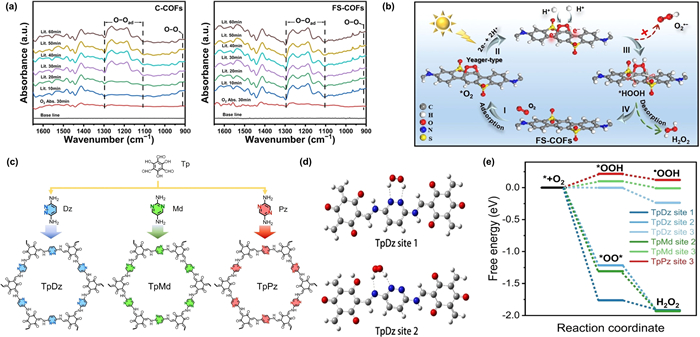
To further elucidate the regulatory mechanism of the ORR pathway and rationally design rich and unique active sites in COFs to achieve one-step 2e− ORR, our previous work introduced sulfone units into the COFs matrix to promote H2O2 photosynthesis from pure water and air [32]. Experimental and theoretical calculations have confirmed that the introduction of sulfone units improved the e−-h+ pairs separation, enhanced the protonation property of FS-COFs, and inhibited the formation of ·O2− (Fig. 4a). The electron-withdrawing effect of the sulfone groups directed O2 adsorption to the two symmetrical carbon atoms on the central benzene ring. This resulted in a Yeager-type conformation with strong binding affinity, achieving high selectivity for O2 reduction via a one-step 2e− ORR process (Fig. 4b).
The well-defined channels of COFs are open and accessible to interface with other nanomaterials [53]. Incorporating specific coordinating groups (such as pyridine, phenanthroline, or salen ligands) into the nodes or linkers of COFs enables precise anchoring of metal atoms or ions through stable coordination bonds at predetermined sites [58–60]. Hou et al. developed a synthetic strategy to immobilize single-atom Fe sites within a novel COF [61]. In this system, the N atoms from the pyridine moieties and imine bonds coordinate with single-atom Fe to form active sites for the ORR process. Porphyrins and phthalocyanines, both featuring large fully conjugated aromatic macrocyclic structures, have emerged as potential platforms for constructing metal-based catalysts [62,63]. Xia et al. synthesized a novel porphyrin-based COF with Ni2+ in the center of porphyrin. This photocatalyst can catalyze the production of H2O2 in the air without sacrificial agents. The incorporation of Ni2+ in the porphyrin center has a positive effect on the adsorption and reduction of O2 molecules while facilitating the charge transfer processes [64].
Introducing nonmetal heteroatoms (e.g., N, S, F) into COFs skeletons via partial replacement of C atoms or bonds can establish new localized energy levels and generate additional charges, effectively enhancing the separation and transfer of photogenerated charge carriers [65,66]. Zeng et al. employed an N-site engineering strategy to systematically regulate H2O2 photosynthesis performance by precisely tuning the type and density of N atoms in the framework. Their work revealed a positive correlation between N density in the framework and H2O2 production rates in these COFs [67]. In a complementary study, Xi et al. designed and synthesized three isomeric COFs functionalized with nitrogen-containing N-heterocycles (diazines) for H2O2 photosynthesis (Fig. 4c) [68]. They investigated how the relative positions of N atoms modulate the photocatalysts’ activity in this reaction. The synergistic configuration of the neighboring N atoms in the pyridazine ring (site 1), combined with the N atoms from the imine bond and other pyridazine ring (site 2), creates multiple O2 adsorption sites for the formation of *OO* (Fig. 4d). This synergistic configuration significantly reduces the Gibbs free energy of the one-step 2e− ORR pathway (Fig. 4e), making it thermodynamically superior to the two-step 2e− ORR pathway, thereby greatly enhancing the selectivity of H2O2 photosynthesis.
The N atoms in the pyridine ring structure provide active sites for binding with H2O molecules through hydrogen bonds. Ma et al. prepared a COF-based photocatalyst incorporating 2,2′-bipyridine units, demonstrating that the bipyridine sites enable the simultaneous adsorption of two H2O molecules. The protonation process at these sites facilitates the Yeager-type adsorption of O2, promoting *OO* intermediates formation and accelerating the one-step 2e− ORR reaction under light irradiation [69]. Similarly, Kim et al. revealed that 2,2′-bipyridine acting as an oxidation center could adsorb and oxidize water molecules under light, leading to bipyridine protonation and ·OH generation [70]. However, due to the presence of imine bonds, some O2 molecules still tend to adsorb onto single N sites, which causes the sequential two-step 2e− ORR process to produce H2O2. Addressing this challenge, Zhao et al. synthesized an imine-based COF (PyIm-COF) using 3,3′-bipyridine. This design creates dual active sites for O2 adsorption through the synergistic combination of the N atom of 3,3′-bipyridine and the adjacent N atom on the imine bond, preferentially forming *OO* intermediates over *OOH species. This strategic configuration effectively suppresses the two-step 2e− ORR pathway at the imine-N sites while enhancing the efficiency of the one-step 2e− ORR process [71].
Nevertheless, the strong polarization of the N atoms in imine linkages can partially hinder charges transfer, leaving opportunities for further improvement of π-delocalization. Sulfur (S) doping presents a promising alternative, as sulfur’s relatively larger atomic radius and lower electronegativity (compared to N and O) significantly alter the electronic structure of materials. These properties make S a strategic heteroatom for tuning local electronic distribution in COFs. Voort et al. synthesized thiazole-linked COFs by combining imine-based COFs with elemental S [72]. The post-sulfurization process expanded the π-conjugation of the COF framework in both the x and y directions, substantially improving e− transport efficiency and significantly enhancing photocatalytic activity.
Halogen atoms, known for their high electronegativity (especially F), can alter the electronic density distribution and surface polarity of COFs upon incorporation. Additionally, dangling halogen groups can stabilize the interlayer stacking of 2D COFs via π-electron interactions, enhancing structural stability and facilitating charge transport. Han et al. rationally engineered the substitution of F atoms on the edge aromatic units of an imine-linked 2D COF with a triazine core (TF-COF) [73]. The F substituents strengthened π-electron interactions, resulting in significantly improved crystallinity and porosity of the partially fluorinated TF50-COF and achieving the highest H2O2 photosynthesis rate.
The linkages in COFs form the fundamental connections between repeating units, profoundly shaping their microenvironment by influencing key properties such as band structure, π-conjugation, hydrophobicity, and charge separation efficiency [74]. This linkage engineering endows the frameworks with tunable crystalline architectures and tailored functionalities. Particularly, certain linkages (e.g., imine and cyano-vinylene) exhibit intrinsic polar asymmetry—their spatial orientation critically modulates the molecular dipole moment distribution, generating an oriented built-in electric field that effectively drives the separation of photogenerated e−-h+ pairs [25]. Furthermore, post-synthetic modification strategies offer additional avenues for constructing optimized COF architectures with enhanced performance.
Imine-based COFs represent the most extensively studied family, offering advantages like facile synthesis conditions (even achievable at room temperature), diverse synthesis methods, and robust chemical stability [75]. Additionally, the p-π conjugation effect of imine bonds contributes to the π-conjugated system within the COFs plane, endowing the material with significant absorption in the UV–visible range. Particularly, the N atom in the imine bond exhibits a certain electron-rich effect, which allows it to act as an oxidation site to adsorb O2 for the two-step 2e− ORR reaction [71].
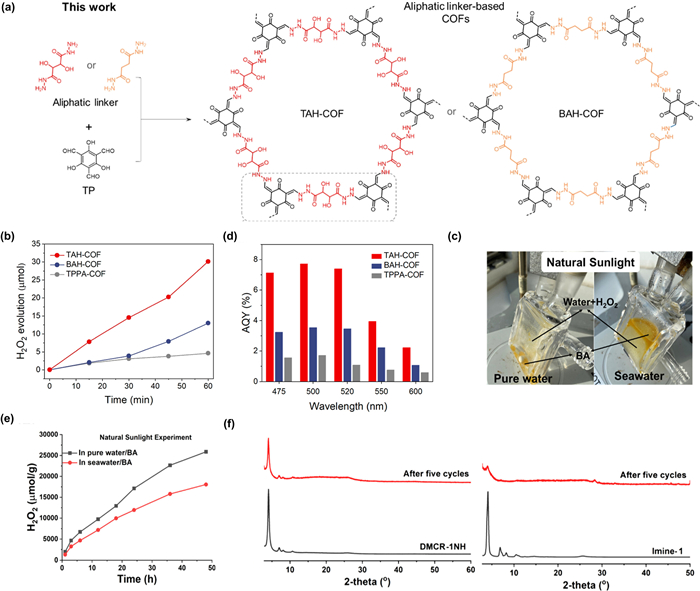
Hydrazone-linked COFs are formed through the condensation reaction between hydrazides and aldehydes. As structural analogues of imine-linked COFs, these COFs have attracted significant attention due to their high physicochemical stability and inherent hydrophilicity [76]. Zhu et al. demonstrated the pioneering example of a hydrazone-linked COF using aliphatic linkers with unprecedented activity for H2O2 photosynthesis (Fig. 5a) [77]. The appropriate proton donors and acceptors on the aliphatic chains form specific multiple hydrogen-bond networks, which restrict the rotational freedom of the chains and enhance the crystallinity of the framework. Compared to the aromatic TPPA-COF without hydrazone linkages, the TAH-COF exhibited stronger affinity for O2 and showed a record-high photocatalytic H2O2 evolution activity (6003 µmol g−1 h−1) without sacrificial agents, coupled with significantly higher apparent quantum yields (AQYs) (Figs. 5b and c).
Azobenzene, an electron-deficient, light-switchable, and light-harvesting organic unit, demonstrates significant potential as a functional linkage in COFs for applications such as proton conductivity and photocatalysis. Zhao et al. transformed an imine COF into an azobenzene-linked COF through a COF-to-COF conversion using 1,4-dinitrobenzene as a linker exchange reagent [78]. Although the transformation involves replacing only one nitrogen atom, the resulting azo linkage significantly broadens light absorption and enhances charge transport. More importantly, it also serves as an active site for oxygen reduction, leading to improved photocatalytic H2O2 production.
Vinyl-linked COFs exhibit exceptional chemical stability and π-delocalization. Han et al. designed different levels of F atoms on the edge units of COFs with triazine cores connected to olefins [79]. The adjacent F atoms-olefinic bond can form p-π conjugation and achieve spin polarization under radiation. This mechanism accelerates triplet excitons generation, activates O2 to 1O2 at the olefinic bond site, and thus generates H2O2 through efficient 2e− ORR photocatalysis. Remarkably, the system simultaneously achieved in situ utilization of the produced H2O2 for efficient oxidation of biomass-derived 5-hydroxymethylfurfural (HMF) to value-added furan compounds with 95% conversion yield.
The electronegativities of the C and N atoms are different, which results in a certain polarity in the C=N bond [38]. Changing the orientation of the imine bond may affect the distribution of the π-electron cloud and the continuity of the conjugated system, thereby influencing the light absorption and e− transfer properties. To systematically investigate the effect of imine bond orientation on the photocatalytic performance of COFs, Yue et al. prepared a pair of structural isomers COFs with reversed imine linkages [80]. This precise control over linkage direction effectively modulated the energy barriers of key redox steps. TB-COF exhibited a larger dipole moment and greater charge separation efficiency along both the ORR and WOR pathways, resulting in significantly enhanced H2O2 photosynthesis performance. It is interesting to note that the system directly utilized the seawater-produced H2O2 for efficient tetracycline degradation, while also achieving 100% selective oxidation of 4-methoxybenzyl alcohol to high-value anisaldehyde when coupled with H2O2 production. Complementing these findings, Zhou et al. explored the intrinsic relationship between imine bond orientation and the activation of molecular O2/H2O [81]. The synthesized PB-PT-COF (the nitrogen of imine is closer to the benzene ring) showed strong H2O interaction, while PT-PB-COF (the nitrogen of imine is closer to the triazine ring) preferentially adsorbed and activated O2. In the H2O2 photoproduction dominated by the ORR pathway, the photocatalytic rate of PT-PB-COF is twice that of PB-PT-COF.
Although some COFs (such as pyridazine COFs) have a relatively high H2O2 photosynthesis activity in pure water, the H2O2 they generate is prone to rapid degradation due to oxidation by photoexcited h+ or ·O2−, which limits their long-term applicability. To achieve high-rate H2O2 photocatalytic production with enhanced stability in COFs, Yu et al. were the first to investigate the impact of imine bond positions on H2O2 photosynthesis [82]. The spatial arrangement of the imine bond dictates the localization of pyridinic nitrogen atoms, creating different types of active sites. Despite having the same composition, o-COF-TpPzda with ortho-positioned imine bonds maintained stable activity after 48 h in pure water. In contrast, the p-COF-TpPzda with imine bonds in the para position achieved a higher initial H2O2 production rate but suffered rapid deactivation within 6 h. This instability stems from preferential photogenerated h+ accumulation in the pyrazine ring region, causing oxidation degradation. This study highlights a critical structure-durability relationship: Strategic imine bond positioning can decouple high activity from long-term stability in photocatalytic COFs design.
The inherent two-orientation isomers of imine bonds may lead to reduced COFs ordering due to the formation of a flat structure [49]. Yaghi et al. showed that the orientation of certain linkers may change even in the solid state [83]. To stabilize the orientation of imine bonds and prevent configurational flipping, converting them into irreversible bond types is an effective strategy [30]. For example, He et al. employed an aza-Diels-Alder cycloaddition reaction to transform dynamic imine bonds into robust quinoline linkages. This post-synthetic modification strategy enhanced the resulting COF’s structural robustness and π-electron delocalization [84]. Expanding on this strategy, Thomas et al. developed synthetic strategies for quinoline-linked COFs and their analogs (DMCR-COFs) [85]. Compared to the imine COF analogue (Imine-1), both quinoline-COFs exhibited improved H2O2 production in natural sunlight and seawater (Figs. 5d and e). Although DMCR-1NH is less conjugated than the aromatic DMCR-1, the -NH functional group provides extra O2 adsorption sites, making the quinoline-COFs perform more efficiently in H2O2 photosynthesis. After 5 cycles, the activity of DMCR-COFs only slightly decreased, whereas Imine-1 lost activity and crystallinity (Fig. 5f).
The azole linkages can also effectively improve the chemical stability and π-conjugation of compounds [86]. Wang et al. converted imine COFs into structurally similar COFs linked by imidazole, oxazole, and thiazole via a linker exchange reaction for the first time [87]. The increase of conjugation along the plane and across the plane collectively enhanced the visible light absorption ability of TZ-COF. Additionally, these azole-based chemical linkages increased the activity near the phenyl rings and facilitated the formation of suitable intermediates in the 2e− ORR, thus promoting the production of H2O2. Their study highlights the important role of the linkage microenvironment in enhancing the photocatalytic performance of azole-linked COFs.
Engineering D–A structures in COFs enables directional charge separation by establishing internal electronic gradients. The electron-rich donor units and electron-deficient acceptor moieties create built-in dipoles, which drive photogenerated electrons toward acceptors while pulling h+ back to donors. This spatial separation suppresses charge recombination and enhances charge mobility [88,89]. In multi-step photocatalytic processes like H2O2 generation, such controlled charge flow is essential for efficient O2 reduction and precise intermediate stabilization.
For H2O2 photosynthesis, the D and A building blocks in COFs can serve as dual active sites for ORR and WOR. The remarkable e− cloud separation between D and A units endows these reaction sites with enhanced activity. Jiang et al. combined the electron-rich hexaphenyl-substituted triphenylene with electron-deficient benzothiadiazole units to synthesize three COFs with different degrees of conjugation [90]. All three COFs have spatially separated D and A, with each D and A unit serving as oxidation and reduction active sites. Notably, in the non-conjugated Hz-TP-BT-COF, the A unit displayed heightened electron-deficient properties while the D unit retained exceptional electron-rich properties. This strategy enabled Hz-TP-BT-COF with ultra-fast photoinduced e− transfer and charge separation from D to A, optimizing photocatalytic efficiency. In a complementary study, Liu et al. designed and synthesized two photoactive tris(triazole)-based COFs through the Schiff-base condensation reaction [91]. The COF-JLU51 demonstrated better D-A characteristics, evidenced by the more pronounced separation of HOMO-LUMO. This enhanced D-A configuration is directly correlated with improved charge separation and photocatalytic performance. In addition, Chen et al. found that the formation of the D-A structure affects the atomic orbitals and the active sites on COFs. By compensating for intrinsic energy barriers in H2O2 formation, their optimized D-A system achieved a 10-fold increase in H2O2 yield [92].
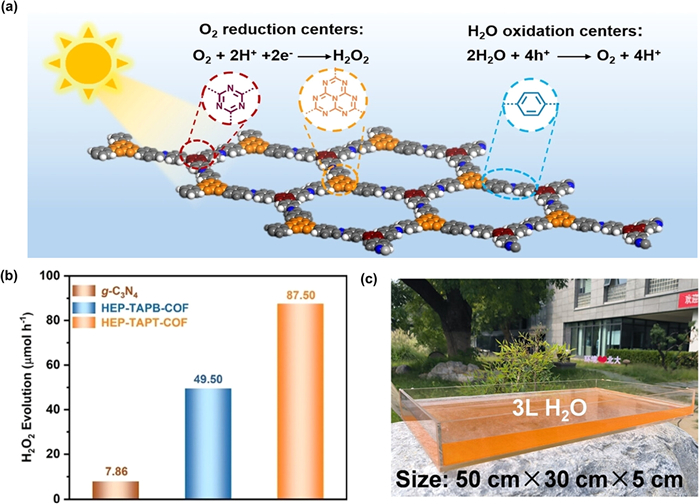
A conventional D-A system typically comprises alternating arrangements of D and A units. Given the fully designable structure of COFs, creating novel D-A COFs with configurations like D1-A-D2 or A1-D-A2 could be engineered to enhance optoelectronic properties and potential photocatalytic applications [89]. These additional D or A units may also offer more active sites. Chen et al. designed and synthesized HEP-COFs with spatially separated redox centers, named HEP-TAPT-COF and HEP-TAPB-COF [28]. Both the heptazine and triazine rings exhibit similar O2 adsorption sites, serving as D units to produce H2O2 via a one-step 2e− ORR pathway (Fig. 6a). Compared with HEP-TAPB-COF, where heptazine acts as the sole O2 reduction center, HEP-TAPT-COF demonstrated superior H2O2 photosynthesis (Fig. 6b).
The electronic landscape of D-A COFs can be further engineered through targeted functionalization and structural hierarchy, enabling precise control over charge transfer pathways. In a further exploration, Ni et al. synthesized three imine-based COFs (COF-0CN, COF-1CN, and COF-2CN) with varying numbers of cyanide groups introduced into the biphenyl units [93]. Cyano groups with strong electron-withdrawing effects can serve as additional A units to connect with D units and provide more WOR catalytic sites for COFs. Notably, as the number of cyanide groups increases, their H2O2 photocatalytic performance continues to improve. The in situ produced H2O2 demonstrated effective antibacterial activity against antibiotic resistance genes and successfully degraded organic pollutants. Besides, under natural sunlight irradiation, COF-2CN still exhibited stable activity and recyclability in the scaled-up reactor (Fig. 6c). In addition, Luo et al. designed two different types of complex cyano-functionalized D-A COFs based on D-A-π-D and D-A-π-A fashion and explored the photocatalytic effect dependent on e− and structures [94]. Compared with the D-A-π-A type, the D-A-π-D type achieved a 1.9-fold increase in H2O2 production efficiency and a 1.3-fold improvement in O2 utilization and conversion rate. Wang et al. designed three benzotrithiophene-based COFs and introduced the concept of a uniport "atom spot-molecular area" via a double D-A method in a periodic framework [95]. When the D-A direction of the imine bond is consistent with the direction of e− transfer between the molecular motifs, a periodic and unobstructed charge transport mode is formed, ultimately achieving the high-yield synthesis of H2O2. Almost 72% of sulfamethoxazole can be directly degraded within 5 min in the produced H2O2 solution through Fenton reactions, demonstrating its immediate applicability for environmental remediation.
Tuning the dimensionality of COFs regulates the spatial arrangement of active sites and mass transport pathways, thereby influencing O2/H2O adsorption and catalytic selectivity. In 3D COFs, interconnected isotropic pores enable co-transport of reactants and stabilize intermediates via spatially distributed active centers. In contrast, 1D COFs confine O2 within linear channels, generating polarized microenvironments and exposing terminal sites for enhanced activation. This dimensional differentiation reveals structure-activity relationships in photocatalysis: dimensional control systematically coordinates the adsorption-catalysis coupling through structural parameters (pore configuration, e− delocalization, and active site distribution), thereby optimizing the efficiency and selectivity of H2O2 photosynthesis.
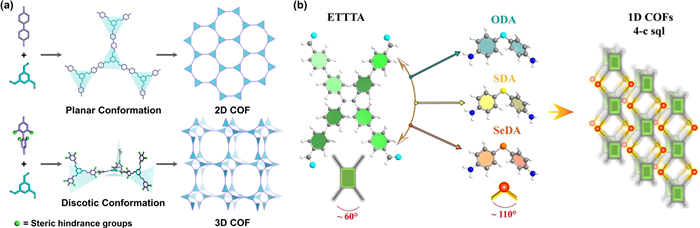
3D COFs have demonstrated unique advantages in regulating microenvironment engineering. Their high porosity and large specific surface area facilitate diverse active site layouts, enhance contact efficiency with O2 and H2O molecules, and effectively promote e− transfer. Zhang et al. designed and synthesized octa-aldehyde difluorobenzene monomer as an 8-connected cubic node for the construction of 3D COF (COF-NUST-16) with tty topology via a bottom-up [8 + 4] reticular strategy for the first time. This framework demonstrated a wide and strong absorbance and narrow bandgap in the visible light region, achieving an H2O2 production rate (1081 µmol g−1 h−1) higher than that of similar 4-linker 2D COF (234 µmol g−1 h−1) [96]. To break the traditional constraint of triangular planar nodes and linear linkers only forming 2D hcb topologies, Gu et al. employed steric hindrance engineering by pre-installing four bulky groups on the linear linker. The resultant intermolecular repulsion causes the planar structure to distort into a spatial disk-like structure, leading to the formation of an extended network and subsequent 3D framework (Fig. 7a) [97]. In photocatalytic reaction pathways, 3D TMP-COFs offer fully exposed active sites and stronger positively charged regions, significantly enhancing H2O2 photocatalytic synthesis performance compared to analogous 2D COFs.
The twisting of flexible units can also be used to transform 2D planar units into 3D structures. Lan et al. constructed 3D COFs with spn topology based on the trigonal antiprism (C3) monomer hexa(4-formyl-phenoxy)cyclotriphosphazene (CTP) [98]. The inherent flexibility of the phosphazene structure building block in CTP induces structural distortions that reconfigure conventional 2D architectures. When combined with a linear C2 configuration monomer, the C6 configuration of CTP tends to form 2D COFs with kgd topology. However, a 3D unit of trigonal antiprism (C3) monomers of CTP will be formed when the CTP twists excessively, which tends to form a 3D COF even connected with planar monomers. This 3D structure enriches active sites and exhibits excellent H2O2 photosynthesis stability.
Studies indicate that 1D COFs possess low-density base sites and numerous exposed edge sites on their skeletons, significantly enhancing active site accessibility and expanding the electrochemically active surface area [99]. In a pioneering advancement, Shen and Zhang et al. systematically designed and eventually synthesized novel VIA group elements (O, S, Se)-bridged 1D COFs with a typical 4-connected sql topology (Fig. 7b) [100]. The 1D structure of EO-COF effectively enhances proton shuttling and O2 activation, and exhibits better visible light absorption, more exposed active sites, more effective charge transfer capabilities, and stronger proton transfer ability. This is the first report of 1D COFs in H2O2 photosynthesis. In addition, Luo et al. used specific functional groups to modulate the edge sites of 1D COFs. By tailoring the electronic states of C atoms on COFs and edge groups, they achieved precise control over photocatalytic performance [101].
To date, research has predominantly focused on constructing novel topological dimensions of COFs, with limited systematic investigation into how dimensional engineering fundamentally enhances photocatalytic performance. Although 3D COFs show great potential for expanding their topological library, precise design and tough synthetic techniques remain significant challenges to their development. Similarly, the application of 1D COFs in topological networks remains underexplored, with only sporadic research addressing their photocatalytic performances. Therefore, optimizing the microenvironment engineering of COFs through multidimensional structural design to improve H2O2 photocatalytic production efficiency is still in its early stages and holds vast potential for future exploration.
Overall, the current strategies for improving the photocatalytic production of H2O2 using COFs predominantly focus on tuning the structure of COFs. Some other factors, such as improving crystallinity and altering topology, have also been reported but remain scarce. For instance, Yan et al. proposed a novel strategy to enhance the crystallinity of a covalent organic framework, TFA-TaPt-COF, connected by imine linkages [102]. By forming an acetal between an aldehyde and ethanol, the aldehyde concentration was reduced, thereby slowing down the reaction rate between the acetal and amine. The TFA-TaPt-COF-E with higher crystallinity had a fivefold higher H2O2 photosynthesis rate than TFA-TaPt-COF-NE. Tang et al. successfully synthesized two novel COFs with similar skeleton compositions but different topologies through the ingenious design of monomer linkage sites [55]. TBD-COF possessing six-arm cores with cpt topology exhibited superior photocatalytic H2O2 performance compared to TBC-COF featuring three-arm cores with hcb topology. Due to the pyridine N-sites acting as potential active centers for both ORR and WOR, the topology of TBD-COF required twice the amount of bipyridine monomers to construct the cpt topology, thereby providing a larger specific surface area and more abundant catalytic active sites. On the other hand, topology modulation lowered the energy barrier for the ROS in the H2O2 formation process via ORR and WOR pathways, further enhancing the photocatalytic performance of TBD-COF.
Studies have shown that organic polymers with highly coplanar frameworks exhibit enhanced π-conjugation, which significantly promotes e−-h+ pairs separation [103]. However, traditional linear linkages (such as C=N, C-N, bipyridine, and biphenyl units) in COFs inevitably introduce structural torsion between building blocks, resulting in dihedral angles that impair coplanarity. To address this limitation, Tan et al. synthesized four CTFs with varying degrees of coplanarity using coplanar triazine rings as linkers combined with planar fused and non-fused ring compounds as building blocks [104]. The near 0° dihedral angles between components in fused CTFs indicate higher coplanarity. Both the e−-h+ separation efficiency and H2O2 photosynthesis performance were improved. Xu et al. demonstrated that by rationally regulating the torsion angle in the basal plane of the as-prepared crystalline CTFs, a remarkable improvement in direct H2O2 photosynthesis can be achieved with a locked-in coplanar conformation [105].
There is no doubt that COFs have an impressive range of properties and great potential in the field of H2O2 synthesis (Table S1 in Supporting information) [106]. While substantial progress has been made, several fundamental and practical challenges remain. Future efforts should aim to deepen the understanding of microenvironment–activity relationships and develop more precise strategies to manipulate the local environments within COFs.
(1) Although current studies demonstrate the influence of functional groups, pore structures, and stacking modes on catalytic performance, the specific correlations between microenvironmental features and reaction intermediates remain elusive. Future work should combine in situ characterization techniques with advanced theoretical calculations to capture the spatial and electronic environment of active sites in real time, and to reveal how nanoconfined environments impact O2 activation and intermediate stabilization.
(2) Most current strategies target static and singular aspects of the microenvironment (e.g., e−-h+ pairs separation efficiency or hydrophilicity). Future studies could explore multi-level regulation, integrating geometric confinement, electrostatic interactions, and dynamic guest-responsive behavior within the same framework. Introducing stimuli-responsive units or reversible host–guest systems may allow adaptive tuning of the microenvironment during catalysis.
(3) 2D COFs have dominated the field, but their dense interlayer stacking can hinder mass transport and limit exposure of active sites. Designing 3D or interpenetrated COFs with hierarchical porosity and open channels could help modulate the microenvironment in three dimensions, enhancing both diffusion and accessibility. Topological control can also enable spatially resolved distributions of functional groups or charge gradients.
(4) To translate COF-based photocatalysts into practical systems, future research should focus on designing microenvironments that not only enhance intrinsic activity but also meet practical requirements such as chemical stability, product selectivity, and ease of separation. An integrated system approach—combining light harvesting, microenvironment modulation, and efficient product recovery—will be essential for scalable H2O2 production. Recent studies have explored various in situ H2O2 utilization strategies [80,107], providing a conceptual framework for future industrial applications (Fig. S3 in Supporting information). Based on this, COF-based photocatalysts must be benchmarked against established industrial processes such as the anthraquinone method, not only considering the intrinsic activity, with careful evaluation not only of their intrinsic activity but also of long-term durability, cost-efficiency, and system-level scalability.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Kun Zhao: Writing – original draft, Methodology, Investigation. Bing Chen: Writing – original draft, Investigation. Luwen Liang: Writing – original draft, Investigation. Yaze Chen: Investigation. Shan Yao: Writing – original draft, Methodology, Investigation, Conceptualization. Qun Peng: Investigation. Yuhao Liu: Writing – original draft, Methodology, Investigation, Conceptualization. Bin Han: Writing – original draft, Supervision, Methodology, Investigation, Conceptualization.
This work was supported by the Basic Science Center Project of the National Natural Science Foundation of China (No. 52388101), the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (No. 2019ZT08L213), and the Natural Science Foundation of Guangdong Province (No. 2023A1515012799).
Supplementary material associated with this article can be found, in the online version, at doi:
R. Ciriminna, L. Albanese, F. Meneguzzo, et al., ChemSusChem 9 (2016) 3374–3381. doi: 10.1002/cssc.201600895
P. Sun, C. Tyree, C.H. Huang, Environ. Sci. Technol. 50 (2016) 4448–4458. doi: 10.1021/acs.est.5b06097
H. Sies, Redox Biol. 11 (2017) 613–619. doi: 10.1016/j.redox.2016.12.035
Z. Li, H. Lv, K. Tong, et al., Appl. Catal. B 345 (2024) 123690. doi: 10.1016/j.apcatb.2024.123690
J.M. Campos-Martin, G. Blanco-Brieva, J.L.G. Fierro, Angew. Chem. Int. Ed. 45 (2006) 6962–6984. doi: 10.1002/anie.200503779
J. García-Serna, T. Moreno, P. Biasi, et al., Green Chem. 16 (2014) 2320. doi: 10.1039/c3gc41600c
Y. Yi, L. Wang, G. Li, et al., Catal. Sci. Technol. 6 (2016) 1593–1610. doi: 10.1039/C5CY01567G
L. Pi, J. Cai, L. Xiong, et al., Chem. Eng. J. 389 (2020) 123420. doi: 10.1016/j.cej.2019.123420
S. Yang, A. Verdaguer-Casadevall, L. Arnarson, et al., ACS Catal. 8 (2018) 4064–4081. doi: 10.1021/acscatal.8b00217
H. Hou, X. Zeng, X. Zhang, Angew. Chem. Int. Ed. 59 (2020) 17356–17376. doi: 10.1002/anie.201911609
X. Wang, Y. Shao, J. Pan, et al., Chem. Eng. J. 490 (2024) 151923. doi: 10.1016/j.cej.2024.151923
S. Chen, T. Luo, X. Li, et al., J. Am. Chem. Soc. 144 (2022) 14505–14516. doi: 10.1021/jacs.2c01194
X. Lu, Y. Chang, S. Wang, et al., Chin. Chem. Lett. 36 (2025) 110277. doi: 10.1016/j.cclet.2024.110277
S. Wu, X. Quan, ACS ES&T Eng. 2 (2022) 1068–1079.
J. Luo, Y. Liu, C. Fan, et al., ACS Catal. 11 (2021) 11440–11450. doi: 10.1021/acscatal.1c03103
J. Cheng, S. Wan, S. Cao, Angew. Chem. Int. Ed. 62 (2023) e202310476. doi: 10.1002/anie.202310476
A. Kulkarni, S. Siahrostami, A. Patel, et al., Chem. Rev. 118 (2018) 2302–2312. doi: 10.1021/acs.chemrev.7b00488
L. Wang, J. Zhang, Y. Zhang, et al., Small 18 (2022) 2104561. doi: 10.1002/smll.202104561
Y. Wang, Y. Yang, Q. Deng, et al., Adv. Funct. Mater. 33 (2023) 2307179. doi: 10.1002/adfm.202307179
H. Lv, Z. Li, P. Yin, et al., Chin. Chem. Lett. 36 (2025) 110457. doi: 10.1016/j.cclet.2024.110457
Z. Li, Y. Zhou, Y. Zhou, et al., Nat. Commun. 14 (2023) 5742. doi: 10.1038/s41467-023-41522-0
U.P. Patil, Chin. Chem. Lett. 35 (2024) 109472. doi: 10.1016/j.cclet.2023.109472
X. Feng, X. Ding, D. Jiang, Chem. Soc. Rev. 41 (2012) 6010–6022. doi: 10.1039/c2cs35157a
C. Liu, X. Liu, B. Chen, et al., Nat. Commun. 16 (2025) 8941. doi: 10.1038/s41467-025-63997-9
S. Liu, M. Wang, Y. He, et al., Coord. Chem. Rev. 475 (2023) 214882. doi: 10.1016/j.ccr.2022.214882
C. Krishnaraj, H. Sekhar Jena, L. Bourda, et al., J. Am. Chem. Soc. 142 (2020) 20107–20116. doi: 10.1021/jacs.0c09684
L. Li, L. Xu, Z. Hu, et al., Adv. Funct. Mater. 31 (2021) 2106120. doi: 10.1002/adfm.202106120
D. Chen, W. Chen, Y. Wu, et al., Angew. Chem. Int. Ed. 62 (2023) e202217479. doi: 10.1002/anie.202217479
J.N. Chang, J.W. Shi, Q. Li, et al., Angew. Chem. Int. Ed. 62 (2023) e202303606. doi: 10.1002/anie.202303606
J.R. Wang, K. Song, T.X. Luan, et al., Nat. Commun. 15 (2024) 1267. doi: 10.1108/heswbl-08-2023-0216
Z. Yu, F. Yu, M. Xu, et al., Adv. Sci. 12 (2025) 2415194. doi: 10.1002/advs.202415194
Y. Luo, B. Zhang, C. Liu, et al., Angew. Chem. Int. Ed. 62 (2023) e202305355. doi: 10.1002/anie.202305355
K. Zhang, L. Tian, J. Yang, et al., Angew. Chem. Int. Ed. 63 (2024) e202317816. doi: 10.1002/anie.202317816
H. Yu, F. Zhang, Q. Chen, et al., Angew. Chem. Int. Ed. 63 (2024) e202402297. doi: 10.1002/anie.202402297
L. Fang, H. Xu, S. Qiu, et al., Angew. Chem. Int. Ed. 64 (2025) e202423220. doi: 10.1002/anie.202423220
X. Chi, Z. Zhang, M. Li, et al., Angew. Chem. Int. Ed. 64 (2024) e202418895.
D. Chen, W. Chen, Y. Wu, et al., Angew. Chem. Int. Ed. 62 (2023) e202217479. doi: 10.1002/anie.202217479
Q. Song, Z. Li, Q. Peng, et al., Appl. Catal. B 378 (2025) 125619. doi: 10.1016/j.apcatb.2025.125619
F. Haase, E. Troschke, G. Savasci, et al., Nat. Commun. 9 (2018) 2600. doi: 10.1038/s41467-018-04979-y
X. Li, C. Zhang, S. Cai, et al., Nat. Commun. 9 (2018) 2998. doi: 10.1038/s41467-018-05462-4
P.J. Waller, S.J. Lyle, T.M. Osborn Popp, et al., J. Am. Chem. Soc. 138 (2016) 15519–15522. doi: 10.1021/jacs.6b08377
A. Kelly, K.M. Knowles, Crystal structures, in: A. Kelly, K.M. Knowles (Eds.), Crystallography and Crystal Defects, John Wiley & Sons, Ltd, Chichester, United Kingdom, 2012, pp. 85–122.
X. Wang, L. Chen, S.Y. Chong, et al., Nat. Chem. 10 (2018) 1180–1189. doi: 10.1038/s41557-018-0141-5
C. Qian, L. Feng, W.L. Teo, et al., Nat. Rev. Chem. 6 (2022) 881–898. doi: 10.1038/s41570-022-00437-y
H.S. Sasmal, A. Kumar Mahato, P. Majumder, et al., J. Am. Chem. Soc. 144 (2022) 11482–11498. doi: 10.1021/jacs.2c02301
S. An, X. Li, S. Shang, et al., Angew. Chem. Int. Ed. 62 (2023) e202218742. doi: 10.1002/anie.202218742
K.S. Rawat, S. Borgmans, T. Braeckevelt, et al., ACS Appl. Nano Mater. 5 (2022) 14377–14387. doi: 10.1021/acsanm.2c02647
Y. Pan, C.H. Ho, F. Paesani, et al., Chem. Mater. 35 (2023) 6235–6245. doi: 10.1021/acs.chemmater.3c00510
F. Haase, B.V. Lotsch, Chem. Soc. Rev. 49 (2020) 8469–8500. doi: 10.1039/d0cs01027h
H.M. Guo, X.Y. Dong, S. Wang, et al., Chin. Chem. Lett. 35 (2024) 108537. doi: 10.1016/j.cclet.2023.108537
E. Jin, J. Li, K. Geng, et al., Nat. Commun. 9 (2018) 4143. doi: 10.1038/s41467-018-06719-8
Y. Wang, Y.Z. Cheng, K.M. Wu, et al., Angew. Chem. Int. Ed. 62 (2023) e202310794. doi: 10.1002/anie.202310794
K. Geng, T. He, R. Liu, et al., Chem. Rev. 120 (2020) 8814–8933. doi: 10.1021/acs.chemrev.9b00550
M.A. Springer, T.J. Liu, A. Kuc, et al., Chem. Soc. Rev. 49 (2020) 2007–2019. doi: 10.1039/c9cs00893d
J.Y. Yue, J.X. Luo, Z.X. Pan, et al., Angew. Chem. Int. Ed. 63 (2024) e202405763. doi: 10.1002/anie.202405763
B. Li, J. Chen, K. Wang, et al., Adv. Energy Mater. 15 (2025) 2404497. doi: 10.1002/aenm.202404497
P. Li, H. Zhao, R. Ji, et al., Catal. Sci. Technol. 14 (2024) 2470–2478. doi: 10.1039/d4cy00298a
L.H. Li, X.L. Feng, X.H. Cui, et al., J. Am. Chem. Soc. 139 (2017) 6042–6045. doi: 10.1021/jacs.7b01523
L.Y. Ai, Q. Wang, X.W. Chen, et al., Aggregate 5 (2024) e582. doi: 10.1002/agt2.582
V. Hasija, S. Patial, P. Raizada, et al., Coord. Chem. Rev. 452 (2022) 214298. doi: 10.1016/j.ccr.2021.214298
Z. Li, X. Shi, H. Cheng, et al., Adv. Energy Mater. 14 (2024) 2302797. doi: 10.1002/aenm.202302797
X. Liu, R. Qi, S. Li, et al., J. Am. Chem. Soc. 144 (2022) 23396–23404. doi: 10.1021/jacs.2c09369
Z. Lei, F.W.S. Lucas, E.Canales Moya, et al., Chin. Chem. Lett. 32 (2021) 3799–3802. doi: 10.1016/j.cclet.2021.04.047
L. Chen, J. Hang, B. Chen, et al., Chem. Eng. J. 454 (2023) 140378. doi: 10.1016/j.cej.2022.140378
H. Wang, Y. Shao, S. Mei, et al., Chem. Rev. 120 (2020) 9363–9419. doi: 10.1021/acs.chemrev.0c00080
Q. Guo, Y. Yang, T. Hu, et al., Chin. Chem. Lett. 36 (2025) 110235. doi: 10.1016/j.cclet.2024.110235
X. Yang, Z.X. Pan, J.Y. Yue, et al., Small 20 (2024) 2405907. doi: 10.1002/smll.202405907
Q. Liao, Q. Sun, H. Xu, et al., Angew. Chem. Int. Ed. 62 (2023) e202310556. doi: 10.1002/anie.202310556
M. Kou, Y. Wang, Y. Xu, et al., Angew. Chem. 134 (2022) e202200413. doi: 10.1002/ange.202200413
T. Kim, D.Y. Lee, E. Choi, et al., Appl. Catal. B 357 (2024) 124264.
W. Wu, Z. Li, S. Liu, et al., Angew. Chem. Int. Ed. 63 (2024) e202404563. doi: 10.1002/anie.202404563
M. Deng, J. Sun, A. Laemont, et al., Green Chem. 25 (2023) 3069–3076. doi: 10.1039/d2gc04459e
H. Wang, C. Yang, F. Chen, et al., Angew. Chem. Int. Ed. 134 (2022) e202202328. doi: 10.1002/ange.202202328
Z. Li, C. Liu, Q. Deng, et al., Adv. Funct. Mater. 34 (2024) 2402676. doi: 10.1002/adfm.202402676
S. Guo, K. Zhao, L. Liang, et al., Angew. Chem. Int. Ed. 64 (2025) e202509141. doi: 10.1002/anie.202509141
C. Li, H. Xie, S. Zhou, et al., Mater. Res. Bull. 173 (2024) 112697. doi: 10.1016/j.materresbull.2024.112697
T. Xu, Z. Wang, W. Zhang, et al., J. Am. Chem. Soc. 146 (2024) 20107–20115. doi: 10.1021/jacs.4c04244
H.H. Sun, Z.-B. Zhou, Y. Fu, et al., Angew. Chem. Int. Ed. 136 (2024) e202409250. doi: 10.1002/ange.202409250
F. Zhang, X. Lv, H. Wang, et al., Adv. Mater. 37 (2025) 2502220. doi: 10.1002/adma.202502220
J.Y. Yue, L.P. Song, Z.X. Pan, et al., ACS Catal. 14 (2024) 4728–4737. doi: 10.1021/acscatal.4c00278
W. Wang, R. Zhang, H. Chu, et al., Small 21 (2024) 2406527.
T. Yang, D. Zhang, A. Kong, et al., Angew. Chem. Int. Ed. 63 (2024) e202404077.
S.J. Lyle, T.M. Osborn Popp, P.J. Waller, et al., J. Am. Chem. Soc. 141 (2019) 11253–11258. doi: 10.1021/jacs.9b04731
Q. Rong, X. Chen, Q. Cheng, et al., ACS Sustainable Chem. Eng. 12 (2024) 13306–13315. doi: 10.1021/acssuschemeng.4c05101
P. Das, G. Chakraborty, J. Roeser, et al., J. Am. Chem. Soc. 145 (2023) 2975–2984. doi: 10.1021/jacs.2c11454
L. Cusin, H. Peng, A. Ciesielski, et al., Angew. Chem. Int. Ed. 60 (2021) 14236–14250. doi: 10.1002/anie.202016667
Y. Mou, X. Wu, C. Qin, et al., Angew. Chem. Int. Ed. 62 (2023) e202309480.
Q. Yang, M. Luo, K. Liu, et al., Appl. Catal. B 276 (2020) 119174.
Y. Xia, W. Zhang, S. Yang, et al., Adv. Mater. 35 (2023) 2301190.
R. Liu, Y. Chen, H. Yu, et al., Nat. Catal. 7 (2024) 195–206. doi: 10.1038/s41929-023-01102-3
X. Liu, Z. Zhang, Q. Zhang, et al., Angew. Chem. Int. Ed. 64 (2024) e202411546.
S. Chai, X. Chen, X. Zhang, et al., Environ. Sci.: Nano 9 (2022) 2464–2469. doi: 10.1039/d2en00135g
Y. Hou, P. Zhou, F. Liu, et al., Angew. Chem. Int. Ed. 63 (2024) e202318562.
L. Guo, L. Gong, Y. Yang, et al., Angew. Chem. Int. Ed. 64 (2024) e202414658.
C. Qin, X. Wu, L. Tang, et al., Nat. Commun. 14 (2023) 5238.
M. Wu, Z. Shan, J. Wang, et al., Chem. Eng. J. 454 (2023) 140121.
R.M. Zhu, Y. Liu, W.K. Han, et al., Angew. Chem. Int. Ed. 64 (2024) e202412890.
J.P. Liao, M. Zhang, P. Huang, et al., ACS Catal. 14 (2024) 3778–3787. doi: 10.1021/acscatal.3c06078
S. An, X. Li, S. Shang, et al., Angew. Chem. Int. Ed. 62 (2023) e202218742.
P. Li, F. Ge, Y. Yang, et al., Angew. Chem. Int. Ed. 63 (2024) e202319885.
Y. Chang, C. Lin, H. Wang, et al., Angew. Chem. Int. Ed. 64 (2024) e202414075.
X. Li, Q. Yang, F. Yi, et al., CrystEngComm 25 (2023) 2995–2999. doi: 10.1039/d3ce00292f
X. Chi, Z.A. Lan, Q. Chen, et al., Angew. Chem. Int. Ed. 62 (2023) e202303785.
R. Sun, X. Yang, X. Hu, et al., Angew. Chem. Int. Ed. 64 (2024) e202416350.
L. Zhang, C. Wang, Q. Jiang, et al., J. Am. Chem. Soc. 164 (2024) 29943–29954. doi: 10.1021/jacs.4c12339
L. Ding, Z. Pan, Q. Wang, Chin. Chem. Lett. 35 (2024) 110125.
Y. Lin, Y. Wang, Z. Weng, et al., Nat. Commun. 15 (2024) 10032.

Figure 2 (a) The ORR process involves multiple complex steps. (b) The process of O2 adsorption in the Pauling-type configuration and (c) Yeager-type configuration for H2O2 photocatalysis.

Figure 4 (a) In-situ DRIFTS spectra vs. illumination time for the photocatalytic system of C-COFs and FS-COFs. (b) Key steps of H2O2 production by FS-COFs. Reproduced with permission [32]. Copyright 2023, Wiley. (c) Pictorial demonstration of chemical structures of DAzCOFs with different relative N locations. (d) Proposed reactive sites of TpDz for activating O2 molecule. (e) Free energy diagram of photocatalytic ORR pathways for TpDz, TpMd, and TpPz. Reproduced with permission [68]. Copyright 2023, Wiley.

Figure 5 (a) Pictorial demonstration of chemical structures of TAH-COF and BAH-COF. (b) Time course of H2O2 evolution for TAH-COF, BAH-COF, and TPPA-COF. (c) AQYs of these three COFs were measured at 475, 500, 520, 550, and 600 nm. Reproduced with permission [77]. Copyright 2024, American Chemical Society. (d) Experimental setup for H2O2 production in natural sunlight in water and seawater and a BA (10:1) mixture. (e) Long-term H2O2 production of DMCR-1NH. (f) PXRD patterns of DMCR-1NH (left) and Imine-1 (right) before and after five cycles. Reproduced with permission [85]. Copyright 2023, American Chemical Society.

Figure 6 (a) The schematic diagram of H2O2 photosynthesis for the HEP-TAPT-COF. (b) Comparison of H2O2 photosynthesis rates of HEP-COFs and g-C3N4. Reproduced with permission [28]. Copyright 2023, Wiley. (c) Photograph of the scaled-up reactor under natural sunlight irradiation. Reproduced with permission [93]. Copyright 2024, Wiley.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载: