Figure 1.
The overall structure schematic illustration of this review.
Covalent organic frameworks-based heterojunctions for photocatalytic hydrogen peroxide production and in-situ application
Zihui Liu , Xiaolin Liu , Lin Tang , Houhe Pan , Xi Liu , Jianzhuang Jiang

Hydrogen peroxide (H2O2), a liquid oxidizing agent renowned for its environmental compatibility and multifunctionality, has been listed among the 100 most crucial chemicals worldwide [1]. Its irreplaceable advantages manifest in diverse applications including wastewater treatment [2-4], organic synthesis [1,5,6], disinfection [7-9], bleaching [10-13], energy-related technologies [14,15], and other fields. Compared to the traditional anthraquinone cycle process characterized by high energy consumption and excessive byproducts, recent attention has focused on green photocatalytic strategies that convert earth-abundant H2O and O2 into H2O2 [10,16]. From a photocatalytic mechanistic perspective, the essence of efficient H2O2 synthesis resides in precise regulation of photogenerated charge carriers: Firstly, catalyst materials must achieve spatial separation of photoinduced electron - hole pairs to prevent energy loss through charge carriers recombination. Secondly, the migrated charge carriers on the catalyst surface require matched redox potentials and favorable reaction kinetics to selectively drive the two-electron oxygen reduction reaction (2e− ORR: O2 + 2H+ + 2e− → H2O2) or the two-electron water oxidation reaction (2e− WOR: 2H2O + 2h+ → H2O2 + 2H+) [17]. On the other hand, the band gap of photocatalysts should remain within an optimal range to ensure efficient utilization of light energy [18,19]. Under these constraints, the ideal band gap is generally considered to fall within the range of 1.8–2.9 eV. Additionally, the limited availability of O2 in aqueous environments necessitates the development of catalysts with high-surface-area architectures. Such structures provide enhanced active sites, which are crucial for promoting oxygen adsorption and activation processes [16].
The high solubility of H2O2 in water renders its separation from aqueous solutions a significant technical challenge. Concurrently, numerous researches have introduced sacrificial agents during synthesis processes to enhance photocatalytic H2O2 yields. However, this approach further restricts the practical application scope of H2O2. Consequently, the strategic integration of generated H2O2 with specific chemical processes, for example pharmaceutical intermediate synthesis and pollutant degradation, has emerged as a critical research frontier. A promising solution involves cascade reaction systems that enable in-situ utilization of H2O2 within the same reactor immediately following photocatalytic synthesis. This integrated methodology streamlines production workflows and enhances the economic feasibility of target compound manufacturing. Furthermore, the in-situ application strategy optimizes solvent utilization efficiency, reduces material waste, lowers energy consumption, and improves overall process sustainability – critical advantages that align with modern low-carbon production requirements.
To date, numerous semiconductor-based photocatalysts have been investigated for photocatalytic H2O2 generation, including titanium dioxide (TiO2) [20], graphite carbon nitride (g-C3N4) [21], metal organic frameworks (MOFs) [22] and others [23-25]. However, most of these semiconductor catalysts exhibit substantial energy gaps between their valence band (VB) and conduction band (CB), resulting in limited visible light absorption. Furthermore, inorganic photocatalysts typified by TiO2 demonstrate rapid charge recombination – a critical factor impairing photocatalytic efficiency – coupled with wide bandgap characteristics (anatase: 3.2 eV, rutile: 3.0 eV) that restrict light absorption to the ultraviolet (UV) spectrum [16,26]. This limitation not only hinders effective solar energy utilization but also promotes UV-induced H2O2 degradation. Notably, the photocatalytic decomposition rate of H2O2 exceeds its formation rate on unmodified TiO2 surfaces. While pristine g-C3N4 possesses favorable band alignment, specific photo-responsive properties, and notable stability, its practical photocatalytic activity remains suboptimal due to pronounced charge recombination, constrained quantum efficiency, and insufficient visible light harvesting [27]. Additionally, many MOFs inherently lack essential photosensitizing components and optimized band structures to efficiently generate photo-induced charge carriers, necessitating reliance on external catalytic systems capable of providing photoelectrons to the MOF matrix. Crucially, the efficiency of photocatalytic systems fundamentally depends on the capacity of photosensitized semiconductors to facilitate charge transfer to catalytic components [27].
Covalent organic frameworks (COFs) represent a class of crystalline porous polymeric materials constructed from organic building blocks interconnected through directional covalent bonds (e.g., boronate ester, imine, and hydrazone linkages) [28]. Leveraging through topology-guided structural rationalization, achieve programmable assembly of organic modules into extended ordered porous architectures, COFs exhibit distinctive performance superiority: (ⅰ) Highly ordered channel structures with precisely tunable pore sizes, facilitating reduced mass transfer resistance and rapid transport of reactants/products; (ⅱ) Extended π-conjugated systems within fully organic skeletons, enabling efficient photogenerated charge carrier migration; (ⅲ) Ultrahigh specific surface areas and porosity; (ⅳ) Exceptional chemical and thermal stability; (ⅴ) Flexible post-synthetic modifiability. Collectively, the structural diversity of COFs building blocks provides opportunities to tailor their physicochemical properties, laying foundation for broad applications. Since the first COF report in 2005 [29], these materials have demonstrated exceptional performance across diverse fields including adsorption/separation [30-32], mass transfer [33-35], luminescence [36-38], sensing [39-41], catalysis [42-44], energy storage [45-48], and biomedicine [49-51]. Harnessing their expansive building block library, COFs employ modular molecular engineering strategies to precisely engineer crystalline frameworks with directional charge-transport channels, enabling simultaneous optimization of pivotal photocatalytic parameters such as light harvesting efficiency, charge-carrier separation dynamics, and surface catalytic reaction kinetics.
Current research strategically focuses on three crucial fronts: innovative building-block design, crystallographic engineering, and multifunctional interface optimization, driving transformative breakthroughs in solar energy conversion technologies across key domains including H2 production, CO2 reduction, and photocatalytic H2O2 generation. In 2020, Krishnaraj et al. pioneered the utilization of TAPD-(OMe)2COFs as photocatalysts for H2O2 generation, marking the first documented application of this material class in solar-driven peroxide synthesis [52]. COFs capitalize on their inherent microporous architecture to orchestrate functional superiority: The interconnected nanopore network not only establishes efficient mass-transfer highways for optimized O2/H2O diffusion kinetics, but also ensures maximal exposure of catalytically active sites. This unique combination of molecular-scale engineering empowers COFs to craft reaction microenvironments with atomic precision for high-efficiency photocatalytic systems, demonstrating promising prospects in the field of solar-driven H2O2 synthesis.
Despite remarkable progress, persistent challenges hinder the performance optimization of COFs-based systems for photocatalytic H2O2 synthesis. The thermodynamic incompatibility between conventional COFs bandgap structures and requisite redox potentials fundamentally limits their capacity to concurrently satisfy the synergistic reaction requirements of ORR and WOR within monolithic architectures, this critical shortfall compelling most reported systems to rely on sacrificial agents for sustaining catalytic cycles [53]. These intrinsic limitations are compounded by elevated interfacial charge transfer resistances that cripple photogenerated carrier separation efficiencies, while the insufficient density of exposed active sites further exacerbates performance bottlenecks. Although COFs possess substantial potential to address these challenges through exploration of novel building blocks or topological engineering via rational permutations of existing modules, the design of COFs-based heterojunctions emerges as a rationally straightforward and effective approach for enhancing photocatalytic performance. The construction strategies and catalytic mechanisms of COFs-based heterojunction architectures have recently emerged as a vibrant research frontier in porous crystalline materials, with seminal reviews systematically elucidating their design principles and application potential across multiple dimensions. Notable contributions include: Li et al. comprehensively reviewed the performance of COFs-based semiconductor heterostructures in various photocatalytic reactions [54]. Lan et al. summarized representative metal-free heterojunction photocatalysts derived from COFs [55]. Xu et al. discussed the capabilities of COFs-based composites in photocatalytic water splitting, CO2 reduction, and pollutant degradation [56]. Jo et al. provided a comprehensive overview of integrating COFs with diverse organic, inorganic, and polymeric materials for photocatalytic hydrogen production [57]. Jiang et al. systematically explores the inherent relationship between COFs-based heterojunction innovations and photocatalytic CO2 reduction [58]. However, a critical knowledge gap persists: no review has yet holistically systematized the rapidly evolving field of COFs-based heterojunction engineering specifically tailored for photocatalytic H2O2 synthesis.
This review comprehensively surveys existing knowledge on COFs-based heterojunction architectures for photocatalytic H2O2 synthesis (Fig. 1). Commences with foundational discussions of photocatalytic H2O2 generation mechanisms and operational principles governing heterojunction material systems in this domain. Subsequent sections systematically examine developments in COFs-based heterojunctions for solar-driven H2O2 synthesis. Then in-situ applications of H2O2 in the context of photosynthesis by COFs-based heterojunctions were discussed. Concluding perspectives outline emerging opportunities and critical challenges in next-generation photocatalyst design, aiming to provide innovative insights for designing COFs-based heterojunction materials with exceptional photocatalytic H2O2 activity.
In a photocatalytic reaction, the catalyst primarily undergoes three critical stages (Fig. 2): (ⅰ) Photoexcitation: When the energy of incident photons (hν) exceeds the bandgap (Eg) of the photocatalyst, the absorption of this energy by the photocatalyst induces the excitation of electrons from the VB to the CB. This process generates negatively charged electrons (e−) in the CB and positively charged holes (h+) in the VB, both possessing certain redox capabilities. Consequently, photogenerated electron-hole pairs with spatial charge separation characteristics are formed. (ⅱ) Photogenerated charge carrier migration: Driven by built-in electric fields or energy band gradients, the excited electrons and resulting holes migrate toward the catalyst surface. This process is constrained by parameters such as carrier lifetime and diffusion length. (ⅲ) Surface redox reactions: Migrated charge carriers participate in redox reactions with chemical species on the photocatalyst surface. However, the majority of photogenerated carriers dissipate through bulk/surface recombination, leaving only a small fraction to drive effective surface reactions (e.g., ORR or WOR). Therefore, optimizing the spectral absorption range, charge carrier separation efficiency, and surface reaction kinetics constitute the central strategy for enhancing photocatalytic performance. Constructing heterojunction systems facilitates multidimensional synergistic optimization, thereby providing a novel approach to overcome the inherent performance limitations of single-component materials.
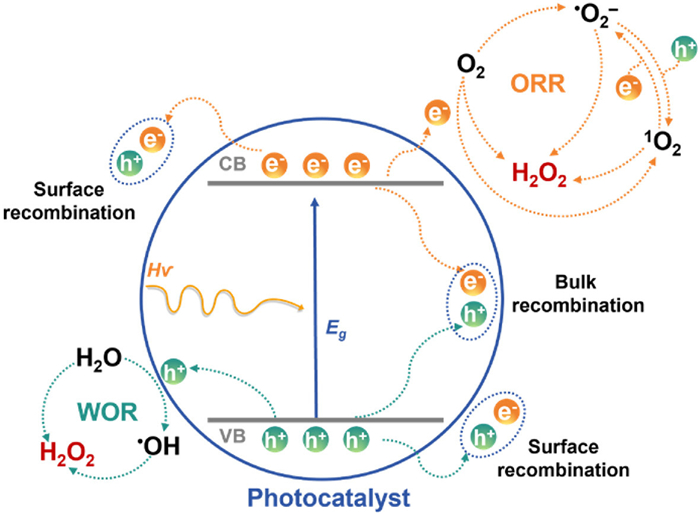
The photosynthesis of H2O2 also adheres to the basic principles of these photocatalytic processes [56]. Currently, it is widely acknowledged that H2O2 can be produced through the ORR pathway or the WOR pathway, as illustrated in Fig. 2. In the ORR pathway, electrons in the CB of the photocatalyst react with adsorbed O2 (Fig. 3). This pathway can be categorized into three distinct scenarios: (ⅰ) Direct one-step two-electron oxygen reduction (O2 → H2O2), (ⅱ) indirect two-step single-electron oxygen reduction (O2 → •O2− → H2O2), and (ⅲ) four-electron oxygen reduction (O2 → H2O) [10]. Compared with the 4e− ORR (+1.23 V), the direct 2e− ORR (+0.68 V) is thermodynamically more favorable (Eq. 1) [17]. In the indirect two-step single-electron pathway, a photogenerated electron is first captured by electrophilic O2 to form a superoxide radical (•O2−) (Eq. 2). This radical subsequently undergoes a proton-coupled electron transfer (PCET) reaction with H+ and e− to form the final H2O2 product (Eq. 3) [10]. From a kinetic perspective, the indirect two-step pathway demonstrates a pronounced advantage due to its only single-electron transfer in each step. Nonetheless, the initial reduction of O2 to •O2− requires a more negative potential (−0.33 V), thus, thermodynamically, a sufficiently negative CB position of the photocatalyst is necessary to satisfy this requirement. Notably, the •O2− can combine with holes to form singlet oxygen 1O2 (•O2− + h+ → 1O2), which can also undergo two-electron reduction to produce H2O2 (1O2 + 2e− + 2H+ → H2O2) [19,59-61]. To validate this pathway, scavengers such as 1,4-benzoquinone for •O2− or ammonium oxalate for h+ are often employed to suppress 1O2 formation. It should be pointed out that the 1O2 also can be interconverted to •O2− by electron reduction (1O2 + e− → •O2−), both processes together to promote the H2O2 formation in a tandem reaction path [62,63].
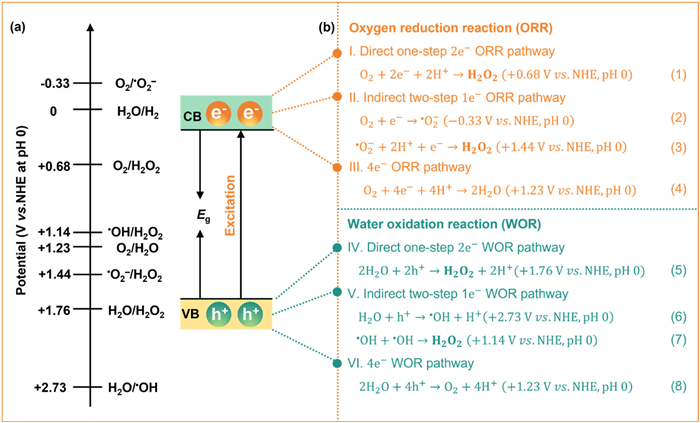
The adsorption configuration of O2 molecules on the photocatalyst surface and their interfacial interaction mechanisms are key regulatory factors governing the selectivity for the formation H2O2. Strong O2 adsorption facilitates the cleavage of the O–O bond, thereby favoring the 4e− ORR pathway with H2O as the final product. Moderate adsorption strength maintains the integrity of the O–O bond, thereby enhancing the selectivity for the 2e− ORR pathway leading to H2O2 formation. Nevertheless, although weak adsorption systems can enhance the selectivity of H2O2, they often lead to a decrease in overall catalytic efficiency due to the increased energy barrier for O2 molecules activation. Therefore, there exists a dynamic equilibrium relationship between adsorption strength and catalytic activity. Studies have demonstrated that adsorption configurations also significantly influence the selectivity for the formation H2O2. The end-on configuration proposed by the Pauling model is able to minimize O–O bond cleavage, promoting the –O–O– intermediate, demonstrating the suppression of 4e− ORR, and thus a highly selective 2e− ORR pathway, which is conducive for H2O2 formation. While the side-on configuration typified by the Yeager model facilitates the cleavage of O–O bonds to form hydroxyl radicals (•OH), an unfavorable process for H2O2 production as the subsequent coupling of •OH to H2O2 faces a high energy barrier [64-66]. However, some literatures also support the superiority of the Yeager model, as it enhances oxygen adsorption and facilitates the direct one-step two-electron ORR pathway over the sequential one-electron process, thereby improving overall catalytic performance [67,68]. This coupling mechanism between adsorption geometry and electron transfer pathways provides critical theoretical guidance for the rational design of high-selectivity ORR catalysts.
In the WOR pathway, holes in the VB of the photocatalyst can oxidize H2O through three distinct routes: direct one-step two-electron water oxidation (H2O → H2O2), indirect two-step single-electron water oxidation (H2O → •OH → H2O2), or four-electron water oxidation (H2O → O2) [16]. In the direct one-step 2e− WOR pathway, two photogenerated holes oxidize H2O to H2O2 (Eq. 5). For the indirect pathway, a single hole oxidizes H2O to generate a •OH, and subsequently, two •OH radicals couple to form H2O2 molecule (Eqs. 6 and 7). From a kinetic perspective, the indirect two-step 1e− WOR pathway is more advantageous compared to the direct one-step 2e− WOR pathway because it requires only one photogenerated hole per step. However, from a thermodynamic standpoint, the direct 2e− WOR pathway (+1.76 V) is more favorable than the indirect 1e− WOR pathway (+2.73 V). Notably, the 4e− WOR pathway (+1.23 V) (Eq. 8) competes with the direct one-step 2e− WOR pathway, thereby reducing the selectivity of H2O2 production. When viewed from a different perspective, the O2 and H+ generated via the 4e− WOR pathway can act as reactants for oxygen reduction reaction pathways, favoring the 2e− ORR pathway and thereby enhancing H2O2 production. The selectivity of the WOR pathway is fundamentally governed by the dynamic equilibrium between adsorption energies and the interactions of key intermediates (e.g., •OH) with active sites [69].
Based on the aforementioned analysis, achieving high-performance photocatalytic H2O2 production requires the spatial separation of photogenerated electrons and holes pairs with their efficient migration to active sites on the catalyst surface, while ensuring these charge carriers possess sufficient redox capability and thermodynamic driving force to enable either the 2e− ORR or 2e− WOR. Notably, the concurrent activation of both ORR and WOR pathways establishes an effective dual-channel strategy for efficient photocatalytic H2O2 synthesis, where photogenerated electrons reduce O2 to H2O2 via the 2e− ORR while hole-mediated 2e− WOR directly oxidizes H2O to H2O2 and supplies protons for the ORR pathway. This overall process (2H2O + O2 → 2H2O2) theoretically achieves 100% atom utilization efficiency [70], yet its substantial positive standard Gibbs free energy change (∆G0 = 117 kJ/mol) renders it a thermodynamically unfavorable uphill reaction (ΔG0 > 0) [10,53]. Consequently, most practical implementations necessitate the addition of sacrificial reagents (e.g., alcohols) to mitigate the inherent thermodynamic barrier and sustain catalytic activity.
Rational design of photocatalysts fundamentally governs the overall efficiency of photocatalytic H2O2 production, as it dictates three pivotal processes: (ⅰ) Photoinduced carrier separation/transport dynamics, (ⅱ) surface redox reaction kinetics, and (ⅲ) selectivity control over H2O2 generation pathways. Although COFs exhibit distinctive advantages in photocatalytic H2O2 synthesis owing to their periodic π-conjugated networks, customizable building blocks, and nanoconfined reaction environments, practical implementation of pristine COFs remains constrained by intrinsic limitations, including rapid carrier recombination and insufficient thermodynamic driving force for selective O2 activation. To overcome these material-level bottlenecks, constructing heterojunction materials through interfacial band engineering presents a paradigm-shifting strategy that enables the optimization of charge dynamics, enhances selectivity, and reinforces operational stability.
The functional superiority of heterojunction photocatalytic materials stems from precisely engineered band alignment and multicomponent synergistic catalytic effects. By regulating interfacial band engineering to precisely control charge separation pathways and spatial distribution of redox-active sites, these materials overcome the thermodynamic limitations inherent to single-component catalysts. Current advances in COFs-based heterojunctions applied in photocatalytic H2O2 synthesis predominantly feature Type Ⅱ, Z-scheme, and S-scheme heterojunctions (Fig. S1 in Supporting information). Among these, Type Ⅱ heterojunctions enhance carrier separation efficiency, while Z-scheme and S-scheme heterojunctions demonstrate superior capacity to preserve robust redox potentials. The fundamental mechanisms governing these three heterojunction types are systematically outlined in Supporting information.
The charge transfer processes in heterojunction photocatalysts are inherently more complex than those in single-component systems. Characterization the type of a heterojunction material and interfacial charge transfer in photocatalysts is a prerequisite for elucidating ultimate catalytic efficiency, providing fundamental insights into photocatalytic mechanisms and enabling rational design of advanced photocatalysts. Six typical approaches are specifically introduced in the following [71-73]:
(Ⅰ) Metal-ion photo-deposition: Application of this technique to heterojunction surfaces enables identification of electron- and hole-enrichment sites through elemental mapping during illumination.
(Ⅱ) In-situ X-ray photoelectron spectroscopy: Permits real-time monitoring of surface chemical states under near-operational conditions.
(Ⅲ) Surface photovoltaic voltage spectroscopy: Probes surface electrochemical properties of semiconductor materials by measuring light-induced voltage changes.
(Ⅳ) Transient absorption spectroscopy: Reveals ultrafast charge carrier dynamics (fs-ns timescales) through transient absorption measurements.
(Ⅴ) Spatially resolved microscopy: Provides direct visualization of spatial charge separation in heterojunction photocatalysts. Two prominent techniques include Kelvin probe force microscopy (KPFM) and single molecule fluorescence microscopy (SMFM). KPFM characterizes surface potential variations, work function differences, and localized charge accumulation by detecting tip-surface interactions. Enables precise mapping of carrier enrichment sites and photoinduced charge dynamics (formation, separation, and transfer). And SMFM employs fluorogenic molecular probes transformed into highly fluorescent products via catalytic reactions to detect photogenerated charges at an individual sample.
(Ⅵ) Density functional theory (DFT) calculations: Simulates spatial distributions of electrons and holes under illumination, offering atomic-level insights into heterojunction configurations.
To date, through strategies such as confined growth, chemical anchoring, or interfacial engineering combined with inorganic semiconductors, quantum dots, and other polymers, diverse COFs-based heterojunctions (including Type Ⅱ, Z-scheme, S-scheme, and other heterojunction configurations) have been successfully developed, demonstrating remarkable efficacy in photocatalytic H2O2 production. With the background, this section provides a comprehensive discussion of recent advances in COFs-based heterojunctions specifically engineered for photocatalytic H2O2 generation.
Conventional photocatalytic H2O2 production systems overwhelmingly rely on continuous exogenous O2 supply to achieve optimal yields. This requirement introduces substantial operational complexity in maintaining low-concentration O2 reaction environments, and significant energy penalties from gas compression/liquefaction processes, fundamentally conflicting with the atomic economy principles central to green chemistry paradigms. These inherent limitations underscore the critical need for advancing oxygen-independent photocatalytic architectures capable of efficient H2O2 generation under ambient air conditions. Yao et al. developed a type-Ⅱ heterojunction photocatalyst (ZnIn2S4/TpPa-1) by integrating ZnIn2S4 nanosheets with TpPa-1 (Fig. S2a in Supporting information) [74]. Leveraging the hierarchical heterostructure, efficient visible-light absorption, and enhanced O2 capture capability of ZnIn2S4/TpPa-1, the system achieved an H2O2 production yield of 516 μmol/L under visible-light irradiation in ambient air after 2 h, using 10 vol% alcohol (EtOH) as a sacrificial agent. This performance corresponds to 3.0-fold and 3.9-fold improvements compared to pristine TpPa-1 and ZnIn2S4, respectively. This work aims to overcome the bottleneck of highly idealized experimental conditions in H2O2 production by efficiently utilizing low-concentration O2, offering insights for enhancing the practical applicability and cost-effectiveness of photocatalytic H2O2 systems. However, the reliance on sacrificial agents limits its economic viability and long-term sustainability.
In the field of photocatalysis, quantum dots (QDs) have garnered significant attention due to their broad spectral response enabled by quantum confinement effects, size-dependent tunable band structures, and efficient photogenerated carrier dynamics. However, their practical application potential is severely constrained by spontaneous aggregation triggered by high-density surface dangling bonds, which leads to active site loss, reduced specific surface area, and accelerated carrier recombination. In this context, the ordered nano-porous architecture of COFs provides an ideal platform for QDs integration, where π-π stacking interactions or covalent anchoring strategies enable precise nanoconfinement of QDs while passivating surface defect states. Concurrently, the periodic conjugated structures of COFs establish directional electron transport channels, forming band matching heterogeneous interface with the excitonic effects of QDs. This synergistic integration significantly enhances the separation efficiency of photogenerated carriers.
Zhou et al. synthesized carbon quantum dots (CQDs) functionalized with both hydroxyl and amino groups on their surfaces [75]. These CQDs were then incorporated into the channels of highly conductive covalent triazine frameworks (CTFs) by the C-O chemical bond at varying mass ratios (ranging from 0.25 wt% to 1 wt%). The incorporation of CQDs enhanced the electrical conductivity of CTFs by approximately 10-fold (at 0.5 wt%) and acted as efficient electron transport mediators, thereby significantly improving charge separation and transfer efficiency during photocatalytic processes. The CQDs not only strengthened oxidative capacity to promote H2O2 generation via the WOR pathway but also increased affinity for H+ ions, thereby facilitating H2O2 production through the ORR pathway. This dual enhancement synergistically improved both WOR and ORR processes. Under visible-light irradiation in pure water without sacrificial agents, the optimized 0.5 CQD-CTF composite achieved an H2O2 production rate of 1036 μmol g−1 h−1, representing a 4.6-fold improvement over the pristine CTF.
The electronic state on the CQDs surface critically dictates charge separation and interfacial charge transfer processes. This electronic state is primarily modulated by surface heteroatom groups, including metal ions, which govern their interfacial interactions with the surrounding chemical environment [76]. Zeta potential analysis of aqueous CQDs dispersions confirmed their inherent negative surface charge. The introduction of monovalent alkali metal ions (Na+, K+, and Cs+) amplified the magnitude of this negative potential, indicative of enhanced electrostatic stabilization. In contrast, multivalent cations (Ca2+, Cu2+, and Ce3+) induced pronounced charge screening effects under identical experimental conditions, thereby attenuating the net negative surface charge of CQDs. Kang et al. first demonstrated that alkali metal ions (e.g., Na+, K+, Cs+) interact with carboxyl groups on the surface of CQDs during their synthesis, thereby enhancing the hole extraction capacity of CTFs (Fig. S2b in Supporting information) [77]. The CDs@CTFs containing Cs+ ions demonstrated an H2O2 production rate of 2464 μmol g−1 h−1, achieving a solar-to-chemical energy conversion efficiency of 0.9% under full-spectrum irradiation and an apparent quantum efficiency (AQE) of 13% at 500 nm. Remarkably, the photocatalyst exhibited exceptional resistance to radical oxidation and retained outstanding stability even after 100 days of storage. The superior performance stems from CQDs extracting photogenerated holes from CTFs through an interfacial built-in electric field, which effectively drives exciton dissociation, acts as a hole reservoir on CTFs, and provides active sites for novel WOR pathways.
The current strategy of COFs-based type-Ⅱ heterojunctions focuses on structural engineering and electronic state modulation, aiming to enhance photogenerated exciton dissociation, broaden environmental adaptability, and minimize dependence on sacrificial agents. These advancements establish fundamental material prerequisites for practical implementation of photocatalytic H2O2 production.
Wang et al. developed a Z-scheme WO3/Tp-TAPB (TAPB: 1,3,5-tris(4-aminophenyl)benzene; Tp: 1,3,5-triformylphloroglucinol) heterojunction that exhibits a substantial specific surface area, providing abundant active sites while WO3 and Tp-TAPB formed a compact chemical interface facilitating interfacial charge separation and transfer [78]. This architecture effectively preserves the strong oxidation capability of WO3 and the reduction capacity of Tp-TAPB. The incorporation of COFs significantly enhances the light-harvesting range and photon absorption efficiency of the WO3/Tp-TAPB composite (Fig. S3a in Supporting information). Under pure aqueous conditions without sacrificial agents, the photocatalytic H2O2 production rate reached 1488.4 μmol g−1 h−1, representing 72.3 times and 2.8 times enhancements over pristine WO3 and Tp-TAPB, respectively. Mechanistic investigations revealed that H2O2 generation proceeds via a dual-channel pathway that fully utilizes the energy of photogenerated electrons and holes during the reaction process. Notably, the composite maintained superior photocatalytic H2O2 production rates when tested in lake water and tap water systems. Practical applications demonstrated that in-situ synthesized H2O2 effectively removed chemical oxygen demand (COD) from real coking wastewater under UV irradiation, achieving over 37% COD removal efficiency through UV/H2O2 advanced oxidation processes.
Huang et al. developed a series of metal-free photocatalysts by covalently conjugating a β-ketoenamine-linked COF, synthesized via solvothermal condensation of 1,3,5-triformylphloroglucinol and 4,4′′-diamino-p-terphenyl, denoted as TpDT-COF (TP), with urease-mimetic perylene diimide-based polymers (UP) through interfacial engineering [79]. The covalent integration established an oriented built-in electric field between TP and UP, which provided an intrinsic driving force for enhanced electron-hole separation. This spatially directed charge migration selectively delivered electrons to adjacent ORR active sites while concentrating holes at WOR centers. Which significantly enhances the activity of the catalytic center on UP toward 4e⁻ WOR, and thus facilitates the overall redox reaction by reducing hole consumption and supplying O2 as a reactant for the ORR pathway (Figs. S3b and S3c in Supporting information). Consequently, in seawater without sacrificial agents, the H2O2 yield can reach up to 3846 μmol g−1 h−1 in seawater. Moreover, the catalyst exhibits remarkable stability, after 13 h consecutive photocatalytic reactions, a 4.7 mmol/L H2O2 aqueous solution can be obtained. This H2O2 solution can be directly utilized for the purification of water contaminated with dyes and phenols.
These studies demonstrate significant advancements in COF-based Z-scheme photocatalysts for H2O2 production, achieving exceptional yields through innovative charge separation strategies. They exhibit strong practical relevance, operating without sacrificial agents in complex environments and enabling real-world applications like pollutant degradation. However, key limitations persist: mechanistic evidence for charge-transfer efficiency remains indirect, scalability is challenged by complex syntheses and costly precursors, long-term stability in real matrices (e.g., biofouling, ion interference) is inadequately addressed, and performance dependencies constrain solar applicability. While representing sophisticated material design, broader viability demands deeper mechanistic validation, visible-light optimization, and rigorous technoeconomic analysis.
S-scheme heterojunctions achieve efficient charge separation while retaining robust redox capacities through a built-in electric field-driven stepwise charge transfer mechanism. However, their performance is highly contingent upon the precise band alignment between the two semiconductor components. Consequently, the rational selection and combination of semiconductor materials with appropriate band structure emerges as the key determinant for constructing high-performance S-scheme heterojunctions. Zhang et al. prepared an imine-based COF (TpPa-Cl) via Schiff-base condensation reaction between 1,3,5-triformylphloroglucinol and 2-chloro-p-phenylenediamine (Pa-Cl), then the suspension of TpPa-Cl was added dropwise into the milk-like dispersion of ZnO nanoparticles, thus engineered an S-scheme ZnO/COF (TpPa-Cl) hybrid by electrostatically self-assembling, the narrow-bandgap TpPa-Cl with strong photocatalytic reduction capability and wide-bandgap ZnO, which exhibits low cost, high stability, and potent oxidation capacity [80]. The optimal band alignment between ZnO and TpPa-Cl enabled the construction of this S-scheme heterostructure, which was systematically investigated for photocatalytic H2O2 production. The S-scheme configuration enhances light absorption, strengthens reactant adsorption capacity, improves redox efficiency, and promotes the effective separation and transfer of photogenerated carriers, thereby significantly elevating H2O2 evolution activity (Fig. S4a in Supporting information). Under simulated solar irradiation, the optimized composite achieved a maximum H2O2 production rate of 2443 μmol g−1 h−1, representing 3.3-fold and 8.7-fold enhancements over pristine ZnO nanoparticles and TpPa-Cl nanoparticles, respectively. Similarly, Yu et al. constructed an S-scheme heterojunction photocatalyst through in-situ growth of COF on ZnO surfaces [81]. The interfacial Zn-N bond formation significantly enhanced charge transfer efficiency while strengthening the internal electric field (IEF) and optimizing band alignment. This synergistic effect substantially improved the overall photocatalytic performance of ZnO/COF heterojunctions for both H2O2 production and isopropanol (IPA) selective oxidation. When tested in 8 mmol/L IPA aqueous solution, the optimized ZCOF20 composite demonstrated exceptional H2O2 generation at 10.56 mmol g−1 h−1. After 4 h of irradiation, this optimal catalyst achieved 77.4% IPA conversion with 96.4% acetone selectivity, corresponding to an acetone yield of 74.6%.
Zhang et al. fabricated an S-scheme photocatalyst for H2O2 production by coupling ZnSe quantum dots (ZnSe QDs) with a flower-like COF (designated as Ta-Dva) via an electrostatic self-assembly method [82]. ZnSe QDs and Ta-Dva COF have a staggered band structure, in terms of energy-band theory, ZnSe QDs are positioned as the reduction photocatalyst and Ta-Dva COF as the oxidation photocatalyst in S-scheme heterojunctions. This configuration enables spontaneous electron transfer from the Ta-Dva COF to ZnSe QDs, effectively separating redox-active carriers while preserving their high redox potentials, thereby significantly improving photocatalytic performance. With 10% ZnSe QDs loading and ethanol as a sacrificial agent, the heterojunction achieves an H2O2 production rate of 1895 μmol g−1 h−1, surpassing the performance of pristine Ta-Dva COF (1215 μmol g−1 h−1) and bare ZnSe QDs (403 μmol g−1 h−1).
Jiang et al. anchored 0D ZnCdS QDs (ZCS) uniformly onto the surface of a 2D conjugated tetrathiafulvalene-based COF (TT-COF), fabricating an S-scheme heterojunction photocatalyst (TT-COF/ZCS) to enhance photocatalytic H2O2 generation efficiency (Fig. S4b in Supporting information) [83]. The formation of the S-scheme heterojunction effectively suppresses ZCS agglomeration, modulates the band structure of TT-COF, and enhances photogenerated carrier migration and redox capability. Under visible-light irradiation in H2O and O2 without sacrificial agents, the optimized heterojunction (denoted as TZ-40) achieves an exceptional H2O2 production rate of 5171 μmol g−1 h−1, significantly surpassing the performance of pristine TT-COF (2520 μmol g−1 h−1) and ZCS (2647 μmol g−1 h−1). Remarkably, even under ambient air conditions, TZ-40 maintains a high H2O2 yield of 3816 μmol g−1 h−1, representing 2.47-times and 2.32-times enhancements over TT-COF and ZCS under identical conditions, respectively.
Substantial advancements have been achieved in developing synthetic protocols for COFs/inorganic semiconductor heterojunctions. Traditional configurations predominantly employ inorganic semiconductors as dual-functional cores, with COFs acting as surface-engineered active layers for targeted functionality. Recent paradigm shifts have witnessed pioneering efforts to reconfigure this architecture, where COF frameworks now serve as nucleation templates for the controlled epitaxial growth of inorganic semiconductor phases, thereby enabling interface customization. For instance, Wang et al. fabricated an S-scheme BiOBr/COF photocatalyst for H2O2 synthesis by in-situ growing BiOBr nanosheets on a Schiff-base COF with an extended π-conjugated structure [84]. The synergistic interplay between the S-scheme heterojunction and unique O2-COF interactions enhances photocatalytic H2O2 generation activity. In an aqueous solution containing 10 vol% EtOH as a sacrificial agent, this photocatalyst achieves a maximum H2O2 production rate of 3749 μmol g−1 h−1, corresponding to 1.85- and 27-times enhancements compared to pristine COF and BiOBr, respectively.
The development of narrow-bandgap photocatalysts to achieve full-spectrum solar energy utilization represents a vital strategy for overcoming energy conversion efficiency bottlenecks and advancing toward practical applications in photocatalysis. Yu et al. fabricated an organic/inorganic S-scheme heterojunction photocatalyst by in-situ growing In2S3 nanosheets on a COF substrate (Fig. S4c in Supporting information) [59]. The coordination between In3+ ions in In2S3 and methoxy/imine groups in the COF generates heterojunction energy levels, narrowing the bandgap while significantly extending the light-harvesting range from visible to near-infrared regions and enhancing visible-light absorption. Benefiting from the broadened spectral response, improved carrier separation efficiency, robust redox capability, and multi-channel H2O2 formation pathways, the optimized heterojunction material achieves an H2O2 production rate of 5713.2 µmol g−1 h−1 in pure water.
Furthermore, precisely controlling the spatial configuration of composite photocatalysts can enhance their photocatalytic efficiency. Peng et al. reported a surfactant-assisted microwave solvothermal strategy, where the anionic surfactant sodium dodecyl sulfate was utilized to occupy the {110} facets of decahedral BiVO4 (D-BVO) [85]. Simultaneously, the electrostatic adsorption between the {010} facets of D-BVO and p-phenylenediamine enabled the selective growth of the covalent organic framework TpPa-1 on the {010} facets, forming TpPa-1/{010}-BVO (denoted as TB-1) (Fig. S4d in Supporting information). Experimental and theoretical analyses revealed that TB-1 generates a stronger built- in electric field compared to TpPa-1/BVO heterojunctions randomly formed on both {010} and {110} facets (denoted as TB-2), significantly favoring the separation of photogenerated carriers. The photocatalytic oxygen reduction reaction activity of TB-1 reached 723 μmol g−1 h−1, corresponding to enhancements of 72.3-times, 5.7-times, and 2.4-times over pristine BiVO4 (10 μmol g−1 h−1), TpPa-1 (127 μmol g−1 h−1), and TB-2 (305 μmol g−1 h−1), respectively. Additionally, the incorporation of TpPa-1 effectively suppresses H2O2 decomposition.
To address the kinetic limitations of the 4e− WOR pathway, traditional transition metal-based cocatalysts can improve reaction kinetics, but their surface-active sites tend to induce catalytic decomposition of H2O2 [86]. This paradoxical mechanism urgently requires resolution through the development of metal-free oxygen evolution active centers to effectively suppress the in-situ degradation of the target product. Zhao et al. developed an S-scheme heterojunction through in-situ integration of triazine-based COF (TD COF) with porous g-C3N4. The synthesis involved introducing pre-synthesized g-C3N4 powder into TAPB and 2,5-divinylterephthalaldehyde (DVA) reaction system. The resulting TD COF/g-C3N4 composite features covalent linkages between -NH2 groups on g-C3N4 and -CHO groups on TD COF. This precisely engineered interface enhanced both efficient H2O2 generation and rhodamine B (RhB) degradation simultaneously [87]. Compared to pristine COF and g-C3N4, the S-scheme COF/g-C3N4 heterojunction exhibited superior charge separation efficiency. Strong photogenerated electrons in the CB of g-C3N4 and robust photogenerated holes in the highest occupied molecular orbital of TD COF were retained, providing sufficient redox potential to enhance photocatalytic activity. Under RhB solution and O2 atmosphere, the 10%-COF/g-C3N4 composite achieved an H2O2 production rate exceeding 2307 μmol g−1 h−1, along with 100% RhB degradation within 45 min. The integration of RhB degradation with H2O2 synthesis in photocatalytic systems represents an innovative strategy for synchronizing pollutant elimination and resource recovery.
Coupling photocatalytic H2O2 production with selective oxidation of organic compounds not only consumes photogenerated holes to yield value-added chemicals but also replaces the sluggish water oxidation reaction, thereby improving H2O2 yield. The work by Yu et al. demonstrated that coupling the photocatalytic H2O2 generation cycle with simultaneous selective furfuryl alcohol (FAL) oxidation. Specifically, they fabricated a core-shell S-scheme TiO2@BTTA photocatalyst by in-situ growing a BTTA COF layer on TiO2 nanofibers (Fig. S4e in Supporting information) [88]. The resulting TiO2@BTTA composite features an extensive interfacial contact area and a reduced carrier migration distance. Moreover, the porous ultrathin BTTA layer endows the composite with abundant active sites and exceptional light-harvesting capacity, while the S-scheme heterojunction effectively promotes carrier separation in space and boosted redox power. The system achieved an H2O2 evolution rate of 740 μmol L−1 h−1, with a coupled oxidation conversion rate of 92% for FAL. Mechanistic studies revealed that H2O2 is formed through a two-step 1e– ORR pathway. Concurrently, FAL is activated by holes into •C5H5O2 radicals, which are further oxidized by holes or •OH radicals to produce furfural (FF) or furoic acid (FAC).
There are significant advancements in achieving high H2O2 yields through innovative S-scheme designs, validating enhanced charge separation and preserved redox capabilities via strategic band alignment, diverse material combinations (ZnO, BiOBr, BiVO4, QDs, etc.), and sophisticated interface engineering (in-situ growth, facet control, covalent linkages, coordination bonds). While notable for enabling coupled organic oxidation, pollutant degradation, and even near-infrared activity, critical limitations persist: Most high-performance systems heavily depend on sacrificial agents, raising practical and economic concerns; H2O2 decomposition suppression remains inadequately addressed; scalability of complex syntheses is challenging; and long-term stability under realistic conditions is rarely demonstrated. Thus, despite impressive lab-scale performance, practical viability hinges on resolving sacrificial agent dependency, ensuring product stability, and proving scalability under real-world operational constraints.
In addition to the heterogeneous junctions mentioned above, there are other special heterojunctions used for the photocatalytic generation of H2O2.
The photocatalytic generation of H2O2 is fundamentally governed by the electron transfer reduction of molecular oxygen following light-induced carrier separation in the catalyst. The electron transfer-mediated reduction of molecular oxygen serves as the rate-determining step for photocatalytic generation of H2O2. Consequently, catalyst design must not only ensure efficient charge separation but also enhance the electron utilization efficiency for oxygen activation, as both aspects are essential to optimizing photocatalytic performance. He et al. synthesized a triazine-based cationic COF with Br− counterions (denoted as TTB-EB) via Schiff-base condensation, and subsequently fabricated a binary PTA@TTB-EB hybrid material by impregnating phosphotungstic acid (PTA) onto the TTB-EB framework [89]. Leveraging rapid and reversible multi-electron redox behavior of PTA under mild conditions, the hybrid system employs PTA as an electron-transfer mediator to construct an efficient electron transport pathway from the CB of COF semiconductor to the adsorbed O2 molecules. The catalytic cycle optimizes reaction pathways, thereby enhancing oxygen capture and reduction kinetics in the ORR. Simultaneously, it synergistically promotes charge separation and modulates the ORR pathway for improved overall performance. The presence of PTA facilitates both the kinetic rate and thermodynamic propensity for O2 conversion to •O2− intermediates, thereby enhancing the photocatalytic activity and selectivity of the sequential two-step 1e− ORR (Fig. S5a in Supporting information). This hybrid achieves an H2O2 production rate of 897.94 μmol L−1 h−1 in pure water without sacrificial agents under visible-light irradiation, representing a 2.2-fold enhancement compared to the pristine TTB-EB. Furthermore, the synergistic interplay between electrostatic interactions of quaternary ammonium cation sites in the COF and PTA, coupled with confinement effects within the porous channels, endows PTA@TTB-EB with exceptional photostability, recyclability, and long-term operational durability.
Chen et al. reported an innovative strategy for in-situ construction of oriented COFs-based PN heterostructure films through monolayer MoS2-induced growth, which significantly enhances photogenerated charge carrier dynamics and facilitates photocatalytic H2O2 synthesis [90]. The fabricated oriented TAPB-DMTA COF/MoS2 film (DMTA: 2,5-dimethoxyterephthalic acid) exhibits a superior photocatalytic H2O2 production rate of 8154 µmol g−1 h−1. This performance represents approximately 2.5- and 4-fold higher than that of non-oriented TAPB-DMTA COF films and powder counterparts, respectively. Mechanistic studies revealed that the oriented architecture of TAPB-DMTA COF crucially promotes the generation and directional transport of photogenerated electron-hole pairs along both the framework axis and through interlayer stacking pathways, thereby amplifying photocatalytic H2O2 generation efficiency (Figs. S5b and c in Supporting information).
Those studies demonstrate innovative heterojunction designs beyond conventional types, effectively addressing the dual requirements of enhanced charge separation and optimized oxygen reduction kinetics for photocatalytic H2O2 generation. While those studies exemplify material-design ingenuity, they share limitations in standardized performance metrics and require further validation of long-term practicality and mechanistic details.
The limited stability of H2O2 under specific reaction conditions (elevated or suboptimal temperatures, extreme pH levels) coupled with its excessive dosage requirements poses significant operational challenges and escalates process costs. Commercially supplied H2O2 is typically transported from centralized production facilities at concentrations substantially exceeding those required for some coupled processes. The mandatory dilution procedure effectively negates the energy previously invested in distillation and concentration steps, resulting in substantial energy wastage. In-situ activation of catalytically generated H2O2 to produce reactive oxygen radicals for chemical transformations presents a promising alternative. This strategy eliminates the need for bulk H2O2 storage and enables on-demand radical generation under milder reaction conditions while maintaining process efficiency.
The photocatalytic generation of H2O2 enables in-situ production of highly redox-active species such as •OH and •O2−, constituting the underlying principle for its autonomous applications [23,91]. Two dominant pathways govern H2O2 utilization: (ⅰ) Self-activation of H2O2 and (ⅱ) H2O2-coupled Fenton/Fenton-like reactions.
(ⅰ) H2O2 self-activation refers to the photolytic decomposition of H2O2 under ultraviolet irradiation (typically 1~280 nm), generating highly reactive •OH radicals through cleavage of the O-O bond. This mechanism has been extensively implemented in water treatment and pollutant degradation systems. Notably, photocatalytic H2O2 production systems inherently face the challenge of concurrent H2O2 decomposition, where photogenerated electrons and holes react with synthesized H2O2, as described in Eqs. 9–11. This dual functionality creates a dynamic equilibrium between H2O2 generation and consumption during photocatalytic processes.
|
|
(9) |
|
|
(10) |
|
|
(11) |
However, the direct decomposition of H2O2 remains kinetically unfavorable due to thermodynamic constraints, resulting in low operational efficiency. This inherent limitation has driven the development of strategic catalyst engineering utilizing transition metal-based materials to circumvent these challenges through in-situ activation or optimized pre-activation protocols [66].
(ⅱ) Fenton/Fenton-like processes coupled with photocatalytically generated H2O2 enable in-situ environmental remediation through activation of H2O2 into •OH and •O2−. In the presence of H2O2, redox cycling of metal ions (e.g., Fe3+/Fe2+ and Cu+/Cu2+) drives the catalytic conversion of H2O2 to •OH (Eqs. 12 and 13). This ion-mediated activation mechanism substantially enhances H2O2 decomposition kinetics and radical yield, achieving exceptional efficiency in pharmaceutical intermediate synthesis and recalcitrant pollutant degradation.
|
|
(12) |
|
|
(13) |
The in-situ H2O2 production-utilization paradigm has gained rapid adoption in sterilization, organic pollutants degradation, biomedical applications, and organic synthesis. By circumventing logistical constraints and low utilization efficiency inherent to conventional H2O2 supply chains, this integrated approach ensures on-demand oxidant availability while minimizing energy losses associated with storage and transportation.
Bacterial contamination poses a critical threat to public health, environmental security, and antimicrobial resistance management, with the global dissemination of drug-resistant bacteria emerging as a paramount challenge in modern healthcare systems. H2O2-based disinfection demonstrates superior sterilization efficacy compared to conventional approaches, offering complete microbial eradication, zero residual toxicity, non-induction of bacterial resistance, and environmentally benign characteristics. These intrinsic advantages position H2O2 as a transformative solution for green antimicrobial strategies, with expanding applications in clinical settings, potable water purification, and public health emergency responses. Photocatalytic H2O2 generation systems further enhance operational practicality through on-demand oxidant production, eliminating hazardous storage and transportation requirements. This technological advancement surpasses conventional disinfection methods in process safety and spatiotemporal controllability, crucially addressing the limitations of traditional bactericidal approaches while maintaining uncompromised antimicrobial performance.
Bi et al. fabricated an inorganic-organic S-scheme SS/TaTp heterojunction photocatalyst by coupling oxidation photocatalyst a specific COF-TaTp and reduction photocatalyst SnS2 (SS) via a facile hydrothermal method (Fig. S6a in Supporting information) [92]. The optimized 10% SS/TaTp demonstrated remarkable H2O2 generation efficiency, achieving 3.45 times and 16.87 times enhancements over pristine TaTp and SS, respectively, with significant performance improvements under both visible and near-infrared light irradiation. The S-scheme charge transfer mechanism was identified to accelerate photogenerated electron migration while reinforcing redox capacity. Notably, the internal electric field intensity of 10% SS/TaTp was calculated to be 2.14- and 4.63-fold stronger than those of TaTp and SS, respectively. When integrated into an in-situ Fenton system, the generated H2O2 achieved complete bacterial eradication (at an initial concentration of 7.0 log10 CFU/mL) within 50 min for Escherichia coli (E. coli) inactivation. Interfacial C-N-S covalent interactions between TaTp and SS were found to fine-tune the band structure and generate hybrid energy levels within the heterojunction, thereby enhancing light-harvesting capacity and catalytic performance across visible and near-infrared spectral regions (Fig. S6b in Supporting information).
Under visible-light irradiation, the CdS-modified star-shaped photocatalyst COF developed by Bi et al., denoted as CdS/TpMA, demonstrated enhanced H2O2 production capability without requiring sacrificial agents. Mechanistic investigations revealed that the ORR pathway dominated the H2O2 generation process, with •O2− serving as the primary intermediate species, while the WOR contributed partially [93]. The superior photocatalytic performance originated from two critical factors: The constructed S-scheme heterojunction between CdS and TpMA significantly promoted the spatial separation of photogenerated charge carriers, and the composite exhibited exceptional oxygen adsorption and activation capabilities. By introducing Fe2+ to activate the in-situ generated H2O2, an efficient Fenton system was established for bacterial inactivation, achieving complete deactivation of Escherichia coli (1 × 107 CFU/mL) within 120 min. Furthermore, a dual-compartment reactor equipped with a semipermeable membrane was designed to spatially separate photocatalysts from microorganisms, enabling a "long-distance" disinfection mechanism while maintaining the inherent advantages of both heterogeneous photocatalysis (facile catalyst recovery) and homogeneous Fenton reactions (Fig. S6c in Supporting information). This innovative configuration simultaneously addresses operational stability and treatment efficiency challenges in practical applications.
Recent advancements in COFs-based heterojunction photocatalytic systems have demonstrated notable progress in the in-situ generation of H2O2 and their antimicrobial applications [92,94,95]. Current research priorities focus on optimizing photo-Fenton synergistic mechanisms, engineering heterojunction interfaces, and innovating reactor designs. These systems exhibit dual advantages of environmentally friendly antibacterial properties and on-demand sterilization capabilities, thereby establishing a strategic foundation for developing next-generation intelligent disinfection systems. However, substantial innovative efforts remain imperative to advance scalable implementation, particularly in addressing critical challenges posed by antibiotic-resistant bacterial proliferation and ensuring robust public health security infrastructure.
Water contamination has emerged as a worldwide issue, presenting a significant risk to ecological systems, public health, and sustainable progress. H2O2 known for its efficiency, eco-friendliness, and safety, holds substantial potential for degrading various pollutants. Notably, generating H2O2 directly in water through photocatalysis and utilizing it in-situ for water treatment is a straightforward and cost-effective approach, eliminating the necessity for external H2O2 supplementation.
Tang et al. synthesized Bi4O5Br2 via hydrothermal method, followed by encapsulating the material within ampules containing 1,3,5-tris(4-formyl-phenyl)triazine and 4-(4′-aminophenyl)-2,6-bis(4′′-aminophenyl)pyridine monomers, ultimately constructing a COFs-based hybrid TTD-COF/Bi4O5Br2 [96]. The BIT6 composite with an optimal 6:1 TTD-COF/Bi4O5Br2 mass ratio exhibited significantly enhanced photocatalytic activity. Under ambient air and sacrificial agent-free conditions in pure water, BIT6 achieved an H2O2 production rate of 5221 μmol g−1 h−1, representing 20- and 1.7-times enhancements compared to pristine Bi4O5Br2 and TTD-COF, respectively. Furthermore, they demonstrated the dual functionality of BIT6 by simultaneously generating H2O2 and degrading antibiotics (tetracycline and doxycycline) in-situ. Under visible-light irradiation for 1 h, the BIT6 photocatalytic system achieved degradation efficiencies of 87% for tetracycline and 82% for doxycycline, alongside H2O2 production rates of 3960 μmol g−1 h−1 and 2456 μmol g−1 h−1, respectively. This sacrificial agent-free design not only demonstrates enhanced practicality through the novel integration of H2O2 photogeneration, but also reveals substrate-specific performance constraints wherein the H2O2 production efficiency exhibits pollutant-dependent variations due to competing interfacial redox reactions. They also proved that H2O2 did not play a key role in antibiotic degradation. Additional experiments with sacrificial agents revealed that holes, electrons, and reactive oxygen species (1O2 and •O2−) all played a role in the process of antibiotic degradation.
The degradation of diverse organic pollutants via H2O2 has been widely recognized as an effective strategy, and numerous related studies have been conducted. However, the degradation, particularly the mineralization of organic contaminants demands stronger oxidative capacity. In-situ Fenton reactions can address these limitations by generating highly reactive •OH. Consequently, researchers have explored in-situ Fenton or Fenton-like processes to enhance degradation efficiency. Therefore, many researchers explore in-situ Fenton or Fenton-like reactions to enhance degradation efficiency.
Tang et al. successfully designed and synthesized a novel Z-scheme MOF/COF heterojunction, name as MT2 (MIL-100(Fe)/TpPa-1 COF heterojunction), coupled with an in-situ Fenton system for tetracycline hydrochloride (TC) removal from aqueous solutions [97]. Experimental results demonstrated that MT2 (0.08 g/L) achieved a 91% TC removal efficiency (20 mg/L, 100 mL) within 120 min, attributable to enhanced charge separation efficiency and synergistic effects among h+, •OH, and •O2− radicals (Fig. S7a in Supporting information). The study revealed that TC exhibited significant inhibitory effects on wheat seedling growth and chlorophyll production, while its degradation intermediates showed negligible phytotoxicity. Notably, the ecological toxicity of TC was substantially mitigated, as evidenced by reduced inhibition zones against both E. coli and Staphylococcus aureus (S. aureus). These findings highlight the practical potential of the developed in-situ photo-Fenton composite material for effective antibiotic remediation.
Similarly, Zhu et al. fabricated a ternary heterojunction by utilizing MXene nanosheets as both a growth platform for in-situ synthesis of ultrafine TiO2 nanoparticles and a confined microenvironment for Schiff-base COF decoration, thereby establishing enhanced 2D/0D/2D interfacial interactions in the COF@TiO2/MXene architecture (denoted as CTMS) [98]. The optimized heterojunction CTMS-2 demonstrated superior visible-light-driven antibiotic degradation performance, achieving maximum reaction kinetics of 0.03423 min−1 with a mineralization efficiency of 69.7%. Owing to the remarkable H2O2 generation capability of the CTMS-2 heterojunction, its photocatalytic self-Fenton activity was systematically investigated through Fe2+-assisted photocatalysis. This synergistic approach further enhanced the antibiotic degradation kinetics to 0.04326 min−1, demonstrating significant improvement in redox efficiency (Fig. S7b in Supporting information).
Current studies have confirmed that COFs-based heterojunctions exhibit efficient, low-toxicity, and environmentally benign characteristics in organic pollutants degradation systems. However, challenges persist in addressing the restricted H2O2 production yield due to competitive reactions with pollutants, optimizing heterojunction stability, and elucidating fundamental reaction mechanisms. In the future, it is necessary to promote the practical application of this technology in complex water quality remediation and industrial wastewater treatment by precisely regulating the electronic structure of the interface, developing stable multifunctional heterojunction materials, and deepening the research on in-situ reaction kinetics.
A large number of diseases caused by drug-resistant bacterial infections have currently become a significant threat to global public health security. It is critically important to develop a novel non-antibiotic antibacterial material that exhibits broad-spectrum antibacterial activity while avoiding the risk of inducing drug resistance. Although recent advancements in antibacterial material research have been promising, these materials generally still encounter the dual challenges of inadequate antibacterial efficacy and insufficient biocompatibility. Consequently, the development of a new type of functional material with both highly efficient, broad-spectrum antibacterial properties and excellent biocompatibility holds substantial scientific significance and practical application value.
Inspired by the antimicrobial properties of natural marine mussels, Lin et al. successfully synthesized an ortho-dihydroxy covalent organic framework hydrogel (Gel@COF) with broad-spectrum antibacterial efficacy and evaluated its wound healing potential in a methicillin-resistant Staphylococcus aureus (MRSA)-infected diabetic mouse model [99]. The authors employed 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (TAPT) and 2,3-dihydroxyterephthalaldehyde (2,3DHA) as monomers to construct the ortho-dihydroxy-functionalized covalent organic framework (TAPT-2,3DHA-COF) through a solvothermal approach. This structure demonstrated exceptional cascade self-oxidation capability for sustained H2O2 generation. At minimum inhibitory concentrations, the material effectively disrupted bacterial membrane integrity, inducing nucleic acid and protein leakage while simultaneously impairing energy metabolism and oxidative stress regulation. These mechanisms conferred superior broad-spectrum antibacterial activity against of six common and drug-resistant bacterial-E. coli, S. aureus, Vibrio alginolyticus (V. alginolyticus), Bacillus cereus (B. cereus), Pseudomonas aeruginosa (P. aeruginosa) and MRSA. Subsequent radical copolymerization at ambient temperature yielded a hybrid polyacrylamide-COF hydrogel (Gel@COF), which maintained potent antibacterial functionality while exhibiting favorable cytocompatibility with L929 cells and minimal hemolytic effects on erythrocytes (Fig. S8a in Supporting information). To validate its clinical potential, the hydrogel’s therapeutic efficacy was systematically investigated in MRSA-infected diabetic wounds. Remarkably, Gel@COF achieved a 99.62% wound closure rate within 12 days, demonstrating exceptional bactericidal and wound-healing properties. The composite hydrogel alleviated the inflammatory microenvironment and enhanced microvascular performance, leading to the promotion of diabetic wound healing via multifaceted synergistic actions. This ultimately resulted in a remarkably high healing rate of approximately 99%. Gel@COF show significant potential for treating bacterial infections in diabetic wounds (Fig. S8b in Supporting information).
Zhang et al. successfully constructed a donor-acceptor architecture metallophthalocyanine-based covalent organic framework (denoted as CuPc-Dha-COF) through solvothermal synthesis using tetra-amine copper phthalocyanine and 2,5-dihydroxy-1,4-benzenedicarboxaldehyde as precursors. Leveraging its well-developed porous structure and tunable functionality, this framework served as an ideal host matrix for silver nanoparticle (AgNP) immobilization [100]. Through precise control of synthetic conditions, the researchers developed a CuPc-Dha-COF@AgNPs heterojunction material featuring uniformly dispersed Cu-N4 single-atom sites and size-controlled AgNPs. At a low concentration of 200 μg/mL, the CuPc-Dha-COF@AgNPs heterojunction achieves near-complete bacterial inactivation (≈100%) through synergistic photothermal/photodynamic effects, in-situ H2O2 generation, and controlled Ag+ ion release. Besides, capitalizing on its enhanced photoelectric conversion efficiency and exceptional enzyme-mimetic activity, the team fabricated an ultra-sensitive photoelectrochemical biosensor. This sensor exhibits a broad linear detection range (1.0 pmol/L-1.0 nmol/L) with an ultralow limit of detection (99 fmol/L) for glutathione, establishing a novel methodology for direct bacterial detection in complex environments (Fig. S8c in Supporting information).
The application of COFs-based heterojunctions for in-situ H2O2 generation in biomedical applications remains in its nascent stage, yet their high-efficiency and controllable nature present a novel strategy to overcome the inherent limitations of conventional H2O2 therapeutic approaches. To advance this technology toward clinical translation, future research must prioritize interdisciplinary collaboration to address critical challenges including biocompatibility optimization, physiological environment compatibility, and scalable fabrication protocols.
The in-situ photocatalytic production of H2O2 presents an excellent strategy for organic chemical synthesis. This approach enables oxidation reactions under mild operating conditions while effectively mitigating the environmental contamination and waste management issues associated with conventional harsh oxidizing agents. Particularly significant in addressing the current energy and environmental challenges holds great significance.
Unspecific peroxygenases (UPOs), act as oxidase, directly utilize H2O2 to form catalytically active oxygen-iron cationic radical complexes. Unlike conventional oxidase systems, these enzymes operate without requiring cofactors for reducing equivalents, enabling efficient and selective oxidation of inert C−H bonds under mild green chemistry conditions. The inherent fragility of the UPO active site necessitates continuous maintenance of low H2O2 concentrations to maximize catalytic efficiency. Wang et al. developed a novel approach through room-temperature synthesis of vinyl-functionalized 2D covalent organic frameworks (TAPB-Vi COF) using 1,4-benzenedicarboxaldehyde and 1,3,5-tris(4-aminophenyl)-benzene [101]. The authors innovatively integrated the photocatalytic in-situ H2O2 generation capability of TAPB-Vi COF with H2O2-dependent UPO activation, establishing a self-contained photo-enzymatic system for one-pot conversion of ethylbenzene to R-1-phenylethanol. Optimal enzyme loading (2.5 μg/mL) achieved peak production of 1.92 μmol R-1-phenylethanol with exceptional enantioselectivity (ee > 99%). This integrated platform streamlines conventional multi-step UPO catalysis into a unified operation, presenting an environmentally benign and operationally simple photo-enzyme cascade system. The methodology demonstrates remarkable success in achieving high-precision C−H bond activation under ambient conditions, advancing sustainable biocatalytic strategies for stereoselective oxidations (Fig. S9 in Supporting information).
Overall, COFs-based heterojunctions demonstrate considerable potential as efficient and environmentally benign platforms in integrated photocatalysis-organic synthesis systems. However, critical scientific challenges remain to be addressed in future investigations, including enhancing material stability and scalability, optimizing multi-reaction synergy, developing greener synthetic pathways, and elucidating fundamental reaction mechanisms. Addressing these key aspects will be essential for driving transformative breakthroughs in this emerging interdisciplinary field.
In recent years, COFs-based heterojunction materials have witnessed significant advancements in the field of photocatalytic H2O2 synthesis (Table S1 in Supporting information). Systematic investigations into the interfacial electronic structures, band alignment, and charge transport dynamics of these heterojunctions have revealed the mechanisms governing the directed migration of photogenerated carriers and the synergistic enhancement of active sites. Building on these insights, the integration of theoretical calculations and precision material design has laid the foundation for developing efficient, stable, and environmentally compatible COFs-based heterojunctions, such as sacrificial agent-free systems and seawater-adaptive architectures. Notably, the construction of heterojunction systems enabling in-situ H2O2 production-utilization paradigm offers innovative strategies to overcome the bottlenecks of traditional photocatalytic systems in terms of H2O2 yield, selectivity, and practical applicability. This review summarizes the fundamental principles of heterojunction photocatalysts for H2O2 production and highlights recent progress in COFs-based heterojunctions in photocatalytic synthesis and in-situ application of H2O2. Despite promising achievements, the field remains in its infancy due to challenges including low H2O2 production rates, insufficient selectivity, and the frequent requirement for additional hole scavengers. Substantial advancements in the engineering of efficient heterojunction photocatalysts are imperative to realize the practical application and commercialization of COFs-based heterojunction systems. Future research directions should prioritize the following aspects (Fig. 4):
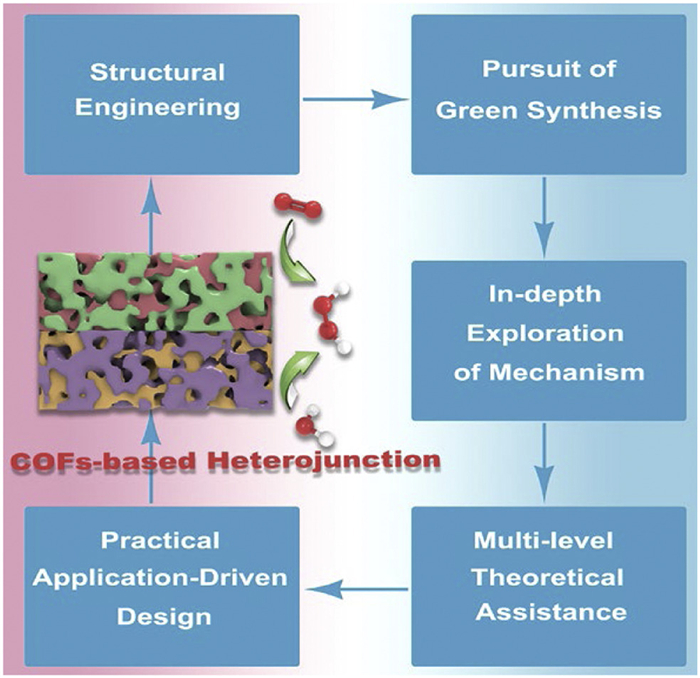
(I) Structural engineering: In COFs-based heterojunctions, the presence of favorable oxygen adsorption/transformation sites, sufficient light-harvesting capability, extensive surface area, and effective substrate interfacial contact collectively enable COFs to primarily serve as active sites for ORR. In contrast, a limited number of heterojunction systems utilize COFs as WOR active sites, typically requiring COFs with elevated valence band positions that exceed the thermodynamic potential for water oxidation. Current optimization strategies focus on structural design modifications, electronic structure tailoring of COF building units, active site engineering, and bandgap modulation to enhance WOR efficiency. Based on these insights, the rational assembly of heterojunctions is pursued through strategic incorporation of semiconductor materials with precisely aligned band structures. Critical challenges persist in heterojunction system design, particularly regarding precise band alignment mechanisms between COFs and other semiconductors. This requires meticulous regulation of VB and CB positions to ensure the VB edge potential exceeds the water oxidation potential while maintaining the CB edge potential below the oxygen reduction potential. Simultaneously, the delicate balance between band offset magnitude and carrier migration barriers must be optimized to prevent excessive offset-induced charge transport impediments. Future research directions should focus on dual-dimensional optimization encompassing both COFs structural design and heterojunction engineering. Primarily, developing structurally stable COFs with enhanced photocatalytic performance remains paramount, necessitating molecular-level innovations in functional building blocks, linkage chemistry, topological optimization, and unit functionalization for precise band structure engineering. Secondary considerations involve heterojunction interface engineering through molecular orbital hybridization for contact barrier modulation, coupled with covalent bonding strategies, defect engineering, and nanoscale interface manipulation to establish gradient band structures enabling directional charge migration.
Moreover, attention should be directed toward broadening the wavelength spectrum responsive to the catalyst, particularly through enhancing its capacity to absorb visible and near-infrared radiation (λ > 600 nm). This strategic modification would improve solar energy utilization efficiency while mitigating H2O2 degradation induced by ultraviolet radiation. Additionally, comprehensive investigations into morphology control, crystallinity optimization, and hierarchical assembly architectures are imperative. The ultimate objective lies in fabricating photocatalytic materials exhibiting robust visible-light absorption, optimized bandgap configurations, superior solar-to-chemical conversion efficiency, exceptional photostability, and scalability for commercial applications.
(II) Pursuit of green synthesis: On the one hand, COFs-based heterojunctions currently lack straightforward, efficient, and cost-effective large-scale preparation methods, significantly limiting their potential to meet practical application requirements for high-performance photocatalysts. On the other hand, current research on photocatalytic H2O2 production has predominantly concentrated on developing 2e− ORR catalysts, while investigations into photocatalysts for the 2e− WOR pathway remain relatively limited. Therefore, enhancing research efforts on 2e− WOR mechanisms and catalyst engineering to achieve dual-channel photocatalytic H2O2 generation could significantly enhance the overall yield and production efficiency. Alternatively, while sacrificial agents significantly enhance the performance of photocatalytic H2O2 production in COFs-based heterojunction systems, their irreversible depletion substantially escalates operational expenditures and critically hinders practical implementation. Consequently, future research must prioritize the development of innovative strategies to accelerate the separation of photogenerated charge carriers and enable directional migration of electron-hole pairs toward targeted reaction sites. This alignment would facilitate efficient synergy between carrier transport and redox reactions. A promising approach involves integrating photocatalytic H2O2 generation with selective oxidation of organic compounds. Such a dual-functional system could replace the kinetically sluggish water oxidation reaction by consuming photogenerated holes to produce high-value chemicals, thereby simultaneously boosting H2O2 production efficiency and achieving solar-to-chemical energy conversion. The ultimate objective is to realize the simultaneous optimization of photocatalytic H2O2 synthesis efficiency and selectivity, advancing the goal of solar-driven green synthesis of peroxides.
(III) In-depth exploration of mechanism: In mechanistic studies of photocatalytic H2O2 synthesis, most of the current studies on charge transfer and adsorption sites are indirect proofs based on theoretical calculations, there remains a lack of systematic structure-activity relationship research connecting the exceptional performance of COFs-based heterojunction materials to their carrier transport mechanisms, active site localization, and interfacial reaction kinetics. To address this, it is imperative to employ advanced dynamic process characterization techniques including but not limited to electron energy loss spectroscopy, synchrotron-based X-ray absorption fine structure, Infrared, and Raman spectroscopy. Above characterization performed under dynamic operational conditions to ensure real-time monitoring of interfacial processes. Complemented by isotopic labeling to unambiguously identify product origins and verify material participation. By integrating multidimensional dynamic characterization data, researchers can achieve real-time tracking of spectral absorption, separation kinetics, key intermediates and migration mechanisms of charge carriers during photocatalytic H2O2 synthesis. Concurrently, precise localization of active sites and in-depth exploration of their coordination structure evolution under operational conditions are essential. This systematic approach enables the establishment of quantitative structure-activity relationship models, thereby providing theoretical foundations for the rational design of photocatalytic interfaces that synergistically enhance both the WOR and ORR.
(IV) Multi-level theoretical assistance: Given the intricate electronic coupling effects and multiphase reaction synergies at heterojunction interfaces, theoretical computations and modeling approaches warrant increased emphasis in the rational design and mechanistic exploration of COFs-based heterojunction systems. This necessitates the establishment of a multiscale computational scheme integrating quantum chemical calculations (e.g., DFT), nonadiabatic molecular dynamics (NAMD), and finite element simulations, complemented by explicit solvation models and microkinetic simulations to unravel PCET processes of water molecules within ordered nanopores. Leveraging machine learning-derived potential functions or artificial intelligence to accelerate the screening of COFs-based heterojunctions with optimal interfacial charge separation efficiency could provide theoretical underpinnings for atomic-level electronic structure modulation of photocatalysts. Such methodologies are indispensable for both synthesizing high-performance photocatalytic materials and advancing fundamental insights into reaction mechanisms.
(V) Practical application-driven design: Advancing the practical implementation of COFs-based heterojunctions necessitates comprehensive investigations into their photocatalytic performance under complex aqueous environments containing inorganic ions and organic pollutants. Critical priorities include determining catalyst pH tolerance windows and elucidating how interfacial competitive adsorption phenomena regulate H2O2 selectivity.
As for the in-situ photocatalytic application of H2O2 generation should prioritize three strategic directions: performance optimization of photocatalysts, innovative design of reaction systems, and breakthroughs in scalable production technologies. Critical emphasis must be placed on ensuring system safety and operational stability while achieving direct solar-driven conversion of water and oxygen into H2O2 for on-demand green synthesis. Enhanced pollutant remediation efficiency can be realized through dynamic response mechanisms and synergistic effects of reactive oxygen species, with potential applications extending to decentralized water purification, medical sterilization, and precision chemical synthesis − thereby significantly mitigating transportation and storage risks associated with conventional H2O2 handling. Systematic optimization should concurrently address fundamental material challenges, process engineering considerations, and life-cycle environmental impacts to ensure technological viability across diverse application scenarios. In addition, optimization of in-situ H2O2 generation-release kinetics is imperative to achieve rapid and sustained microbial inactivation, pollutants degradation, biomedical applications or organic synthesis. Concurrently, systematic evaluations of economic viability and ecological impacts must be conducted, emphasizing the development of catalysts devoid of noble metals (e.g., Pt, Au) and transition metals (e.g., Fe, Cu). Strategic integration of natural organic ligands as functional units could further enhance material biodegradability. Furthermore, the advancement of photocatalytic technology necessitates transcending the limitations of mono-energy-driven systems through synergistic integration with multi-catalytic platforms, such as piezoelectric and thermal catalytic mechanisms, to establish enhanced cooperative energy conversion paradigms, or construction of gas-liquid-solid triple-phase interfaces to enhance the concentration of gaseous reactants thereby potentially improving the photocatalytic performance of COFs-based heterojunctions.
Through the aforementioned systematic design, this approach ensures photocatalytic efficiency while providing a comprehensive solution to facilitate the transition of photocatalytic H2O2 technology from laboratory research to practical applications in environmental remediation and medical sterilization. Despite current challenges, the progress achieved in H2O2 production using COFs-based heterojunction photocatalysts demonstrates their promising potential for practical implementation. This review systematically consolidates critical design principles to advance the development of COFs-based heterojunction materials with optimized photocatalytic performance.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Zihui Liu: Writing – original draft, Investigation. Xiaolin Liu: Writing – review & editing, Writing – original draft, Project administration, Funding acquisition, Formal analysis. Lin Tang: Validation. Houhe Pan: Writing – review & editing, Visualization. Xi Liu: Supervision, Conceptualization. Jianzhuang Jiang: Supervision, Project administration.
The authors are grateful for the support and funding from the National Natural Science Foundation of China (Nos. 22405028, 22235001, 22175020, and 52403085), Natural Science Foundation of Chongqing (Nos. CSTB2025NSCQ-GPX0997 and CSTB2025NSCQ-LZX0117), Science and Technology Research Program of Chongqing Municipal Education Commission (No. KJQN202400523), and the Fundamental Research Funds for the Central Universities (Nos. FRF-IDRY-23-021 and FRF-TP-24-014A). Chongqing Normal University and University of Science and Technology Beijing are gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
K. Sato, M. Aoki, R. Noyori, Science 281 (1998) 1646–1647. doi: 10.1126/science.281.5383.1646
C. Chu, Z. Chen, D. Yao, et al., Angew. Chem. Int. Ed. 63 (2024) e202317214. doi: 10.1002/anie.202317214
X. Zhang, D. Huang, R. Luo, et al., J. Hazard. Mater. 490 (2025) 137835. doi: 10.1016/j.jhazmat.2025.137835
Y. Wang, P. Liu, R. Lv, et al., J. Energy Chem. 106 (2025) 455–474. doi: 10.1016/j.jechem.2025.02.034
M. Borrell, M. Costas, J. Am. Chem. Soc. 139 (2017) 12821–12829. doi: 10.1021/jacs.7b07909
Y. Bu, R. Ma, Y. Wang, et al., Adv. Mater. 36 (2024) 2412670. doi: 10.1002/adma.202412670
D. Poppendieck, H. Hubbard, R.L. Corsi, Environ. Sci. Technol. Lett. 8 (2021) 320–325. doi: 10.1021/acs.estlett.0c00948
H. Choi, P. Chatterjee, E. Lichtfouse, et al., Environ. Chem. Lett. 19 (2021) 1945–1951. doi: 10.1007/s10311-021-01180-4
A. Pal, S. Suresh, A. Khan, et al., Sci. Adv. 11 (2025) eads4711. doi: 10.1126/sciadv.ads4711
H. Hou, X. Zeng, X. Zhang, Angew. Chem. Int. Ed. 59 (2020) 17356–17376. doi: 10.1002/anie.201911609
D. An, Q. Xiao, C. Zhang, et al., Food Hydrocolloids 113 (2021) 106520. doi: 10.1016/j.foodhyd.2020.106520
Y. Fredes, G. Álvaro, M. Guillén, et al., J. Environ. Chem. Eng. 13 (2025) 115902. doi: 10.1016/j.jece.2025.115902
H.N. Fernandez-Escamilla, J. Guerrero-Sanchez, E. Contreras, et al., Adv. Energy Mater. 11 (2021) 2002459. doi: 10.1002/aenm.202002459
K. Mase, M. Yoneda, Y. Yamada, et al., Nat. Commun. 7 (2016) 11470. doi: 10.1038/ncomms11470
B. Chen, A. Gsalla, A. Gaur, et al., ACS Appl. Nano Mater. 2 (2019) 4143–4149. doi: 10.1021/acsanm.9b00593
A. Hayat, Z. Ajmal, A.Y.A. Alzahrani, et al., Coord. Chem. Rev. 522 (2025) 216218. doi: 10.1016/j.ccr.2024.216218
L. Ding, Z. Pan, Q. Wang, Chin. Chem. Lett. 35 (2024) 110125. doi: 10.1016/j.cclet.2024.110125
Z. Chen, D. Yao, C. Chu, et al., Chem. Eng. J. 451 (2023) 138489. doi: 10.1016/j.cej.2022.138489
J. Luo, Y. Liu, C. Fan, et al., ACS Catal. 11 (2021) 11440–11450. doi: 10.1021/acscatal.1c03103
Y. Zhu, J. Li, Y. Lai, et al., Environ. Res. 279 (2025) 121785. doi: 10.1016/j.envres.2025.121785
T. Gao, D. Zhao, S. Yuan, et al., Carbon Energy 6 (2024) e596. doi: 10.1002/cey2.596
Q. Hu, S. Liu, J. Liu, et al., ACS Energy Lett. 9 (2024) 3252–3260. doi: 10.1021/acsenergylett.4c01336
G. Chen, C. Lin, F. Han, et al., Mater. Horiz. 12 (2025) 5492–5512. doi: 10.1039/d5mh00295h
Y. Wu, Y. Yang, M. Gu, et al., Chin. J. Catal. 53 (2023) 123–133. doi: 10.1016/S1872-2067(23)64514-0
X. Zheng, R. Yanagi, Z. Pan, et al., Chem Catal. 5 (2025) 101238.
S. Banerjee, S.C. Pillai, P. Falaras, et al., J. Phys. Chem. Lett. 5 (2014) 2543–2554. doi: 10.1021/jz501030x
L. Chen, Y. Wang, S. Cheng, et al., App. Catal. B: Environ. 303 (2022) 120932. doi: 10.1016/j.apcatb.2021.120932
H. Dong, C. Qu, C. Li, et al., Chin. J. Catal. 70 (2025) 142–206. doi: 10.3390/a18030142
A.P. Côté, A.I. Benin, N.W. Ockwig, et al., Science 310 (2005) 1166–1170. doi: 10.1126/science.1120411
Z. Zhou, T. Ma, H. Zhang, et al., Nature 635 (2024) 96–101. doi: 10.1038/s41586-024-08080-x
F. Jin, E. Lin, T. Wang, et al., J. Am. Chem. Soc. 144 (2022) 5643–5652. doi: 10.1021/jacs.2c01058
J. Wang, S. Zhuang, Coord. Chem. Rev. 400 (2019) 213046. doi: 10.1016/j.ccr.2019.213046
W. Lee, H. Li, Z. Du, et al., Chem. Soc. Rev. 53 (2024) 8182–8201. doi: 10.1039/d4cs00409d
N. He, Y. Zou, C. Chen, et al., Nat. Commun. 15 (2024) 3896. doi: 10.1038/s41467-024-48160-0
D. Naberezhnyi, S. Park, W. Li, et al., Small 17 (2021) 2104392. doi: 10.1002/smll.202104392
N. Keller, T. Bein, Chem. Soc. Rev. 50 (2021) 1813–1845. doi: 10.1039/d0cs00793e
M. Yuan, F. Ma, X. Dai, et al., Angew. Chem. Int. Ed. 60 (2021) 21250–21255. doi: 10.1002/anie.202108650
S.M. Jing, Z.G. Gu, J. Zhang, J. Am. Chem. Soc. 147 (2025) 8948–8958. doi: 10.1021/jacs.5c00947
Q. Ouyang, Y. Rong, G. Xia, et al., Adv. Sci. 12 (2025) 2411621. doi: 10.1002/advs.202411621
X. Liu, D. Huang, C. Lai, et al., Chem. Soc. Rev. 48 (2019) 5266–5302. doi: 10.1039/c9cs00299e
X. Yao, Y. Zhang, Y. Qiu, et al., J. Am. Chem. Soc. 147 (2025) 9665–9675. doi: 10.1021/jacs.4c17776
S.S.A. Shah, M.S. Javed, T. Najam, et al., Mater. Today 67 (2023) 229–255. doi: 10.1016/j.mattod.2023.05.023
X. Han, W. Li, B. Yang, et al., Adv. Mater. 37 (2025) 2415574. doi: 10.1002/adma.202415574
I.E. Khalil, P. Das, A. Thomas, Acc. Chem. Res. 57 (2024) 3138–3150. doi: 10.1021/acs.accounts.4c00491
J. Li, X. Jing, Q. Li, et al., Chem. Soc. Rev. 49 (2020) 3565–3604. doi: 10.1039/d0cs00017e
D.G. Wang, T. Qiu, W. Guo, et al., Energy Environ. Sci. 14 (2021) 688–728. doi: 10.1039/d0ee02309d
J. Chen, D. Yuan, Y. Wang, Adv. Funct. Mater. 33 (2023) 2304071. doi: 10.1002/adfm.202304071
L. Wang, J. Li, S. Ji, et al., Energy Environ. Sci. 17 (2024) 8482–8528. doi: 10.1039/d4ee03704a
Y.H. Wijesundara, T.S. Howlett, S. Kumari, et al., Chem. Rev. 124 (2024) 3013–3036. doi: 10.1021/acs.chemrev.3c00409
M. Li, S. Qiao, Y. Zheng, et al., J. Am. Chem. Soc. 142 (2020) 6675–6681. doi: 10.1021/jacs.0c00285
M. Feng, C. Xing, Y. Jin, et al., J. Am. Chem. Soc. 146 (2024) 32883–32905. doi: 10.1021/jacs.4c09259
C. Krishnaraj, H. Sekhar Jena, L. Bourda, et al., J. Am. Chem. Soc. 142 (2020) 20107–20116. doi: 10.1021/jacs.0c09684
Y. Zhang, C. Pan, J. Li, et al., Acc. Mater. Res. 5 (2024) 76–88. doi: 10.1021/accountsmr.3c00187
K. Chen, A. Cai, T.T. Li, ChemSusChem 16 (2023) e202300021. doi: 10.1002/cssc.202300021
T. Zhou, Y. Ma, H. Feng, et al., Adv. Funct. Mater. 34 (2024) 2409396. doi: 10.1002/adfm.202409396
S. Wei, R. Hou, Q. Zhu, et al., InfoMat 7 (2025) e12646. doi: 10.1002/inf2.12646
D.E. Lee, A. Ali, K.T. Kang, et al., Mater. Sci. Eng. R Rep. 161 (2024) 100858. doi: 10.1016/j.mser.2024.100858
Q. Li, X. Li, B. Zhang, et al., Adv. Funct. Mater. 35 (2025) 2506421. doi: 10.1002/adfm.202506421
J. Qiu, K. Meng, Y. Zhang, et al., Adv. Mater. 36 (2024) 2400288. doi: 10.1002/adma.202400288
J. Cheng, S. Wan, S. Cao, Angew. Chem. Int. Ed. 62 (2023) e202310476. doi: 10.1002/anie.202310476
Z. Jiang, J. Zhang, B. Cheng, et al., Small 21 (2025) 2409079. doi: 10.1002/smll.202409079
Q. Zhang, J. Zhou, H. Zhang, et al., Adv. Funct. Mater. 34 (2024) 2401579. doi: 10.1002/adfm.202401579
H. Lv, Z. Li, P. Yin, et al., Chin. Chem. Lett. 36 (2025) 110457. doi: 10.1016/j.cclet.2024.110457
K. Zhang, L. Tian, J. Yang, et al., Angew. Chem. Int. Ed. 63 (2024) e202317816. doi: 10.1002/anie.202317816
Z. Teng, Q. Zhang, H. Yang, et al., Nat. Catal. 4 (2021) 374–384. doi: 10.1038/s41929-021-00605-1
S. Zhang, D. Zeng, H. Wang, et al., Chem. Eur. J. 30 (2024) e202402767. doi: 10.1002/chem.202402767
J. Chen, S. Yan, F. Wang, et al., Angew. Chem. Int. Ed. 64 (2025) e202500924. doi: 10.1002/anie.202500924
D. Jiao, C. Ding, M. Xu, et al., Adv. Funct. Mater. 35 (2025) 2416753. doi: 10.1002/adfm.202416753
J. Li, C.A. Triana, W. Wan, et al., Chem. Soc. Rev. 50 (2021) 2444–2485. doi: 10.1039/d0cs00978d
H. Cheng, J. Cheng, L. Wang, et al., Chem. Mater. 34 (2022) 4259–4273. doi: 10.1021/acs.chemmater.2c00936
J. Xue, J. Bao, Surf. Interf. 25 (2021) 101265. doi: 10.1016/j.surfin.2021.101265
Z. Han, Y. Song, Y. Jia, et al., Adv. Mater. Interfaces 12 (2025) 2500191. doi: 10.1002/admi.202500191
X. Zhao, S. Xiao, B. Yao, et al., J. Chem. Theory Comput. 20 (2024) 9770–9786. doi: 10.1021/acs.jctc.4c01051
G. Xia, J. Qiu, L. Zhang, et al., Colloid Surf. Physicochem. Eng. Asp. 664 (2023) 131124. doi: 10.1016/j.colsurfa.2023.131124
Y. Yang, Q. Guo, Q. Li, et al., Adv. Funct. Mater. 34 (2024) 2400612. doi: 10.1002/adfm.202400612
C. Rosso, G. Filippini, M. Prato, ACS Catal. 10 (2020) 8090–8105. doi: 10.1021/acscatal.0c01989
W. Ren, N. Li, Q. Chang, et al., Chin. J. Catal. 62 (2024) 178–189. doi: 10.1016/S1872-2067(24)60050-1
Y. Yang, Y. Li, X. Ma, et al., Catal. Sci. Technol. 13 (2023) 5599–5609. doi: 10.1039/d3cy00878a
X. Shi, Y. You, L. Huang, et al., Adv. Funct. Mater. 35 (2025) 2414755. doi: 10.1002/adfm.202414755
Y. Zhang, J. Qiu, B. Zhu, et al., Chem. Eng. J. 444 (2022) 136584. doi: 10.1016/j.cej.2022.136584
K. Meng, J. Zhang, B. Zhu, et al., Adv. Mater. 37 (2025) 2505088. doi: 10.1002/adma.202505088
Y. Zhao, C. Yang, S. Zhang, et al., Chin. J. Catal. 63 (2024) 258–269. doi: 10.1016/S1872-2067(24)60069-0
X. Ma, S. Li, Y. Gao, et al., Adv. Funct. Mater. 34 (2024) 2409913. doi: 10.1002/adfm.202409913
H. Zhang, J. Liu, Y. Zhang, et al., J. Mater. Sci. Technol. 166 (2023) 241–249. doi: 10.1016/j.jmst.2023.05.030
Z. Tong, D.K. Ma, J. Wu, et al., Chem. Eng. J. 496 (2024) 154262. doi: 10.1016/j.cej.2024.154262
D. Huang, J. Zhao, Chem 6 (2020) 1512–1514. doi: 10.1016/j.chempr.2020.06.008
Y. Zhao, Y. Zhang, L. Wang, et al., J. Mater. Sci. Technol. 229 (2025) 213–222. doi: 10.1016/j.jmst.2024.12.040
Y. Yang, J. Liu, M. Gu, et al., Appl. Catal. B: Environ. 333 (2023) 122780. doi: 10.1016/j.apcatb.2023.122780
Q. Rong, X. Chen, S. Li, et al., ACS Appl. Mater. Interfaces 16 (2024) 5758–5768. doi: 10.1021/acsami.3c14870
M. Liu, Y. Xu, Y. Liu, et al., Angew. Chem. Int. Ed. 64 (2025) e202505491. doi: 10.1002/anie.202505491
Y. Nosaka, A.Y. Nosaka, Chem. Rev. 117 (2017) 11302–11336. doi: 10.1021/acs.chemrev.7b00161
X. Xu, S. Dong, J. Lv, et al., Appl. Surf. Sci. 689 (2025) 162489. doi: 10.1016/j.apsusc.2025.162489
Y. He, J. Zhao, Y.T. Sham, et al., ACS Sustain. Chem. Eng. 11 (2023) 17552–17563. doi: 10.1021/acssuschemeng.3c06421
L. Zhuo, S. Dong, Y.T. Sham, et al., npj Clean Water 8 (2025) 5. doi: 10.1038/s41545-025-00437-7
H. Chen, S. Gao, G. Huang, et al., App. Catal. B: Environ. 343 (2024) 123545. doi: 10.1016/j.apcatb.2023.123545
J.Y. Yue, Z.X. Pan, P. Yang, et al., ACS Mater. Lett. 6 (2024) 3932–3940. doi: 10.1021/acsmaterialslett.4c01273
Q. Li, M. Deng, J. Gao, et al., Sep. Purif. Technol. 362 (2025) 131722. doi: 10.1016/j.seppur.2025.131722
Y. Zhang, F. Zhou, X. Ding, et al., Chem. Eng. J. 508 (2025) 160891. doi: 10.1016/j.cej.2025.160891
Y. Xue, X. Chen, F. Wu, et al., Adv. Funct. Mater. 35 (2025) 2505669. doi: 10.1002/adfm.202505669
J. Liu, C. Guo, Z. Liu, et al., Chem. Eng. J. 494 (2024) 153139. doi: 10.1016/j.cej.2024.153139
C.X. Liu, Z.W. Zhou, C.X. Cai, et al., ACS Appl. Mater. Interfaces 17 (2025) 6347–6356. doi: 10.1021/acsami.4c19081

Figure 3 (a) Graphical representation of photocatalytic H2O2 generation techniques, as well as energy graphs and (b) reaction pathways and corresponding energy diagrams for ORR and WOR pathways.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载: