
Scheme 1.
Selected polycyclic cembranoid and norcembranoid natural products, prior total syntheses, and our retrosynthetic analysis of sinugyrosanolide A.
Asymmetric total synthesis of (–)-14-epi-sinugyrosanolide A
Hongjin Xu , Jinghua Wu , Hui Wang , Huanfeng Jiang , Zhiqiang Ma

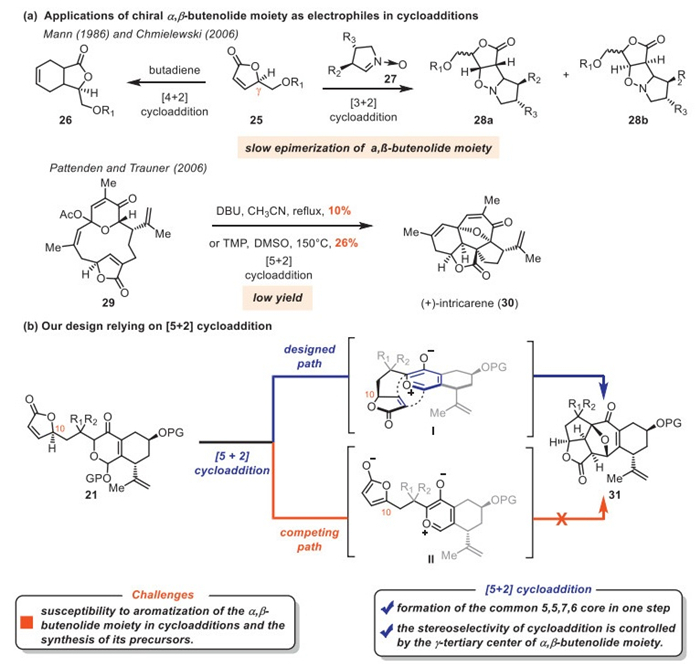
The prevalence of γ-butyrolactone moiety in natural products and biologically active molecules has fueled the demand for effective synthetic strategies to enable their enantioselective synthesis [1–3]. While the synthesis of relatively simple ring systems containing the γ-butyrolactone moiety has been extensively studied, access to complex polycyclic frameworks remains scarce [4–9]. The utilization of α,β-butenolide moieties as electrophiles in cycloadditions offers the most straightforward and efficient approach to γ-butyrolactones [10–17]. However, the tendency of α,β-butenolide moiety toward aromatization poses significant challenges for its practical applications as electrophiles [10,11,18,19].
The soft coral species have yielded a variety of structurally novel and bioactive polycyclic cembranoid and norcembranoid diterpenes featuring the γ-butyrolactone moiety (Scheme 1a, highlighted in blue) [20,21]. Many of these natural products, such as sinugyrosanolide A (1) [22], rameswaralide (2) [23], ineleganolide (3) [24], sinuscalide C (4) [25], scabrolide B (5) [26], and fragilolide A (6) [27] share a common 5,5,7,6 tetracyclic core. Some possess a 5,5,6,7 ring system decorated with unique oxygenation patterns, as exemplified by scabrolide A (7) [28], sinulochmodin C (8) [29] and yonarolide (9) [30]. As the prototypical representative of the former class, 1 was isolated from Sinularia gyrosa by Duh and co-workers in 2014 [22]. Sinugyrosanolide A features a 5,5,7,6 all cis-fused framework with seven stereogenic centers and a unique γ-butyrolactone moiety, exhibiting moderate cytotoxicity against P-388 mouse lymphocytic leukemia cells (EC50 = 11.8 µmol/L).
Owing to their challenging structures and potential bioactivities, polycyclic cembranoids and norcembranoids containing 5,5,7,6 core have drawn much attention from the synthetic community (Scheme 1b) [31–35]. In 2022, the Romo group [31] utilized a ring expansion to build the 5,5,7-tricycle core and assembled the key D ring via Stille coupling and intramolecular Michael addition, enabling the first racemic total synthesis of rameswaralide (2). Subsequently, the Wood group [32] completed the first total synthesis of ineleganolide (3), featuring a Nozaki−Hiyama−Kishi cyclization/transannular Michael reaction sequence. The Stoltz group [33] reported the total synthesis of 3 using a Michael addition/aldol cascade to rapidly establish pentacyclic framework and a unique semipinacol rearrangement to furnish the central seven-membered ring. In 2024, Fürstner and co-workers [34] disclosed a highly efficient intramolecular alkenylation to construct the central cycloheptene ring and completed the total synthesis of 3, sinuscalide C (4), scabrolide B (5), and horiolide. Very recently, the Sarlah group [35] achieved an elegant synthesis of 5 and realized its transformation to 3, 4, and 6 via sequential Mukaiyama−Michael/aldol reactions.
Our interest in sinugyrosanolide A (1) stems from its characteristic 5,5,7,6 ring system shared by several Sinularia diterpenoids and its potential as a synthetic platform for accessing related natural products and analogs. Moreover, cembranoid and norcembranoid diterpenes featuring the 5,5,7,6 system generally exhibit greater bioactivity than those with the 5,5,6,7 core, which typically display weak or negligible activity [20–30]. With our constant interest in the synthesis of bioactive polycyclic terpenoids and steroids [36,37], we report herein the construction of the common 5,5,7,6 ring system via an acetic acid-promoted intramolecular [5 + 2] cycloaddition of an oxidopyrylium ylide with an α,β-butenolide moiety, culminating in the asymmetric total synthesis of (–)-14-epi-sinugyrosanolide A.
Our retrosynthetic analysis for the enantioselective total synthesis of sinugyrosanolide A (1) is depicted in Scheme 1c. We envisioned that 1 could arise from 20 through selective reductive cleavage of the C–O bond and reduction of the tetrasubstituted olefin. Having simplified our target to 20, we sought to identify an efficient strategy for the rapid assembly of its synthetically challenging 5,5,6,7 core. Cycloaddition reactions, such as [4 + 3] [38–41] and [5 + 2] cycloadditions [42–59], have emerged as powerful strategies for synthesizing polycyclic natural products containing seven-membered ring. After careful consideration, we opted for [5 + 2] cycloaddition as the way to form the congested central seven-membered system. The tetracyclic core of 20 could be efficiently assembled from 21 via an intramolecular [5 + 2] cycloaddition. Our original strategy for synthesizing key precursor 21 involved nucleophilic addition of furan 23 to α,β-butenolide 22, followed by an Achmatowicz reaction [60,61]. Further retrosynthetic simplification of 23 gave lactone 24 through a series of functional group transformations.
The α,β-butenolide moiety, due to the acidity at its γ-position, readily forms the corresponding enolate, enabling its use as a nucleophile in aldol, Mannich, and Mukaiyama-Michael reactions [5]. It also serves as an electrophile in Michael additions and cycloadditions [10–17,19]. In these processes, the chirality and functionality of the α,β-butenolide moiety are frequently exploited to control stereo- and regioselectivity. However, the susceptibility of the α,β-butenolide moiety to aromatization has limited its application in cycloadditions. Although the stereochemical integrity of the α,β-butenolide moiety was maintained in [2 + 2] and some [4 + 2] cycloadditions, these reactions usually occurred under neutral conditions [15–17]. The Chmielewski [11] and Mann groups [10] studied [3 + 2] and [4 + 2] cycloadditions, respectively, involving α,β-butenolide moiety 25 as electrophiles, wherein slow epimerization of α,β-butenolide moiety was observed (Scheme 2a). The Pattenden [12,14] and Trauner [13] groups subsequently disclosed a biomimetic total synthesis of intricarene via [5 + 2] cycloaddition reaction between an oxidopyrylium ylide and α,β-butenolide moiety. However, the significantly low yield (10%−26%) compromised the synthetic efficiency. In our designed strategy, compound 21 could be converted to the oxidopyrylium ylide intermediate Ⅰ (Scheme 2b). Owing to the chirality at C10, the oxidopyrylium ylide was expected to approach the α,β-butenolide moiety from its β face and undergo an intramolecular [5 + 2] cycloaddition to deliver 31. This single-step transformation would rapidly construct the 5,5,7,6-tetracyclic framework. However, conventional [5 + 2] cycloadditions are generally carried out under harsh conditions, such as high temperature and base. Consequently, a key challenge is to suppress aromatization of the α,β-butenolide moiety to prevent formation of the dienol intermediate Ⅱ. Additionally, synthesizing the cycloaddition precursor 21 bearing the fragile α,β-butenolide itself presents another significant obstacle. In brief, employing the α,β-butenolide moiety as an electrophile in cycloadditions within structurally complex systems (e.g., 21) remains challenging.
To explore the proposed synthetic strategy, we first investigated the synthesis of related cycloaddition precursors. Initially, the conventional method [54,55] was employed, which involved nucleophilic addition of lithiated furan 32 to 22 bearing an α,β-butenolide moiety, followed by the Achmatowicz reaction (Scheme 3a). However, despite screening various conditions, this transformation could not be achieved. We speculated that the deprotonation at C10 in 22 might compete with the nucleophilic addition, quenching the newly generated lithiated furan. Consequently, the reaction sequence was adjusted. Furan 35 first underwent an Achmatowicz reaction to afford dihydropyranone acetal derivative 36, which then participated in an aldol reaction with aldehyde 37, a transformation not previously reported. However, epimerization of the α,β-butenolide moiety occurred during this process, affording a pair of diastereomers 38a and 38b. Subsequent endeavors to inhibit the aromatization were unsuccessful. Therefore, we further altered the reaction sequence. First, the dihydropyranone acetal derivative 45 underwent aldol reaction with an aldehyde lacking the α,β-butenolide moiety (e.g., 46, Scheme 3b); the resulting product then underwent a ring-closing metathesis (RCM) to ultimately reveal the α,β-butenolide moiety required for cycloaddition.
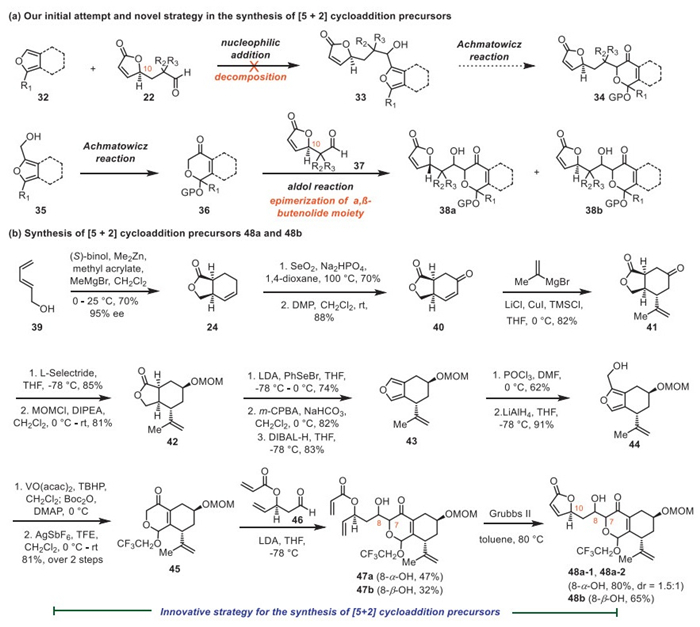
The synthesis commenced with the preparation of known lactone 24, which was obtained in 70% yield (95% ee) via an intermolecular Diels-Alder reaction developed by Ward and Santos et al. (Scheme 3b) [62,63]. An allylic oxidation of lactone 24 with SeO2 [64], followed by Dess-Martin periodinane (DMP) oxidation of the desired secondary alcohol, led to enone 40. Subjecting 40 to CuX3Li2-catalyzed conjugate addition [65] delivered ketone 41 in 82% yield with exclusive facial selectivity. The stereochemistry influence of the cis-fused ring system directed the addition of the isopropenyl group selectively from the less hindered convex face. Selective reduction of ketone-carbony of 41 gave the corresponding alcohol, which was protected with MOM to provide ester 42. The subsequent formation of desired furan 43 involved a three-step sequence: α-selenation of 42 to standard conditions, followed by oxidation with m-CPBA (3-chloroperbenzoic acid) afforded enone, which was ultimately transformed into furan 43 via reduction and aromatization [66,67]. Finally, furan 43 was formylated [68], and a follow-up LiAlH4 reduction yielded alcohol 44.
With ample quantities of 44 in hand, we then focused on the synthesis of the cycloaddition precursor. An Achmatowicz reaction of 44 with VO(acac)2/TBHP, followed by Boc-protection of the anomeric hydroxyl group in the same pot, furnished the desired ring expansion product. However, this intermediate failed to undergo coupling with aldehyde 46 (see Supporting information for preparation of 46). In 2019, the Li group reported the use of a trifluoroethoxyl group as a leaving group in a [5 + 2] cycloaddition [56]. Inspired by this work, the intermediate underwent trifluoroethoxylation to afford 45 as a single diastereomer in 81% yield over two steps. Fortunately, aldol reaction of compound 45 and aldehyde 46 led to 47a (8-α-OH; a pair of inseparable diastereomers, dr = 1.5:1 at C7) and 47b (8-β-OH; a pair of inseparable diastereomers, dr = 1.6:1 at C7). Subsequent RCM reaction [69] furnished three separable diastereomers (48a-1, 48a-2 and 48b) as cycloaddition precursors on a multigram scale. Critically, no epimerization at C10 was observed. Moreover, for intramolecular oxidopyrylium-mediated [5 + 2] cycloadditions, literature precedents typically employ furan as the tethering fragment prior to an Achmatowicz reaction to access cycloaddition precursors. To the best of our knowledge, this work represents the first example: coupling of a dihydropyranone acetal and another fragment to access [5 + 2] cycloaddition precursor. This strategy offers an alternative approach for preparing structurally complex [5 + 2] cycloaddition precursors bearing sensitive functional groups.
To examine the feasibility of the [5 + 2] cycloaddition reaction, compound 48a-1 was selected as the test substrate. While previous reports typically generate the oxidopyrylium ylide intermediate under heating or basic conditions for [5 + 2] cycloadditions, these conditions proved ineffective for the present substrate 48a-1 (Table 1, entries 1 and 2, see Table S1 in Supporting information for details). Consequently, we turned to acid-promoted [5 + 2] cycloadditions, despite limited precedents [50–53]. In 1999, the Magnus group reported a trifluoroacetic acid (CF3CO2H)-promoted [5 + 2] cycloaddition [50]. The Li group used acid-promoted [5 + 2] reaction to investigate the synthesis of the natural product bufogargarizin C [51], phomarol [52] and venezuelaene B [53]. Our initial treatment of 48a-1 with CF3CO2H in CH2Cl2 still failed to yield the desired product. Fortunately, heating this intermediate in MeCN at 100 ℃ afforded the desired 5,5,7,6-tetracyclic framework 49a in 10% yield (entry 3). The relative configuration of 49a was unambiguously confirmed by X-ray crystallography. Direct heating of 48a-1 in CF3CO2H/CH3CN at 100 ℃ slightly improved the yield (entry 4). Subsequent screening of Lewis and Brønsted acids revealed acetic acid (AcOH) as optimal (entry 5). Further optimization of temperature and equivalents of AcOH led to lower yields. Remarkably, switching the solvent from CH3CN to toluene significantly enhanced the yield to 48% (entry 6), prompting a systematic solvent screening. Ultimately, heating 48a-1 with AcOH in mesitylene at 150 ℃ delivered 49a in an excellent yield of 82% (entry 7). Scaling up the reaction with 48a-1 to gram scale gave slightly lower yield (75%, entry 8). Meanwhile, subjecting 48a-2 to the optimized conditions provided 49a in 82% yield, indicating that the C7 stereochemistry does not significantly influence the reaction outcome (entry 9). Using precursor 48b under identical conditions provided tetracycle 49b in 66% yield on gram scale (entry 10). The absolute configuration of 49b was confirmed by X-ray analysis.
 DownLoad:
CSV
DownLoad:
CSV
 |
|||
| Entry | Substrate | Conditionsa | Isolated yield (%) |
| 1 | 48a-1 | PhCN, 170 ℃ | 49a, 0 |
| 2 | 48a-1 | TMP, PhCN, 150 ℃ | Decomposed |
| 3 | 48a-1 | CF3CO2H, CH2Cl2, 0 ℃ – 60 ℃; then CF3CO2H, CH3CN, 100 ℃ | 49a, 10 |
| 4 | 48a-1 | CF3CO2H, CH3CN, 100 ℃ | 49a, 13 |
| 5 | 48a-1 | AcOH, CH3CN, 150 ℃ | 49a, 19 |
| 6 | 48a-1 | AcOH, toluene, 150 ℃ | 49a, 48 |
| 7 | 48a-1 | AcOH, mesitylene, 150 ℃ | 49a, 82 |
| 8 | 48a-1 | AcOH, mesitylene, 150 ℃b | 49a, 75 |
| 9 | 48a-2 | AcOH, mesitylene, 150 ℃ | 49a, 82 |
| 10 | 48b | AcOH, mesitylene, 150 ℃b | 49b, 66 |
| a Reaction scale: 0.008 mmol. b Performed on gram scale. |
|||
Having established the key 5,5,7,6-tetracyclic skeleton, we sought to install the final carbon of sinugyrosanolide A via nucleophilic addition (Scheme 4). We postulated that the methyl Grignard reagent would preferentially attack the C8 carbonyl group, as this C8 position is less sterically hindered than the C6 carbonyl. Moreover, due to the 5,5,7-cis-fused ring framework, the methyl Grignard reagent would approach from the less hindered α-face, thereby affording the desired stereoselectivity. In practice, oxidation of 49a and 49b with DMP furnished ketone 50 in 84% and 89% yield, respectively. Gratifyingly, exposure of 50 to methylmagnesiumbromide afforded desired product 51 in 70% yield with excellent regio- and stereoselectivity. The structure of 51 was unambiguously confirmed by X-ray crystallography.
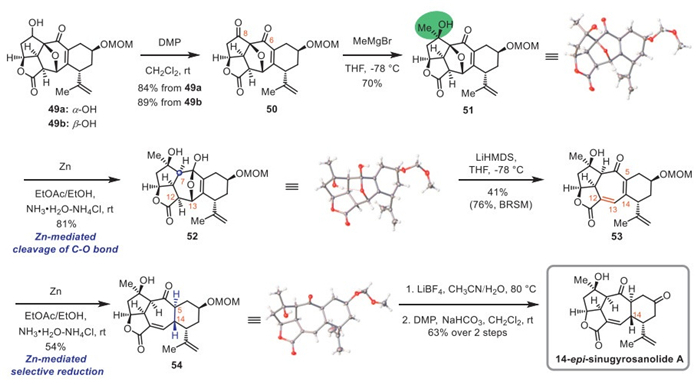
Owing to the highly constrained caged structure of compound 51, cleavage of the C–O bond proved to be quite challenging. Although SmI2-mediated reactions achieved cleavage [70,71], the resulting stereochemistry at C7 was opposite to that of natural sinugyrosanolide A (Table S2 in Supporting information). After a wide variety of conditions were screened, the following solution was identified. Treatment of 51 with Zn in a mixture of ethyl acetate (EtOAc), ethyl alcohol (EtOH) and a buffered solution of NH3•H2O–NH4Cl provided hemiketal 52 possessing the desired C7 stereochemistry as a single isomer in 81% yield [72]. The structure of 52 was confirmed by single-crystal X-ray analysis.
Next, our focus turned to construct the Δ12,13 olefin via dehydration of hemiketal 52. Under conventional dehydration conditions, the tertiary hydroxy group at C8 or its protected analogs preferentially underwent elimination. Inspired by the base-mediated cleavage of the ether bridge [73], we proposed that compound 52 would undergo a retro-Michael reaction under basic conditions to establish the Δ12,13 olefin, forming a ketal intermediate that hydrolyzes to afford target product. Gratifyingly, treatment of hemiketal 52 with LiHMDS (lithium bis(trimethylsilyl)amide) at 0 ℃ gave enone 53, albeit in a very low yield. Precooling 52 to −78 ℃ and adding LiHMDS in one portion increased the yield to 41% (76% based on recovered starting material (BRSM)).
With compound 53 in hand, the next step required the regio- and stereoselective reduction of the tetrasubstituted Δ5,14 olefin. Initial attempts (e.g., NaBH4/NiCl2, NaSeH and NaHTe) failed to afford the desired regioselectivities (Table S3 in Supporting information). Hydrogenation also proved ineffective. We therefore turned our attention to single-electron reduction conditions. Fortunately, the reduction of tetrasubstituted Δ5,14 olefin was accomplished using zinc under basic conditions, affording compound 54 as a single diastereomer in 54% yield [72]. The structure of 54 was confirmed by single crystal X-ray diffraction. Unfortunately, the stereochemistry at C14 was opposite to that of natural sinugyrosanolide A. Subsequent efforts to invert this C14 stereogenic center by controlling temperature or altering solvents proved unsuccessful. Additionally, similar results were observed by using SmI2-mediated reactions [70,71]. Reversible radical epimerization strategies (e.g., BIN3 and TBADT/(4-ClPhS)2) [74,75] led only to complex reaction mixtures without discernible product. Moving forward, a mild Lewis acid LiBF4 was used to cleave the MOM protective group of 54 [76], followed by oxidation with DMP, which completed the asymmetric total synthesis of (–)-14-epi-sinugyrosanolide A, a stereoisomer of the natural product 1, in a 63% yield over two steps.
In conclusion, we have accomplished the asymmetric total synthesis of (–)-14-epi-sinugyrosanolide A via an acid-promoted intramolecular [5 + 2] cycloaddition between an oxidopyrylium ylide and an α,β-butenolide moiety. This key step effectively suppressed the inherent tendency of the α,β-butenolide toward aromatization and rapidly constructed the tetracyclic 5,5,7,6-fused ring system, the core skeleton in several polycyclic cembranoid and norcembranoid natural products. A unique reaction sequence involving aldol condensation of dihydropyranone acetal derivatives and aldehyde and subsequent RCM provided the key precursor for cycloaddition. The efficient strategy, involving fragment coupling of dihydropyranone acetal derivatives with aldehyde, followed by RCM and intramolecular [5 + 2] cycloaddition, may provide a platform for access to structurally complex molecules containing the γ-butyrolactone moiety. Investigations based on this approach are currently underway in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Hongjin Xu: Writing – original draft, Validation, Methodology, Investigation, Formal analysis, Data curation. Jinghua Wu: Methodology, Formal analysis, Investigation. Hui Wang: Writing – review & editing, Investigation. Huanfeng Jiang: Supervision. Zhiqiang Ma: Writing – review & editing, Supervision, Project administration, Formal analysis, Conceptualization, Funding acquisition.
This work was supported by the National Natural Science Foundation of China (Nos. 22471078, 22271099 and 22071064) and the Basic and Applied Basic Research Foundation of Guangdong Province (No. 2024A1515012297). We also thank Dr. Xu W. from the Public Research Platform of the School of Pharmacy at Jinan University for testing and data analysis of all single crystals in this work.
Supplementary material associated with this article can be
found, in the online version, at doi:
S.R.M. Ibrahim, G.A. Mohamed, A.I.M. Khedr, Nat. Prod. Commun. 12 (2017) 791–800.
S.K. Sartori, M.A.N. Diaz, G. Diaz-Muñoz, Tetrahedron 84 (2021) 132001–132039. doi: 10.1016/j.tet.2021.132001
J. Hur, J. Jang, J. Sim, Int. J. Mol. Sci. 22 (2021) 2769–2817. doi: 10.3390/ijms22052769
M. Neumeyer, R. Brückner, Eur. J. Org. Chem. 2016 (2016) 5060–5087. doi: 10.1002/ejoc.201600520
B. Mao, M. Fañanas-Mastral, B.L. Feringa, Chem. Rev. 117 (2017) 10502–10566. doi: 10.1021/acs.chemrev.7b00151
K.J.R. Murauski, A.A. Jaworski, K.A.A. Scheidt, Chem. Soc. Rev. 47 (2018) 1773–1782. doi: 10.1039/c7cs00386b
B. Wei, K.W. Li, Y.C. Wu, et al., Synthesis 52 (2020) 3855–3865. doi: 10.1055/s-0040-1707835
Q. Zhang, J. Pang, T.Z. Wang, et al., Chin. Chem. Lett. 34 (2023) 108121. doi: 10.1016/j.cclet.2022.108121
L. Chu, X. Zhang, J. Li, et al., Chin. Chem. Lett. 35 (2024) 108896. doi: 10.1016/j.cclet.2023.108896
M.G.B. Drew, J. Mann, A. Thomas, J. Chem. Soc., Perkin Trans. 1 (1986) 2279–2285.
S. Stecko, K. Pasniczek, M. Jurczak, et al., Tetrahedron: Asymmetry 17 (2006) 68–78. doi: 10.1016/j.tetasy.2005.11.027
B. Tang, C.D. Bray, G. Pattenden, Tetrahedron Lett. 47 (2006) 6401–6404. doi: 10.1016/j.tetlet.2006.06.150
P.A. Roethle, P.T. Hernandez, D. Trauner, Org. Lett. 8 (2006) 5901–5904. doi: 10.1021/ol062581o
B. Tang, C.D. Bray, G. Pattenden, Org. Biomol. Chem. 7 (2009) 4448–4457. doi: 10.1039/b910572g
K. Takao, H. Kai, A. Yamada, et al., Angew. Chem. Int. Ed. 58 (2019) 9851–9855. doi: 10.1002/anie.201904404
M. Breunig, P. Yuan, T. Gaich, Angew. Chem. Int. Ed. 59 (2020) 5521–5525. doi: 10.1002/anie.201912613
P.D. Scesa, L.M. West, S.P. Roche, J. Am. Chem. Soc. 143 (2021) 7566–7577. doi: 10.1021/jacs.1c03336
P. Duret, B. Figadere, R. Hocquemiller, et al., Tetrahedron Lett. 38 (1997) 8849–8852. doi: 10.1016/S0040-4039(97)10390-2
I. Panfil, W. Abramski, M. Chmielewski, J. Carbohydr. Chem. 17 (1998) 1395–1403. doi: 10.1080/07328309808002361
R.A. Craig-Ⅱ, B.M. Stoltz, Chem. Rev. 117 (2017) 7878–7909. doi: 10.1021/acs.chemrev.7b00083
M.J. Palframan, G. Pattenden, Eur. J. Org. Chem. 2020 (2020) 2330–2349. doi: 10.1002/ejoc.201901438
S.Y. Cheng, N.L. Shih, C.T. Chuang, et al., Bioorg. Med. Chem. Lett. 24 (2014) 1562–1564. doi: 10.1016/j.bmcl.2014.01.073
P. Ramesh, N. Srinivasa Reddy, Y. Venlmteswarlu, Tetrahedron Lett. 39 (1998) 8217–8220. doi: 10.1016/S0040-4039(98)01879-6
C.Y. Duh, S.K. Wang, M.C. Chia, et al., Tetrahedron Lett. 40 (1999) 6033–6035. doi: 10.1016/S0040-4039(99)01194-6
J. Liu, Q. Tang, J. Huang, et al., J. Org. Chem. 87 (2022) 9806–9814. doi: 10.1021/acs.joc.2c00784
Y. Du, L. Yao, X. Li, et al., Chin. Chem. Lett. 34 (2023) 107512. doi: 10.1016/j.cclet.2022.05.026
W. Cheng, M. Ji, X. Li, et al., Tetrahedron 73 (2017) 2518–2528. doi: 10.1016/j.tet.2017.03.037
J.H. Sheu, A.F. Ahmed, R.T. Shiue, et al., J. Nat. Prod. 65 (2002) 1904–1908. doi: 10.1021/np020280r
Y. Ju. Tseng, A.F. Ahmed, C.F. Dai, et al., Org. Lett. 7 (2005) 3813–3816. doi: 10.1021/ol051513j
K. Iguchi, K. Kajiyama, Y. Yamada, Tetrahedron 36 (1995) 8807–8808. doi: 10.1016/0040-4039(95)01867-H
N.J. Truax, S. Ayinde, J.O. Liu, et al., J. Am. Chem. Soc. 144 (2022) 18575–18585. doi: 10.1021/jacs.2c08245
J.P. Tuccinardi, J.L. Wood, J. Am. Chem. Soc. 144 (2022) 20539–20547. doi: 10.1021/jacs.2c09826
B.M. Gross, S.J. Han, S.C. Virgil, et al., J. Am. Chem. Soc. 145 (2023) 7763–7767. doi: 10.1021/jacs.3c02142
D.S. Lin, G. Späth, Z. Meng, et al., J. Am. Chem. Soc. 146 (2024) 24250–24256. doi: 10.1021/jacs.4c09467
E.J. Simmons, D.B. Ryffel, D.A. Lopez, et al., J. Am. Chem. Soc. 147 (2025) 130–135. doi: 10.1021/jacs.4c16629
P. Chen, C. Wang, R. Yang, et al., Angew. Chem. Int. Ed. 60 (2021) 5512–5518. doi: 10.1002/anie.202013881
H. Wang, L. Wei, J. Huang, et al., Angew. Chem. Int. Ed. 64 (2025) e202507961. doi: 10.1002/anie.202507961
F. Lopez, J.L. Mascarenas, Chem. Soc. Rev. 43 (2014) 2904–2915. doi: 10.1039/C4CS00024B
Z. Yin, Y. He, P. Chiu, Chem. Soc. Rev. 47 (2018) 8881–8924. doi: 10.1039/c8cs00532j
W. Tan, J.Y. Zhang, C.H. Gao, et al., Sci. China Chem. 66 (2023) 966–992. doi: 10.1007/s11426-022-1471-2
Y. Que, W. Lei, Y. Fang, et al., Green Synth. Catal. 5 (2024) 270–276.
V. Singh, U. Murali Krishna, Vikrant, Tetrahedron 64 (2008) 3405–3428. doi: 10.1016/j.tet.2008.01.049
K.E.O. Ylijoki, J.M. Stryker, Chem. Rev. 113 (2013) 2244–2266. doi: 10.1021/cr300087g
X. Liu, Y.J. Hu, J.H. Fan, et al., Org. Chem. Front. 5 (2018) 1217–1228. doi: 10.1039/C7QO01123G
H. Pellissier, Adv. Synth. Catal. 360 (2018) 1551–1583. doi: 10.1002/adsc.201701379
L.P. Bejcek, R.P. Murelli, Tetrahedron 74 (2018) 2501–2521. doi: 10.1016/j.tet.2018.04.006
K. Gao, Y.G. Zhang, Z.M. Wang, et al., Chem. Commun. 55 (2019) 1859–1878. doi: 10.1039/c8cc09077g
L. Min, X. Liu, C.C. Li, Acc. Chem. Res. 53 (2020) 703–718. doi: 10.1021/acs.accounts.9b00640
N.A. Harry, U.S. Mohanan, Curr. Org. Chem. 26 (2022) 735–744. doi: 10.2174/1385272826666220510152025
P. Magnus, L. Shen, Tetrahedron 55 (1999) 3553–3560. doi: 10.1016/S0040-4020(98)01162-4
J.H. Fan, Y.J. Hu, Q. Guo, et al., Org. Chem. Front. 6 (2019) 22–26. doi: 10.1039/C8QO01089G
J.H. Fan, J.J. Wang, F.F. Li, et al., CCS Chem. 3 (2021) 348–357. doi: 10.31635/ccschem.021.202000721
L. Chen, C.H. Zhang, J. Liu, et al., Org. Lett. 27 (2025) 3517–3520. doi: 10.1021/acs.orglett.5c00301
M. Zhang, N. Liu, W. Tang, J. Am. Chem. Soc. 135 (2013) 12434–12438. doi: 10.1021/ja406255j
B. Chen, X. Liu, Y.J. Hu, et al., Chem. Sci. 8 (2017) 4961–4966. doi: 10.1039/C7SC01341H
L. Min, X. Lin, C.C. Li, J. Am. Chem. Soc. 141 (2019) 15773–15778. doi: 10.1021/jacs.9b08983
X. Liu, J. Liu, J. Wu, et al., J. Am. Chem. Soc. 141 (2019) 2872–2877. doi: 10.1021/jacs.8b12647
L.X. Li, L. Min, T.B. Yao, et al., J. Am. Chem. Soc. 144 (2022) 18823–18828. doi: 10.1021/jacs.2c09548
Y.P. Zou, Z.L. Lai, M.W. Zhang, et al., J. Am. Chem. Soc. 145 (2023) 10998–11004. doi: 10.1021/jacs.3c03755
O.J. Achmatowicz, P. Bukowski, B. Szechner, et al., Tetrahedron 27 (1971) 1973–1996. doi: 10.1016/S0040-4020(01)98229-8
T.L. Ho, S.G. Sapp, Synth. Commun. 13 (1983) 207–211. doi: 10.1080/00397918308065990
D.E. Ward, M.S. Santos, Org. Lett. 7 (2005) 3533–3536. doi: 10.1021/ol051262e
L. Chen, K.B. Riaz Ahmed, P. Huang, et al., Angew. Chem. Int. Ed. 52 (2013) 3446–3449. doi: 10.1002/anie.201209300
A.W. Schuppe, Y. Zhao, Y. Liu, et al., J. Am. Chem. Soc. 141 (2019) 9191–9196. doi: 10.1021/jacs.9b04508
M.T. Reetz, A. Kindler, J. Organomet. Chem. 502 (1995) 5–7. doi: 10.1515/9783112400050-001
I.T. Chen, I. Baitinger, L. Schreyer, et al., Org. Lett. 16 (2014) 166–169. doi: 10.1021/ol403156r
M. Deng, X. Li, Z. Zhang, et al., Org. Lett. 21 (2019) 1493–1496. doi: 10.1021/acs.orglett.9b00285
H.D. Hao, D. Trauner, J. Am. Chem. Soc. 139 (2017) 4117–4122. doi: 10.1021/jacs.7b00234
S. Michaelis, S. Blechert, Org. Lett. 7 (2005) 5513–5516. doi: 10.1021/ol052332k
M. Szostak, M. Spain, D.J. Procter, J. Org. Chem. 79 (2014) 2522–2537. doi: 10.1021/jo4028243
X. Just-Baringo, D.J. Procter, Acc. Chem. Res. 48 (2015) 1263–1275. doi: 10.1021/acs.accounts.5b00083
R. Marder, J. Dubois, D. Guenard, et al., Tetrahedron 51 (1995) 1985–1994. doi: 10.1016/0040-4020(94)01078-E
J.H. Sohn, Bull. Korean Chem. Soc. 31 (2010) 1841–1842. doi: 10.5012/bkcs.2010.31.7.1841
Y. Wang, X. Hu, C. Morales-Rivera, et al., J. Am. Chem. Soc. 140 (2018) 9678–9684. doi: 10.1021/jacs.8b05753
Y.A. Zhang, V. Palani, A.E. Seim, et al., Science 378 (2022) 383–390. doi: 10.1126/science.add6852
J.A. Marshall, G.P. Luke, J. Org. Chem. 58 (1993) 6229–6234. doi: 10.1021/jo00075a017

Scheme 1 Selected polycyclic cembranoid and norcembranoid natural products, prior total syntheses, and our retrosynthetic analysis of sinugyrosanolide A.

Scheme 2 Previous studies of α,β-butenolide moiety in cycloadditions and our design relying on [5 + 2] cycloaddition.
Table 1. Evaluation of [5 + 2] cycloaddition reaction conditions.
|
|||
| Entry | Substrate | Conditionsa | Isolated yield (%) |
| 1 | 48a-1 | PhCN, 170 ℃ | 49a, 0 |
| 2 | 48a-1 | TMP, PhCN, 150 ℃ | Decomposed |
| 3 | 48a-1 | CF3CO2H, CH2Cl2, 0 ℃ – 60 ℃; then CF3CO2H, CH3CN, 100 ℃ | 49a, 10 |
| 4 | 48a-1 | CF3CO2H, CH3CN, 100 ℃ | 49a, 13 |
| 5 | 48a-1 | AcOH, CH3CN, 150 ℃ | 49a, 19 |
| 6 | 48a-1 | AcOH, toluene, 150 ℃ | 49a, 48 |
| 7 | 48a-1 | AcOH, mesitylene, 150 ℃ | 49a, 82 |
| 8 | 48a-1 | AcOH, mesitylene, 150 ℃b | 49a, 75 |
| 9 | 48a-2 | AcOH, mesitylene, 150 ℃ | 49a, 82 |
| 10 | 48b | AcOH, mesitylene, 150 ℃b | 49b, 66 |
| a Reaction scale: 0.008 mmol. b Performed on gram scale. |
|||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: