A facile entry toward multi-substituted chiral cyclic nitrones and N-heterocycles via Pd-catalyzed enantioselective cyclization coupling of alkenyl oxime
Citation:
Dongyan Ju, Hui Wang, Chuang Zhao, Jiahang Li, Luoluo Li, Xiaoyan Liu, Jie Tao, Junliang Zhang, Jinbo Zhao. A facile entry toward multi-substituted chiral cyclic nitrones and N-heterocycles via Pd-catalyzed enantioselective cyclization coupling of alkenyl oxime[J]. Chinese Chemical Letters,
2026, 37(7): 112000.
doi:
10.1016/j.cclet.2025.112000
A facile entry toward multi-substituted chiral cyclic nitrones and N-heterocycles via Pd-catalyzed enantioselective cyclization coupling of alkenyl oxime
English
A facile entry toward multi-substituted chiral cyclic nitrones and N-heterocycles via Pd-catalyzed enantioselective cyclization coupling of alkenyl oxime
Received Date:
24 April 2025 Accepted Date:
21 October 2025 Revised Date:
11 October 2025 Available Online:
15 July 2026
Abstract:
Multi-substituted pyrrolidine and piperidine N-heterocycles are the top scaffolds of clinical drugs, but syntheses of these scaffolds are far from trivial. Herein, we report a highly enantioselective protocol for the synthesis of 5- and 6-membered cyclic nitrones via Pd-catalyzed cyclization coupling of alkenyl oximes with aryl, alkenyl and alkynyl (pseudo)halides enabled by chiral sulfonamidephosphine (SadPhos) ligands. These cyclic nitrones products, besides their significance per ser, can be facilely converted to multi-substituted pyrrolidines and piperidines by simple operations. This study also represents the first example of two-component catalytic asymmetric synthesis of cyclic nitrones and provides important insights for further development of enantioselective heterocyclization reaction and processes involving multiple selectivity issues.
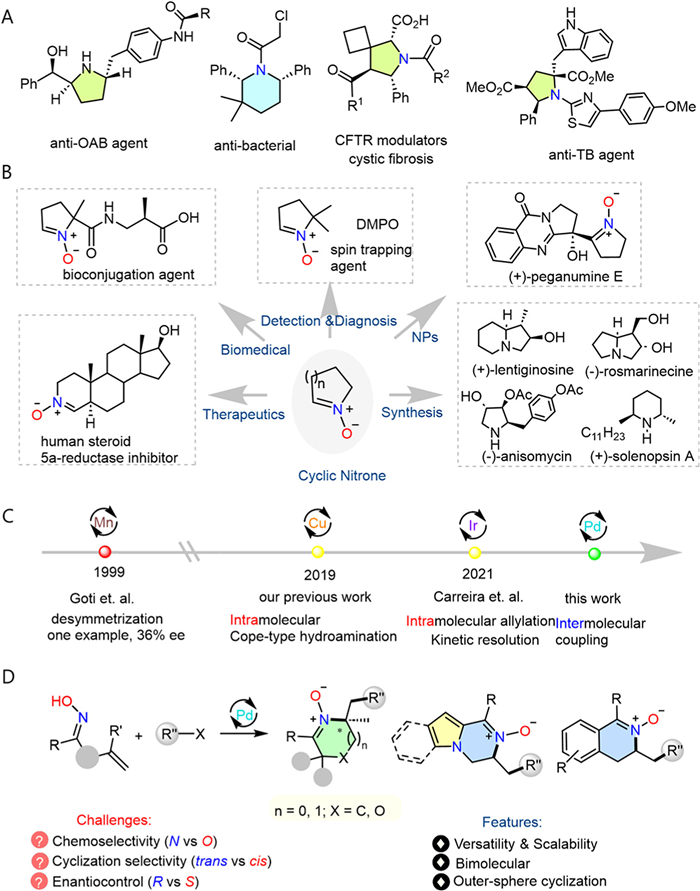
N-Heterocycles are essential to drug development. Among the N-heterocyclic pharmacophores, piperidines and pyrrolidines figure prominently and rank No. 2 and 3 respectively in the FDA approved drugs in the past decade [1]. While a large percentage of drugs and natural products contain a multiply (>2) substituted piperidine or pyrrolidine core (Fig. 1A), efficient protocols to assemble these cores are nonetheless nontrivial. For instance, synthesis of vibegron, an anti-overactive bladder agent, requires a long sequence of >10 steps. This multi-substitution issue has significantly limited the exploration of piperidine and pyrrolidine chemical space for drug development campaigns.
Figure 1
Figure 1.
Background and research synopsis. (A) The multi-subsitution conudrum in 5- and 6-membered N-heterocycle. (B) Cyclic nitrone-based functional molecules. (C) Catalytic asymmetric protocols for cyclic nitrone synthesis. (D) Two-component asymmetric cyclic nitrone synthesis via Pd catalysis (this work).
The cyclic nitrone scaffold is found in many important functional molecules, including immuno-spin trapping agents [2,3], biorthogonal probe [4], natural products, therapeutic agents [5–7], and acts as a highly useful 1,3-dipole in dipolar cycloaddition reactions and a key intermediate to important 5- and 6-membered N-heterocyclic natural products (Fig. 1B) [8–13]. In contrast to their prominent role, methods for enantioselective synthesis of nitrones are strikingly underdeveloped. During the past few years, Zhang and Zhao [14], the Kobayashi group [15] and the Breit group [16] have made progresses on transition-metal and Lewis acid catalyzed asymmetric intermolecular addition of oxime to olefins. However, catalytic asymmetric synthesis of cyclic nitrones, a key subset of nitrone family [9], remains highly underdeveloped. The first report on catalytic asymmetric synthesis of cyclic nitrones was reported by Goti and coworkers on the oxidative desymmetrization of 5-membered cyclic hydroxyamine [17], which obtained only 68/32 er in a single example (Fig. 1C). Zhang and Zhao et al. reported in 2019 the first highly enantioselective synthesis of chiral cyclic nitrones via Cu-catalyzed intramolecular Cope-type hydroamination, but only 5-membered carbocyclic nitrones succumbed to high asymmetric induction [18]. The Carreira group subsequently reported an elegant Ir-catalyzed highly enantioselective intramolecular allylation of hydroxy-latedalkenyl oximes to deliver a series of 5-, 6- and 7-membered cyclic nitrones with high levels of enantiocontrol (Fig. 1C) [19]. To date, however, all enantio-selective syntheses of cyclic nitrones are limited to unimolecular intramolecular versions. Practical, adaptable, and highly enantioselective methods for diversified nitrone synthesis are still in urgent demand.
Pd-catalyzed heterocyclization of N- and O-nucleophiles onto pendant alkene has been established as an important tool for the syntheses of various important N- and O-heterocycles [20–22]. While significant progresses have been made in enantioselective aza-Wacker cyclization [23,24] and carboamination [22,25,26] of alkenyl amides in the past decade, the related process of N-cyclization of alkenyl oxime toward chiral nitrone synthesis remains elusive. Due to the challenging chemoselectivity issue caused by the oxime functionality (N vs. O), as well as the thermodynamic unfavourability of nitrone due to charge-separation, development of enantioselective synthesis of cyclic nitrone via alkenyl oxime poses additional challenges as compared to alkenyl amides. For instance, competitive O-cyclization can intervene, for which the highly enantioselective versions have been realized recently by the Zhao [27] and Zhang groups [28]. In contrast, enantiocontrol in N-cyclization toward nitrone formation remains an unsolved challenge [29]. Furthermore, an additional challenge comes from competing Heck reaction of the alkene with the electrophile. Herein we disclose the development of highly enantioselective synthesis of 5- and 6-membered nitrones from Pd-catalyzed cyclization coupling of γ,δ- and δ,ε-alkenyl oximes with (hetero)aryl and alkenyl electrophiles (Fig. 1D). This protocol enables a facile access to multi-substituted piperidines and pyrrolidines upon facile reductive N‒O bond scission and subsequent nucleophilic addition.
Our study commenced by optimizing the Pd-catalyzed asymmetric cyclization of γ,δ-alkenyl oxime 1a with 4-tolyl iodide. Initial scouting experiments revealed that a suitable combination of base and media is critical for the enantiocontrol, with Cs2CO3 and methyl tert-butyl ether (MTBE) being optimal (Tables S1 and S2 in Supporting information). Reactions with a series of commercial monodentate and bidentate ligands were then examined, but were found unsatisfactory with respect to enantiocontrol (entries 2–5, Table 1). In contrast, a type of Sadphos ligand developed by the Zhang group [31] turned out to be highly competent, retrieving cyclic nitrone 3aa in 96.7/3.3 er. Notably, using Pd(dba)2 as precatalyst appreciably facilitated the conversion, allowing the reaction to be completed at 50 ℃ within the same time span with concomitant improvement of enantioselection (entries 6–9, Table 1). This suggests that reductive elimination should be turnover limiting. We substituted PhBr or PhOTf for PhI, and the results were not as satisfactory (entries 10 and 11, Table 1).
Table 1
Table 1.
Optimization of Pd-catalyzed asymmetric coupling cyclization of γ,δ-alkenyl oxime.a
a Reaction conditions: 1a (0.20 mmol), 2a (0.24 mmol), Pd catalyst (0.01 mmol), ligand (0.012 mmol), Cs2CO3 (0.24 mmol), MTBE (2 mL), Ar, 60 ℃, 12 h. b Isolated yield. c Determined on a chiral stationary phase.
Our attention then shifted to the coupling of δ,ε-alkenyl oxime to deliver 6-membered cyclic nitrones. The above standard conditions for 5-membered nitrone synthesis with L1 was found to give poor enantioselectivity for 4na (entry 1, Table 2). After extensive trials (Table S3 in Supporting information), we found that SadPhos ligands bearing a bulkier PAd2 (Ad = 1-admantyl) analogues [31] could enable the reaction of oxime 1n to deliver the 6-membered nitrone 4na in higher enantioselectivities compared to its PCy2 counterpart, but in a low yield (entry 2, Table 2). Moreover, we found that changing the Ar moiety posited α to the nitrogen from 3,5-di-t-Bu-4-MeOC6H2- (L6) to 3,5-di-t-Bu-4-n-BuOC6H2- (L7) had a positive effect on the enantioselectivity (entry 2 vs. 3). When ArI was replaced with ArBr and the temperature was lowered to 90 ℃, the yield was not improved (entry 4). To our surprise, the yield can be significantly improved by increasing the catalyst loading (entries 5 and 6, Table 2), which we attributed to appreciable product sequestration (see DFT studies, vide infra). Eventually, 4 mol% catalyst loading enabled to produce the product 4na in a good enantiomeric ratio (95/5 er) and a high yield of 90% at 80 ℃ with aryl bromide as coupling partner (entry 7, Table 2).
Table 2
Table 2.
Optimization of Pd-catalyzed arylative cyclization of δ,ε-alkenyl oxime.a
a Reaction conditions: 1n (0.20 mmol), 2a (0.3 mmol), Pd catalyst (4 µmol), ligand (0.02 mmol), Cs2CO3 (0.24 mmol), Et2O (2 mL), Ar, 100 ℃, 12 h. b Isolated yield. c Determined on a chiral stationary phase.d PhBr instead of iodide 2a.
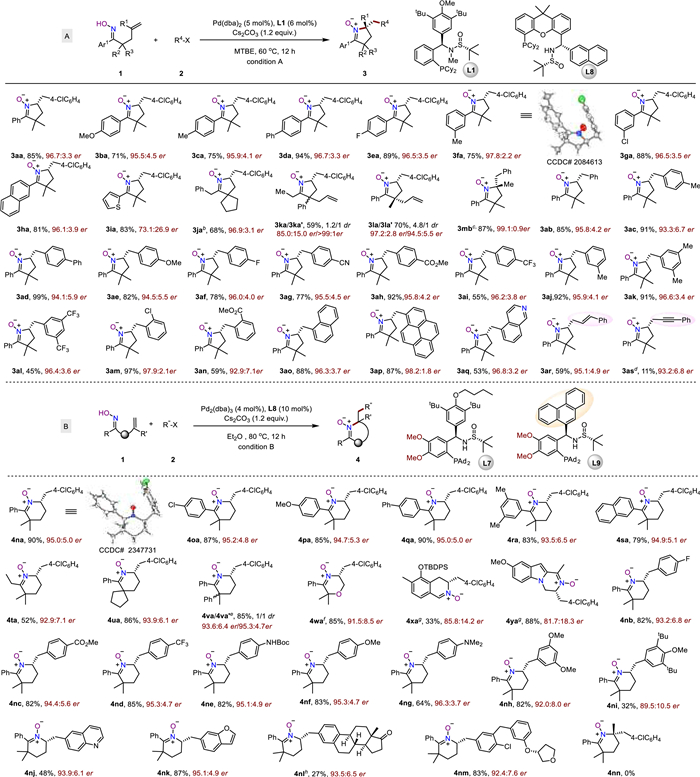
The scope of the enantioselective synthesis of 5-membered nitrones summarized in Fig. 2A showcased the robustness of the conditions to accommodate substituents of both electronic and steric nature. Substitution at various positions of the aryl groups of the alkenyl oxime have little effect on the efficiency or enantioselectivity (3ba-3ia), except for 2-thienyl, which led to a compromised 73.1/26.9 er likely due to competitive coordination with the catalyst which changed the ratio of anti/syn aminopalladation route (vide infra). Single crystals of 3fa were obtained which enabled determination of its absolute configuration to be (R) by X-ray diffraction studies. Spirocyclic nitrone 3ja was obtained in a good yield and 96.9/3.1 er. The reaction of α,α-disubstituted oximes led to 3ka and 3la in high ers. To our delight, the reaction of the prochiral bis-homoallylated oxime retrieved the corresponding cyclization products 3la/3la' in 4.8:1 dr as readily separated diastereomers, both with high enantioselectivity. Alkyl substitution at the internal carbon of the alkene did not affect the reactivity, and remarkably, a higher level of enantiocontrol was realized (3mb).
Figure 2
Figure 2.
Scope of the enantioselective nitrone synthesis. a Conditions A: 1 (0.20 mmol), 2 (0.24 mmol), Pd(dba)2 (5 mol%), L1 (6 mol%), Cs2CO3 (1.2 equiv.), MTBE (2 mL), Ar, 60 ℃, 12 h. Conditions B: 1 (0.20 mmol), 2 (0.3 mmol), Pd2(dba)3 (4 mol%), L7 (10 mol%), Cs2CO3 (1.2 equiv.), Et2O (2 mL), Ar, 80 ℃, 12 h. b 50 ℃. c PhI was used. dL8 (6 mol%), hexane, 50 ℃, 24 h. e 100 ℃. fL9 (10 mol%), 100 ℃. g 50 ℃, 24 h. h Triflate was used.
Next, aryl halides bearing electronically distinct groups were examined and found compatible (3ab-3al). Remarkably, the sterically congested ortho-substitution is also tolerated (3am and 3an). Fused aromatic hydrocarbons such as naphthalene and pyrene (3ao-3ap), and heterocycle such as quinoline can also be tolerated (3aq). Alkenyl halides were also found competent, as showcased in the comparable reactivity and selectivity in nitrone 3ar (59%, 95/5 er). The use of 1-alkynyl bromide as the electrophile retrieved nitrone 3as in a low yield with only nominal enantioselectivity, indicating the importance of an electron-rich π-system for the enantiocontrol. In this case, switching to a XantPhos-hybrid ligand, PC-Phos L8 [30] enabled high enantiocontrol, albeit the yield still needs further optimization (Table S4 in Supporting information).
We then explored the scope of the 6-membered nitrones with a panel of substrates (Fig. 2B). Firstly, δ,ε-alkenyl oximes with different aryl moieties substituted at the para and/or meta position were well-tolerated, affording the desired products in good yields and excellent enantioselectivities (4oa-4sa). The absolute configuration of the products was determined to be (S) by the crystal structure of 4na, which is the same sense of enantiocontrol with the 5-membered nitrones. The alkyl substituted alkenyl oxime 1t can also deliver the corresponding nitrone 4ta in a moderate yield and comparable level of enantiocontrol. Likewise, spirocyclic nitrone 4ua was obtained in a good yield and higher er of 93.9/6.1. Racemic oxime 1v gives the diastereoisomers 4va/4va' with 85% yield in 1:1 dr, both were formed in a high enantio-selectivity. The O-tethered substrate was also competent, affording tetrahydro-1,4-oxazine N-oxide 4wa in a high yield and 92/8 er. Dihydroisoquinoline N-oxide 4xa, featuring an aldehyde-derived nitrone moiety, was also successfully synthesized in 33% yield and a good enantiomeric ratio of 86/14. Indole-derived nitrone 4ya can also be successfully prepared in a good yield yet with inferior enantiocontrol as compared to carbon-tethered ones. The relatively poor performance of these heterocycle-tethered and benzo-derivatives can be attributed to the rigidity of the tethers that prevent a better alignment of the two approaching moieties (oxime and alkene) in the cyclization transition structures, which may make the inner-sphere migratory insertion mechanism more favorable. Next, aryl halides bearing substituents of distinct electronic properties were tested under the optimized conditions, and the corresponding products (4nb-4ni) were obtained in 33%~85% yields with excellent enantioselectivities (89.5/10.5 ~ 96.5/3.5 er). Electron-rich heteroaromatic halides are also tolerated (4nj and 4nk). The aryl triflate derived from estrone proceeded smoothly, and the desired product 4nl was furnished in 93.5/6.5 er, albeit with a low yield due to competing Heck reaction. The aryl bromide with 3,4-disubstitution and a chiral tetrahydrofuran moiety also showed excellent reactivity, and the corresponding nitrone 4nm was afforded in 92.4/7.6 er. Unfortunately, the substrate bearing an alkene moiety with a substituent at the internal carbon did not result in formation of the expected 6-membered nitrone 4nn, probably due to unfavored cyclization step.
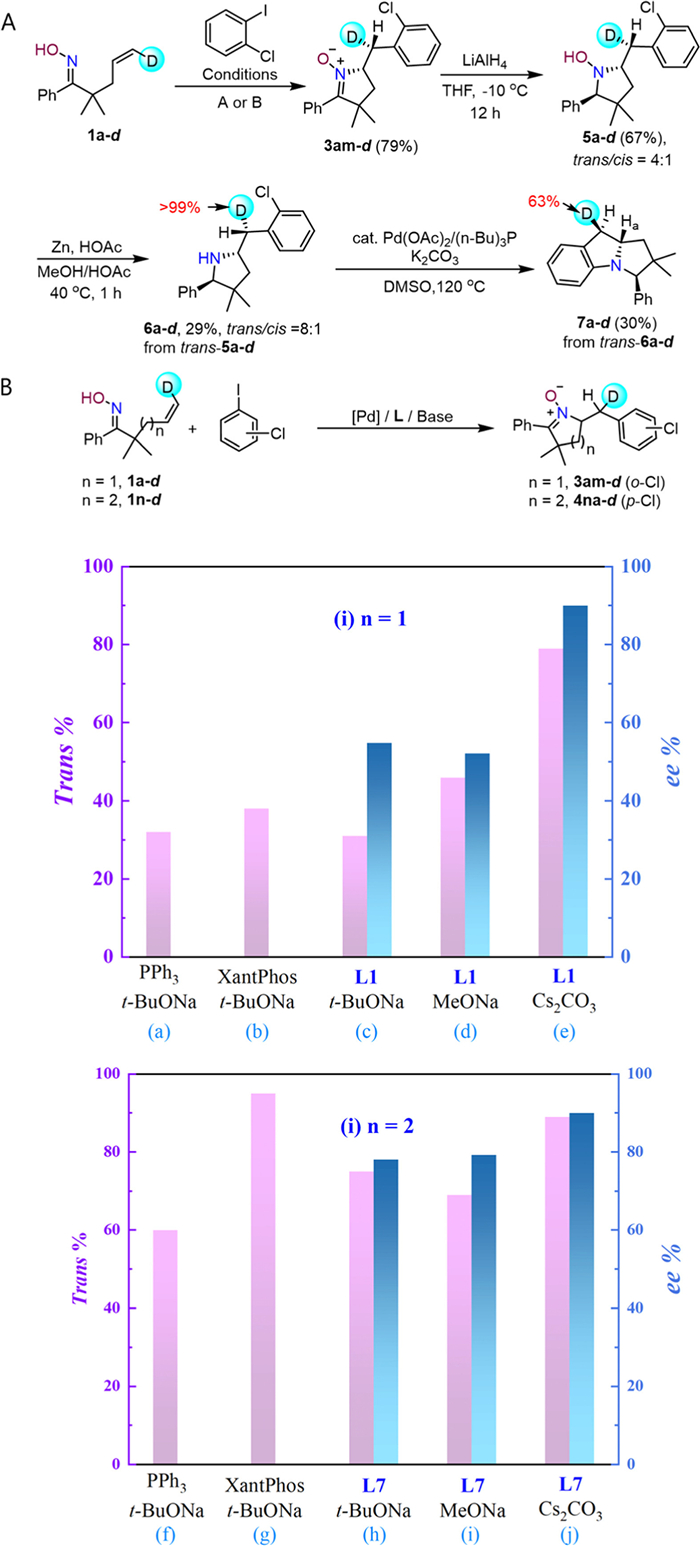
Revelation of the stereochemical course of the cyclization is key to development of highly enantioselective protocols, as syn- and anti-type cyclization processes usually exert distinct requirements for chiral discrimination. Mechanistic studies on Pd-catalyzed cyclization of alkenyl amides revealed that both cis-amino-palladation and aza-Wacker type anti-aminopalladation are possible [28]. To discriminate these two pathways, stereo-defined deuterium labeled alkenyl oxime 1a-d was prepared and subjected to the optimized reaction conditions (Fig. 3A). The product, deuterated nitrone 3am-d, was subsequently reduced by LiAlH4 to the corresponding trans-configured hydroxyamine, which underwent reductive N‒O cleavage to secondary amine 6a-d. Partial configurational scrambling was observed at this step, likely due to elimination to the imine followed by reduction. Finally, a Pd0-catalyzed C‒N coupling delivered the cyclization product, benzopyrrolizidine derivative 7a-d. An inner-sphere process is expected to result in deuterated benzopyrrolizidine 7a-d with the deuterium atom posited trans to Hα, while the outer-sphere mechanism would lead to a cis-relationship, and vice versa. Comparison of the 1H NMR of 7a and 7a-d, as well as the 2D NOESY spectrum of 7a indicated that 7a-d has a trans-arrangement of D and Hα, suggesting that the reaction underwent exclusive trans-aminopalldation via outer-sphere mechanism (Figs. S1 and S2 in Supporting information). The deuterated substrate 1a-d was then used as a probe to examine the stereochemical outcome of the cyclization under different conditions. As shown in Fig. 3B, the nature of base and ligands play an important role in the cyclization pathway.
Figure 3
Figure 3.
Experimental mechanistic studies. (A) Determination of cyclization stereochemistry by (un-optimized) product derivatization with isotope-labeled substrates. (B) The stereochemical outcome with 1a-d and 1n-d as probes revealed condition-dependent pathway selectivities that translate to the difference in enantiocontrol.
For 5-membered cyclic nitrone, when the strong base tBuONa was used, the reaction proceeds through both inner-sphere migratory insertion pathway and outer-sphere anti-aminopalldation, with the former being slightly more favored, regardless of the ligand being used (slots a~c, Fig. 3B). Under the catalytic conditions of Pd/L1, the reactions under different bases show that as the basicity of the base decreases, consistently higher percentage of outer-sphere anti-aminopalldation took place, which is also in line with the increase of enantiocontrol (slots c~e, Fig. 3B). In contrast, for the synthesis of 6-membered cyclic nitrone, the pathway selectivity favors outer-sphere anti-aminopalldation even with NaOt-Bu as the base, and ligand also plays a role (slots f~h, Fig. 3B). Yet, under the catalytic conditions of Pd/L6, use of a weaker base also increased the anti/syn cyclization selectivity, albeit to a smaller extent (slots h~j, Fig. 3B).
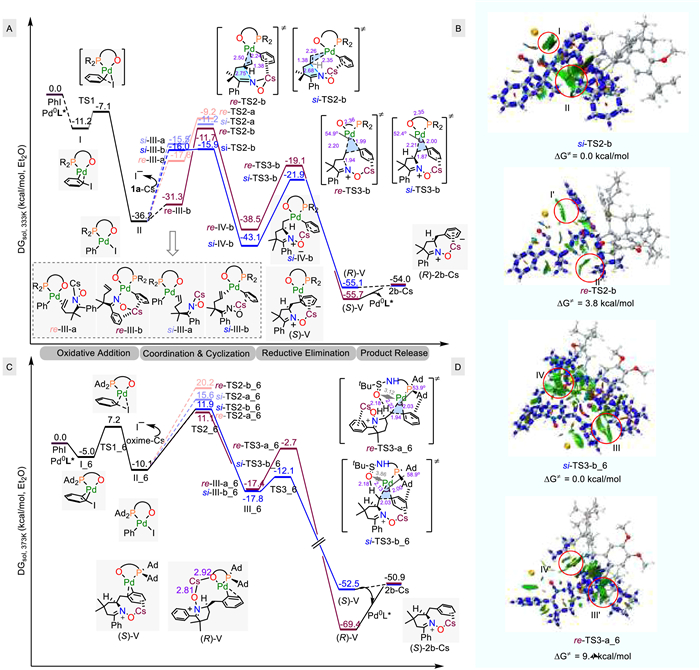
To gain insights into the mechanism regarding the origin of enantiocontrol and rate-determining step(s), DFT calculation was performed first to explore the potential energy surface of the 5-membered nitrone formation reaction (Fig. 4A). Our calculation reveals that oxidation addition of Pd0L* (L* = L1) with PhI has a low barrier (4.1 kcal/mol) and a net free energy downhill of 25.0 kcal/mol. For the subsequent ligand exchange of Ph-PdIL* with alkene moiety of the Cs-countered oxime anion, we sampled the trajectories of the alkene via both Re- and Si-attack patterns, and for both attacking patterns, two representative approaching poses of the oxime were evaluated (denoted by a and b; wherein the poses having the oxime Cs part being proximal to the Pd‒O moiety are named with a, and those having oxime Cs part being proximal to the Pd-Ph moiety are named with b). The four cyclization transition states TS2 feature long C‒N bonds (2.2–2.8 Å, Fig. S6 in Supporting information), among which the Si-b pathway is most energetically favored with an activation energy of 19.3 kcal/mol (Si-TS2-b), leading to (S)-configured cyclization product Si-Ⅳ-b. In comparison, the lowest energy Re-face attacking transition state (Re-TS2-b) is disfavored by 4.2 kcal/mol. The irreversible nature of the cyclization step, as inferred from the 27.2 kcal/mol activation energy of the reverse event (Si-TS2-b vs Si-Ⅳ-b), suggests that the cyclization step is enantio-determining. As it is experimentally observed that electron deficient dba (dibenzylidene acetone) facilitates the reaction (vide supra), the reductive elimination step, with a slightly higher barrier of 21.2 kcal/mol (vs. 20.3 for the cyclization), should be involved in the rate-limiting step(s). In addition, the reductive elimination step of the Re-b pathway also has a higher barrier compared to the Si-b pathway (Re-TS3-b vs. Si-TS3-b). Furthermore, the transition structures of the two cis-aminopalladation pathways were also located and found to be energetically less favored than the above two lowest trans-aminopalladation TSs, and are very close in energy (Fig. S5 in Supporting information), consistent with experimental observation that the more cis-aminopalladation is involved in the cyclization, the lower the enantiocontrol was observed.
Figure 4
Figure 4.
DFT studies. Free energy profiles of Pd-catalyzed asymmetric cyclization/coupling reaction of alkenyl oxime to form 5-membered (A) and 6-membered (C) nitrone, and the interaction region indicator (IRI) analysis of the enantio-determining TSs (B, D). Level of theory: SMD(Et2O)-PBE0-D3(BJ)/def2-tzvpp//PBE0-D3(BJ)/6–31G(d)/SDD (Pd, I, Cs). IRI analysis isosurface value: 0.5.
Interaction region indicator (IRI) analysis [31] revealed key differences in the weak interaction regions within the two cyclization TSs arising from the two interacting fragments, i.e., the oxime Cs and the Ph-PdL* moiety (Fig. 4B). The two notable regions are the cation···π interaction between the Cs+ and the Pd-Ph, which is present in both TSs but looks stronger in Si-TS2-b (regions Ⅰ/Ⅰ'), as well as a dispersion interaction region between the alkyl tether and the ligand's P-Cy moiety which is much more extensive in Si-TS2-b but only sporadic in Re-TS2-b (regions Ⅱ/Ⅱ').
The free energy profile of the reaction of the 6-membered nitrone formation with Ligand L7 revealed a significantly different scenario (Fig. 4C). The large P-substituents makes the cyclization step not only basically non-selective, but also even slightly favor Re-face attack (which leads to the un-favored R-product antipode). In contrast, the reductive elimination step shows a strong preference for the Si-b pathway (Re-TS3-a_6 vs. Si-TS3-b_6), leading to the experimentally favored product antipode. At the experimental temperature (373 K), the cyclization step should be more reversible, resulting in both cyclization and the subsequent reductive elimination step to be engaged in the enantio-control, while cyclization is likely rate-determining. In contrast to the 5-membered nitrone formation, both reductive elimination TSs feature a broken Pd‒O bond, which may expedite the reductive elimination. In Si-TS3-b_6, the distance between Pd and O atoms is significantly longer (3.86 Å vs. 3.12 Å in Re-TS3-a_6), which may play a role in facilitating the reductive elimination. It is also noted that the (R)-product, albeit being the unfavored stereoisomer, forms a much more stable complex with the catalyst, (R)-V, which may rationalize the requirement of higher catalyst loading (entries 5~7, Table 2).
IRI analysis of the two selectivity-determining reductive elimination TSs (Fig. 4D) shows that the oxygen is enclosed in a dispersion region between the aliphatic linker of the oxime and the t-Bu of the ligand's bulky aryl chiral side arm in Si-TS3-b_6 which does not have a counterpart in Re-TS3-a_6 (regions Ⅲ/Ⅲ'). In addition, the C—H···π interaction between one of the P-adamantly moieties and the Pd-pH adopts head-on pose in Si-TS3-b_6, which could be more efficient compared to the side-on pose in Re-TS3-a_6 (regions Ⅳ/Ⅳ'). These two interaction regions may contribute to the large energy difference for the enantio-differentiation. It is thus likely that, as inferred in Fig. 4B, ii, the lower overall enantiocontrol in formation of the 6-membered nitrone may be caused by poor pathway selectivity, or a more pronounced sabotage by the minor non-selective inner-sphere pathway. Combined with the information in 5-membered nitrone formation, the calculation agrees well with experimental observation that the aryl moiety of the aryl halide electrophile is important for the enantiocontrol.
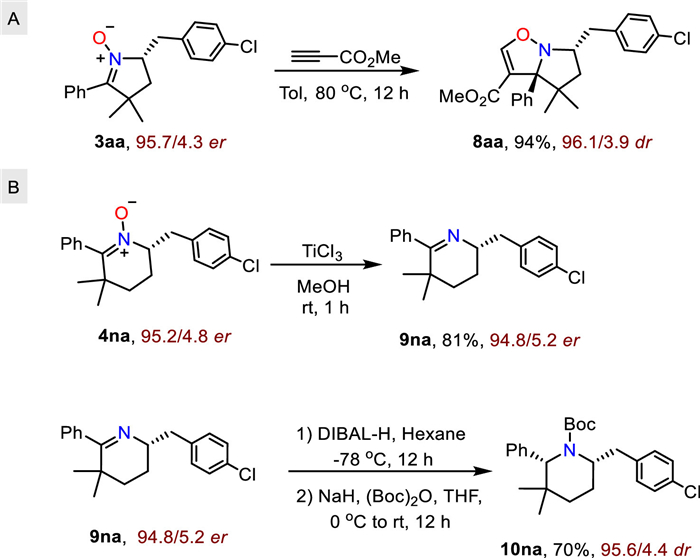
We then performed preliminary downstream elaboration of the chiral cyclic nitrone products. First, 5-membered nitrone 3aa undergoes dipolar cycloaddition with methyl propionate to afford bicyclic dihydroisooxazole 8aa in 94% yield and high diastereoselectivity (Scheme 1A). In addition, the 6-membered nitrone 4na can be facilely deoxygenated with TiCl3 and reduced to the chiral pipyridine 10na in an overall yield of 70% with no encroachment of stereoselectivity (Scheme 1B).
In summary, we have developed a highly enantioselective synthesis of various 5- and 6-membered nitrones via Pd-catalyzed coupling cyclization of γ,δ- and δ,ε-alkenyl oximes with (hetero)aryl, alkenyl and alkynyl oximes with tailored sulfinamidephosphine ligands. The products could be facilely transformed to important multi-substituted chiral N-heterocycles, such as bicyclic dihydroisooxazole and cyclic imines. Reactions with isotope-labeled substrate revealed a highly base and solvent dependent cyclization pathway distribution which significantly impacts enantiocontrol. Under the optimized conditions, the anti-aminopalladation cyclization prevails and the bidentate ligand exerts high differentiation for the enantiocontrol while the non-selective cis-aminopalladation is minimized. DFT calculations revealed a more demanding cyclization step as moved from 5-membered annulation to the 6-membered one, and provided insights into the factors for the enantiocontrol in both senarios. The addressing of the multiple selectivity issues for enantiocontrol has significant implication for related heterocyclization processes and asymmetric cyclization reactions at large.
Declaration of competing interest
The authors declare the following competing interests: a provisional Chinese patent application has been filed wherein J. Z. and D. J. were listed as inventors (application No. CN 120441469 A).
Financial support from National Natural Science Foundation of China (Nos. 22471024, 21831045) and Natural Science Foundation of Jilin Province (Nos. 20240101151JC and JJKH20250700BS) is gratefully acknowledged. We thank Prof. Wei-Bo Yang of Shanghai Institute of Material Medica, CAS for valuable suggestions on manuscript preparation.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.112000.
Figure 3
Experimental mechanistic studies. (A) Determination of cyclization stereochemistry by (un-optimized) product derivatization with isotope-labeled substrates. (B) The stereochemical outcome with 1a-d and 1n-d as probes revealed condition-dependent pathway selectivities that translate to the difference in enantiocontrol.
Figure 4
DFT studies. Free energy profiles of Pd-catalyzed asymmetric cyclization/coupling reaction of alkenyl oxime to form 5-membered (A) and 6-membered (C) nitrone, and the interaction region indicator (IRI) analysis of the enantio-determining TSs (B, D). Level of theory: SMD(Et2O)-PBE0-D3(BJ)/def2-tzvpp//PBE0-D3(BJ)/6–31G(d)/SDD (Pd, I, Cs). IRI analysis isosurface value: 0.5.

 DownLoad:
DownLoad:



 下载:
下载:






 下载:
下载:
