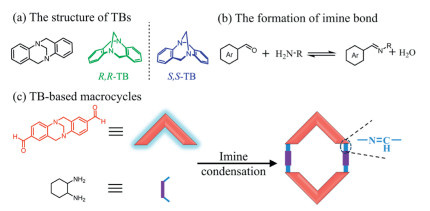
Figure 1.
Schematic diagram of the TB-based macrocycles: (a) The structure of Tröger's base. (b) The formation of imine bond. (c) TB-based macrocycles.
Reaction-driven crystallization: Gram-scale synthesis of chiral macrocycles bearing Tröger's base with self-sorting
Heng Qiu , Wang Xie , Mingxia Ye , Conghao Shi , Juli Jiang , Zhouyu Wang , Wim Dehaen , Leyong Wang

Chirality is an important feature of biological systems, with well-known examples the DNA double helix and protein folding [1,2], relying on the precise arrangement of chiral molecules. The investigation of chirality is of paramount importance in elucidating the intricate mechanisms underlying biological processes and deciphering the enigmatic pathways that govern biological functions [3,4]. Supramolecular science plays a very important role in chiral studies, utilizing the concept of self-assembly and dynamically reversible chemical bonding to mimic molecular recognition and assembly processes in living organisms [5-7]. As a main research object of supramolecular science, macrocycles provide a reliable platform for chiral investigation, and a wide variety of chiral macrocycles have been synthesized with the vigorous development of macrocyclic chemistry in recent years [8-10]. These macrocyclic hosts with special structures and properties have unique applications in the fields of mechanically interlocked molecules (MINs) [11,12], chiral recognition [13-15], sensing [16,17], drug delivery [18,19], adsorption-separation [20] and so on. However, the synthesis of macrocycles has been presenting significant challenges, including low yields for one-pot procedures, complex precursor requirements, and difficulties in production at larger scale. Moreover, the precise structural elucidation of macrocycles relied on X-ray crystallography, therefore, developing macrocycles that balance synthetic efficiency and easy crystallinity is one of challenging tasks in supramolecular chemistry.
Dynamic covalent chemistry (DCC) offers a promising thought process to develop novel macrocycles [21,22]. The reversible formation and breaking of covalent bonds provide the emerging structure a high degree of flexibility and adaptability. The imine bond is formed by the reversible condensation of an amino group with a carbonyl group and is a one of the most renown dynamic covalent bonds (Fig. 1b) [23]. Its rich stimulus-responsive properties make it widely used in the construction of smart materials and self-repairing materials [24,25]. For example, Li's group constructed molecular cages with circularly polarized luminescence (CPL) properties through imine bonds [26]. Also, Feringa's group recently developed the transformation between molecular cages and macrocycles by utilizing the dynamic reversibility of the imine bonds [27]. Meanwhile, the multiple imine bonds attenuate the hydrolyzability of imine bonds, which makes that the resulting molecules exhibit high stability [28].
Tröger's base (TB), a rigid V-shaped skeleton with a 90° clamp angle (Fig. 1a) [29-31], provides a broad platform for the development of unique chiral materials [32-34]. TB was one of the earliest molecules to have nitrogen chiral centres. The synthesis of a number of TB-based macrocycles has been reported in previous work [35-38]. For example, Yang's group has achieved chiral self-sorting in during macrocycle synthesis and highly selective chiral recognition with TB [39]. The TB skeleton exhibited excellent capabilities in these investigations. Therefore, integrating DCC with TB may synergistically address the aforementioned challenges by balancing synthetic accessibility and structural characterization.
Herein, we report a versatile strategy for the gram-scale synthesis of chiral macrocycles with tuneable cavities based on Tröger's base (TBCHMs), utilizing enantiopure TB aldehyde derivatives of varying lengths and 1,2-diaminocyclohexane (CHDA) in imine condensation reactions (Fig. 1c). Different from macrocycles synthesized by other methods, these TB-based macrocycles could crystallize directly from the reaction mixture in large quantities. The synergistic effect of multiple imine bonds confers excellent hydrolytic stability to the macrocycles. TBCHMs achieve chiral self-sorting in acetonitrile at crystalline state, which is primarily driven by the energy difference between diastereomers and their selective precipitation. In addition, benefiting from the combination of TB and DCC, the macrocycle system undergoes reversible transformations between the macrocycle and its TB precursor upon acid-base regulation in the solid state.
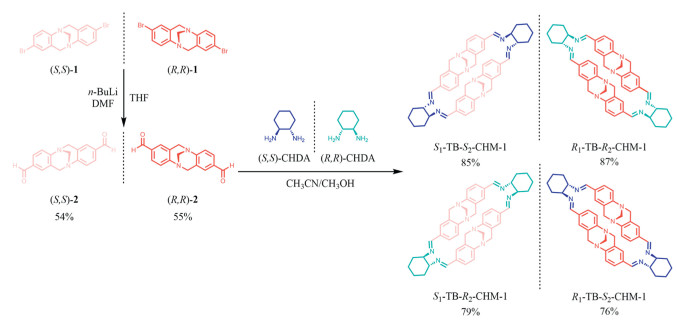
Aiming to simultaneously utilize the rigid V-shaped skeleton of TB and the dynamic reversibility of the imine bonds, the macrocycles were synthesized as shown in Scheme 1. To circumvent the complexities of chiral systems, we initiated our preparation of chiral TB-based macrocycles from the enantiomerically pure compound 1. The synthesis and chiral resolution of compound dibromide TB 1 is shown in Schemes S1 and S2 (Supporting information) [40,41], which was nearly 100% ee HPLC (Figs. S37-S39 in Supporting information). Electrophilic substitution via lithium-halogen exchange and reaction with dimethyl formamide (DMF) gave the macrocyclic precursor TB dialdehyde compound 2 in 55% yield [42]. A condensation reaction was performed with enantiopure CHDA and TB dialdehyde compound 2 in a solution of an acetonitrile/methanol mixture (10/1, v/v) at 60 ℃ without stirring. White crystals were gradually appearing, and after 18 h, the reaction was cooled to room temperature slowly, and the crystalline TB-based macrocycles (TBCHMs) were obtained after filtration in gram quantities with yields of 85%, 87%, 79% and 76%, respectively for the different combinations of stereoisomeric building blocks. The crystals of R1-TB-R2-CHM-1 and S1-TB-S2-CHM-1 were cubic in shape, while those of R1-TB-S2-CHM-1 and S1-TB-R2-CHM-1 were elongated. All of the macrocycles showed a molecular ion peak in their mass spectrum (calcd. m/z 713.4075, Figs. S9-S12 in Supporting information), corresponding to [2 + 2] macrocycles.
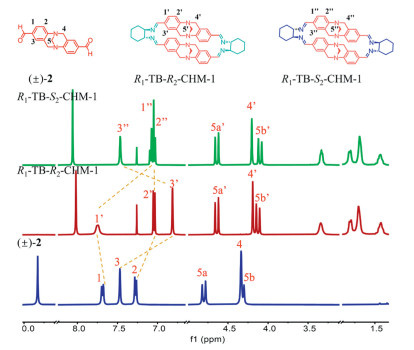
Compared to the precursor compound 2, the macrocycles themselves exhibited an apparent downfield shift of the TB skeleton protons in the 1H NMR spectra (Fig. 2), revealing that the TB group was directed outside the macrocycle cavity. The 1H NMR spectra of R1-TB-R2-CHM-1 revealed three kinds of aromatic proton peaks between 6.8 ppm and 7.8 ppm while the R1-TB-S2-CHM-1 showed the corresponding signals at 7.0–7.5 ppm, showing that the peaks at 6.80–7.03 ppm exhibited apparent downfield shifts and the peak at 7.71 ppm exhibited an upfield shift, which suggested that the hydrogens on the benzene ring of R1-TB-R2-CHM-1 and R1-TB-S2-CHM-1 experienced different shielding effects because of the change in the chirality of CHDA. On the other hand, similar chemical shift values were observed for all hydrogen atoms on the TB's diazocine core and CHDA groups in all macrocycles. The difference of 13C NMR spectra for above macrocycles R1-TB-R2-CHM-1 and R1-TB-S2-CHM-1 also supported their different shielding effects (Figs. S5-S8 in Supporting information). The ability of the TB skeleton to maintain stability under the reaction conditions provides a reliable basis for the growth of macrocycle crystals.
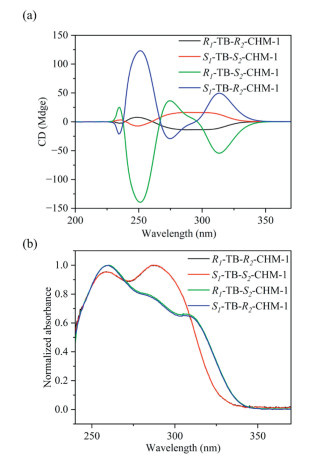
In addition, the enantiomers of both homochiral macrocycles were characterized by circular dichroism (CD) spectroscopy, exhibiting the expected mirroring CD signals (Fig. 3). R1-TB-R2-CHM-1 revealed Cotton effects at 236, 249, and 284–300 nm, while the enantiomer S1-TB-S2-CHM-1 demonstrated opposite Cotton effects at the same wavelengths. Similarly, R1-TB-S2-CHM-1 and S1-TB-R2-CHM-1 showed Cotton effects at 234, 251, 275 and 314 nm. In reference to the CD spectrum of compound 1 (Fig. S13 in Supporting information), peaks at a longer wavelength were caused by the chiral skeleton of TB, and the introduction of CHDA results in peaks at shorter wavelengths. R1-TB-S2-CHM-1 and S1-TB-R2-CHM-1 demonstrated stronger Cotton effects obviously, which suggested that similar chirality of TB with CHDA would weaken the overall chirality of the macrocycle.
The chemical structures of the macrocycles were definitively elucidated by SC-XRD (Single crystal X-ray diffraction) analysis. As shown in Fig. 4, the TB skeletons in R1-TB-R2-CHM-1 (Fig. 4a) and S1-TB-S2-CHM-1 (Fig. 4b) were stretched, which lead to their manifestation of a rhombohedral morphology in the crystalline state. The dihedral angle between the two benzene rings in the TB units shifted from 90° to 111° [43], demonstrating the adaptability of the TB skeleton. The two opposing benzene rings were almost parallel, and the distance between them was in both cases 8.6 Å. Compared to R1-TB-R2-CHM-1, the clamping angle of the TB skeleton in R1-TB-S2-CHM-1/S1-TB-R2-CHM-1 (98°) (Figs. 4c and d) was similar to that of 2 (92°), making them approximately square. Differently from those in R1-TB-R2-CHM-1, the two opposing benzene rings lost their parallelism. They demonstrated a tendency to converge towards each other, whereas at the opposite end, they diverged. As a result, the TB units alternated up and down, visually forming an X-shape, while R1-TB-R2-CHM-1 exhibited a planar structure (Fig. S22 in Supporting information). This observation aligned with the directional variations in chemical shifts of aromatic protons observed in the 1H NMR spectrum. Beyond structural distinctions, divergent packing arrangements were clearly observed in these molecular single crystals. R1-TB-R2-CHM-1 showed a parallel stacking, whereas the R1-TB-S2-CHM-1 formed an alternating structure with a 90° rotation between the two layers, explaining the difference in the single-crystal morphology of these two diastereoisomers.
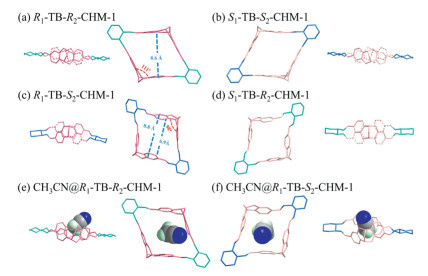
Despite morphological variations, both sets of diastereomeric macrocycles possessed similarly sized cavities, and acetonitrile was intriguingly found to penetrate the macrocyclic crystals, signifying that all of the macrocycles have an electronegative cavity which was large enough to accommodate the acetonitrile molecule (Figs. 4e and f). Surface electrostatic potential and Independent Gradient Model based on Hirshfeld partition (IGMH) analysis were performed to substantiate our deduction [44-46]. The structural diagrams of acetonitrile containing macrocycles showed that there are multiple interactions between the two species, including CH-π interactions and electrostatic interactions (Figs. S31 and S32 in Supporting information).
Additionally, compared to those of R1-TB-R2-CHM-1 and S1-TB-S2-CHM-1, the crystals of R1-TB-S2-CHM-1 and S1-TB-R2-CHM-1 were small (~100 µm), suspended and aggregated like cotton wool. Slight perturbation or prolonged immersion would result in a remarkable change in morphology of the crystals, fine crystals merged to form larger crystals up to 2 mm in 10 h (Figs. S29 and S30 in Supporting information). X-ray diffraction exhibited that the stacking in the R1-TB-S2-CHM-1 crystals was altered, with one layer of molecules flipping over by 90° to be perpendicular to the other layer, forming the "parallel-perpendicular-parallel stacking". This increased the distance between layers, expanding the size of the crystal. Surface electrostatic potential of the R1-TB-S2-CHM-1 showed that the TB skeleton is rich in electrons, and the imine bond possess positive charge (Fig. S33 in Supporting information). Such charge-complementary arrangement stabilized the crystals, resulting in the formation of larger dimensions.
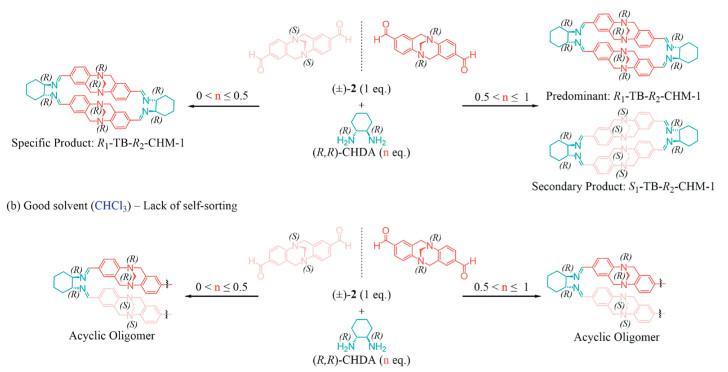
When carrying out reactions between racemic building blocks, chiral self-sorting emerged, and the formation of macrocycles could be controlled by adjusting the feed ratio and the reaction solvent. In the case of reactions between 1 equiv. of (±)-2 and R,R-CHDA in CH3CN/CH3OH (Scheme 2a), crystals of two different-morphologies would appear in the system, with an approximately equal proportion between the two isomers (1:1). These crystals could be separated via vibration screening method, perfectly matched with the two macrocyclic compounds: R1-TB-R2-CHM-1 and R1-TB-S2-CHM-1 respectively. It is interesting that reducing CHDA below 0.5 equiv. would exclusively yield the crystalline product R1-TB-R2-CHM-1. When the amount of CHDA increases to between 0.5 equiv. and 1 equiv., R1-TB-R2-CHM-1 become the predominant product, while R1-TB-S2-CHM-1 formation increases proportionally with CHDA concentration. This trend was unequivocally confirmed by 1H NMR analysis, showing a continuous increase in the imine proton signal intensity corresponding to R1-TB-S2-CHM-1 (Figs. S14-S17 in Supporting information). This phenomenon of chiral self-sorting suggested that the macrocycle R1-TB-R2-CHM-1 was the thermodynamically favoured product in this system.
To validate this deduction, minimized energy structure and single-point energy of these diastereoisomers were calculated based on density functional theory (DFT). In comparison with the crystal structure, only one of the TB skeletons was stretched in the theoretical calculation structure of R1-TB-R2-CHM-1 and the structure was kite shaped, while the R1-TB-S2-CHM-1 was similar to the actual crystal structure. Calculations of the relative energies of the two diastereoisomers were performed at the B3LYP/6–31G(d) level. The result showed that R1-TB-R2-CHM-1 exhibited lower energy than R1-TB-S2-CHM-1 (ΔE = −1.24 kcal/mol). The superior thermodynamic stability enabled the preferential reaction of R,R-2 and R,R-CHDA in reaction system, forming R1-TB-R2-CHM-1. This product crystallized and precipitated due to solubility changes. Following depletion of R,R-2, R1-TB-S2-CHM-1 begins to form, triggering chiral self-sorting in the crystalline state (Figs. S34-S36 and Table S6 in Supporting information).
However, this chiral self-sorting phenomenon was not observed for reactions of racemic TB with R,R- or S,S-CHDA in CDCl3 which was a good solvent for the macrocycles (Scheme 2b). 1H NMR spectra showed that protons of the TB skeleton experienced multiple spin-spin splitting, regarded as an asymmetric TB [47], suggesting the formation of oligomers rather than macrocycles (Fig. S18 in Supporting information). For comparison purposes, enantiomerically pure 2 was treated with CHDA in CDCl3, yielding macrocyclic compounds, as evidenced by ¹H NMR analysis, along with incompletely cyclized intermediates which could not be purified due to their imine bonds (Fig. S17 in Supporting information).
This solvent-dependent chiral self-sorting phenomenon was the result of the dynamic reversibility of the imine bonds in the macrocyclization process. In a poor solvent, the formed macrocycles crystallized, where the packing amplified the energy difference, contributing to preferential formation of the more stable macrocycle. Once one configuration of 2 was completely consumed, the other macrocycle naturally formed. The lack of precipitation in good solvents wherein the two enantiomers od 2 underwent simultaneously imine condensation reactions with R,R-CHDA during the macrocyclization process, caused insufficient energy disparities for achieving chiral self-sorting.
Although the imine bond is prone to hydrolysis, R1-TB-R2-CHM-1 and S1-TB-S2-CHM-1 demonstrate excellent stability (Table S5 in Supporting information). The single crystals of these macrocycles exhibited great structural stability even after immersing in water or 1 mol/L NaOH over 1 week at room temperature. It was worth noting that even when soaked in a weak acid, the crystals remained unchanged. Only when exposed to acidic solutions with pH ≤ 2, hydrolysis of the macrocycles began. Mixing crystals of R1-TB-R2-CHM-1 with a HCl solution (pH 2), the solution turned green immediately. As monitored by 1H NMR, less than 10% of macrocycles were hydrolysed within the first 1 h, while approximately 70% was hydrolysed in 24 h, and the process was completed within 48 h. For R1-TB-S2-CHM-1, the hydrolysis was very rapid and completed almost at the moment of mixing. Even when exposed to atmospheric moisture (40%−50% relative humidity, RH), hydrolysis occurred (Figs. S24-S26 in Supporting information). According to crystallographic and theoretical analysis, we propose that R1-TB-R2-CHM-1 exhibited significantly greater hydrolytic stability than R1-TB-S2-CHM-1, owing to its close parallel packing (which restricts water access), shorter imine bonds, and higher thermodynamic stability.
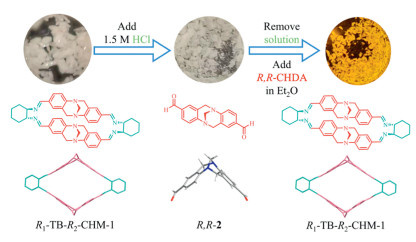
On this basis, the solid-state conversion R1-TB-R2-CHM-1 to its precursor was firstly investigated (Fig. 5). After placing the macrocycle crystals in HCl (pH 1.5) for 3 h, the crystals underwent a change, transitioning from being colourless to white while diminishing in thickness. Fortunately, the resultant crystals maintained diffraction grade. X-ray diffraction proved that the new crystal was building block R,R-2 (Figs. S27 and S28 in Supporting information). Although the crystals gradually cracked into powder as the immersion time extended, most of the crystals were able to remain intact. After moving the obtained crystals into an ether solution of R,R-CHDA and gentle heating to about 30 ℃, the crystals transformed back to R1-TB-R2-CHM-1 within 5 h, with a transformation yield of 90% evidenced by 1H NMR. Heating 12 h could complete the transformation of all 2, constituting a macrocycle-to-precursor transformation cycle. The re-obtained macrocycle crystals could be subjected again to the above cycle. No matter how the hydrolysis conditions were adjusted, neither R1-TB-S2-CHM-1 nor S1-TB-R2-CHM-1 could achieve crystal retention. Also, the transformation from 2 to these macrocycles in the crystalline state was not achieved, and the aldehyde only partially reacts to produce imine, without any macrocycle formation. Comparing the structures of the diastereoisomers and the results of theoretical calculations (Figs. S22 and S34 in Supporting information), it was speculated that interleaved TB units are detrimental to the stabilization of molecules and crystals.
In conclusion, this work affords an efficient synthesis that delivers gram-scale quantities of novel, crystalline supramolecular hosts directly from the reaction mixture, representing the first imine-linked Tröger's base-based macrocycles (TBCHMs). This access enables precise modulation of macrocycle chirality and cavity size. Notably, leveraging the inherent properties of the TB skeleton, chiral self-sorting was achieved in acetonitrile/methanol. Furthermore, system expansion and functionalization/recognition studies are planned for future work. Hence, this work establishes a robust platform for constructing supramolecular hosts based on dynamic covalent chemistry (DCC) and Tröger's base. These macrocycles would hold significant promise for applications in chiral recognition, asymmetric catalysis, circularly polarized luminescence, and related fields.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Heng Qiu: Writing – original draft, Investigation, Formal analysis, Data curation, Conceptualization. Wang Xie: Formal analysis. Mingxia Ye: Investigation. Conghao Shi: Methodology. Juli Jiang: Writing – review & editing. Zhouyu Wang: Writing – review & editing. Wim Dehaen: Writing – review & editing. Leyong Wang: Writing – review & editing, Funding acquisition, Conceptualization.
We gratefully thank the financial support of the Innovation Support Program of Jiangsu Province (No. BZ2023055), and the National Natural Science Foundation of China (No. 22471124), and the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (No. SN-ZJU-SIAS-006).
Supplementary material associated with this article can be found, in the online version, at doi:
P. Cintas, Angew. Chem. Int. Ed. 41 (2002) 1139–1145. doi: 10.1002/1521-3773(20020402)41:7<1139::AID-ANIE1139>3.0.CO;2-9
H. Shigemitsu, T. Fujisaku, W. Tanaka, et al., Nat. Nanotechnol. 13 (2018) 165–172. doi: 10.1038/s41565-017-0026-6
S. He, Z. Jiang, X. Dou, et al., ChemPlusChem 88 (2023) e202300226. doi: 10.1002/cplu.202300226
P. Peluso, B. Chankvetadze, Chem. Rev. 122 (2022) 13235–13400. doi: 10.1021/acs.chemrev.1c00846
G. Liu, M.G. Humphrey, C. Zhang, Y. Chao, Chem. Soc. Rev. 52 (2023) 4443–4487. doi: 10.1039/d2cs00476c
D.Y. Zhang, Y. Sang, T.K. Das, et al., J. Am. Chem. Soc. 145 (2023) 26791–26798. doi: 10.1021/jacs.3c08966
Q. Zhou, X. Dong, G. Chi, et al., J. Am. Chem. Soc. 146 (2024) 22405–22412. doi: 10.1021/jacs.4c05376
Z. Sun, H. Tang, L. Wang, D. Cao, Chem. Eur. J. 31 (2024) e202404217.
Y. Yu, Y. Hu, C. Ning, et al., Angew. Chem. Int. Ed. 63 (2024) e202407034. doi: 10.1002/anie.202407034
S. Bleus, W. Dehaen, Coord. Chem. Rev. 509 (2024) 215762. doi: 10.1016/j.ccr.2024.215762
Y. Wang, J. Gong, X. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202210542. doi: 10.1002/anie.202210542
Z. Yang, K. Fu, W. Yu, et al., Chin. Chem. Lett. 36 (2025) 110842. doi: 10.1016/j.cclet.2025.110842
N. Liu, X.N. Han, H. Ma, et al., Org. Lett. 26 (2024) 7239–7243. doi: 10.1021/acs.orglett.4c02709
H. Li, Z. Li, J. Jiang, et al., Org. Chem. Front. 11 (2024) 6358–6366. doi: 10.1039/d4qo01498g
H. Li, Z. Li, C. Lin, et al., Nat. Commun. 15 (2024) 5315. doi: 10.1038/s41467-024-49540-2
Y. Mao, Y. Li, S. Davis, L. Pu, Eur. J. Org. Chem. 28 (2025) e202401250. doi: 10.1002/ejoc.202401250
X. Zhang, Z. Lu, T. Ren, et al., Chin. Chem. Lett. 36 (2025) 110946. doi: 10.1016/j.cclet.2025.110946
L. Yue, C. Gao, J. Li, et al., Adv. Mater. 35 (2023) e2211626. doi: 10.1002/adma.202211626
C.D. Zhao, H. Yao, S.Y. Li, et al., Chin. Chem. Lett. 35 (2024) 108879. doi: 10.1016/j.cclet.2023.108879
T. Pausch, S. Clopot, D.N. Jordan, et al., Angew. Chem. Int. Ed. 63 (2024) e202418877. doi: 10.1002/anie.202418877
F.B.L. Cougnon, A.R. Stefankiewicz, S. Ulrich, Chem. Sci. 15 (2024) 879–895. doi: 10.1039/d3sc05343a
J.M. Lehn, Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 4763–4768.
M.E. Belowich, J.F. Stoddart, Chem. Soc. Rev. 41 (2012) 2003–2024. doi: 10.1039/c2cs15305j
C. Qian, L. Feng, W.L. Teo, et al., Nat. Rev. Chem. 6 (2022) 881–898. doi: 10.1038/s41570-022-00437-y
J. Li, S.S. Hii, X. Zhu, et al., Chin. Chem. Lett. (2026) 111151. doi: 10.1016/j.cclet.2025.111151
C. Ge, W. Shang, Z. Chen, et al., Angew. Chem. Int. Ed. 63 (2024) e202408056. doi: 10.1002/anie.202408056
M. Ovalle, C.N. Stindt, B.L. Feringa, J. Am. Chem. Soc. 146 (2024) 31892–31900. doi: 10.1021/jacs.4c11206
Y. Lei, Q. Chen, P. Liu, et al., Angew. Chem. Int. Ed. 60 (2021) 4705–4711. doi: 10.1002/anie.202013045
M.A. Spielman, J. Am. Chem. Soc. 57 (1935) 583–585. doi: 10.1021/ja01306a060
J. Tröger, J. Prakt. Chem. 36 (1887) 225–245. doi: 10.1002/prac.18870360123
E.C. Wagner, J. Am. Chem. Soc. 57 (1935) 1296–1298. doi: 10.1021/ja01310a037
E. Benchimol, H.M. O'Connor, B. Schmidt, et al., Angew. Chem. Int. Ed. 64 (2024) e202421137.
M. Carta, M. Croad, J.C. Jansen, et al., Polym. Chem. 5 (2014) 5255. doi: 10.1039/C4PY00607K
S. Shanmugaraju, C. Dabadie, K. Byrne, et al., Chem. Sci. 8 (2017) 1535–1546. doi: 10.1039/C6SC04367D
C. Shi, G. Xu, H. Qiu, et al., Chem. Commun. 61 (2025) 2450–2467. doi: 10.1039/d4cc05134c
Ö.V. Rúnarsson, J. Artacho, K. Wärnmark, Eur. J. Org. Chem. 2012 (2012) 7015–7041. doi: 10.1002/ejoc.201201249
H. Feng, Y. Chen, R. Wang, et al., Chin. Chem. Lett. 34 (2023) 108038. doi: 10.1016/j.cclet.2022.108038
P. Niu, C. Shi, J. Jiao, et al., Chem. Commun. 59 (2023) 10960–10963. doi: 10.1039/d3cc02804f
X. Liang, T. Zhao, Y. Shen, et al., Angew. Chem. Int. Ed. 64 (2025) e202416975. doi: 10.1002/anie.202416975
D.L. Jameson, T. Field, M.R. Schmidt, et al., J. Org. Chem. 78 (2013) 11590–11596. doi: 10.1021/jo401873x
Z.Q. Liang, X.M. Wang, Y.Y. Zhou, et al., Asian J. Chem. 26 (2014) 1781–1785. doi: 10.14233/ajchem.2014.17354
Y.H. Chu, Y. Wan, Z. Liu, et al., Tetrahedron Lett. 56 (2015) 7046–7052. doi: 10.1016/j.tetlet.2015.11.024
M.A. Spielman, J. Am. Chem. Soc. 57 (2002) 583–585.
T. Lu, J. Chem. Phys. 161 (2024) 082503. doi: 10.1063/5.0216272
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580–592. doi: 10.1002/jcc.22885
T. Lu, Q. Chen, J. Comput. Chem. 43 (2022) 539–555. doi: 10.1002/jcc.26812
S. Sergeyev, D. Didier, V. Boitsov, et al., Chem. Eur. J. 16 (2010) 8181–8190. doi: 10.1002/chem.201000216

Figure 1 Schematic diagram of the TB-based macrocycles: (a) The structure of Tröger's base. (b) The formation of imine bond. (c) TB-based macrocycles.

Figure 2 1H NMR (400 MHz, 298 K) spectra of R1-TB-R2-CHM-1 (red), R1-TB-S2-CHM-1 (green) and (±)-2 (blue) in CDCl3.

Figure 4 Crystal structure side view and top view of (a) R1-TB-R2-CHM-1, (b) S1-TB-S2-CHM-1, (c) R1-TB-S2-CHM-1, (d) S1-TB-R2-CHM-1, (e) CH3CN@R1-TB-R2-CHM-1, and (f) CH3CN@R1-TB-S2-CHM-1.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:




 下载:
下载: