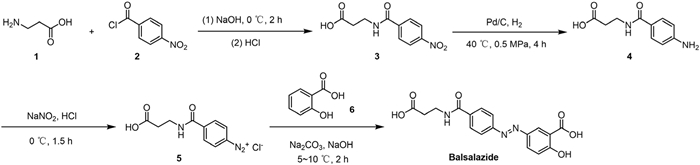
Scheme 1.
The synthetic route of balsalazide in batch method.
A continuous-flow process for the synthesis of balsalazide disodium
Zhen Zhang , Rui Su , Xiongjun Xiao , Jiaxin Yang , Yuwei Li , Tiemin Sun , Yang Liu , Chengjun Wu

Ulcerative colitis (UC) is a lifelong inflammatory disease with an increasing prevalence worldwide [1,2]. Balsalazide disodium is a 5-amino-salicylic acid (5-ASA) prodrug that is broken down in the intestine by azoreductase to produce 5-ASA [3]. 5-ASA alleviates inflammation by blocking the production of arachidonic acid metabolites in the colon [4]. Balsalazide disodium was developed by Astra Corporation and was first approved to improve mildly to moderately active UC in the United Kingdom in 1997 [5]. Compared to other 5-ASA drugs, balsalazide disodium has high efficacy, low toxicity, and fewer adverse effects [5].
The reported synthetic routes all start from β-alanine and sequentially proceed through condensation, reduction, diazotization and azocoupling reactions to obtain balsalazide (Scheme 1) [6,7]. These routes have several disadvantages, including unavoidable side reactions (such as hydrolysis), a large amount of waste liquids, and low yield and conversion.
Microreactors have the advantages of fast heat exchange, high reaction efficiency and enhanced safety, making them suitable for the preparation of azo compounds [8–11]. High reaction efficiency and precise temperature management keep the diazonium salt stable under milder conditions, which is conducive to the subsequent coupling reaction [12,13]. In the micro-packed bed reactor, the continuous hydrogenation reaction significantly improves the selectivity and efficiency of the process [14–18]. Response surface methodology (RSM) is an effective method for process optimization and has been applied in various fields such as dye adsorption, pharmaceuticals, and materials [19–22]. With the aid of statistical modeling, RSM identifies the optimal process parameters with fewer experiments [23,24]. Therefore, we applied flow chemistry to the synthesis of balsalazide and developed a continuous route for industrial production.
The water-only solubility of 3-aminopropanoic acid makes the preparation of 3 prone to hydrolysis of the chloride. In the microchannel, its weak nucleophilicity amplified the formation of p-nitrobenzoic acid (9). Therefore, we tried to replace the carboxyl group with a methyl ester group to provide a stronger nucleophilic ability for the amino group (Scheme 2).
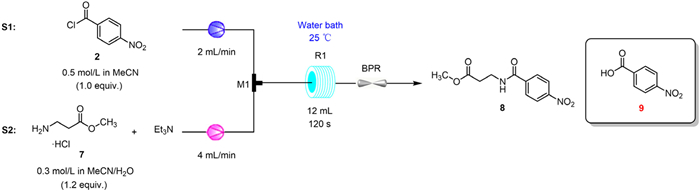
In mixed solvents, organic bases are used to free methyl 3-aminopropanoate hydrochloride and catalyze condensation reactions as deacidifying agents. When pyridine was used as deacidifying agent, the selectivity of 8 was only 77.3% in a residence time of 60 s (Table 1, entry 1). When increasing the alkalinity of the deacidifying agent, both Et3N and DIPEA provided over 99% selectivity for intermediate 8. Compared to DIPEA, the inexpensive Et3N offered a higher isolated yield (Table 1, entries 2 and 3). In addition, we found that the ratio of MeCN to H2O in the mixed solvent had little effect on selectivity. The substrates were well mixed in MeCN without significant side reactions (Table 1, entries 4 and 5). When MeCN was replaced with acetone, a large solid precipitate formed, leading to clogging of the line (Table 1, entry 6). Although THF also provided good selectivity, significant delamination of the solution was observed after the reaction. The lower reaction yields may have been caused by poorer mixing within the microchannels (Table 1, entry 7). Additionally, we tried to adjust the retention time and reaction temperature to improve the reaction yield. Reaction yields showed an increasing trend with increasing residence time (Table 1, entries 8-11). When the reaction temperature was in the range of 10–40 ℃, the reaction yield remained relatively stable (Table 1, entries 12 and 13). Finally, the optimal conditions for step 1 were determined as follows: reaction temperature of 25 ℃, residence time of 120 s, and 2.20 equiv. of Et3N with a selectivity of 99.7%.
 DownLoad:
CSV
DownLoad:
CSV
| Entry | Solvent | Base | T (℃) | tR1 (s) | 9 (%)b | 8 (%)b | Yield (%)c |
| 1 | 1:1 | Pyridine | 25 | 60 | 20.8 | 77.3 | 61.5 |
| 2 | 1:1 | Et3N | 25 | 60 | <0.1 | >99 | 94.0 |
| 3 | 1:1 | DIPEA | 25 | 60 | <0.1 | >99 | 92.7 |
| 4 | 2:1 | Et3N | 25 | 60 | <0.1 | >99 | 94.9 |
| 5 | 3:1 | Et3N | 25 | 60 | <0.1 | >99 | 93.9 |
| 6d | 1:1 | Et3N | 25 | 60 | − | − | − |
| 7e | 1:1 | Et3N | 25 | 60 | <0.1 | >99 | 83.7 |
| 8 | 1:1 | Et3N | 25 | 40 | <0.1 | >99 | 75.9 |
| 9 | 1:1 | Et3N | 25 | 90 | <0.1 | >99 | 92.8 |
| 10 | 1:1 | Et3N | 25 | 120 | <0.1 | >99 | 95.8 |
| 11 | 1:1 | Et3N | 25 | 150 | <0.1 | >99 | 91.6 |
| 12 | 1:1 | Et3N | 10 | 120 | <0.1 | >99 | 92.9 |
| 13 | 1:1 | Et3N | 40 | 120 | <0.1 | >99 | 93.5 |
| a Reaction conditions: S1: 0.5 mol/L in MeCN; S2: 0.3 mol/L in MeCN/H2O. b The content was determined by HPLC. c Isolated yield. d Acetone as organic solvent. e THF as organic solvent. | |||||||
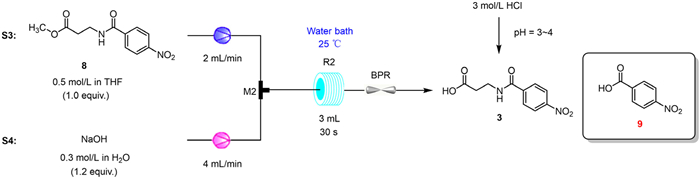
Subsequently, we systematically investigated the continuous process of methyl ester hydrolysis (Scheme 3). We screened three process parameters: reaction temperature, residence time, and equivalents of NaOH. At a reaction temperature of 40 ℃ and 2.5 equiv. of NaOH, we observed the generation of the hydrolysis product for the amide (Table 2, entry 1). This phenomenon was not found in the batch experiments. In general, hydrolysis of the amide bond is more difficult than that of the ester bond; however, high temperature and strong alkalinity may facilitate it. To avoid the side reaction, we reduced the reaction temperature and the equivalents of NaOH (Table 2, entries 2-6). The experimental results showed that the milder reaction conditions sufficiently inhibited the hydrolysis reaction. As the equivalent of NaOH was reduced to 1.0, HPLC detected a small amount of intermediate 8 remaining (Table 2, entry 5). This reveals the incompleteness of the continuous reaction. Compared to 25 ℃, increasing the reaction temperature showed no obvious advantages (Table 2, entries 7 and 8). Changes in residence time by 30 s had no effect on the selectivity of 3, but the isolated yield declined (Table 2, entries 9 and 10). Furthermore, the use of other solvents for the reaction would result in the introduction of unknown impurities or clogging of the pipelines (Table 2, entries 11-13). Therefore, the optimal conditions for step 2 were determined, as follows: reaction temperature of 25 ℃, residence time of 30 s, and 1.2 equiv. of NaOH with a selectivity of 99.6%.
 DownLoad:
CSV
DownLoad:
CSV
| Entry | Solvent | NaOH (equiv.) | T (℃) | tR2 (s) | 9 (%)b | 3 (%)b | Yield (%)c |
| 1 | THF | 2.5 | 40 | 30 | 7.3 | 91.2 | 76.8 |
| 2 | THF | 2.0 | 25 | 30 | 1.9 | 97.4 | 84.7 |
| 3 | THF | 1.5 | 25 | 30 | <0.1 | >99 | 85.3 |
| 4 | THF | 1.2 | 25 | 30 | <0.1 | >99 | 92.4 |
| 5 | THF | 1.0 | 25 | 30 | <0.1 | 95.6 | 86.1 |
| 6 | THF | 0.8 | 25 | 30 | <0.1 | 76.8 | 81.2 |
| 7 | THF | 1.2 | 40 | 30 | <0.1 | >99 | 91.7 |
| 8 | THF | 1.2 | 10 | 30 | <0.1 | >99 | 83.7 |
| 9 | THF | 1.2 | 25 | 60 | <0.1 | >99 | 90.9 |
| 10 | THF | 1.2 | 25 | 20 | <0.1 | >99 | 71.3 |
| 11 | MeOH | 1.2 | 25 | 30 | − | − | − |
| 12 | MeCN | 1.2 | 25 | 30 | − | − | − |
| 13 | Acetone | 1.2 | 25 | 30 | <0.1 | 69.3 | 75.6 |
| a Reaction conditions: S3: 0.5 mol/L in THF; S4: 0.3 mol/L in H2O. b The content was determined by HPLC. c Isolated yield. | |||||||
In the continuous hydrogenation of 3, we systematically investigated four factors: Gas flow rate, temperature, gas pressure, and liquid flow rate (Scheme 4). The gas flow rate may enhance gas‒liquid mass transfer, which in turn affects the conversion and selectivity of the product [14]. Therefore, gradient experiments at different gas flow rates were performed (Fig. 1a). When the hydrogen flow rate was gradually increased to 30 mL/min, the conversion of 3 increased to 98.4%. A higher gas flow rate might shorten the residence time and lead to incomplete reactions. Therefore, a gas flow rate of 30 mL/min was selected for subsequent conditions screening.

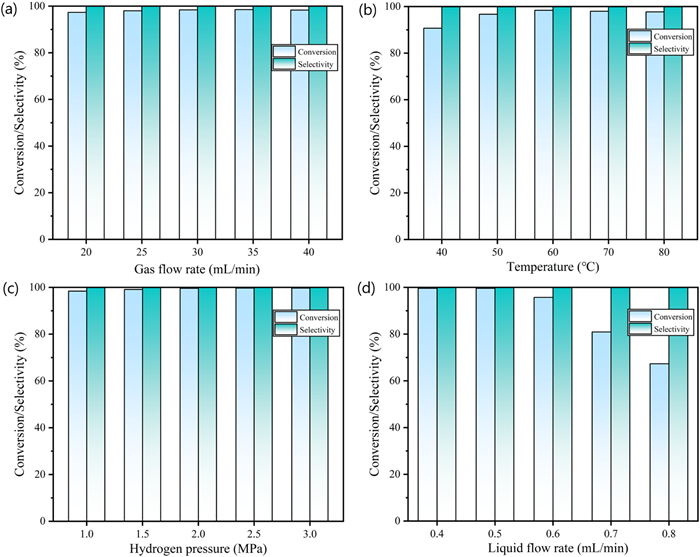
Too low a reaction temperature may lead to insufficient reduction while too high a temperature may increase impurity content. Therefore, we investigated the effect of temperature on the reduction activity under a gas pressure of 1.0 MPa, a liquid flow rate of 0.4 mL/min, and a gas flow rate of 30 mL/min. The conversion of 3 increased with rising temperature, reaching a maximum at 60 ℃ (Fig. 1b). No significant impurities were detected. Hydrogen pressure is another important factor affecting the hydrogenation of intermediate 3 to intermediate 4 via Raney Ni in the micro-hydrogenation reactor. In the range of 1.0–3.0 MPa, the effect of hydrogen pressure on the hydrogenation reaction was carefully investigated (Fig. 1c). Within that range, the selectivity of intermediate 4 remained at almost 100%. The conversion of intermediate 3 gradually increased with hydrogen pressure and reached a maximum at 2.0 MPa. The conversion has been increased to 99.6%. When the hydrogen pressure was above 2.0 MPa, the conversion of the reaction no longer changed significantly, even with further increases in pressure. To save energy, the hydrogen pressure was determined to be 2.0 MPa.
Variations in liquid flow rate significantly affected the residence time of the reaction which influences the completion of the reduction. When the residence time was too short, the feedstock flowed out of the reactor without being sufficiently reduced by the hydrogen on the catalyst, resulting in lower conversion. However, excessive residence time would reduce productivity and increase impurity content. As shown in Fig. 1d, the conversion of 3 was higher than 99% at a liquid flow rate of 0.5 mL/min. Higher liquid flow rates significantly reduced the residence time, resulting in a substantial decrease in the conversion of 3. To improve the conversion and productivity, 0.5 mL/min was determined to be the optimal condition. Based on the above experimental results, the optimal process parameters for the continuous hydrogenation were determined as follows: gas flow rate of 30 mL/min, reaction temperature of 60 ℃, hydrogen pressure of 2.0 MPa, and liquid flow rate of 0.5 mL/min. At a concentration of 0.2 mol/L, the isolated yield of intermediate 4 was 95.6% with a purity of 99.3%.
The diazonium salt of balsalazide had good stability and was not easily decomposed at 0–5 ℃. However, it became turbid with poor solubility when left for a long time. A small amount of solid precipitation was observed, which could lead to the embedding of some reactants. We attempted to improve this phenomenon by regulating the temperature. The generation of suspended solids significantly decreased at 10 ℃, and the reaction solution became clear and yellow at 15 ℃. However, a deeper color of the solution was observed at 30 ℃ and a few bubbles were produced. These results suggest that elevated temperatures can accelerate the rate of diazotization and improve solubility, but with a risk of deterioration.
Given the good stability of diazonium salts at 15 ℃, we screened the types and equivalents of acids and diazotization reagents (Scheme 5). Protonic acid plays the role of dissolving the feedstock, catalyzing the reaction, and maintaining the stability of the diazonium salt during diazotization. In terms of chemical equilibrium, diazotization requires only 2.0 equiv. of protons; however, at least 3.0 equiv. are often needed in actual reactions. The equivalent of acid was chosen to be 3.2 (in terms of H+) to examine the commonly used acids (Table 3, entries 1-4). During the examination of the equivalents of NaNO2, we found that diazotization must be carried out with a slight excess of nitrite (Table 3, entry 5 and 6). Excessive amounts may induce a self-coupling reaction and affect the selectivity of the azo reaction. Under identical conditions, isopentyl nitrite provided a lower isolated yield than NaNO2. The economic costs of large-scale production are relatively high (Table 3, entries 7-9). Therefore, 3.2 equiv. of HCl and 1.05 equiv. of NaNO2 were chosen as the optimal conditions.
 DownLoad:
CSV
DownLoad:
CSV
| Entry | Acid (equiv.) | Diazotization reagent | Reagent (equiv.) | Yield (%)b |
| 1 | HCl (3.0) | NaNO2 | 1.05 | 82.5 |
| 2 | HCl (3.2) | NaNO2 | 1.05 | 88.2 |
| 3 | HCl (3.4) | NaNO2 | 1.05 | 86.7 |
| 4 | H2SO4 (1.6) | NaNO2 | 1.05 | 83.8 |
| 5 | HCl (3.2) | NaNO2 | 1.00 | 78.9 |
| 6 | HCl (3.2) | NaNO2 | 1.10 | 87.0 |
| 7 | HCl (3.2) | Isoamyl nitrite | 1.05 | 82.3 |
| 8 | HCl (3.2) | Isoamyl nitrite | 1.10 | 80.4 |
| 9 | H2SO4 (1.6) | Isoamyl nitrite | 1.05 | 79.7 |
| a Reaction conditions: S6: 0.2 mol/L in H2O; S8: 0.2 mol/L in H2O. tR3: 35 s; tR4: 41 s; reaction temperature: 15 ℃. b Isolated yield. | ||||
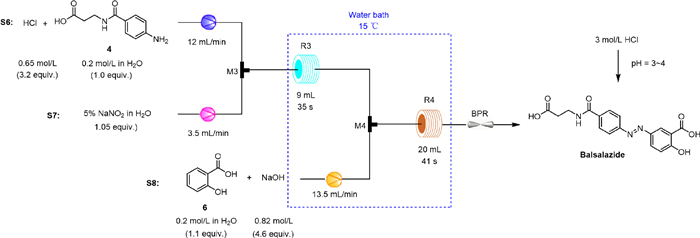
To ensure that the diazonium salt was fully reacted, at least 1.05 equiv. of salicylic acid needed to be added. In the batch reaction, only part of the base was used to prepare the salicylic acid solution. The remaining base was added in batches to maintain a stable pH during the azo reaction. The addition of mixed bases enabled the formation of a buffer system of
Although microchannels allowed for a good azo coupling reaction, a new problem was identified during continuous experiments. The ratio between the two reaction fluids in the T-mixer is often unknown before the reaction system stabilizes. When the acidic solution of diazonium salt met the basic solution of salicylic acid, there is a possibility that an excess of acid results in the conversion of
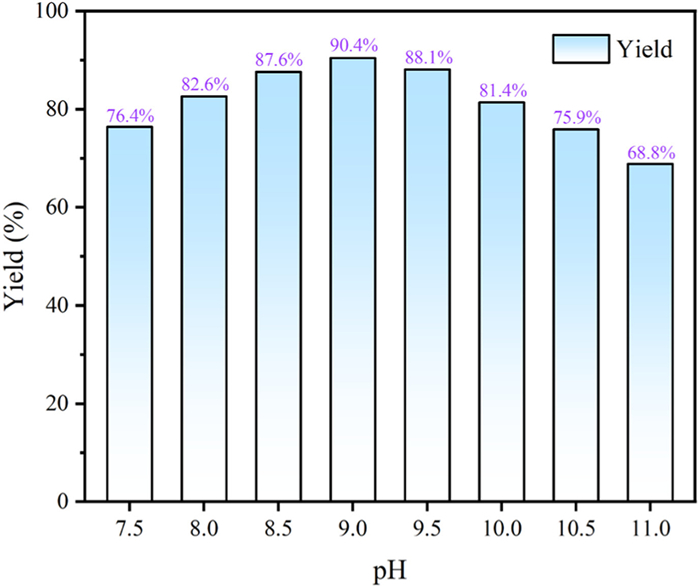
The yield of balsalazide gradually increased with the equivalents of NaOH in the range of 4.1–4.6 (Table 4, entries 1-3). Using 4.6 equiv. of NaOH maintained the pH of the reaction at 9.2, providing an optimal catalytic effect. The activity of the azo-coupling reaction gradually decreased with further increase in the equivalents of NaOH (Table 4, entries 4-6). When the equivalents were increased to 5.5, the pH of the environment reached 11.3, and significant amounts of large polar impurities were observed. The properties of the crude product gradually changed from yellow to reddish brown as the pH increased from 9.2 to 11.3. Furthermore, a certain excess of salicylic acid facilitated the complete conversion of the amines but could be introduced as an impurity at 1.2 equiv. (Table 4, entries 7-9). Finally, the equivalents of NaOH and salicylic acid were determined to be 4.6 and 1.1, respectively, with a selectivity of 96.59%.
DownLoad:
CSV
| Entry | NaOH (equiv.) | Salicylic acid (equiv.) | Yield (%)b |
| 1 | 4.1 | 1.05 | 71.8 |
| 2 | 4.4 | 1.05 | 85.5 |
| 3 | 4.6 | 1.05 | 87.1 |
| 4 | 4.9 | 1.05 | 83.6 |
| 5 | 5.2 | 1.05 | 69.4 |
| 6 | 5.5 | 1.05 | 64.5 |
| 7 | 4.6 | 1.00 | 84.5 |
| 8 | 4.6 | 1.10 | 89.2 |
| 9 | 4.6 | 1.20 | 70.9 |
| a Reaction conditions: S6: 0.2 mol/L in H2O; S8: 0.2 mol/L in H2O; tR3: 35 s; tR4: 41 s; reaction temperature: 15 ℃. b Isolated yield. | |||
In the entire continuous synthesis, the diazotization and azo coupling reactions were controlled within a single temperature zone. Regulating the temperature and residence time within the microchannel may reduce the decomposition of the diazonium salt and the formation of the 3-site coupling impurity of salicylic acid. In the range of 10–30 ℃, the reaction temperature showed a weak effect on yield (Table 5, entries 1-5). We obtained an isolation yield of 91.6% at a reaction temperature of 20 ℃, with a tR3 of 35 s, and a tR4 of 50 s (Table 5, entry 3). In addition, an appropriate increase in the residence time was beneficial for improving the selectivity and yield of balsalazide (Table 5, entries 6-12). Shortening the residence time resulted in the reaction mixture flowing out of the reactor without complete conversion. However, a significant change in yield was observed when tR4 was elevated to 70 s (Table 5, entry 12). Therefore, the process parameters for step 4 were adjusted as follows: reaction temperature of 20 ℃, tR3 of 45 s, and tR4 of 55 s with a selectivity of 96.6%.
DownLoad:
CSV
| Entry | T (℃) | tR3 (s) | tR4 (s) | Yield (%)a |
| 1 | 10 | 35 | 50 | 89.3 |
| 2 | 15 | 35 | 50 | 90.7 |
| 3 | 20 | 35 | 50 | 91.6 |
| 4 | 25 | 35 | 50 | 88.9 |
| 5 | 30 | 35 | 50 | 84.7 |
| 6 | 20 | 25 | 40 | 75.6 |
| 7 | 20 | 40 | 50 | 91.2 |
| 8 | 20 | 45 | 50 | 91.9 |
| 9 | 20 | 50 | 50 | 89.4 |
| 10 | 20 | 45 | 55 | 92.5 |
| 11 | 20 | 45 | 60 | 90.5 |
| 12 | 20 | 45 | 70 | 84.9 |
| a Isolated yield. | ||||
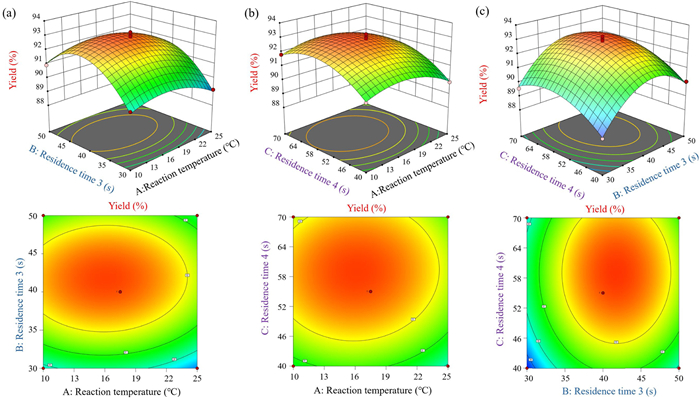
To further investigate the optimal conditions for the continuous synthesis of balsalazide, we carried out a response surface analysis for the continuous process with yield as the response value (Y) and reaction temperature (A), residence time of diazotization (B), and residence time of the azo-coupling reaction (C) as the independent variables.
Fig. 3 illustrate the interactive effects of temperature, diazotization time, and azo-coupling time on yield. An increased residence time for both reactions at 15–25 ℃, significantly enhanced the yield (Figs. 3a and b). However, a decline in yield caused by a decrease in the stability of the diazonium salt was observed above 25 ℃. At a fixed diazotization time of 35–45 s, there is a parabolic trend in yield (Fig. 3c). When the reaction temperature of 18.2 ℃, diazotization time of 41.7 s, and azo coupling time of 58.9 s were employed, the model predicted a maximum yield of 93.0%. Experimental validation at 18.2 ℃ with adjusted times confirmed these results (diazotization: 42 s; azo coupling reaction: 59 s). Prolonged reaction durations improve completeness, but excessive temperatures may destabilize the intermediate. Based on the response surface analysis, we obtained the optimized reaction conditions. To further verify the experimental results and determine the optimal reaction yield, three experiments were conducted under these conditions (Table 6). The experimental results were close to the predicted yield with low deviation. The maximum yield of the reaction increased to 93.4% under optimal conditions.
 DownLoad:
CSV
DownLoad:
CSV
| Entry | T (℃) | tR3 (s) | tR4 (s) | Balsalazide (%)b | Yield (%)c |
| 1 | 18.2 | 42 | 59 | 97.9 | 93.4 |
| 2 | 18.2 | 42 | 59 | 97.7 | 93.2 |
| 3 | 18.2 | 42 | 59 | 97.9 | 92.7 |
| a Optimal reaction conditions predicted by RSM. b The purity of crude product was determined by HPLC. c Isolated yield. | |||||
In summary, a stable and effective method for the preparation of balsalazide disodium was developed using an innovative combination of continuous flow chemistry and response surface analysis. Compared to the traditional batch method, the method presented in this work significantly improves the safety and efficiency of the process, reduces side reactions, and is more suitable for industrial production. The total yield is 66.1%, and the purity of balsalazide disodium is 99.63%.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Zhen Zhang: Writing – original draft. Rui Su: Data curation. Xiongjun Xiao: Software. Jiaxin Yang: Methodology. Yuwei Li: Validation. Tiemin Sun: Writing – review & editing. Yang Liu: Writing – review & editing, Funding acquisition. Chengjun Wu: Writing – review & editing.
We thank the Education Department of Liaoning (No. 2020LJC06). We are also gratitude to Dalian Wanfu Pharmaceutical Co., Ltd. for their support in process testing.
Supplementary material associated with this article can be found, in the online version, at doi:
C. Le Berre, S. Honap, L. Peyrin-Biroulet, Lancet 402 (2023) 571–584. doi: 10.1016/S0140-6736(23)00966-2
L. Du, C. Ha, Gastroenterol. Clin. N. 49 (2020) 643–654. doi: 10.1016/j.gtc.2020.07.005
S.A. Patil, A.C. Moss, Expert Rev. Gastroent. 2 (2008) 177–184. doi: 10.1586/17474124.2.2.177
J. Zhao, B. Zhang, Q. Mao, et al., J. Med. Chem. 65 (2022) 4926–4948. doi: 10.1021/acs.jmedchem.1c02166
A. Prakash, C.M. Spencer, Drugs 56 (1998) 83–89. doi: 10.2165/00003495-199856010-00008
P.K. Chan, in: Patent, GB2080796, 1982.
P.K. Chan, in: Patent, US4412992, 1983.
M. Movsisyan, E.I.P. Delbeke, J.K.E.T. Berton, et al., Chem. Soc. Rev. 45 (2016) 4892–4928. doi: 10.1039/C5CS00902B
H. Jin, Q. Cai, P. Liu, et al., Chin. Chem. Lett. 35 (2024) 108721. doi: 10.1016/j.cclet.2023.108721
Y. Liu, X. Yang, T. Lin, et al., Chin. Chem. Lett. 36 (2025) 109747. doi: 10.1016/j.cclet.2024.109747
T. Gong, F. Ji, Y. Shi, et al., Org. Process Res. Dev. 29 (2025) 146–154. doi: 10.1021/acs.oprd.4c00405
D. Polterauer, K.M.P. van Eeten, W. Stam, et al., Org. Process Res. Dev. 28 (2024) 1903–1909. doi: 10.1021/acs.oprd.3c00448
Y. Mao, C. Zhou, C. Wang, et al., Chin. Chem. Lett. 34 (2023) 108061. doi: 10.1016/j.cclet.2022.108061
X. Duan, X. Wang, X. Chen, et al., Org. Process Res. Dev. 25 (2021) 2100–2109. doi: 10.1021/acs.oprd.1c00164
Y. Zhang, M. Xiao, R. Feng, et al., Org. Process Res. Dev. 28 (2024) 1675–1682. doi: 10.1021/acs.oprd.3c00323
Y. Liu, D. Wang, Y. Chen, et al., Org. Process Res. Dev. 29 (2025) 909–919. doi: 10.1021/acs.oprd.5c00006
H. Pang, J. Huang, J. Wang, et al., J. Flow Chem. 14 (2024) 427–435. doi: 10.1007/s41981-024-00310-7
J. Tu, L. Sang, H. Cheng, et al., Org. Process Res. Dev. 24 (2020) 59–66. doi: 10.1021/acs.oprd.9b00416
S.K. Komati, A. Manda, S. Vasam, et al., Org. Process Res. Dev. 29 (2025) 155–172. doi: 10.1021/acs.oprd.4c00416
A. Afshar, M. Rahimi, P. Salehi, et al., Org. Process Res. Dev. 29 (2024) 3171–3181. doi: 10.1021/acs.oprd.4c00086
A. Kraš, D. Kramar, I. Milošev, et al., Corros. Sci. 242 (2025) 112551. doi: 10.1016/j.corsci.2024.112551
M. Khan, A. Abdulhameed, H. Alshahrani, et al., Int. J. Biol. Macromol. 261 (2024) 129964. doi: 10.1016/j.ijbiomac.2024.129964
F.Y. Zhang, H.Y. Zhou, J.Q. Yuan, et al., Org. Process Res. Dev. 26 (2022) 3051–3066. doi: 10.1021/acs.oprd.2c00132
A. Nagababu, D.S. Reddy, G.V.K. Mohan, J. Ind. Eng. Chem. 114 (2022) 377–390. doi: 10.1016/j.jiec.2022.07.027

Figure 3 Response surface analysis of three-factor interaction: (a) Reaction temperature and diazotization time, (b) reaction temperature and azo-coupling reaction time, and (c) diazotization time and azo-coupling time.
Table 1. Optimization of reaction conditions for 8.a
| Entry | Solvent | Base | T (℃) | tR1 (s) | 9 (%)b | 8 (%)b | Yield (%)c |
| 1 | 1:1 | Pyridine | 25 | 60 | 20.8 | 77.3 | 61.5 |
| 2 | 1:1 | Et3N | 25 | 60 | <0.1 | >99 | 94.0 |
| 3 | 1:1 | DIPEA | 25 | 60 | <0.1 | >99 | 92.7 |
| 4 | 2:1 | Et3N | 25 | 60 | <0.1 | >99 | 94.9 |
| 5 | 3:1 | Et3N | 25 | 60 | <0.1 | >99 | 93.9 |
| 6d | 1:1 | Et3N | 25 | 60 | − | − | − |
| 7e | 1:1 | Et3N | 25 | 60 | <0.1 | >99 | 83.7 |
| 8 | 1:1 | Et3N | 25 | 40 | <0.1 | >99 | 75.9 |
| 9 | 1:1 | Et3N | 25 | 90 | <0.1 | >99 | 92.8 |
| 10 | 1:1 | Et3N | 25 | 120 | <0.1 | >99 | 95.8 |
| 11 | 1:1 | Et3N | 25 | 150 | <0.1 | >99 | 91.6 |
| 12 | 1:1 | Et3N | 10 | 120 | <0.1 | >99 | 92.9 |
| 13 | 1:1 | Et3N | 40 | 120 | <0.1 | >99 | 93.5 |
| a Reaction conditions: S1: 0.5 mol/L in MeCN; S2: 0.3 mol/L in MeCN/H2O. b The content was determined by HPLC. c Isolated yield. d Acetone as organic solvent. e THF as organic solvent. | |||||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Optimization of reaction conditions for 3.a
| Entry | Solvent | NaOH (equiv.) | T (℃) | tR2 (s) | 9 (%)b | 3 (%)b | Yield (%)c |
| 1 | THF | 2.5 | 40 | 30 | 7.3 | 91.2 | 76.8 |
| 2 | THF | 2.0 | 25 | 30 | 1.9 | 97.4 | 84.7 |
| 3 | THF | 1.5 | 25 | 30 | <0.1 | >99 | 85.3 |
| 4 | THF | 1.2 | 25 | 30 | <0.1 | >99 | 92.4 |
| 5 | THF | 1.0 | 25 | 30 | <0.1 | 95.6 | 86.1 |
| 6 | THF | 0.8 | 25 | 30 | <0.1 | 76.8 | 81.2 |
| 7 | THF | 1.2 | 40 | 30 | <0.1 | >99 | 91.7 |
| 8 | THF | 1.2 | 10 | 30 | <0.1 | >99 | 83.7 |
| 9 | THF | 1.2 | 25 | 60 | <0.1 | >99 | 90.9 |
| 10 | THF | 1.2 | 25 | 20 | <0.1 | >99 | 71.3 |
| 11 | MeOH | 1.2 | 25 | 30 | − | − | − |
| 12 | MeCN | 1.2 | 25 | 30 | − | − | − |
| 13 | Acetone | 1.2 | 25 | 30 | <0.1 | 69.3 | 75.6 |
| a Reaction conditions: S3: 0.5 mol/L in THF; S4: 0.3 mol/L in H2O. b The content was determined by HPLC. c Isolated yield. | |||||||
下载: 导出CSV
Table 3. Optimization of diazotization.a
| Entry | Acid (equiv.) | Diazotization reagent | Reagent (equiv.) | Yield (%)b |
| 1 | HCl (3.0) | NaNO2 | 1.05 | 82.5 |
| 2 | HCl (3.2) | NaNO2 | 1.05 | 88.2 |
| 3 | HCl (3.4) | NaNO2 | 1.05 | 86.7 |
| 4 | H2SO4 (1.6) | NaNO2 | 1.05 | 83.8 |
| 5 | HCl (3.2) | NaNO2 | 1.00 | 78.9 |
| 6 | HCl (3.2) | NaNO2 | 1.10 | 87.0 |
| 7 | HCl (3.2) | Isoamyl nitrite | 1.05 | 82.3 |
| 8 | HCl (3.2) | Isoamyl nitrite | 1.10 | 80.4 |
| 9 | H2SO4 (1.6) | Isoamyl nitrite | 1.05 | 79.7 |
| a Reaction conditions: S6: 0.2 mol/L in H2O; S8: 0.2 mol/L in H2O. tR3: 35 s; tR4: 41 s; reaction temperature: 15 ℃. b Isolated yield. | ||||
下载: 导出CSV
Table 4. Optimization of azo coupling reaction.a
| Entry | NaOH (equiv.) | Salicylic acid (equiv.) | Yield (%)b |
| 1 | 4.1 | 1.05 | 71.8 |
| 2 | 4.4 | 1.05 | 85.5 |
| 3 | 4.6 | 1.05 | 87.1 |
| 4 | 4.9 | 1.05 | 83.6 |
| 5 | 5.2 | 1.05 | 69.4 |
| 6 | 5.5 | 1.05 | 64.5 |
| 7 | 4.6 | 1.00 | 84.5 |
| 8 | 4.6 | 1.10 | 89.2 |
| 9 | 4.6 | 1.20 | 70.9 |
| a Reaction conditions: S6: 0.2 mol/L in H2O; S8: 0.2 mol/L in H2O; tR3: 35 s; tR4: 41 s; reaction temperature: 15 ℃. b Isolated yield. | |||
下载: 导出CSV
Table 5. Overall optimization of the reaction system.
| Entry | T (℃) | tR3 (s) | tR4 (s) | Yield (%)a |
| 1 | 10 | 35 | 50 | 89.3 |
| 2 | 15 | 35 | 50 | 90.7 |
| 3 | 20 | 35 | 50 | 91.6 |
| 4 | 25 | 35 | 50 | 88.9 |
| 5 | 30 | 35 | 50 | 84.7 |
| 6 | 20 | 25 | 40 | 75.6 |
| 7 | 20 | 40 | 50 | 91.2 |
| 8 | 20 | 45 | 50 | 91.9 |
| 9 | 20 | 50 | 50 | 89.4 |
| 10 | 20 | 45 | 55 | 92.5 |
| 11 | 20 | 45 | 60 | 90.5 |
| 12 | 20 | 45 | 70 | 84.9 |
| a Isolated yield. | ||||
下载: 导出CSV
Table 6. Recommended conditions for optimization of the reaction yield.a
| Entry | T (℃) | tR3 (s) | tR4 (s) | Balsalazide (%)b | Yield (%)c |
| 1 | 18.2 | 42 | 59 | 97.9 | 93.4 |
| 2 | 18.2 | 42 | 59 | 97.7 | 93.2 |
| 3 | 18.2 | 42 | 59 | 97.9 | 92.7 |
| a Optimal reaction conditions predicted by RSM. b The purity of crude product was determined by HPLC. c Isolated yield. | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们







 下载:
下载: