Received Date:
12 May 2025 Accepted Date:
12 October 2025 Revised Date:
09 October 2025 Available Online:
15 July 2026
Abstract:
The pathological protein α-synuclein (α-Syn) aggregates are considered a key toxic substance responsible for the degeneration of dopaminergic neurons in Parkinson's disease (PD). Clearing these pathological aggregates can potentially control PD progression at its source. However, conventional drug delivery systems face significant challenges, including the blood-brain barrier (BBB) and lack of tissue-specific targeting. To address this, we developed a core-shell hybrid system, named RExo-si-TN-T10, for the co-delivery of triptolide (T10) and small interfering RNA targeting α-Syn (siSNCA). This system comprises two components: an outer shell of exosomes (RExo) derived from BV2 microglial cells and modified with rabies virus glycoprotein peptide (RVG), and an inner core of a nanomicelle composed of phenylboronic acid derivatives: 4-Nitrophenyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) benzyl carbonate (NBC) and trimethyl chitosan (TMC) for co-delivering gene and small-molecule drugs. Furthermore, we utilized cellular nanoporation (CNP) technology to significantly enhance exosome yield and transfection efficiency. This innovative nano-scavenger effectively eliminates α-Syn aggregates and reduces their cytotoxic effects in PD-affected neurons. Following treatment with RExo-si-TN-T10, we observed significant improvement in the motor behavior of PD mice. Our findings suggest that RExo-si-TN-T10 holds promise as a therapeutic platform for PD.
Parkinson's disease (PD) is one of the most common neurodegenerative disorders, primarily characterized by motor symptoms such as tremors, rigidity, and bradykinesia [1-3]. The pathological hallmark of PD is the accumulation of insoluble aggregates of α-synuclein (α-Syn), known as Lewy bodies, which contribute to the progressive degeneration of dopaminergic neurons in the substantia nigra [4-6]. Current therapeutic strategies, including pharmacological management, surgical interventions, and stem cell therapies, offer symptomatic relief but fall short of halting or reversing disease progression. Therefore, there is an urgent need to develop novel therapeutic approaches that target the underlying pathological mechanisms of PD.
In recent years, RNA interference (RNAi) technology has emerged as a promising tool for gene silencing in neurological diseases. Small interfering RNA (siRNA) targeting α-Syn (siSNCA), the gene encoding α-Syn, holds potential for reducing the production of pathological α-Syn [7]. Similarly, natural compounds such as triptolide (T10), derived from Tripterygium wilfordii, exhibit potent anti-inflammatory and neuroprotective effects and have been shown to promote the clearance of α-Syn aggregates [8,9]. The combination of siRNA and T10 offers a synergistic strategy for PD treatment by simultaneously inhibiting α-Syn synthesis and promoting the degradation of existing aggregates. However, the clinical application of siRNA is hampered by its poor stability, rapid clearance, and inability to cross the blood-brain barrier (BBB) [10-14]. T10 suffers from poor solubility, low bioavailability, and non-specific distribution. Therefore, the lack of an efficient delivery system that can overcome the BBB, achieve neuronal targeting, and enable controlled drug release remains a major obstacle.
To address these challenges, we developed a core-shell hybrid nanoparticle system for co-delivery of siSNCA and T10, named RExo-si-TN-T10. The system is composed of reactive oxygen species (ROS)-responsive nanomicelles trimethyl chitosan (TMC)-4-nitrophenyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl carbonate (NBC) (TN) made of phenylboronic acid derivatives NBC and Chitosan derivatives TMC encapsulating T10 and adsorbing siSNCA by electrostatic interaction to form a core [15,16]. The outer shell is composed of BV2 microglia-derived exosomes and is functionalized by rabies virus glycoprotein peptide (RVG) to enhance BBB penetration and neuronal targeting [17-23]. To overcome the limitations of low exosome yield and inefficient transfection associated with conventional methods [24], we employed cellular nanoporation (CNP) technology, which significantly increases exosome production and mRNA packaging efficiency [25]. The resulting nanocomposite, RExo-si-TN-T10, demonstrates excellent stability, biocompatibility, and the ability to release drugs in response to the high ROS environment characteristic of PD lesions [26,27].
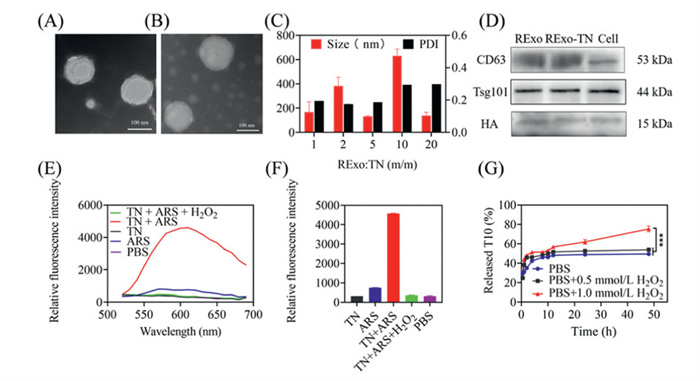
Here, we propose the development of RExo-si-TN-T10, a gene–chemical hybrid nanoparticle engineered to cross the BBB. This system enables ROS-responsive drug release in the high ROS microenvironment characteristic of PD injury, thereby achieving targeted synergistic therapy for PD in the brain. The RExo-si-TN-T10 was prepared in two main steps: first, the synthesis of ROS-responsive drug-loaded micelles (si-TN-T10), and second, the production of RVG-modified exosomes (RExo). The ROS-responsive micelle TN was prepared by conjugating TMC with the ROS-sensitive compound NBC. Successful synthesis was confirmed by Fourier transform infrared spectroscopy (FT-IR) and nuclear magnetic resonance hydrogen spectroscopy (1H NMR) (Figs. S1–S4 in Supporting information). TMC and NBC self-assembled into micelles with a hydrophobic core and hydrophilic shell. T10 was encapsulated into the micelles via sonication, achieving an encapsulation efficiency of 86.56% ± 1.40% as determined by high performance liquid chromatography (HPLC). The resulting TN-T10 micelles had a hydrodynamic size of 116.36 ± 0.19 nm, a polydispersity index (PDI) of 0.265, and a zeta potential of 12.83 ± 0.76 mV (Figs. S5A and B in Supporting information), indicating a positive surface charge suitable for adsorbing negatively charged siSNCA via electrostatic interactions. Agarose gel electrophoresis confirmed complete siSNCA adsorption by TN-T10 at a nitrogen-phosphorus (N/P) ratio of 8/1 (Fig. S6 in Supporting information). For RExo preparation, BV2 microglial cell-derived exosomes were isolated from day-7 culture supernatant via differential centrifugation. To address low yield and targeting efficiency, CNP was employed to introduce the RVG-Lamp2b plasmid, significantly enhancing exosome production and functionalization [26]. RExo exhibited a size of ~126 ± 1.85 nm (PDI = 0.280 ± 0.013) and a zeta potential of −8.11 ± 1.03 mV, enabling adsorption onto positively charged TN. TEM imaging revealed that RExo exhibited a cup-shaped morphology with sizes between 30 and 150 nm, while RExo-si-TN-T10 appeared as spherical particles with a core-shell structure and sizes of 100–200 nm (Figs. 1A and B). RExo-si-TN-T10 were formed via extrusion, with a final size of 175.36 ± 1.19 nm (PDI = 0.263 ± 0.01) and zeta potential of −5.24 ± 1.21 mV. The charge reversal confirmed successful RExo coating (Fig. S5B). Optimization based on RExo: TN mass ratios showed that a 5:1 ratio yielded nanocomposites with a size of 132.24 ± 2.87 nm and PDI of 0.186 ± 0.019, optimal for drug delivery (Fig. 1C). Western blot (WB) confirmed the presence of exosomal markers (CD63, Tsg101) and HA-tagged RVG on both RExo and RExo-TN, demonstrating successful modification and integrity after encapsulation (Fig. 1D).
Figure 1
Figure 1.
Preparation and characterization of RExo-si-TN-T10. Transmission electron micrograph of RExo (A) and RExo-TN (B). Scale bar: 100 nm. (C) Particle size and PDI of REXo-si-TN-T10 obtained at different mass ratios. (D) Characterisation of Exo and Exo-TN surface signature proteins. (E, F) The fluorescence intensity of ARS in different conditions (Em: 500–700 nm in E; Em: 600 nm in F). (G) The release profile of T10 from RExo-si-TN-T10 in different conditions. Data are presented as mean ± standard deviation (SD) (n = 3). *** P < 0.001.
ROS-responsive drug release was evaluated using alizarin red S (ARS). Fluorescence intensity increased significantly upon ARS binding to phenylboronic acid groups in TN, but pre-treatment with H2O2 (1.0 mmol/L) markedly reduced fluorescence, confirming NBC degradation under oxidative conditions (Figs. 1E and F). HPLC analysis showed that T10 release from TN-T10 was enhanced under high H2O2 concentrations, with cumulative release reaching 75.34% at 48 h under 1.0 mmol/L H2O2, compared to 49.33% without H2O2 (Fig. 1G). The nanocomposites showed good stability at 4 ℃, with minimal changes in size, PDI, zeta potential, and encapsulation efficiency. Stability was reduced at room temperature, and freeze-thawing and lyophilization also maintained acceptable stability (Figs. S7A–F in Supporting information). In summary, we successfully developed a targeted, ROS-responsive exosome-based nanoparticle system capable of co-delivering siSNCA and T10 with favorable physicochemical properties.
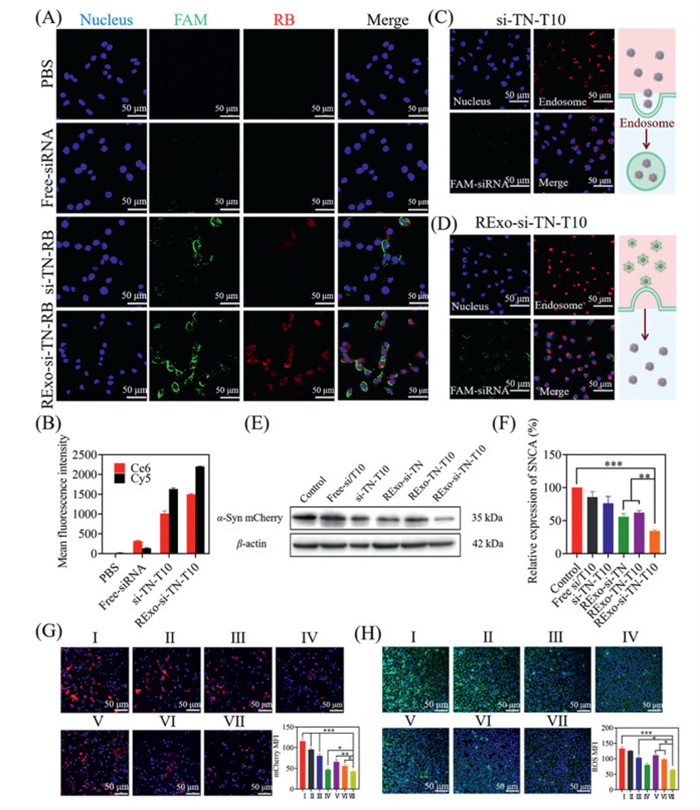
Cellular uptake is critical for drug delivery efficacy. To evaluate this, T10 was labeled with rhodamine B (RB, red), siSNCA with FAM (green), and nuclei with Hoechst (blue). SH-SY5Y cells were incubated for 4 h with FAM-siSNCA, FAM-si-TN-T10-RB, or RExo-FAM-si-TN-T10-RB. Confocal microscopy (Fig. 2A) revealed negligible green fluorescence from free FAM-siSNCA due to degradation. Micelles without exosome coating (si-TN-T10) showed weak fluorescence, while RExo-coated micelles exhibited strong red and green fluorescence near nuclei, with clear co-localization, indicating enhanced cellular uptake. Flow cytometry (Fig. 2B) further confirmed significantly higher mean fluorescence intensity (MFI) for the nanocomposite group, demonstrating improved stability and uptake of both T10 and siRNA.
Figure 2
Figure 2.In vitro drug delivery. RExo-si-TN-T10 cell uptake and cell quantification: (A) CLSM images of SH-SY5Y cells after 4 h treatment with different formulations. (B) The mean fluorescence intensity of Ce6 and Cy5-siRNA was quantified by flow cytometry after incubation for 4 h. (C) CLSM images of lysosomal escape with si-TN-T10 after 4 h. (D) CLSM images of lysosomal escape with RExo-si-TN-T10 after 4 h. (E) WB analysis of α-Syn protein. (F) Quantitative analysis of α-Syn protein. (G) Confocal image of α-Syn-mCherry in SH-SY5Y cells after incubation with different nanopreparations for 72 h. The quantification of mCherry fluorescence. (H) ROS fluorescence images in α-Syn-mCherry-SH-SY5Y cells after incubation with different nanopreparations for 72 h. Ⅰ: control, Ⅱ: free-si/T10, Ⅲ: si-TN-T10, Ⅳ: Exo-si-TN-T10, Ⅴ: RExo-TN-T10, Ⅵ: RExo-si-TN, Ⅶ: RExo-si-TN-T10. Scale bar: 50 µm. Data are presented as mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001.
Lysosomal escape is essential to avoid degradation of nucleic acids. Lysosomal labeling was performed using LysoTracker Red. We observed that free siRNA degraded rapidly, while si-TN-T10 showed improved stability but strong lysosomal co-localization (Fig. 2C). In contrast, RExo-si-TN-T10 displayed intense green fluorescence with separation from lysosomal signals (Fig. 2D), indicating facilitated lysosomal escape. This is likely because exosome coating shifts cellular entry from clathrin-mediated endocytosis to membrane fusion, enabling direct cytosolic delivery. The overlap coefficient decreased from 0.94 for si-TN-T10 to 0.62 for RExo-si-TN-T10, confirming reduced lysosomal accumulation. Since the free siRNA is not protected by the carrier, it degrades quickly after entering the cell, so there is almost no fluorescence (Fig. S8 in Supporting information).
We assessed the biocompatibility of si-TN-T10 and RExo-si-TN-T10 using cell counting kit-8 (CCK-8) assays. Both formulations maintained cell viability above 80% across various N/P ratios, with an optimal ratio of 8:1 yielding 98.85% viability (Fig. S9A in Supporting information). Without exosome coating, cell viability decreased with increasing TN concentration, whereas RExo coating significantly reduced cytotoxicity (Fig. S9B in Supporting information), likely due to charge reversal and the innate biocompatibility of exosomes.
As shown in Fig. S10 (Supporting information), to overcome the BBB, we employed an in vitro Transwell model with bEnd.3 cells (Fig. S10A). After confirming barrier integrity (TEER ≥ 200 Ω cm2), we observed higher fluorescence in the lower chamber for RExo-coated formulations, particularly with RVG targeting, indicating enhanced BBB penetration (Fig. S10B). Furthermore, we verified the RVG targeting mechanism via competitive inhibition and receptor blockade. Pretreatment with free RVG or α-bungarotoxin reduced transmembrane penetration by 73.62% and 76.84%, respectively (Fig. S10C). Flow cytometry confirmed decreased binding to endothelial cells (Fig. S10D), demonstrating that RVG facilitates BBB penetration through nAChR receptor-dependent mechanisms.
We established an α-Syn-overexpressing dopaminergic neuron model (α-Syn-mCherry-SH-SY5Y) and treated the cells with various formulations for 72 h. α-Syn aggregation was assessed by confocal laser scanning microscopy (CLSM). As shown in Fig. 2G, all drug treatments reduced mCherry fluorescence compared to phosphate buffered saline (PBS). The free siSNCA and T10 mixture showed minimal effect due to poor stability and low cellular uptake. In contrast, the exosome-coated nanocomposite (RExo-si-TN-T10) led to significantly reduced fluorescence compared to the micelle-based carrier (si-TN-T10), which can be attributed to enhanced cellular uptake and protection of nucleic acids. Furthermore, RExo-si-TN-T10 outperformed single-drug treatments, indicating synergistic action. WB analysis corroborated these findings (Figs. 2E and F). The exosome-coated group showed substantial α-Syn downregulation, unlike the micelle group or free drugs, highlighting the role of exosomes in promoting siRNA stability and delivery. The dual-drug nanocomposite yielded more pronounced protein reduction than single-drug formulations, demonstrating effective synergy between siSNCA and T10. To elucidate the synergy mechanism, we measured SNCA mRNA and α-Syn protein levels under single and combined treatments. Quantitative real-time polymerase chain reaction (qPCR) results indicated that siSNCA alone or in combination reduced SNCA mRNA by approximately 70%, while T10 alone had no effect (Fig. S10E). WB analysis showed that T10 and siSNCA monotherapies reduced α-Syn by 20% and 30%, respectively, whereas the combination led to an 80% reduction (Figs. S10F and G). This suggests that the synergy arises from source inhibition (siRNA) enhanced by effective clearance (T10), particularly at the protein level.
We also evaluated intracellular ROS levels as an indicator of anti-inflammatory effects. CLSM imaging revealed that all drug treatments reduced ROS, with the exosome-coated group exhibiting the strongest reduction (Fig. 2H). These results indicate that the developed nanocomposites can effectively attenuate the inflammatory response in PD pathological regions, thereby contributing to the alleviation of PD symptoms.
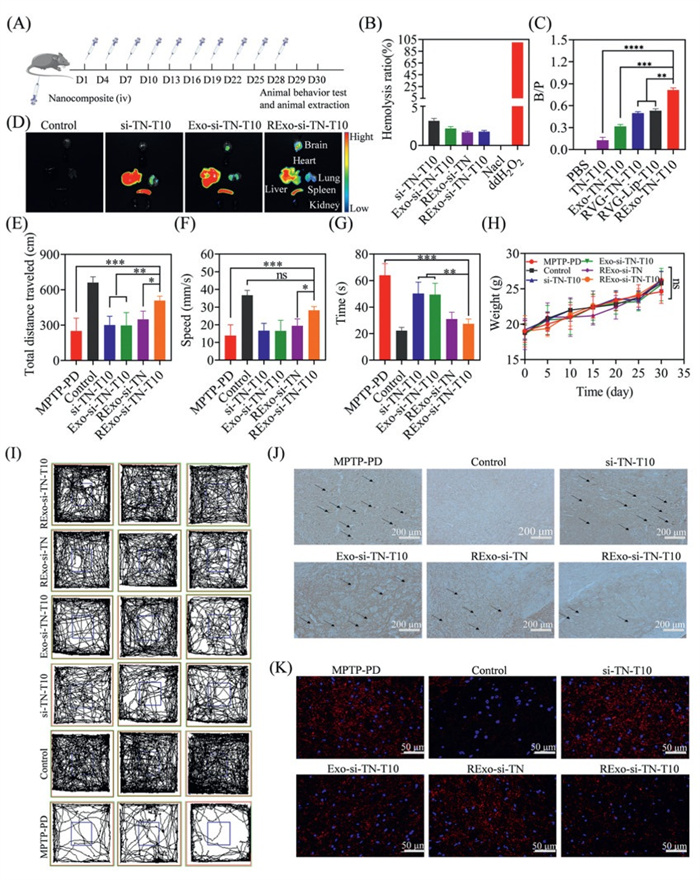
To evaluate the in vivo efficacy and safety of the nanocomplex, we established a PD mouse model via intraperitoneal injection of 1-methyl-4-phenyl-1,2,3,6-te-trahydropyridine(MPTP) hydrochloride (30 mg/kg) for 9 consecutive days (Fig. S12 in Supporting information). All experimental procedures involving animals were performed in compliance with relevant guidelines and were approved by the Ethics Committee of Chongqing University of Technology. Hemocompatibility was assessed through hemolysis assay, confirming good biocompatibility with a hemolysis rate below 5% at 1000 µg/mL. We then investigated the brain targeting efficiency and biodistribution of various formulations, including PBS, TN-T10, DSPE-PEG-RVG-TN-T10, Exo-TN-T10, RExo-TN-T10, and DSPE-PEG-RVG-Lip-T10, by measuring the brain-to-plasma ratio (B/P) and using Cy5.5-labeled nanocomplexes. Motor functions were evaluated via open field test (OFT) and pole test. Immunohistochemistry and immunofluorescence were performed to assess α-Syn aggregation. Additionally, in vivo safety was evaluated by histopathological examination of major organs, body weight monitoring, liver and kidney function tests, and cytokine level measurements. The specific treatment plan was shown in Fig. 3A.
Figure 3
Figure 3.
Treatment of PD mice. (A)Administer the medication every three days for a total of ten doses. (B) Hemolysis experiment of nanomaterials. (C) Brain plasma ratio after administration of different preparations (n = 3). (D) Distribution of the fluorescently labeled nanoparticle in various organs of mice after administration by tail vein. (E) Total field experiment movement distance (n = 5). (F) Average speed of movement of experimental mice in the mine field (n = 5). (G) Pole climbing experiment: time taken to climb to the top (n = 5). (H) Changes in body weight of mice during the administration period (n = 5). (I) Field experiment movement track. (J) Immunohistochemical staining of α-Syn in sections of the substantia nigra region of the mouse brain after treatment with different drug groups. Scale bar: 200 µm. (K) Immunofluorescence staining of α-Syn in sections of the substantia nigra region of the mouse brain after treatment with different drug groups. Scale bar: 50 µm. Data are presented as mean ± SD. P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, no significance.
The hemolysis rate was below 5%, indicating good biocompatibility, which was further improved by exosome coating (Fig. 3B). Among all formulations, RExo-TN-T10 showed the highest B/P value (0.81), which was 6.2-fold higher than that of TN-T10 (0.13), demonstrating superior brain targeting due to RVG peptide and exosome-mediated BBB penetration. Exo-TN-T10 (B/P = 0.32) also exhibited significantly higher brain accumulation than synthetic carriers (Fig. 3C). In vivo imaging confirmed enhanced brain delivery and reduced off-target accumulation for RExo-si-TN-T10 (Fig. 3D). Image J was used to quantitatively analyze the fluorescence intensity of different tissues (Fig. S13 in Supporting information).
Behavioral tests revealed that PD mice treated with RExo-si-TN-T10 showed significant improvements in motor function, including increased total distance, average speed, and center zone entries in OFT, as well as reduced descent time in the pole test (Figs. 3E–G and I). Neither si-TN-T10 nor Exo-si-TN-T10 produced significant improvements. Immunostaining results indicated that RExo-si-TN-T10 effectively reduced α-Syn aggregation in the brain, outperforming both non-targeted and single-treatment groups, suggesting synergistic effects between siSNCA and T10 (Figs. 3J and K).
No significant body weight changes or pathological abnormalities in major organs were observed in the RExo-si-TN-T10 group (Fig. 3H and Fig. S15 in Supporting information). Liver and kidney function markers, as well as cytokine levels, remained normal, indicating excellent biosafety (Figs. S16A–H in Supporting information). In contrast, si-TN-T10 induced mild hepatorenal toxicity and elevated inflammatory factors, likely attributable to the positive charge of the micelles. Exosome encapsulation effectively shielded this positive charge and enhanced biocompatibility. Furthermore, RVG modification improved brain targeting and reduced peripheral exposure, thereby further increasing the biosafety of the nanocomposites.
In summary, to improve the pathological progression of PD, ameliorate degeneration of dopaminergic neurons, restore motor function, and overcome delivery challenges for combined gene and small-molecule therapies, we developed a targeted nanocomposite leveraging the natural transport properties of exosomes and the high-ROS microenvironment in PD lesions. Using CNP technology, we isolated abundant RVG-modified exosomes from BV2 cells. Concurrently, we synthesized a ROS-responsive nanomicelle for co-loading siSNCA and T10. The system exhibits high drug-loading capacity, efficient cellular uptake, stable BBB penetration, and neuron-specific targeting, resulting in precise microenvironment-responsive release and synergistic therapeutic effects. The ability of RExo-si-TN-T10 to cross the BBB and downregulate α-Syn was validated both in vitro and in vivo. By delivering siSNCA and T10 directly to affected dopaminergic neurons, this approach reduces α-Syn aggregation, offering a targeted strategy for controlling PD at its pathological source and presenting a promising therapeutic breakthrough.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 82100253, 22307012), the Chongqing Natural Science Foundation Postdoctoral Science Fund Project (No. CSTB2023 NSCQ-BHX0210), the Science and Technology Research Program of Chongqing Education Commission of China (Nos. KJQN202201168, KJZD-M202501106), the Science and Technology Innovation Key R&D Program of Chongqing (No. 2024CCZ096), the Graduate Scientific Research and Innovation Foundation of Chongqing, China (Nos. CYS240721, CYS23701, CYS25768), Applied Basic Research Project of Chongqing University of Technology (No. 2024TBZ036).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111948.
L.Y. Liu, Y. Li, H. Peng, et al., Sci. Adv. 6 (2020) eaba3967. doi: 10.1126/sciadv.aba3967
Figure 1
Preparation and characterization of RExo-si-TN-T10. Transmission electron micrograph of RExo (A) and RExo-TN (B). Scale bar: 100 nm. (C) Particle size and PDI of REXo-si-TN-T10 obtained at different mass ratios. (D) Characterisation of Exo and Exo-TN surface signature proteins. (E, F) The fluorescence intensity of ARS in different conditions (Em: 500–700 nm in E; Em: 600 nm in F). (G) The release profile of T10 from RExo-si-TN-T10 in different conditions. Data are presented as mean ± standard deviation (SD) (n = 3). *** P < 0.001.
Figure 2In vitro drug delivery. RExo-si-TN-T10 cell uptake and cell quantification: (A) CLSM images of SH-SY5Y cells after 4 h treatment with different formulations. (B) The mean fluorescence intensity of Ce6 and Cy5-siRNA was quantified by flow cytometry after incubation for 4 h. (C) CLSM images of lysosomal escape with si-TN-T10 after 4 h. (D) CLSM images of lysosomal escape with RExo-si-TN-T10 after 4 h. (E) WB analysis of α-Syn protein. (F) Quantitative analysis of α-Syn protein. (G) Confocal image of α-Syn-mCherry in SH-SY5Y cells after incubation with different nanopreparations for 72 h. The quantification of mCherry fluorescence. (H) ROS fluorescence images in α-Syn-mCherry-SH-SY5Y cells after incubation with different nanopreparations for 72 h. Ⅰ: control, Ⅱ: free-si/T10, Ⅲ: si-TN-T10, Ⅳ: Exo-si-TN-T10, Ⅴ: RExo-TN-T10, Ⅵ: RExo-si-TN, Ⅶ: RExo-si-TN-T10. Scale bar: 50 µm. Data are presented as mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001.
Figure 3
Treatment of PD mice. (A)Administer the medication every three days for a total of ten doses. (B) Hemolysis experiment of nanomaterials. (C) Brain plasma ratio after administration of different preparations (n = 3). (D) Distribution of the fluorescently labeled nanoparticle in various organs of mice after administration by tail vein. (E) Total field experiment movement distance (n = 5). (F) Average speed of movement of experimental mice in the mine field (n = 5). (G) Pole climbing experiment: time taken to climb to the top (n = 5). (H) Changes in body weight of mice during the administration period (n = 5). (I) Field experiment movement track. (J) Immunohistochemical staining of α-Syn in sections of the substantia nigra region of the mouse brain after treatment with different drug groups. Scale bar: 200 µm. (K) Immunofluorescence staining of α-Syn in sections of the substantia nigra region of the mouse brain after treatment with different drug groups. Scale bar: 50 µm. Data are presented as mean ± SD. P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. ns, no significance.

 DownLoad:
DownLoad:

 下载:
下载:




 下载:
下载:
