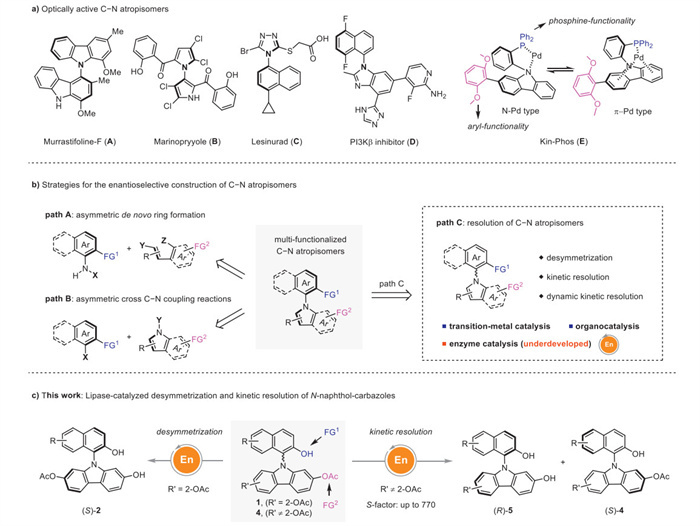
Scheme 1.
Construction of C−N atropisomers: state-of-the-art and this work.

C−N axially chiral compounds [1-11] have attracted more and more attention in recent years, due to the fact that these types of skeletons have found wide applications in natural products and drug molecules (Scheme 1a), such as Murrastifoline-F (A) [12], an alkaloid isolated from Murraya euchrestifolia; Marinopyrrole (B) [13], a natural product isolated from the marine origin of Streptomyces; a drug molecule Lesinurad (C) [14], which is an active drug for the treatment of gout-associated hyperuricemia by inhibiting urate reabsorption transporter (URAT1); and phosphatidylinositol 3-kinase β (PI3Kβ) inhibitor (D) [15]. Moreover, Kin-Phos (E) is a chiral mono-phosphine ligand based on the C−N axially chiral skeleton, and exhibits excellent enantioselectivity control in promoting asymmetric coupling reactions to construct axial chiral backbones [16]. It is interesting to observe a characteristic pattern in coordination of Kin-Phos (E) with palladium, that is an equilibrium between N−Pd and π−Pd type coordination (Scheme 1a). Therefore, methodologies to access C−N axially chiral compounds are highly desirable, especially those towards multi-functionalized C−N atropisomers (Scheme 1b). Conventional strategies for the enantioselective construction of C−N atropisomers are based on asymmetric de novo ring formation (path A, Scheme 1b) [17-32] and asymmetric cross C−N coupling reactions (path B, Scheme 1b) [33-38]. However, the challenge would be the preservation of extra functional groups (FG1, FG2) through these methods, given the fact that the control of chemoselectivity becomes difficult with coupling partners bearing pre-installed functional groups (X, Y, or Z) [39-42]. At this stage, the resolution strategy becomes uniquely advantageous (path C, Scheme 1b), which starts from multi-functionalized C−N atropisomer substrates in a racemic form or bearing a plane of symmetry. Recently, desymmetrization methodologies to generate C−N axially chiral N-arylcarbazoles has been emerged with functionalization occurring at C1-position on the carbazole ring [43]. Transition-metal catalysis and organocatalysis have been demonstrated as efficient tools [44-49], while enzyme catalysis, generally endowed with high efficiency and selectivity still remains underdeveloped [50-54]. Enzyme catalysis represents a powerful tool for asymmetric synthesis, however their application in the construction of C−N axially chiral biaryls is still unprecedented. Herein, we disclose our recent development on the lipase-catalyzed desymmetrization and kinetic resolution of N-naphthol-carbazoles (Scheme 1c), producing enantioenriched C−N atropisomers with good functional group tolerance and enantioselectivity. Moreover, the functionality of two hydroxyl groups at the 2-position on the naphthene ring and the 2-position on the carbazole ring reserve reactive sites for late-stage transformations.
With this concept in mind, we start to investigate the asymmetric hydrolysis process by using a symmetric diacetate substrate, 9-(2-hydroxynaphthalen-1-yl)-9H-carbazole-2,7-diyl diacetate 1a. The resolution capability of various commercially available lipases was evaluated with methanol as the proton donor in toluene at 40 ℃ for 24 h (Table 1, entries 1–5). To our delight, enantioselective hydrolysis of 1a could be detected to deliver the desirable C−N axially chiral product (S)-2a with Lipozyme-RM (72% yield, 80% ee) and LPL311-powder (60% yield, 71% ee) respectively, while other lipase catalysts failed to promote such transformation. To further enhance the enantioselectivity of lipase-catalyzed hydrolysis, we tried to immobilize the well-performing catalyst, LPL311-powder to improve its activity and stability in the organic system. The following optimizations revealed that the immobilized LPL311 on Celite (LPL311-Celite) provided an effective control over the enantioselectivity in producing (S)-2a (60% yield, 94% ee), while minimizing the formation of byproduct 3a (3%) via two-step hydrolysis (entry 6). To our surprise, when LPL311 was immobilized onto methacrylate-based polymer resins (LPL311-polyester), the desirable product (S)-2a was further improved to 80% yield with 96% ee (entry 7). Further studies on solvent effects indicated that non-polar aromatic solvents (benzene and toluene) are more suitable for lipase-catalyzed resolution (entries 8–11). In contrast, the hydrolysis reaction hardly occurred in a polar solvent, e.g., THF, CH3CN, or DCM, which tends to block the lipase's active pockets for the substrate (entries 12–14, and Table S2 in Supporting information).
 DownLoad:
CSV
DownLoad:
CSV
 |
||||||
| Entry | Lipase | Solvent | Yield of (S)-2a (%)b | ee of (S)-2a (%)c | Yield of 3a (%)b | Recovery of 1a (%)b |
| 1 | Lipase AK/PS/PPL/CALB | Toluene | N.D. | – | – | – |
| 2 | Lipase-TL | Toluene | Trace | – | – | 98 |
| 3 | Lipase-AYS | Toluene | Trace | – | – | > 99 |
| 4 | Lipozyme-RM | Toluene | 72 | 80 | 7 | 18 |
| 5 | LPL311-powderd | Toluene | 60 | 71 | 22 | 15 |
| 6 | LPL311-Celite | Toluene | 60 | 94 | 3 | 37 |
| 7 | LPL311-polyester | Toluene | 80 | 96 | Trace | 18 |
| 8 | LPL311-polyester | PhCF3 | 77 | 89 | Trace | 17 |
| 9 | LPL311-polyester | PhCl | 76 | 92 | 3 | 22 |
| 10 | LPL311-polyester | Benzene | 82 | 94 | – | – |
| 11 | LPL311-polyester | Anisole | Trace | 82 | – | 94 |
| 12 | LPL311-polyester | CH3CN | N.D. | – | – | – |
| 13 | LPL311-polyester | THF | Trace | – | – | – |
| 14 | LPL311-polyester | DCM | Trace | – | – | – |
| a The reaction was carried out under an air atmosphere with N-naphthol-carbazole 1a (0.10 mmol), lipase (3 w/w), and CH3OH (1.0 mmol) in indicated solvent (1.0 mL) at 40 ℃ for 24 h. b The yield was determined by 1H NMR analysis using CH2Br2 as the internal standard. c Enantiomeric excess (ee) was determined by chiral HPLC. d The reaction was carried out with N-naphthol-carbazole 1a (0.10 mmol), lyophilized powder LPL311 (5%) and CH3OH (1.0 mmol) in toluene (1.0 mL) at 40 ℃ for 24 h. |
||||||
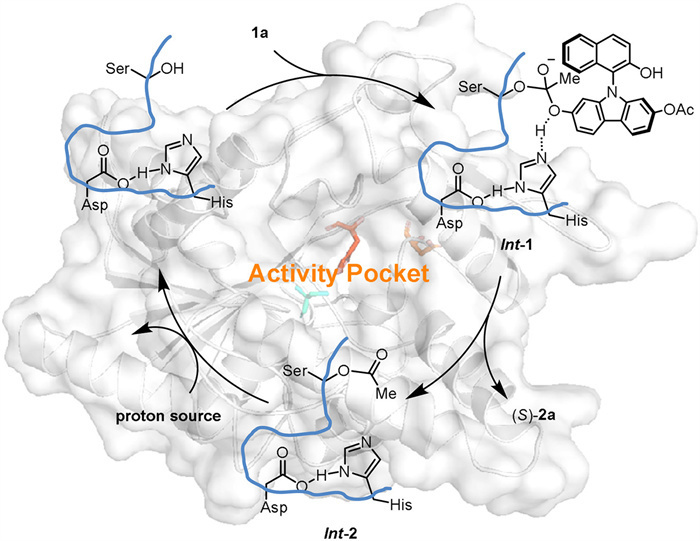
The enantioselective hydrolysis catalyzed by LPL-311 is proposed to proceed via a classic serine hydrolase mechanism [55,56]. This mechanism involves a three-step pathway involving Int-1 and Int-2. The enantioselective hydrolysis of esters unit in 1a occurs inside the active pocket of LPL311, and the hydrolysis process is generally considered to proceed with the assistance of three key amino acids: serine, aspartic acid, and histidine (Scheme 2 and Scheme S1 in Supporting information).
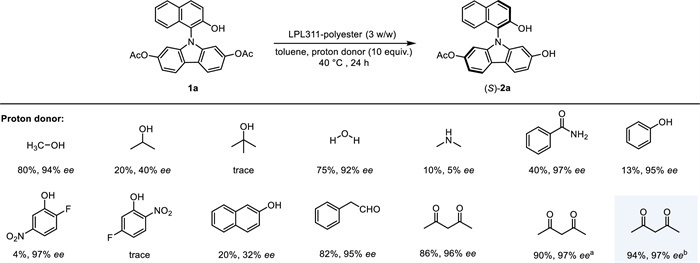
Subsequently, screening of proton sources was carried out for the hydrolytic desymmetrization of symmetric diacetate 1a under the catalysis of LPL311-polyester in toluene at 40 ℃ for 24 h (Scheme 3). Small molecules containing an active proton, such as alcohols and amines, can serve as proton sources. However, when these molecules are equipped with a bulky group, the reaction typically provided a significantly lower conversion with decreased enantioselectivity. Water was able to hydrolyze diacetate 1a directly, leading to C−N axially chiral product (S)-2a in 75% yield with 92% ee. However, when phenol and its derivatives were employed, the hydrolysis process became inefficient, probably due to the low solubility of phenolic derivatives in toluene. To our delight, when carbonyls were employed as the proton donor, e.g., phenylacetaldehyde and acetylacetone, enzymatic asymmetric hydrolysis of 1a was able to yield mono-hydrolyzed product (S)-2a efficiently, while further hydrolysis to access byproduct 3a could be suppressed. It is noteworthy to mention that the active proton in phenylacetaldehyde and acetylacetone probably comes from the hydroxyl group in their enolate tautomers. Finally, further reaction outcomes demonstrate that an additive of base such as Na2CO3 or NaHCO3 could promote the reaction, thereby increasing the yield to 94% with 97% ee in the presence of NaHCO3 (Scheme 3 and Table S3 in Supporting information).
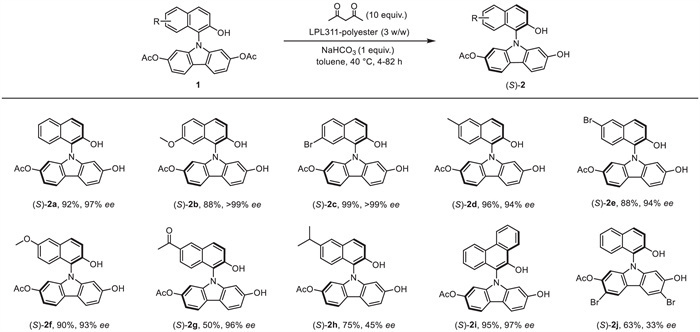
With the optimal conditions in hand, we set out to study the scope of the lipase-catalyzed hydrolytic desymmetrization of N-naphthol-carbazole diacetates 1 (Scheme 4). We first examined substrates bearing a substituent at the C6- or C7-position on the naphthol moiety, such as a bromo, methyl, or methoxy group, affording the desired C−N axially chiral products (S)-2a-f in 88%−99% yields with excellent enantioselectivity (93%−99% ee). However, when an acetyl group was introduced at the C6-position on the naphthalene ring, the yield of (S)-2g dropped to 50% with 96% ee. Notably, when a bulky group (i-Pr) was installed at the C6-position on the naphthalene, the reaction exhibited reduced enantioselectivity control, and a similar phenomenon could be found with two bromo groups on the carbazole ring, as shown by the formation of (S)-2j in 63% yield, with only 33% ee. The hydrolytic reaction ran equally well when phenanthracol-based carbazole 1i was used as the substrate, yielding the corresponding product (S)-2i in 95% yield with 97% ee.
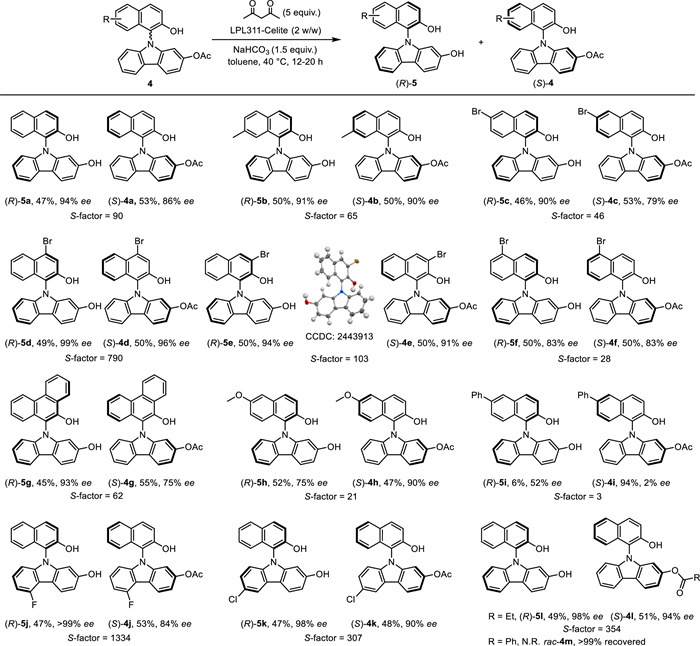
After successfully realizing the lipase-catalyzed chemo- and enantioselective desymmetrization process, we turned to investigate the kinetic resolution (KR) of C1-symmetric N-naphthol-carbazoles 4 (Scheme 5). Given the high catalytic activity of LPL311-polyester bringing obstacles to determine the reaction endpoint, we opted for LPL311-Celite to achieve enantioselective hydrolysis of C1-symmetric substrates 4 (for more information of condition optimizations, see Table S4 in Supporting information). Gratifyingly, the KR strategy turned out to be effective for most substrates with high selectivity (S-factor ≥ 21). When a methyl group was introduced at the C7-position on the naphthene ring, the KR of 4b provided a S-factor of 65, delivering (R)-5b in 50% yield with 91% ee, while (S)-4b was recovered in 50% yield with 90% ee. Notably, substrates bearing a bromo substituent at the C3-, C4, -C5, or C6-position were well tolerated as shown by the KR outcomes using 4e, 4d, 4f, and 4c with good S-factors (28–1334). N-Phenanthracol-carbazole 4 g, bearing one more fused benzene ring, was conducted under the standard conditions, leading to (R)-5g in 45% yield with 93% ee, and (S)-4g in 55% yield with 75% ee. The absolute configuration of (R) for the hydrolyzed enantioenriched N-naphthol-carbazoles 5 was determined by the single-crystal X-ray diffraction study of (R)-5d Methoxyl group was also proven to be compatible at the C6-position (S-factor = 21), however the detected S-factor would be significantly decreased to 3 when installing one phenyl group at the same position. It is worth emphasizing that, a fluor substituent was involved to the carbazole ring to favor the enantioselectivity as shown by the formation of (R)-5j in 47% yield with > 99% ee, and (S)-4j in 53% yield with 84% ee, providing the highest S-factor of 1334. The substrate of rac-4k bearing a chlorine atom on the carbazole ring turned out to be compatible with excellent enantioselectivity (S-factor = 307). However, introducing an isopropyl group at the same place leads to a significant decrease on the obtained S-factor (< 5 for rac-4k'). A similar phenomenon was observed in the reaction of rac-4l, bearing one more carbon in the acyl group. However, the KR of 4m, which bears a benzoyl group becomes inactive under enzyme catalysis, and 4m was quantitively recovered (99%) in a racemic form.
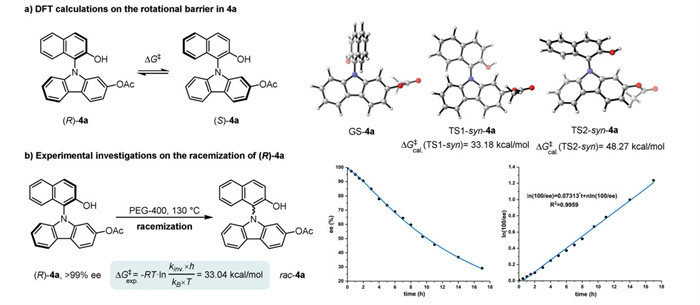
To further elucidate the racemization mechanism, we employed density functional theory (DFT) calculations [57-59] on the rotational barrier in 4a. Through structural optimizations, we identified two transition states (Scheme 6a), i.e., TS1 (planar) and TS2 (sandwich-like), both of which are the key transition states in the racemization of 4a. Notably, the rotational barrier (ΔG‡) via the planar TS1 was calculated to be 33.18 kcal/mol, while another one via the dearomatized TS2 exhibited a much higher energy level (48.27 kcal/mol). To investigate the nature of C−N axially chiral biaryls, experimental investigations on the racemization of (R)-4a (> 99% ee) was subsequently operated (Scheme 6b). The rotational barrier (ΔG‡) was tested to be 33.04 kcal/mol according to the Eyring Equation [60]. The experimental results are highly consistent with that disclosed by the DFT calculations via the planar transition state (TS1), reflecting the reliability of the mechanism of racemization.
In summary, we have developed an enzymatic tool for the construction of C−N atropisomers via lipase-catalyzed desymmetrization and kinetic resolution of N-naphthol-carbazoles. Acetylacetone was found as an efficient proton donor under LPL311-polyester, an immobilized lipase catalyst onto methacrylate-based polymer resins. The enantioselective deacylation process has been demonstrated with good functional group tolerance, offering a metal-free approach to directly provide C−N skeletons with good to excellent enantioselectivity (S-factors up to 1334). The rotational barrier in 4a was calculated to be 33.18 kcal/mol via the planar transition state (TS1), which is highly consistent with experimental data (33.04 kcal/mol). The two hydroxyl groups in enantioenriched difunctionalized C−N atropisomers provide feasibility for subsequent transformations. Finally, the findings presented herein offer novel insights into the design of dynamic kinetic resolution (DKR) [61-67] by merging with an efficient and compatible racemization system. Further studies on the DKR development are currently underway in our laboratory [68,69].
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Dingkai Lou: Writing – original draft, Methodology, Investigation. Haozhe Wu: Methodology, Investigation. Can Zhu: Writing – review & editing, Supervision, Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Conceptualization.
We acknowledge the National Natural Science Foundation of China (No. 22271054), the "1000-Youth Talents Plan", Sinopec Seeding Program (No. ZC0607-0258), and Fudan University (start-up grant) for financial support. The project was also supported by Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University, and Open Project of State Key Laboratory of Synergistic Chem-Bio Synthesis.
Supplementary material associated with this article can be found, in the online version, at doi:
E. Kumarasamy, R. Raghunathan, M.P. Sibi, J. Sivaguru, Chem. Rev. 115 (2015) 11239–11300. doi: 10.1021/acs.chemrev.5b00136
Y.B. Wang, B. Tan, Acc. Chem. Res. 51 (2018) 534–547. doi: 10.1021/acs.accounts.7b00602
J.K. Cheng, S.H. Xiang, S. Li, L. Ye, B. Tan, Chem. Rev. 121 (2021) 4805–4902. doi: 10.1021/acs.chemrev.0c01306
G.J. Mei, W.L. Koay, C.Y. Guan, Y. Lu, Chem 8 (2022) 1855–1893. doi: 10.1016/j.chempr.2022.04.011
A.M. Faisca Phillips, A.J.L. Pombeiro, Symmetry 15 (2023) 1261. doi: 10.3390/sym15061261
J. Moon, S. Kim, S. Lee, et al., ChemCatChem 16 (2024) e202400690. doi: 10.1002/cctc.202400690
L.H. Bock, R. Adams, J. Am. Chem. Soc. 53 (1931) 374–376. doi: 10.1021/ja01352a055
O. Kitagawa, H. Izawa, T. Taguchi, M. Shiro, Tetrahedron Lett. 38 (1997) 4447–4450. doi: 10.1016/S0040-4039(97)00928-3
T. Hata, H. Koide, N. Taniguchi, M. Uemura, Org. Lett. 2 (2000) 1907–1910. doi: 10.1021/ol0001057
I. Abdellah, N. Debono, Y. Canac, L. Vendier, R. Chauvin, Chem. Asian J. 5 (2010) 1225–1231. doi: 10.1002/asia.200900663
G. Bringmann, T. Gulder, B. Hertlein, Y. Hemberger, F. Meyer, J. Am. Chem. Soc. 132 (2010) 1151–1158. doi: 10.1021/ja9097687
G. Bringmann, S. Tasler, H. Endress, et al., J. Am. Chem. Soc. 123 (2001) 2703–2711. doi: 10.1021/ja003488c
K.C. Nicolaou, N.L. Simmons, J.S. Chen, N.M. Haste, V. Nizet, Tetrahedron Lett. 52 (2011) 2041–2043. doi: 10.1016/j.tetlet.2010.09.059
J. Wang, W. Zeng, S. Li, et al., ACS Med. Chem. Lett. 8 (2017) 299–303. doi: 10.1021/acsmedchemlett.6b00465
J. Chandrasekhar, R. Dick, J. Van Veldhuizen, et al., J. Med. Chem. 61 (2018) 6858–6868. doi: 10.1021/acs.jmedchem.8b00797
K.B. Gan, R.L. Zhong, Z.W. Zhang, et al., J. Am. Chem. Soc. 144 (2022) 14864–14873. doi: 10.1021/jacs.2c06240
N. Ototake, Y. Morimoto, A. Mokuya, et al., Chem. Eur. J. 16 (2010) 6752–6755. doi: 10.1002/chem.201000243
G. Onodera, M. Suto, R. Takeuchi, J. Org. Chem. 77 (2012) 908–920. doi: 10.1021/jo202083z
L. Zhang, J. Zhang, J. Ma, D.J. Cheng, B. Tan, J. Am. Chem. Soc. 139 (2017) 1714–1717. doi: 10.1021/jacs.6b09634
Y.B. Wang, S.C. Zheng, Y.M. Hu, et al., Nat. Commun. 8 (2017) 15489. doi: 10.1038/ncomms15489
Y. Kwon, A.J. Chinn, B. Kim, S.J. Miller, Angew. Chem. Int. Ed. 57 (2018) 6251–6255. doi: 10.1002/anie.201802963
X. Fan, X. Zhang, C. Li, Z. Gu, ACS Catal. 9 (2019) 2286–2291. doi: 10.1021/acscatal.8b04789
Y. Kwon, J. Li, J.P. Reid, et al., J. Am. Chem. Soc. 141 (2019) 6698–6705. doi: 10.1021/jacs.9b01911
L. Wang, J. Zhong, X. Lin, Angew. Chem. Int. Ed. 58 (2019) 15824–15828. doi: 10.1002/anie.201909855
N. Man, Z. Lou, Y. Li, et al., Org. Lett. 22 (2020) 6382–6387. doi: 10.1021/acs.orglett.0c02214
Q.J. An, W. Xia, W.Y. Ding, et al., Angew. Chem. Int. Ed. 60 (2021) 24888–24893. doi: 10.1002/anie.202111251
Z.S. Liu, P.P. Xie, Y. Hua, et al., Chem 7 (2021) 1917–1932. doi: 10.1016/j.chempr.2021.04.005
L. Sun, H. Chen, B. Liu, et al., Angew. Chem. Int. Ed. 60 (2021) 8391–8395. doi: 10.1002/anie.202012932
P. Zhang, X.M. Wang, Q. Xu, et al., Angew. Chem. Int. Ed. 60 (2021) 21718–21722. doi: 10.1002/anie.202108747
V. Thönnißen, I.L. Atodiresei, F.W. Patureau, Chem. Eur. J. l 29 (2023) e202300279. doi: 10.1002/chem.202300279
P. Zhang, C.Q. Guo, W. Yao, et al., ACS Catal. 13 (2023) 7680–7690. doi: 10.1021/acscatal.3c00732
S.P. Zhu, W.Y. Wang, K. Fang, et al., Chin. Chem. Lett. 25 (2014) 229–233. doi: 10.1016/j.cclet.2013.10.022
M.E. Diener, A.J. Metrano, S. Kusano, S.J. Miller, J. Am. Chem. Soc. 137 (2015) 12369–12377. doi: 10.1021/jacs.5b07726
J. Rae, J. Frey, S. Jerhaoui, et al., ACS Catal. 8 (2018) 2805–2809. doi: 10.1021/acscatal.7b04343
W. Xia, Q.J. An, S.H. Xiang, et al., Angew. Chem. Int. Ed. 59 (2020) 6775–6779. doi: 10.1002/anie.202000585
J. Frey, A. Malekafzali, I. Delso, et al., Angew. Chem. Int. Ed. 59 (2020) 8844–8848. doi: 10.1002/anie.201914876
U. Dhawa, T. Wdowik, X. Hou, et al., Chem. Sci. 12 (2021) 14182–14188. doi: 10.1039/d1sc04687j
L.K. Verdhi, N. Fridman, A.M. Szpilman, Org. Lett. 24 (2022) 5078–5083. doi: 10.1021/acs.orglett.2c01860
P. Rodríguez-Salamanca, R. Fernández, V. Hornillos, J.M. Lassaletta, Chem. Eur. J. 28 (2022) e202104442. doi: 10.1002/chem.202104442
Y.J. Wu, G. Liao, B.F. Shi, Green Synth. Catal. 3 (2022) 117–136.
J. Feng, C.J. Lu, R.R. Liu, Acc. Chem. Res. 56 (2023) 2537–2554. doi: 10.1021/acs.accounts.3c00419
C.J. Lu, Q. Xu, J. Feng, et al., Angew. Chem. Int. Ed. 62 (2023) e202216863. doi: 10.1002/anie.202216863
J. Huang, H. Yang, X. Chen, et al., Chem 11 (2025) 102439. doi: 10.1016/j.chempr.2025.102439
J. Zhang, Q. Xu, J. Wu, J. Fan, M. Xie, Org. Lett. 21 (2019) 6361–6365. doi: 10.1021/acs.orglett.9b02243
S. Zhang, Q.J. Yao, G. Liao, et al., ACS Catal. 9 (2019) 1956–1961. doi: 10.1021/acscatal.8b04870
U. Dhawa, T. Wdowik, X. Hou, et al., Chem. Sci. 12 (2021) 14182–14188. doi: 10.1039/d1sc04687j
L.K. Verdhi, N. Fridman, A.M. Szpilman, Org. Lett. 24 (2022) 5078–5083. doi: 10.1021/acs.orglett.2c01860
M. Ishida, R. Adachi, K. Kobayashi, et al., Chem. Commun. 60 (2024) 678–681. doi: 10.1039/d3cc05447k
V. Thönnißen, J. Westphäling, I.L. Atodiresei, F.W. Patureau, Chem. Eur. J. 30 (2024) e202304378. doi: 10.1002/chem.202304378
T. Zhang, C. Zhu, Synlett 35 (2023) 1170–1174. doi: 10.3390/e25081170
K. Wang, W. Wang, D. Lou, et al., ACS Cent. Sci. 10 (2024) 2099–2110. doi: 10.1021/acscentsci.4c01370
J. Zhang, Z. Zheng, C. Zhu, Chin. Chem. Lett. 35 (2024) 109160. doi: 10.1016/j.cclet.2023.109160
P. Xue, Z.Z. Kang, X.Y. Lai, G.Q. Qu, Y.Y. Li, Chin. Chem. Lett. 24 (2013) 1112–1114. doi: 10.1016/j.cclet.2013.07.005
H.J. Wen, Q. Chen, G.J. Zheng, Chin. Chem. Lett. 26 (2015) 1431–1434. doi: 10.1016/j.cclet.2015.07.005
K.K. Kim, H.K. Song, D.H. Shin, K.Y. Hwang, S.W. Suh, Structure 5 (1997) 173–185. doi: 10.3348/jkrs.1997.37.1.173
K. Kasama, H. Aoyama, S. Akai, Eur. J. Org. Chem. 2020 (2020) 654–661. doi: 10.1002/ejoc.201901583
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580–592. doi: 10.1002/jcc.22885
T. Lu, Q. Chen, Comput. Theor. Chem. 1200 (2021) 113249. doi: 10.1016/j.comptc.2021.113249
T. Lu, J. Chem. Phys. 161 (2024) 082503. doi: 10.1063/5.0216272
H. Eyring, J. Chem. Phys. 3 (1935) 107–115. doi: 10.1063/1.1749604
A.L.E. Larsson, B.A. Persson, J.E. Bäckvall, Angew. Chem. Int. Ed. 36 (1997) 1211–1212. doi: 10.1002/anie.199712111
O. Verho, J.E. Bäckvall, J. Am. Chem. Soc. 137 (2015) 3996–4009. doi: 10.1021/jacs.5b01031
J. Berreur, B.S.L. Collins, J. Clayden, Dynamic kinetic resolution and Dynamic kinetic asymmetric transformations of atropisomers, in: J.E. Bäckvall, M.C. Bagley (Eds.), Science of Synthesis, Georg Thieme Verlag, Stuttgart, 2023, pp. 441–483.
J. Zhang, K. Wang, C. Zhu, JACS Au 4 (2024) 502–511. doi: 10.1021/jacsau.3c00623
J. Zhang, X. Huo, Y. Liu, C. Zhu, Chem Catal. 5 (2025) 101329.
Y. Wu, C. Zhu, Chem. Commun. 61 (2025) 5228–5233. doi: 10.1039/d5cc01026h
R.C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, et al., Chem. Soc. Rev. 42 (2013) 6290–6307. doi: 10.1039/C2CS35231A
A. Zhou, M.M.K. Amer, Q. Yin, Chin. Chem. Lett. 37 (2026) 111929. doi: 10.1016/j.cclet.2025.111929
H. Gong, H. Zhuang, Y. Xiao, et al., Chin. J. Chem. 43 (2025) 2318-2324. doi: 10.1002/cjoc.70120

Scheme 3 Hydrolytic desymmetrization of N-naphthol-carbazole diacetate 1a: screening of proton sources. The reaction was carried out under an air atmosphere with N-naphthol-carbazole 1a (0.10 mmol), LPL311-polyester (3 w/w), and proton source (1.0 mmol) in toluene (1.0 mL) at 40 ℃ for 24 h. The yield was determined by 1H NMR analysis using CH2Br2 as the internal standard; enantiomeric excess (ee) was determined by chiral HPLC. a Na2CO3 (0.10 mmol) was added. b NaHCO3 (0.10 mmol) was added.

Scheme 4 Substrate scope of the lipase-catalyzed hydrolytic desymmetrization of diacetates 1. The reaction was carried out under an air atmosphere with Cs–sym N-naphthol-carbazole 1 (0.15 mmol), LPL311-polyester (3 w/w), NaHCO3 (0.15 mmol) and acetylacetone (1.5 mmol) in toluene (1.5 mL) at 40 ℃ for 4–82 h. Enantiomeric excess (ee) was determined by chiral HPLC.

Scheme 5 Substrate scope of the enzymatic kinetic resolution of 4. The reaction was carried out under an air atmosphere with C1-symmetric N-naphthol-carbazole 3 (0.20 mmol), LPL311-Celite (2 w/w), NaHCO3 (0.30 mmol), and acetylacetone (1.0 mmol) in toluene (2.0 mL) at 40 ℃ for 12–20 h. Enantiomeric excess (ee) was determined by chiral HPLC. S-factors were calculated according to the equation: S = ln[(1-c) × (1-ees)]/ln[(1-c) × (1+ees)], in which c = ees/(ees + eep). N.R. = no reaction.

Scheme 6 (a) Structure and the rotational barrier of 4a computed at B3LYP-D3/6-31G(d,p) (scrf = 1,2-ethanediol)//B3LYP-D3/def2-TZVP. (b) Experimental investigations on the racemization of (R)-4a.
Table 1. Hydrolytic desymmetrization of N-naphthol-carbazole diacetate 1a: Optimization of reaction conditions.a
|
||||||
| Entry | Lipase | Solvent | Yield of (S)-2a (%)b | ee of (S)-2a (%)c | Yield of 3a (%)b | Recovery of 1a (%)b |
| 1 | Lipase AK/PS/PPL/CALB | Toluene | N.D. | – | – | – |
| 2 | Lipase-TL | Toluene | Trace | – | – | 98 |
| 3 | Lipase-AYS | Toluene | Trace | – | – | > 99 |
| 4 | Lipozyme-RM | Toluene | 72 | 80 | 7 | 18 |
| 5 | LPL311-powderd | Toluene | 60 | 71 | 22 | 15 |
| 6 | LPL311-Celite | Toluene | 60 | 94 | 3 | 37 |
| 7 | LPL311-polyester | Toluene | 80 | 96 | Trace | 18 |
| 8 | LPL311-polyester | PhCF3 | 77 | 89 | Trace | 17 |
| 9 | LPL311-polyester | PhCl | 76 | 92 | 3 | 22 |
| 10 | LPL311-polyester | Benzene | 82 | 94 | – | – |
| 11 | LPL311-polyester | Anisole | Trace | 82 | – | 94 |
| 12 | LPL311-polyester | CH3CN | N.D. | – | – | – |
| 13 | LPL311-polyester | THF | Trace | – | – | – |
| 14 | LPL311-polyester | DCM | Trace | – | – | – |
| a The reaction was carried out under an air atmosphere with N-naphthol-carbazole 1a (0.10 mmol), lipase (3 w/w), and CH3OH (1.0 mmol) in indicated solvent (1.0 mL) at 40 ℃ for 24 h. b The yield was determined by 1H NMR analysis using CH2Br2 as the internal standard. c Enantiomeric excess (ee) was determined by chiral HPLC. d The reaction was carried out with N-naphthol-carbazole 1a (0.10 mmol), lyophilized powder LPL311 (5%) and CH3OH (1.0 mmol) in toluene (1.0 mL) at 40 ℃ for 24 h. |
||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: