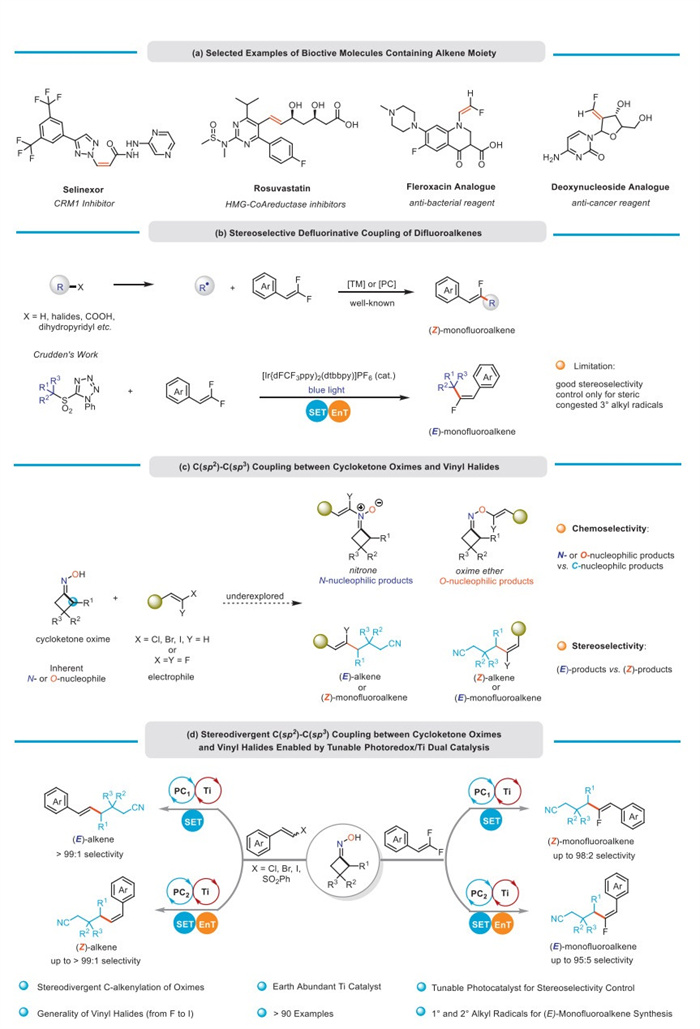
Scheme 1.
Stereodivergent C(sp2)–C(sp3) coupling between cycloketone oximes and vinyl halides enabled by tunable photoredox/Ti dual catalysis.
Stereodivergent reductive C(sp2)-C(sp3) coupling between cycloketone oximes and vinyl halides enabled by tunable photoredox/Ti dual catalysis
Huai-Gui Li , Can-Ming Zhu , Weidong Yuan , Hongyi Chen , Zhengyuan Bo , Chao Deng , Yingguang Zhu , Kang Chen , Qing-Yuan Meng

Alkenes are ubiquitous structural units in organic molecules, including natural products, pharmaceuticals, and feedstock chemicals [1,2]. They are also recognized as privileged synthetic building blocks that can be readily converted into various functional groups [3-5]. Since the different configurations of alkenes significantly impact their bioactivity (Scheme 1a) [6-9], it is of great value for a precise manipulation of alkene configurations in both organic synthesis and pharmaceutical discovery. Vinyl halides are widely employed as important electrophilic coupling reagents in alkenylation reactions, predominantly yielding thermodynamically stable E-alkenes [10,11]. However, the stereodivergent synthesis of alkenes from vinyl halides, which combine the coupling with vinyl halides and the E/Z photoisomerization of alkene products [12-20], is still inadequately explored to date.
Particularly, gem–difluoroalkenes, a unique class of vinyl halides, are synthetically useful substrates for the synthesis of thermodynamically stable Z-monofluoroalkenes through defluorinative couplings with diverse radical precursors [21-29]. In contrast, the formation of thermodynamically unfavorable E-selective isomers has been rarely reported. An exemplary case of selective E-monofluoroalkene synthesis through defluorinative coupling between gem–difluoroalkenes and redox-active sulfone derivatives has been presented by Crudden and colleagues (Scheme 1b) [30]. Nonetheless, achieving high stereoselectivity for E-monofluoroalkenes in this transformation has been limited to sterically congested 3° alkyl radical precursors as coupling partners. Given that monofluoroalkenes serve as bioisosteres of the amide functional group [31], enhancing the bioactivity and metabolic stability of organic molecules, there is a strong demand for developing novel methods that enable stereodivergent synthesis of monofluoroalkenes from gem–difluoroalkenes under mild conditions. Cycloketone oxime esters and ethers are frequently utilized as crucial precursors for constructing cyano-substituted scaffolds in a cyanide-free manner [32-37]. While the prefunctionalization of cycloketone oximes with redox-active auxiliaries can promote N–O homolysis to initiate the ring-opening process, directly using unprotected cycloketone oximes as substrates would significantly reduce waste generation, enhancing step- and atom-economy in these transformations. However, in the absence of redox-active auxiliaries, cycloketone oximes possess highly negative reduction potentials (Ered < −3.0 V vs. SCE), rendering N–O homolysis via a single electron transfer (SET) reduction process impractical. Furthermore, oximes inherently function as N- and O-nucleophiles, potentially producing nitrones (N-nucleophilic products) and oxime ethers (O-nucleophilic products) as byproducts when reacting with electrophilic coupling reagents [38]. To achieve stereodivergent coupling between cycloketone oximes and vinyl halides, an optimal catalytic system should not only generate nucleophilic C-centered radicals from cycloketone oximes in high chemoselectivity, but also facilitate the facile modulation of E/Z products through minor adjustments of the catalytic components (Scheme 1c).
To address aforementioned issues, we pay our attention to the earth-abundant early transition metal Ti [39,40]. As known, the TiⅣ compound could be readily converted to TiⅢ species under photoinduced SET reduction [41-45]. Subsequently, the highly oxophilic TiⅢ species would facilitate the N–O homolysis of cycloketone oximes (BDE: Ti–O bond 112 kcal/mol; N–O bond 44 kcal/mol) [46,47], which eventually gives the desired cyanoalkyl radicals. In recent years, the cooperative metallaphotoredox catalysis has significantly enriched the chemistry of stereodivergent alkene synthesis. Despite well-established catalytic systems involving late transition metals (such as Pd [48,49], Ni [50-54], and Cu [55-57]), the early transition metal catalyzed stereodivergent synthesis of alkenes, especially for monofluoroalkenes, still remains underdeveloped. Herein, we report a stereodivergent C(sp2)–C(sp3) coupling between cycloketone oximes and vinyl halides via tunable photoredox/Ti dual catalysis (Scheme 1d). The stereoselectivity of products is precisely controlled by tuning photocatalysts with different triplet energies for the triplet-triplet energy transfer (EnT). This methodology features mild reaction conditions, broad scope of vinyl halides, and good functional group tolerance, providing a unique and general platform for constructing cyano(fluoro)alkenes in both configurations.
We commenced our investigations into the photocatalytic stereodivergent protocol by using cyclic ketoxime 1a and vinyl bromide 2a as model substrates (Table 1). Combing Cp2TiCl2 as the titanocene catalyst and N,N-diisopropylethylamine (DIPEA) as the stoichiometric reductant, we were delighted to find that the exposure of reaction mixture in a DCE solution to a 9 W blue LED lamp in the presence of 4CzIPN (ET = 58.3 kcal/mol) as the photocatalyst afforded 40% yield of 3a with exclusive E selectivity (E/Z > 99:1) (Table 1, entry 1). Other photocatalysts including 4DPAIPN, [Ir(dFCF3ppy)2dtbbpy]PF6 and fac-Ir(ppy)3, resulted in lower yields and E selectivity (Table 1, entries 2–4). The substitution of titanocene catalyst with Cp2Ti(OTf)2, Cp2Ti(TFA)2, Cp*TiCl3 or CpTiCl3 led to diminished yields (Table 1, entries 5–8). After a series of modulation in solvents, catalyst loading, irradiation time, and the equivalent ratio of substrates (Table 1, entries 9–12, for more details see Table S3 and S4 in Supporting information), we finally obtained the coupling product 3a in an optimal yield (72%) and excellent E selectivity (E/Z > 99:1). Later on, further investigations on the Z-alkene formation revealed that Ir photocatalyst with lower triplet energy such as [Ir(ppy)2dtbbpy]PF6 (ET = 51.0 kcal/mol) and [Ir(dFppy)2dtbbpy]PF6 (ET = 57.1 kcal/mol) were difficult to drive the Z/E isomerization (Table 1, entries 13 and 14). To our delight, a reversal of Z/E ratio in the coupling product (4a) was obtained as the main product when [Ir(dFCF3ppy)2dtbbpy]PF6 (ET = 61.8 kcal/mol) was employed as the photocatalyst under the prolonged irradiation time (Table 1, entry 15). It was noted that different ratios of DCE and MeOH mainly influenced the yield of 4a, while the Z/E selectivity between 3a and 4a remained unchanged (Table 1, entries 16–18). Control experiments indicated the absence of photocatalyst would significantly decrease the reaction efficiency (Table 1, entry 19), and titanocene catalyst, reductant, and light irradiation were all essential for the product generation (Table 1, entries 20–22).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||||
| Entry | PC | Ti catalyst | Solvent | Time (h) | Yield (%) (E:Z) c |
| 1 | 4CzIPN | Cp2TiCl2 (10 mol%) | DCE | 24 | 40 (> 99:1) |
| 2 | 4DPAIPN | Cp2TiCl2 (10 mol%) | DCE | 24 | 34 (> 99:1) |
| 3 | [Ir(dFCF3ppy)2dtbbpy]PF6 | Cp2TiCl2 (10 mol%) | DCE | 24 | 31 (74:26) |
| 4 | fac-Ir(ppy)3 | Cp2TiCl2 (10 mol%) | DCE | 24 | 11 (75:25) |
| 5 | 4CzIPN | Cp2Ti(OTf)2 (10 mol%) | DCE | 24 | 30 (> 99:1) |
| 6 | 4CzIPN | Cp2(TFA)2 (10 mol%) | DCE | 24 | 32 (> 99:1) |
| 7 | 4CzIPN | Cp*TiCl3 (10 mol%) | DCE | 24 | 34 (> 99:1) |
| 8 | 4CzIPN | CpTiCl3 (10 mol%) | DCE | 24 | 38 (> 99:1) |
| 9 | 4CzIPN | Cp2TiCl2 (10 mol%) | DCE: MeOH (9:1) | 24 | 47 (> 99:1) |
| 10 | 4CzIPN | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 24 | 53 (> 99:1) |
| 11 | 4CzIPN | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 48 | 62 (> 99:1) |
| 12b | 4CzIPN | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 48 | 72 (> 99:1) |
| 13b | [Ir(ppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 72 | 68 (> 99:1) |
| 14b | [Ir(dFppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 72 | 59 (50:50) |
| 15b | [Ir(dFCF3ppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 72 | 59 (30:70) |
| 16b,d | [Ir(dFCF3ppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 72 | 56 (22:78) |
| 17b,d | [Ir(dFCF3ppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (4:1) | 72 | 72 (22:78) |
| 18b,d | [Ir(dFCF3ppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (3:1) | 72 | 62 (22:78) |
| 19b | — | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 48 | 26 (> 99:1) |
| 20b | 4CzIPN | — | DCE: MeOH (9:1) | 48 | n.d. |
| 21b, e | 4CzIPN | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 48 | n.d. |
| 22b, f | 4CzIPN | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 48 | n.d. |
| a Reactions were performed with 1a (0.40 mmol), 2a (E:Z = 92:8) (0.20 mmol), photocatalyst (2.0 mol%), Cp2TiCl2 (10 mol%), and DIPEA (0.24 mmol) in solvent at room temperature under N2 atmosphere and the irradiation of a 9 W blue LED lamp. Isolated yield based on 2a. n.d.= not detected. b Reactions were performed with 1a (0.20 mmol) and 2a (0.40 mmol). c The E/Z ratio of products were determined by 1H NMR analysis of the crude reaction mixture. d 15 W blue LED lamp was used. e No DIPEA. f In the dark. |
|||||
With the optimal conditions in hand, we further explored the substrate scope of stereodivergent reductive C(sp2)-C(sp3) coupling between cycloketone oximes and vinyl halides enabled by tunable photo/Ti dual catalysis. As outlined in Scheme 2, vinyl electrophiles with various leaving groups (Br, Cl, I and PhSO2) are all suitable for this transformation, providing the (E)-alkene 3a in moderate to good yields (Method A). A broad range of vinyl bromides bearing both electron-rich and electron-deficient aromatic groups at para- or ortho-position on the aromatic rings successfully underwent the desired reductive C(sp2)-C(sp3) coupling, yielding structurally diverse cyano-functionalized alkenes with synthetically useful yields and excellent E selectivity (3b-3h, 50%−69% yields, E/Z > 99:1). Substrates containing di-substituted aryl groups, naphthalene ring, and heteroaryl motifs were all well-tolerated under standard conditions (3i-3q, 50%−77% yields, E/Z > 99:1). Gratifyingly, this protocol was adaptable to the late-stage functionalization of complex molecules derived from pharmaceutical compounds such as diflunisal and febuxostat (3r and 3s). In addition, the diphenyl-substituted vinyl bromide could smoothly participate in this coupling reaction (3t). Next, the scope of cycloketone oximes were explored with bromostyrene as the coupling reagent. Oximes featuring various heteroatoms, as well as diverse functional groups at a- or β-positions were all viable partners for this reaction, delivering the corresponding products with promising yields (3u-3z, 40%−70% yields, E/Z > 99:1). Either spiroring-containing oxime or bicyclic oxime could also efficiently participate in this transformation (3aa and 3ab). Furthermore, the less strained five-membered dihydrofuranone oxime could undergo such a ring-opening reductive C(sp2)-C(sp3) coupling, affording the desired product 3ac in a moderate yield.
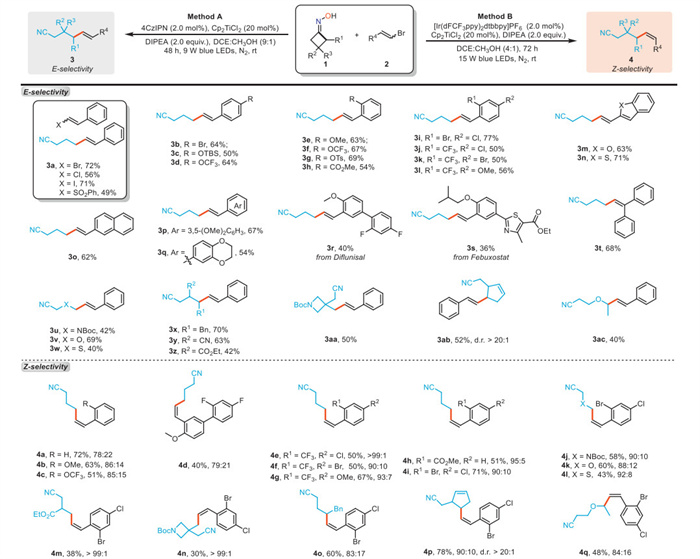
We subsequently examined the substrate scope with respect to Z-selectivity using an Ir-based photocatalyst, which facilitated a reaction pathway involving both C(sp2)-C(sp3) coupling and triplet-triplet energy transfer induced Z/E isomerization (Method B). Vinyl bromides with electron-donating substituents at ortho-position exhibited moderate Z- selectivity in the coupling reaction (4b-4d). Notably, bromostyrenes with electron-withdrawing groups such as CF3, ester, and halogen atoms mainly afforded the corresponding cross-coupling products 4e-4i in synthetically useful yields and good to excellent Z-selectivity (50%−71% yields, Z/E = 90:10 to > 99:1). The coupling products of oximes bearing various different heteroatoms reserved good Z selectivity in this transformation (4j-4l). The β-substituted oximes displayed excellent stereoselectivity under recent conditions (4m and 4n, Z/E > 99:1). Oximes which generated secondary alkyl radicals via ring opening worked smoothly in this transformation as well (4o-4q).
The stereodivergent defluorinative C(sp2)-C(sp3) coupling between gem–difluoroalkenes and cyclic ketoximes was further investigated (Scheme 3). Under modified conditions (Method C, see Table S7 and S8 in Supporting information for detailed screening), a broad array of gem–difluoroalkenes bearing substituents with different electronic properties managed to afford thermodynamically stable Z-monofluoroalkenes in moderate to good stereoselectivities (6a-6o, Z/E = 84:16 to 97:3). Mono-substituted aryl gem–difluoroalkenes bearing either electron-donating or electron-withdrawing group generally exhibited good Z-selectivity (6a-6f). Particularly, for 2,4-disubstuted substrates, the combination of substituents with different electronic properties would present good Z-selectivity (6h-6j). However, two electron-withdrawing groups at both ortho- and para-sites in substrates would diminish the stereoselectivity (6g and 6k). Notably, a scale-up reaction managed to produce 6h in 60% yield and 97:3 Z/E ratio. A variety of functional groups such as ester, ether, sulfonyl group, CF3 group, halogen atoms, and heteroaryl rings were all compatible in this reaction system. The late-stage functionalization of gem–difluoroalkenes derived from difunisal and febuxostat was also readily achieved (6p and 6q). Meanwhile, the difluoroalkene bearing two aryl rings worked quite smoothly (6r). The effects of substitution patterns on the cycloketone oximes were also examined, confirming their suitability for this reaction. A variety of cycloketone-derived oximes bearing functionalities at either α- or β-positions were successfully transformed into the desired Z-monofluoroalkenes in moderate to good yields and stereoselectivity (6s-6w, Z/E = 83:17 to 92:8). Particularly, oximes with bicyclic or spiroring motifs as well as five-membered ring exhibited high Z-selectivity (6x-6z, Z/E > 90:10).

Finally, we turned our attention to evaluating the scope of gem–difluoroalkenes and cycloketone oximes for E-selective monofluoroalkene synthesis (Method D, see Table S9 in Supporting information for detailed screening). The presence of ortho-electron-withdrawing substituents such as -Br, -CF3, -Cl, and -CO2Me on the aromatic ring of difluoroalkenes was crucial for achieving thermodynamically unfavored E-monofluoroalkenes, giving rise to the corresponding products in moderate to good stereoselectivity (7a-7e, 39%−72% yields, E/Z = 82:18 to 90:10). Oximes that generated more sterically hindered secondary cyanoalkyl radicals via ring opening demonstrated superior E-selectivity control compared to those produced primary radicals (7f-7k). To our delight, the probenecid-derived oxime underwent defluorinative coupling smoothly, delivering the E-monofluoroalkene in excellent selectivity (7l, 52% yield, E/Z = 95:5).
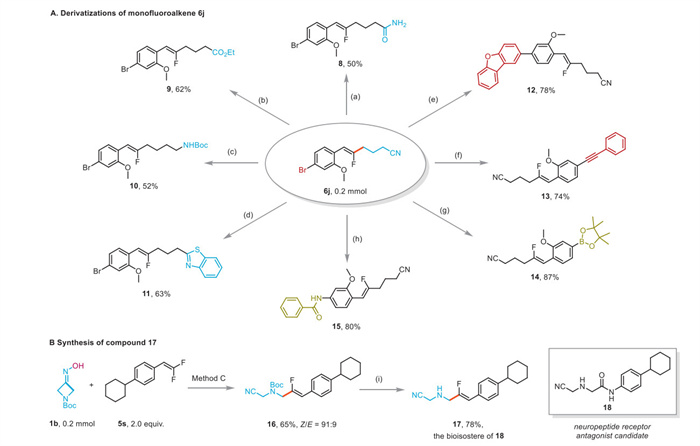
To demonstrate the synthetic application of monofluoroalkenes, the compound 6j was employed as a versatile synthetic building block in various transformations (Scheme 4). The cyano group in 6j was efficiently transformed into either an amide (8) or ester (9) under acidic conditions. Boc-protected amide 10 was obtained from 6j in a moderate yield via a tandem Ni-mediated reduction/Boc protection. The synthesis of benzothizole-containing product 11 from 6j was also achieved via Cu catalysis. In addition, the compound 6j was able to participate in Pd-catalyzed cross-coupling reactions to forge new C–C bonds, affording arylation product 12 and alkynylation product 13 in good yields, respectively. Furthermore, the success of Pd-catalyzed Miyaura borylation (14) and Cu-catalyzed Ullmann-type amidation (15) from 6j allowed for the efficient installation of hetero-atom-containing functional groups on the aryl moiety. Finally, this methodology was applied in the facile synthesis of monofluoroalkene 17, which was considered as the bioisostere of a neuropeptide receptor antagonist candidate [58].
Phytopathogenic fungi have posed a significant threat to both the quantity and quality of food production, highlighting the urgent need for novel antifungal agrochemicals. Given the widespread presence of alkene structural motifs in bioactive compounds, we conducted an in vitro evaluation of cyano-containing alkene derivatives, using the commercially available fungicide boscalid as a positive control. As outlined in Table S10 (see Supporting information for details), thermodynamically stable (E)-alkenes 3e and 3i exhibited superior inhibition rates against R. solani and C. orbiculare compared to boscalid. Conversely, their (Z)-isomers 4b and 4i exhibited slightly lower inhibition rates, indicating that the configuration of alkenes might influence their biological activity. Moreover, the diflunisal-derived alkene 3r displayed moderate inhibitive activity, while its monofluoro-substituted analog 6p showed enhanced antifungal activities, particularly against C. orbiculare. These findings revealed that the installation of fluorine atom can significantly modulate the biological activity of organic molecules. Overall, the preliminary in vitro antifungal activities of cyano-containing alkenes suggest their potential for further scaffold optimization.
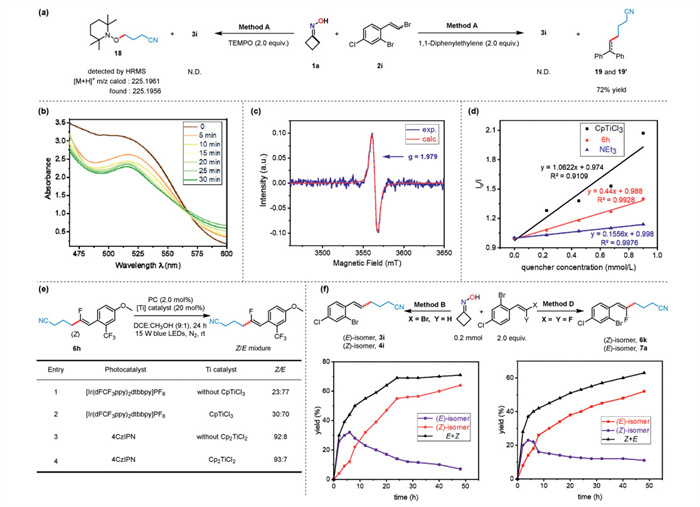
To gain deep insights into the mechanism of this stereodivergent coupling reaction, a series of mechanistic investigations were performed. The addition of radical scavengers such as TEMPO or 1, 1-diphenylethylene into reaction system completely inhibit the generation of coupling product 3i, and the corresponding radical-trapping adducts 18 and 19 were observed (Scheme 5a). These results indicated that the cyanoalkyl radical generated from the ring opening of cycloketone oximes is likely a plausible intermediate in this reaction. Upon blue light irradiation, the UV–vis absorption of a mixed solution of Ir photocatalyst, Cp2TiCl2 and i-Pr2NEt significantly decreased around 520 nm within 30 min (Scheme 5b), indicating the consumption of Cp2TiCl2 occurred. In addition, the DCE solution of 4CzIPN/Cp2TiCl2 exposed to blue light was EPR active, showing a signal consistent with the simulation of Cp2TiⅢCl (Scheme 5c), which provided additional evidence for the generation of Cp2TiⅢCl species via a single electron transfer from photocatalyst 4CzIPN to Cp2TiCl2. The Stern-Volmer quenching experiments (Scheme 5d and Fig. S7 in Supporting information) demonstrated that TiⅣ catalysts effectively quenched both excited 4CzIPN and [Ir(dFCF3ppy)2dtbbpy]PF6, indicating the coupling reaction was initiated by the SET reduction of TiⅣ catalysts. Notably, the thermodynamically stable product 6h served as a week quencher for the excited Ir photocatalyst, revealing that the energy transfer (EnT) from the triplet Ir photocatalyst (ET = 61.8 kcal/mol) to 6h (ET = 63.4 kcal/mol) might be involved in the reaction pathway. In addition, the quenching rate of [Ir(dFCF3ppy)2dtbbpy]PF6 quenched by 6h was Kq = 1.9 × 108 L mol−1 s−1, which were faster than that of 4CzIPN quenched by 6h (Kq = 2.7 × 107 L mol−1 s−1 (for more details see Fig. S7 in Supporting informatoin). These kinetic data supported that [Ir(dFCF3ppy)2dtbbpy]PF6 was a more efficient EnT photocatalyst than 4CzIPN for the E/Z isomerization of 6h. It was interesting to find that the presence of Ti catalysts in control reactions had no significant effect on the E/Z ratio of monofluoroalkenes in the isomerization of a stereochemically pure (Z)-monofluoroalkene 6h (Scheme 5e), despite the fact that Ti catalysts were shown to be more competitive quenchers than 6h. Furthermore, kinetic profiles clearly unveiled the thermodynamically stable (E)-alkene 3i (ET = 62.1 kcal/mol) and (Z)-monofluoroalkene 6k (ET = 62.2 kcal/mol) were observed as predominant products in the early stage of coupling reactions (Scheme 5f). As the reaction progressed, the amounts of corresponding thermodynamically unstable (Z)-alkene 4i and (E)-monofluoroalkene 7a increased, leading to a reversal of the E/Z ratio of products. These observations suggest that the E/Z isomerization of products can be ascribed to the energy transfer process catalyzed by the photoactive Ir complex.
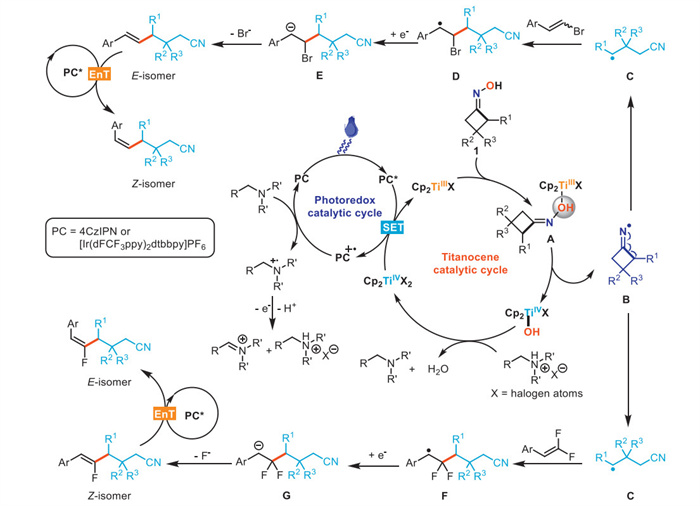
On the basis of mechanistic studies, we depicted the plausible reaction mechanism as shown in Scheme 6. Initially, the titanocene catalyst undergoes single electron transfer (SET) reduction by excited photocatalyst, resulting in the generation of a TiⅢ species. The highly oxophilic TiⅢ species would facilitate N–O homolysis of cycloketone oximes to give iminyl radical intermediate B, which is subsequently converted into cyanoalkyl radical C via a facile ring opening process. Then, the intermediate C was captured by vinyl bromides and gem–difluoroalkenes to form the benzylic radical intermediate D and F, respectively. D and F are further reduced to corresponding carboanions E and G, leading to the formation of thermodynamically favored alkene products via β-halide elimination. The photoinduced E/Z isomerization of alkenes eventually yields less thermodynamically stable isomers via triplet-triplet energy transfer between alkenes and the Ir photocatalyst.
In conclusion, we have realized a tunable photoredox/Ti dual catalyzed stereodivergent C(sp2)–C(sp3) coupling between cycloketone oximes and vinyl halides, providing a facile access to a broad scope of cyano-substituted (fluoro)alkenes with high efficiency and controlled E/Z selectivity. The success of product derivatization underscores the utility of this protocol in synthetic organic chemistry. Mechanistic investigations have revealed the generation of TiⅢ species and the origin of observed E/Z stereoselectivity. Additionally, several cyano-substituted (fluoro)alkene compounds exhibited antifungal activities, suggesting potential for further agrochemical development.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Huai-Gui Li: Writing – review & editing, Writing – original draft, Investigation, Formal analysis, Conceptualization. Can-Ming Zhu: Investigation, Formal analysis. Weidong Yuan: Investigation, Formal analysis. Hongyi Chen: Investigation. Zhengyuan Bo: Investigation. Chao Deng: Writing – review & editing, Supervision. Yingguang Zhu: Writing – review & editing, Supervision, Formal analysis. Kang Chen: Writing – review & editing, Writing – original draft, Supervision, Formal analysis, Conceptualization. Qing-Yuan Meng: Writing – review & editing, Supervision, Formal analysis, Conceptualization.
We thank the National Natural Science Foundation of China (Nos. 22101135, 22271293 and 21502096), the Natural Science Foundation of Jiangsu Province (Nos. BK20210381 and BK20150652), the Fundamental Research Funds for the Central Universities (Nos. KYQN2022058, KJQN201629, and XUEKEN2022032), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB0960000), CAS Project for Young Scientists in Basic Research (No. YSBR-050), the Chinese Academy of Sciences and the "333 High Level Talent Project" of Jiangsu Province for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
C. Dugave, L. Demange, Chem. Rev. 103 (2003) 2475–2532. doi: 10.1021/cr0104375
C. Thirsk, A. Whiting, J. Chem. Soc., Perkin Trans. 1 (2002) 999–1023.
T. Takeda, Modern Carbonyl Olefination: Methods and Applications, Wiley-VCH, 2004.
S.W.M. Crossley, C. Obradors, R.M. Martinez, R.A. Shenvi, Chem. Rev. 116 (2016) 8912–9000. doi: 10.1021/acs.chemrev.6b00334
O.M. Ogba, N.C. Warner, D.J. O'Leary, R.H. Grubbs, Chem. Soc. Rev. 47 (2018) 4510–4544. doi: 10.1039/c8cs00027a
L. Jian, R.U. Zscherp, X. Shen, et al., J. Med. Chem. 66 (2023) 11940–11950. doi: 10.1021/acs.jmedchem.3c00549
N. Vempala, S.V. Rao, A.J. Shree, B.S. Pradhan, Synthesis 48 (2016) 4167–4174. doi: 10.1055/s-0035-1562787
Y. Asahina, K. Iwase, F. Iinuma, M. Hosaka, T. Ishizaki, J. Med. Chem. 48 (2005) 3194–3202. doi: 10.1021/jm0402061
J.R. McCarthy, D.P. Matthews, D.M. Stemerick, et al., J. Am. Chem. Soc. 113 (1991) 7439–7440. doi: 10.1021/ja00019a061
S.K. Pagire, T. Föll, O. Reiser, Acc. Chem. Res. 53 (2020) 782–791. doi: 10.1021/acs.accounts.9b00615
Y. Gong, J. Hu, C. Qiu, H. Gong, Acc. Chem. Res. 57 (2024) 1149–1162. doi: 10.1021/acs.accounts.3c00810
K. Singh, S.J. Staig, J.D. Weaver, J. Am. Chem. Soc. 136 (2014) 5275–5278. doi: 10.1021/ja5019749
J.B. Metternich, R. Gilmour, J. Am. Chem. Soc. 137 (2015) 11254–11257. doi: 10.1021/jacs.5b07136
J.B. Metternich, R. Gilmour, J. Am. Chem. Soc. 138 (2016) 1040–1045. doi: 10.1021/jacs.5b12081
A. Singh, C.J. Fennel, J.D. Weaver, Chem. Sci. 7 (2016) 6796–6802. doi: 10.1039/C6SC02422J
J.J. Molloy, J.B. Metternich, C.G. Daniliuc, A.J.B. Watson, R. Gilmour, Angew. Chem. Int. Ed. 57 (2018) 3168–3172. doi: 10.1002/anie.201800286
J.J. Molloy, M. Schäfer, M. Wienhold, et al., Science 369 (2020) 302–306. doi: 10.1126/science.abb7235
J. Xu, N. Liu, H. Lv, et al., Green Chem. 22 (2020) 2739–2743. doi: 10.1039/c9gc04303a
X. Shen, C. Huang, X.A. Yuan, S. Yu, Angew. Chem. Int. Ed. 60 (2021) 9672–9679. doi: 10.1002/anie.202016941
T. Neveselý, M. Wienhold, J.J. Molloy, R. Gilmour, Chem. Rev. 122 (2022) 2650–2694. doi: 10.1021/acs.chemrev.1c00324
J. Xie, J. Yu, M. Rudolph, F. Rominger, A.S. Hashmi, Angew. Chem. Int. Ed. 55 (2016) 9416–9421. doi: 10.1002/anie.201602347
X. Lu, Y. Wang, B. Zhang, et al., J. Am. Chem. Soc. 139 (2017) 12632–12637. doi: 10.1021/jacs.7b06469
J. Li, Q. Lefebvre, H. Yang, Y. Zhao, H. Fu, Chem. Commun. 53 (2017) 10299–10302. doi: 10.1039/C7CC05758J
H.W. Du, J. Sun, Q.S. Gao, et al., Org. Lett. 22 (2020) 1542–1546. doi: 10.1021/acs.orglett.0c00134
M.Z. Lu, J. Goh, M. Maraswami, et al., Chem. Rev. 122 (2022) 17479–17646. doi: 10.1021/acs.chemrev.2c00032
Z. Wang, Y. Sun, L.Y. Shen, et al., Org. Chem. Front. 9 (2022) 853–873. doi: 10.1039/D1QO01512E
L.V. Hooker, J.S. Bandar, Angew. Chem. Int. Ed. 62 (2023) e202308880. doi: 10.1002/anie.202308880
F. Wu, X. Li, J. Chang, D. Bai, Chin. Chem. Lett. 35 (2024) 109155. doi: 10.1016/j.cclet.2023.109155
C. Liu, Y. Song, W. Ju, et al., Chin. Chem. Lett. 37 (2026) 111167. doi: 10.1016/j.cclet.2025.111167
M. Nambo, K. Ghosh, J.C.H. Yim, et al., ACS Catal. 12 (2022) 9526–9532. doi: 10.1021/acscatal.2c02233
S. Kumari, A.V. Carmona, A.K. Tiwari, P.C. Trippier, J. Med. Chem. 63 (2020) 12290–12358. doi: 10.1021/acs.jmedchem.0c00530
E.M. Dauncey, S.P. Morcillo, J.J. Douglas, N.S. Sheikh, D. Leonori, Angew. Chem. Int. Ed. 57 (2018) 744–748. doi: 10.1002/anie.201710790
X.Y. Yu, J.R. Chen, P.Z. Wang, et al., Angew. Chem. Int. Ed. 57 (2018) 738–743. doi: 10.1002/anie.201710618
J. Chen, Y.J. Liang, P.Z. Wang, et al., J. Am. Chem. Soc. 143 (2021) 13382–13392. doi: 10.1021/jacs.1c06535
D.M. Whalley, J. Seayad, M.F. Greaney, Angew. Chem. Int. Ed. 60 (2021) 22219–22223. doi: 10.1002/anie.202108240
X.Y. Yu, J.R. Chen, W.J. Xiao, Chem. Rev. 121 (2021) 506–561. doi: 10.1021/acs.chemrev.0c00030
K.A. Rykaczewski, E.R. Wearing, D.E. Blackmun, C.S. Schindler, Nat. Synth. 1 (2022) 24–36. doi: 10.1038/s44160-021-00007-y
D.S. Bolotin, N.A. Bokach, M.Y. Demakova, V.Y. Kukushkin, Chem. Rev. 117 (2017) 13039–13122. doi: 10.1021/acs.chemrev.7b00264
M. Manβen, L.L. Schafer, Chem. Soc. Rev. 49 (2020) 6947–6994. doi: 10.1039/d0cs00229a
X. Wu, Y. Chang, S. Lin, Chem 8 (2022) 1805–1821. doi: 10.1016/j.chempr.2022.06.005
Z. Zhang, R.B. Richrath, A. Gansäuer, ACS Catal. 9 (2019) 3208–3212. doi: 10.1021/acscatal.9b00787
A. Gualandi, F. Calogero, M. Mazzarini, et al., ACS Catal. 10 (2020) 3857–3863. doi: 10.1021/acscatal.0c00348
Z. Zhang, T. Hilche, D. Slak, et al., Angew. Chem. Int. Ed. 59 (2020) 9355–9359. doi: 10.1002/anie.202001508
M. Parasram, B.J. Shields, O. Ahmad, T. Knauber, A.G. Doyle, ACS Catal. 10 (2020) 5821–5827. doi: 10.1021/acscatal.0c01199
F. Li, S. Lin, Y. Chen, et al., Angew. Chem. Int. Ed. 60 (2021) 1561–1566. doi: 10.1002/anie.202010780
W. Yuan, A. Qu, Y. Li, et al., Adv. Synth. Catal. 364 (2022) 3932–3940. doi: 10.1002/adsc.202200827
H. Li, Y. Li, W. Yuan, et al., Green Synth. Catal. 5 (2024) 159–164. doi: 10.61935/acetr.2.1.2024.p159
H. Zhang, X. Huang, Adv. Synth. Catal. 358 (2016) 3736–3742. doi: 10.1002/adsc.201600704
J. Corpas, M.T. Quirós, P. Mauleón, R.G. Arrayás, J.C. Carretero, ACS Catal. 9 (2019) 10567–10574. doi: 10.1021/acscatal.9b02768
C. Zhu, H. Yue, B. Maity, et al., Nat. Catal. 2 (2019) 678–687. doi: 10.1038/s41929-019-0311-x
F. Song, F. Wang, L. Guo, et al., Angew. Chem. Int. Ed. 59 (2020) 177–181. doi: 10.1002/anie.201909543
X. Li, M. Yuan, F. Chen, et al., Chem 9 (2023) 154–169. doi: 10.1016/j.chempr.2022.09.020
J. Qin, Z. Zhang, Y. Lu, S. Zhu, L. Chu. Chem. Sci. 14 (2023) 12143–12151. doi: 10.1039/d3sc04645a
F. Ye, S. Tong, W. Yuan, ACS Catal. 14 (2024) 9655–9661. doi: 10.1021/acscatal.4c02602
M.M. Mastandrea, S. Cañellas, X. Caldentey, M.A. Pericàs, ACS Catal. 10 (2020) 6402–6408. doi: 10.1021/acscatal.0c01742
H. Zhang, C. Huang, X.A. Yuan, S. Yu, J. Am. Chem. Soc. 144 (2022) 10958–10967. doi: 10.1021/jacs.2c04040
H. Zhang, X. He, X.A. Yuan, S. Yu, ACS Catal. 13 (2023) 2857–2866. doi: 10.1021/acscatal.2c06183
R.D. Connell, T.G. Lease, G.H. Ladouceur, M.H. Osterhout, Patent, US006048900A, 2000.

Scheme 1 Stereodivergent C(sp2)–C(sp3) coupling between cycloketone oximes and vinyl halides enabled by tunable photoredox/Ti dual catalysis.

Scheme 2 Stereodivergent coupling between cycloketone oximes and vinyl halides. Reaction conditions: 1 (0.20 mmol), 2 (0.40 mmol), PC (2.0 mol%), Ti catalyst (20 mol%), and reductant (0.40 mmol) in DCE:CH3OH at room temperature under N2 atmosphere and the irradiation of a blue LED lamp unless otherwise noted. Isolated yields based on 1 were reported. The Z/E ratio of products were determined by 1H NMR analysis of the crude reaction mixture.

Scheme 3 Stereodivergent coupling between cycloketone oximes and gem–difluoroalkenes. Reaction conditions: 1 (0.20 mmol), 5 (0.40 mmol), PC (2.0 mol%), Ti catalyst (20 mol%), and reductant (0.40 mmol) in DCE:CH3OH at room temperature under N2 atmosphere and the irradiation of a blue LED lamp unless otherwise noted. Isolated yields based on 1 were reported. The Z/E ratio of products were determined by 1H NMR analysis of the crude reaction mixture. a5.0 mmol scale reaction.

Scheme 4 Synthetic applications. (a) HCl, 40 ℃, 0.5 h. (b) TMSCl, EtOH, 60 ℃, 12 h. (c) NiCl2, (Boc)2O, NaBH4, MeOH, r.t., 36 h. (d) Cu(OAc)2, Et3N, 2-aminobenzenethiol, EtOH, 75 ℃, 24 h. (e) Pd(PPh3)4, dibenzofuran-2-ylboronic acid, K2CO3, THF:H2O (10:1), 80 ℃. (f) Pd(PPh3)4, CuI, ethynylbenzene, Et3N, 80 ℃, 24 h. (g) Pd(PPh3)4, B2pin2, CH3COOK, DMSO, 80 ℃, 24 h, N2. (h) (1R, 2R)-(-)-1,2-diaminocyclohexane, CuI, benzamide, K3PO4, 110 ℃, dioxane. (i) CF3COOH, r.t., 2 h, then NaOH (aq.).

Scheme 5 Mechanistic investigations. (a) Radical trapping reactions. (b) UV–vis absorption spectrum of Cp2TiCl2 in presence of Ir photocatalyst and i-Pr2NEt. (c) EPR experiment. (d) Stern-Volmer quenching experiments. (e) Control experiments. (f) Kinetic profiles for E/Z isomerization of coupling products.
Table 1. Optimization of reaction conditions.a
|
|||||
| Entry | PC | Ti catalyst | Solvent | Time (h) | Yield (%) (E:Z) c |
| 1 | 4CzIPN | Cp2TiCl2 (10 mol%) | DCE | 24 | 40 (> 99:1) |
| 2 | 4DPAIPN | Cp2TiCl2 (10 mol%) | DCE | 24 | 34 (> 99:1) |
| 3 | [Ir(dFCF3ppy)2dtbbpy]PF6 | Cp2TiCl2 (10 mol%) | DCE | 24 | 31 (74:26) |
| 4 | fac-Ir(ppy)3 | Cp2TiCl2 (10 mol%) | DCE | 24 | 11 (75:25) |
| 5 | 4CzIPN | Cp2Ti(OTf)2 (10 mol%) | DCE | 24 | 30 (> 99:1) |
| 6 | 4CzIPN | Cp2(TFA)2 (10 mol%) | DCE | 24 | 32 (> 99:1) |
| 7 | 4CzIPN | Cp*TiCl3 (10 mol%) | DCE | 24 | 34 (> 99:1) |
| 8 | 4CzIPN | CpTiCl3 (10 mol%) | DCE | 24 | 38 (> 99:1) |
| 9 | 4CzIPN | Cp2TiCl2 (10 mol%) | DCE: MeOH (9:1) | 24 | 47 (> 99:1) |
| 10 | 4CzIPN | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 24 | 53 (> 99:1) |
| 11 | 4CzIPN | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 48 | 62 (> 99:1) |
| 12b | 4CzIPN | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 48 | 72 (> 99:1) |
| 13b | [Ir(ppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 72 | 68 (> 99:1) |
| 14b | [Ir(dFppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 72 | 59 (50:50) |
| 15b | [Ir(dFCF3ppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 72 | 59 (30:70) |
| 16b,d | [Ir(dFCF3ppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 72 | 56 (22:78) |
| 17b,d | [Ir(dFCF3ppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (4:1) | 72 | 72 (22:78) |
| 18b,d | [Ir(dFCF3ppy)2dtbbpy]PF6 | Cp2TiCl2 (20 mol%) | DCE: MeOH (3:1) | 72 | 62 (22:78) |
| 19b | — | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 48 | 26 (> 99:1) |
| 20b | 4CzIPN | — | DCE: MeOH (9:1) | 48 | n.d. |
| 21b, e | 4CzIPN | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 48 | n.d. |
| 22b, f | 4CzIPN | Cp2TiCl2 (20 mol%) | DCE: MeOH (9:1) | 48 | n.d. |
| a Reactions were performed with 1a (0.40 mmol), 2a (E:Z = 92:8) (0.20 mmol), photocatalyst (2.0 mol%), Cp2TiCl2 (10 mol%), and DIPEA (0.24 mmol) in solvent at room temperature under N2 atmosphere and the irradiation of a 9 W blue LED lamp. Isolated yield based on 2a. n.d.= not detected. b Reactions were performed with 1a (0.20 mmol) and 2a (0.40 mmol). c The E/Z ratio of products were determined by 1H NMR analysis of the crude reaction mixture. d 15 W blue LED lamp was used. e No DIPEA. f In the dark. |
|||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: