State Key Laboratory of Elemento-Organic Chemistry, Research Institute of Elemento-Organic Chemistry, Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai University, Tianjin 300071, China
* Corresponding author. E-mail address: wangqm@nankai.edu.cn (Q. Wang). 1 These authors contributed equally to this work.
Received Date:
21 June 2025 Accepted Date:
09 October 2025 Revised Date:
24 September 2025 Available Online:
15 July 2026
Abstract:gem–Difluorovinyl compounds and silicon-containing compounds are valuable structural units in organic and medicinal chemistry. However, they are mostly found separately in most compounds. In this study, we present a photocatalytic approach for the synthesis of gem–difluorovinylsilanes, using readily accessible silylboronic pinacol esters as reagents. Through a radical transfer strategy, this method overcomes the limitation of their high oxidation potential. Biological assays further confirmed that several target compounds exhibited moderate to potent antifungal activity against plant pathogens, including Botrytis cinerea, Setosphaeria turcica and Rhizoctonia solani.
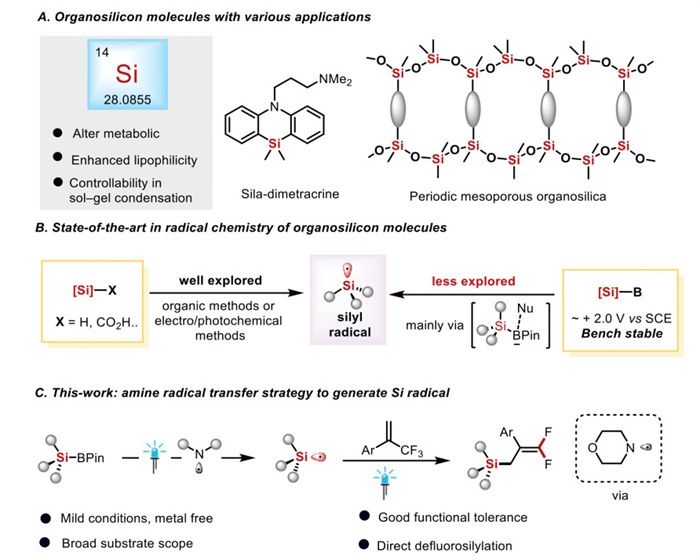
Silicon, belonging to Group Ⅳ elements, ranks as the second most abundant element in both the Earth's crust surface and soils [1-3]. Silicone–based organosilicon compounds possess distinct advantages such as controllable sol–gel condensation, lipophilicity, and low surface tension owing to their unique structure [4-6]. As a result, silicone compounds, serving as crucial raw materials, have assumed an indispensable role in materials science, polymer chemistry, and agrochemistry (Fig. 1A) [7-12]. With the development of organic photo/electrocatalysis, chemoselective silylation via silyl radicals holds great potential in the industrial realm (Fig. 1B, left) [13,14]. Typically, silyl radicals are generated through the direct activation of Si–H bonds [15-23]. Silylboronic pinacol esters, containing Si–B bonds, possess advantages such as good stability, commercial availability, and ease of preparation [24,25], and they have the potential to generate silane radicals. Nevertheless, silylboronic pinacol esters have a high oxidation potential (approximately +2.0 V vs. SCE). This high oxidation potential makes it more challenging to cleave the Si–B bond under photoredox conditions [26]. The conventional method of activating Si–B bonds usually involve introducing nucleophilic reagents to coordinate with boron in order to lower the redox potential (Fig. 1B, right) [26]. Amine radicals, frequently used as radical transfer reagents in photocatalysis, have been demonstrated to drive C–B bond activation to generate carbon-radicals [27-32]. We believe that they also hold excellent synthetic potential for use in the activation of silylboronic pinacol esters.
Figure 1
Figure 1.
From inspiration to reaction design. (A) Organosilicon molecules with various applications. (B) State-of-the-art in radical chemistry of organosilicon molecules. (C) This-work: amine radical transfer strategy to generate Si radical.
Fluorine–containing compounds endow drug molecules with advantages such as enhanced lipophilicity, specific conformation, and improved bioavailability [33-40]. In recent years, with the continuous maturation of synthetic techniques, the number of fluorine–containing drugs and pesticides featuring complex structures has been on the rise [40]. gem–Difluorovinyl compounds have attracted extensive interest from researchers due to their isosteric relationship with carbonyl groups [41-48].
Given the unique properties of silicon–containing organic compounds, we postulate that introducing both "Si" atoms and gem–difluoroalkene fragments into the same molecule may offer medicinal chemists a novel perspective. Inspired by the research advancements, we devise a radical transfer strategy for generating silane radicals. Under visible–light irradiation, the catalyst transforms the amine into amine radicals, which in turn induce the cleavage of the Si–B bond to generate silyl radicals. This approach successfully enables the defluorination of trifluoroolefins for the synthesis of fluorine–containing organosilicon compounds under mild reaction conditions (Fig. 1C). This study synthesized novel difluoromethylsilane compounds, showing potent fungicidal activity (inhibition rates > 60%) against major plant pathogens at 50 μg/mL. Their EC50 values compared favorably with commercial fungicides. DFT and MEP analyses linked activity to molecular orbital distributions and electrostatic properties.
In our initial experiments, we selected 1-(benzyloxy)-4-(3,3,3-trifluoroprop-1-en-2-yl)benzene (1a) and triethyl(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)silane (2a) as model substrates (Table 1). A solution of these substrates in dry acetonitrile ([1a] = 0.1 mol/L) was prepared, containing 4CzIPN (2 mol%) as a photocatalyst, morpholine (1.5 equiv.) as an activation reagent, and K3PO4 (2 equiv.) as a base. This solution was irradiated with a 456 nm LED at room temperature (~25 ℃) under an argon atmosphere for 24 h. As a result, the desired addition product 3 was obtained in an 85% yield (entry 1).
Table 1
Table 1.
Optimization of conditions for addition reaction between 1-(benzyloxy)-4-(3,3,3-trifluoroprop-1-en-2-yl)benzene (1a) with triethyl(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)silane 2a.a
a Standard conditions: 1a (0.2 mmol), 2a (0.4 mmol), photocatalyst (PC, 0.004 mmol), morpholine (0.3 mmol), K3PO4 (0.4 mmol), MeCN (2 mL), Ar, 456 nm LED, r.t., 24 h. b Yields were determined by 19F NMR spectroscopy with fluorobenzene as an internal standard. NR = no reaction. c Isolated yield.
We tested various other photocatalysts, such as Ir[dF(CF3)ppy]2(dtbbpy)PF6 (entry 2; for more detailed information, please refer to Supporting information for details). However, 4CzIPN was found to be the optimal choice. Different activation reagents were also evaluated. Piperidine and n–butylamine led to lower yields of product 3 (31% and trace amounts, respectively; entries 3 and 4). Solvent screening indicated that dichloromethane gave slightly lower yields of 3 compared to acetonitrile (compare entry 1 with entry 5), while toluene gave a relatively low yield, with only trace amounts detected. The other solvents tested yielded < 45% (see Supporting information for details). When we replaced K3PO4 with Na2CO3 or Cs2CO3 as the base, the yields of 3 decreased to 65% and 82%, respectively (entries 7 and 8). Control experiments confirmed that the activation reagent, the photocatalyst, light, and the exclusion of oxygen were all essential for this transformation (entries 9–11).
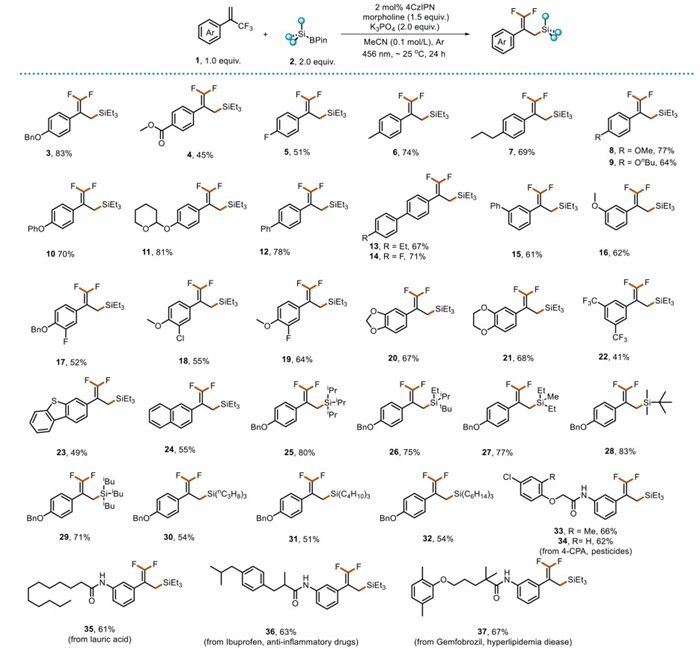
After identifying the optimized conditions, we explored the substrate scope for the synthesis of fluorinated organosilicon compounds through the defluorination functional-group reaction between α-trifluoromethyl arylalkene and silylboronic pinacol esters derivatives (Fig. 2). These conditions were widely applicable to 1 and demonstrated excellent selectivity and functional-group compatibility. Electron–withdrawing groups attached to the phenyl ring were well–tolerated, resulting in good yields (45%‒51%) of the corresponding gem–difluoroalkenes 4–5. We observed that when the para-position of the phenyl ring in the arylalkene was substituted with electron–donating groups such as methyl, n-propyl, or methoxy, the corresponding products 6–14 were produced with yields ranging from moderate to excellent (64%–81%).
However, when the meta–position was substituted with an aryl ring or a methoxy group, the yields decreased slightly (15–16, 61%–62%). By placing bisubstituted groups at different positions on the benzene ring, the desired products 17–21 were obtained with moderate yields (52%–68%). Alkenes with trifluoromethyl groups located on the benzene ring were poorly tolerated in this reaction system, yielding the product in only 41% (22). When α-trifluoromethyl alkenes containing heterocycles such as dibenzo[b, d]thiophene-4-yl and naphthalene-2-yl moieties were used, the corresponding products 23–24 were obtained in moderate yields (49%−55%). Next, we explored the reaction scope of silylboronic pinacol esters using compound 1a as the free–radical acceptor (Fig. 2). As shown, various silylboronic pinacol esters bearing different sterically diverse substituents, including triisopropyl, dimethylethyl, and dimethyl–tert–butyl groups, as well as some primary silylboronic pinacol esters, successfully reacted with α-trifluoromethyl arylalkene 1a. This reaction afforded the desired products 25–32 in yields ranging from 51% to 83%. Subsequently, for further evaluating the applicability of this reaction, various complex α-trifluoromethyl arylalkene-derived from natural products 33–37 were subjected to the methodology.
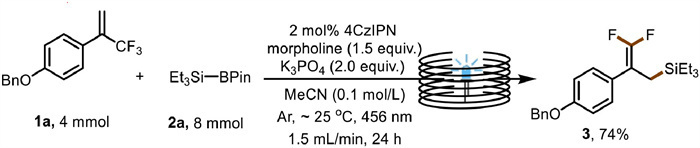
To further demonstrate the synthetic potential of our method, we carried out a gram-scale continuous-flow reaction to synthesize the difluoroketone compound (Fig. 3). With a flow rate of 1.5 mL/min and a residence time of 24 h, we were able to scale up the reaction to 4 mmol.
Plant-pathogenic fungi pose a serious threat to global food security and ecosystems. Chemical control is an effective means of preventing and managing fungal diseases [49,50]. However, the repeated long-term use of fungicides with a single mode of action can lead to resistance issues. An effective approach to developing new fungicides is the construction of new fungicide skeletons through organic synthesis methodology [51-54]. Compounds containing silicone that are environmentally compatible and exhibit synergistic effects have attracted considerable attention in the field of drug discovery and development [1-12]. Using this method, we successfully synthesized a series of novel gem–difluoroallylsilane compounds and investigated their fungicidal properties.
The mycelial growth rate method was used to test the fungicidal activity of the gem–difluoroallylsilane skeleton compounds against 10 pathogenic fungi at a concentration of 50 μg/mL. Commercial fungicides pyraclostrobin and boscalid were selected as positive controls (Table S6 in Supporting information). Some of the target compounds exhibited potent fungicidal activity against Botrytis cinerea, Setosphaeria turcica and Rhizoctonia solani. Most of these compounds exhibited inhibitory activity against B. cinerea of greater than 60%, with compounds 13–15 inhibiting it by > 75%. Compounds 3, 8, 11, 18 and 19 exhibited an inhibition rate of over 70% against S. turcica, while compounds 13, 14, 15, 23, 26 and 33 demonstrated an inhibition rate of over 60% against R. solani. Compound 13 exhibited relatively broad-spectrum antifungal activity, with inhibition rates exceeding 60% against four phytopathogenic fungi. These results suggest that the gem–difluoroallylsilane skeleton has fungicidal properties. Compounds with an inhibition rate greater than 75% were selected for EC50 activity determination (Table 2). For B. cinerea, the EC50 values of compounds 13–15 ranged from 2.39 μg/mL to 3.09 μg/mL, which are lower than those of the commercial fungicides pyraclostrobin (1.30 μg/mL) and pydiflumetofen (0.62 μg/mL). Compound 8 (2.58 μg/mL) exhibited comparable activity to boscalid (2.97 μg/mL) against S. turcica, but was less effective than pyraclostrobin (1.77 μg/mL). Compounds 8 and 13 showed good antifungal activity and could serve as lead compounds for further structural optimization and mode-of-action studies. For R. solani, the EC50 values of compound 14 and 33 were determined to be 14.64 and 8.31 μg/mL, respectively. Although both compound 14 and 33 demonstrated inhibitory effects against R. solani, their potency was lower than that of the commercial fungicides boscalid (1.63 μg/mL) and pyraclostrobin (0.03 μg/mL). This class of compounds can serve as lead fungicides for structural optimization and mechanism-of-action studies.
Table 2
Table 2.In vitro EC50 values of selected compounds.
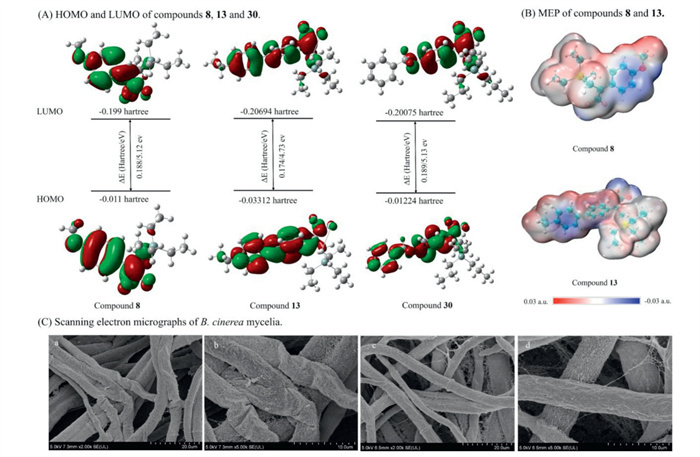
To further investigate the structural properties underlying the antifungal activity of target compounds, density functional theory (DFT) calculations were performed on the high-activity compounds 8 and 13, as well as the low-activity compound 30. The electron distributions of the HOMO and LUMO for the highly active compounds 8 and 13 were similar. In contrast, the LUMO of the low-activity compound 30 was mainly distributed over the benzene ring and the gem–difluoroalkene moiety. This suggested that variations in substituents on the benzene ring significantly impact biological activity (Fig. 4A). Fig. 4B shows that, for compounds 8 and 13, the MEP maps indicate that the gem–difluoroalkenyl group is located in the negative electrostatic potential region (blue), suggesting nucleophilic characteristics. Meanwhile, the silyl group and aromatic ring are situated in the positive potential region (red), indicating electrophilic properties. Optimizing the electrostatic potential around the active core region (e.g., the aromatic ring) facilitates effective interaction with fungal receptors or enzyme active sites. These results imply that variations in factors such as energy levels, molecular orbital distribution and MEP characteristics may account for differences in antifungal activity by affecting receptor interactions.
Figure 4
Figure 4.
Frontier molecular orbitals (HOMO and LUMO) and molecular electrostatic potential (MEP) surfaces of target compounds. (A) HOMO and LUMO of compounds 8, 13 and 30. (B) MEP of compounds 8 and 13. (C) Scanning electron micrographs of B. cinerea mycelia: (a, b) Compound 13, treated with 0.5% DMF plus the compound 13 at 4.78 μg/mL; (c, d) control, treated with 0.5% DMF.
Subsequently, the effect of active compound 13 on the mycelial morphology of B. cinerea was observed using scanning electron microscopy. The results were shown in Fig. 4C. After treatment with compound 13, the mycelia exhibited obvious collapse and shrinkage, while the mycelial structure in blank control maintained smooth and regular. This result indicated that compound 13 could disrupt the mycelial morphology, thereby interfering with the normal growth of B. cinerea.
To gain insight into the reaction pathway, several control experiments were performed. We conducted a light/dark experiment (see Supporting information for details); the results revealed that in the absence of light, the reaction halted. This suggests that continuous visible-light irradiation is essential for this amination reaction to proceed. A series of cyclic voltammetry measurements were carried out with Ag/AgCl as a reference electrode in CH3CN using tetrabutylammonium perchlorate as a supporting electrolyte. The purpose was to determine the possible changes in oxidation-reduction potential after adding morpholine. The cyclic voltammetry results showed that the oxidation peak potential of morpholine (E = +0.95 V vs. SCE) was lower than that of the silylboronic pinacol esters (E = +1.94 V vs. SCE), indicating that morpholine would be preferentially oxidized (see Supporting information for details). Furthermore, adding morpholine to the silylboronic pinacol esters did not result in a shift in the NMR signal of [¹¹B], suggesting that no direct complex was formed between morpholine and the silylboronic pinacol esters (see Supporting information for details).
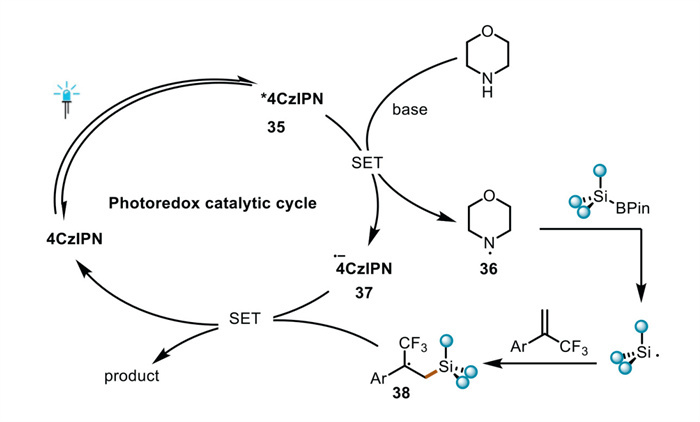
On the basis of the above-mentioned experiments and literature reports, we propose the reaction mechanism (Fig. 5): Initially, photocatalyst is excited by light irradiation giving excited state of photocatalyst 35. morpholine quenches the excited-state photocatalyst to generate an amine radical 36via the SET process and lead to the transformation from 35 to 37. Then amine radical 36 interacts with the boron species to liberate the corresponding silyl radical. Subsequently, the silyl radical undergoes a Michael addition reaction with the α-trifluoromethyl alkenes to generate the CF3-styrene carbon radical 38, Single-electron reduction of the CF3-styrene carbon radical by 37 generates the CF3-styrene carbanion intermediate, which undergoes a β-fluoride elimination reaction to afford the target product and complete the photoredox cycle.
In conclusion, we have developed a photochemical method to generate silyl radicals from readily available silylboronic pinacol esters through homolytic substitution reactions of the boron group with amine radicals. Under mild conditions, this method excels in forming silicon-containing difluoroalkene compounds and can be scaled up to the gram level. Antifungal activity showed that at 50 μg/mL, some compounds showed > 60% inhibition against B. cinerea, S. turcica and R. solani, with EC50 values comparable to or lower than commercial fungicides boscalid and pyraclostrobin. DFT and MEP analyses revealed that high-activity compounds like 8 and 13 had distinct molecular orbital distributions and electrostatic properties compared to low-activity ones. Scanning electron microscopy indicated that compound 13 effectively inhibited the mycelial growth of B. cinerea. These compounds serve as promising leads for developing new fungicides with optimized structures and mechanisms.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We are grateful to the National Natural Science Foundation of China (No. 22271166) and the Frontiers Science Center for New Organic Matter, Nankai University (No. 63181206), for generous financial support.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111940.
Figure 1
From inspiration to reaction design. (A) Organosilicon molecules with various applications. (B) State-of-the-art in radical chemistry of organosilicon molecules. (C) This-work: amine radical transfer strategy to generate Si radical.
Figure 4
Frontier molecular orbitals (HOMO and LUMO) and molecular electrostatic potential (MEP) surfaces of target compounds. (A) HOMO and LUMO of compounds 8, 13 and 30. (B) MEP of compounds 8 and 13. (C) Scanning electron micrographs of B. cinerea mycelia: (a, b) Compound 13, treated with 0.5% DMF plus the compound 13 at 4.78 μg/mL; (c, d) control, treated with 0.5% DMF.
Table 1.
Optimization of conditions for addition reaction between 1-(benzyloxy)-4-(3,3,3-trifluoroprop-1-en-2-yl)benzene (1a) with triethyl(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)silane 2a.a
Entry
Deviation from standard conditions
Yield (%)b
1
None
85 (83c)
2
Ir[dF(CF3)ppy]2(dtbbpy)PF6 as PC
77
3
Piperidine as activation reagent
30
4
n-Butylamine as activation reagent
NR
5
Dichloromethane as solvent
70
6
Toluene as solvent
NR
7
Na2CO3 as base
65
8
Cs2CO3 as base
82
9
No morpholine
NR
10
No PC/light
NR
11
Under air
30
a Standard conditions: 1a (0.2 mmol), 2a (0.4 mmol), photocatalyst (PC, 0.004 mmol), morpholine (0.3 mmol), K3PO4 (0.4 mmol), MeCN (2 mL), Ar, 456 nm LED, r.t., 24 h. b Yields were determined by 19F NMR spectroscopy with fluorobenzene as an internal standard. NR = no reaction. c Isolated yield.

 DownLoad:
DownLoad:


 下载:
下载:






 下载:
下载:
