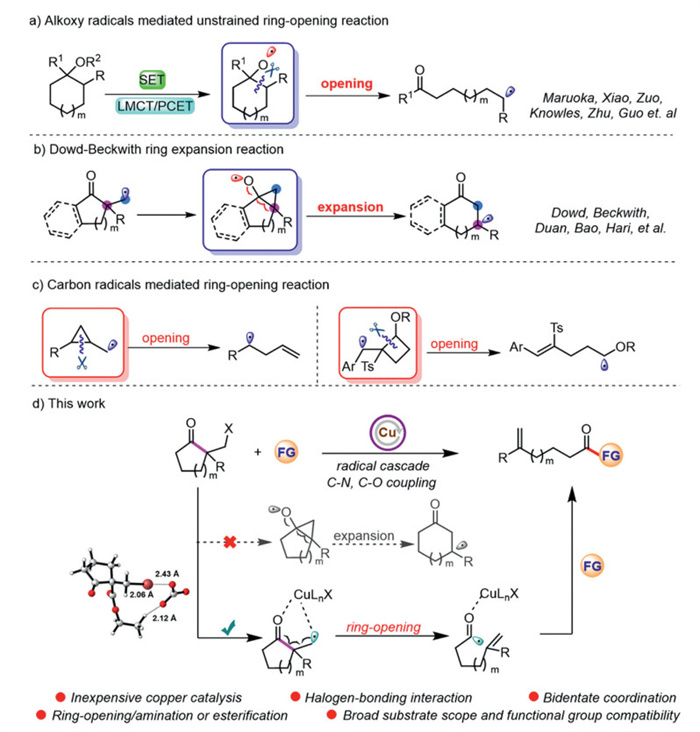
Scheme 1.
Radical-mediated C‒C bond cleavage.
An unusual carbon radical-mediated ring-opening/amination or esterification cascade of unstrained α-haloalkyl cycloketone derivatives
Hong Xin , Shutao Wang , Li-Na Guo , Cheng Guan , Jinyi Liao , Zi-Hang Yuan , Zhengze Zhang , Xin-Hua Duan

The direct activation/functionalization of carbon-carbon σ bond has always been considered as one of the hot topics in organic synthesis [1-7]. In this field, the cleavage of carbon-carbon bonds in unstrained cyclic ketone has long been challenging and has attracted considerable attention from chemists [8-12]. In recent years, alkoxy radical-mediated cyclic C‒C bond cleavage has significantly advanced. To access reactive alkoxy radical intermediates, various strategies have been reported, such as the direct activation of O‒H and O-heteroatom bonds in alcohol and their derivatives (Scheme 1a) [13-23]. Additionally, the radical addition of carbon-centered radicals to the carbonyl group provided another strategy [24]. Among these, the most famous example is the Dowd-Beckwith ring expansion reaction (Scheme 1b) [25-31]. Recently, our group [32,33], Hari's group [34] and Bao's group [35] have successfully achieved the Dowd-Beckwith ring expansion/coupling reaction under the conditions of precious metal catalysis (Pd, Ir) or radical initiator, respectively, which produced the medium cyclic ketones with various functional groups.
As we know, the C‒C bond cleavage initiated by the carbon-centered radical is a classical transformation in radical chemistry [36-39]. In particular, the ring-opening reactions of cyclopropylmethyl radicals have been commonly used to verify a radical pathway for mechanistic investigation. In addition, Zhang and co-workers [40,41] reported the carbon-centered radical mediated ring-opening/functionalization of 4-membered ring by strain release, providing a facile approach to the γ,δ-unsaturated aldehydes of ketones (Scheme 1c). However, the 5- and 6-membered ring analogues did not undergo such a transfer probably due to the lower ring strain and the rapid reverse cyclization [42,43]. We wonder whether unstrained ring opening could be facilitated through carbon-centered radical-mediated strategy. Herein, we disclose an unusual copper-catalyzed ring opening/amination and esterification of unstrained α-haloalkyl cycloketone derivatives, which provides a variety of distal unsaturated amides and esters in good yields (Scheme 1d). Mechanistic studies suggest a radical pathway for this transformation and halogen-bonding interaction is critical for initiating the reaction. Moreover, we speculate that copper engaged in bidentate coordination with both the carbonyl group and the carbon radical to form a bicyclic Cu-complex intermediate, which not only effectively inhibits ring expansion, but also activates the pre-cleavage carbon-carbon bond. Furthermore, the formation of stable acyl radicals and α,β-unsaturated fragments are important driving forces for this C‒C bond cleavage. Remarkably, this protocol can be applied to the modification of drug molecules and fragments.
Initially, the ethyl 1-(bromomethyl)-2-oxocyclopentane-1- carboxylate 1a and indazole 2a were chosen as model substrates to find the optimal reaction conditions (Table 1). To our delight, it was found that the reaction proceeded smoothly in the presence of Cu(MeCN)4PF6 (10 mol%) and Cs2CO3 (2 equiv.) in DMF for 24 h, yielding the ring opening product 3a in 83% yield (entry 1). Other copper salts, such as CuBr, Cu(OTf)2 and CuBr2 were all efficient catalysts for the reaction, but gave lower yields than Cu(MeCN)4PF6 (entries 2–4). The use of Fe(OTf)2 as catalyst also afforded the product 3a in 68% yield, while Lewis acid ZnCl2 resulted in only a trace amount of 3a (entries 5 and 6). A variety of organic and inorganic bases were tested, with Cs2CO3 proving to be the best base (entries 7–9). Solvent screening revealed that polar solvents were more suitable for this transformation, and DMF was still the optimal one (entries 10–12, for other solvents see Supporting information). Fortunately, increasing the amount of Cs2CO3 from 2.0 equiv. to 3.0 equiv. improved the yield of 3a up to 94% (entry 13). In addition, the reaction yield was poor when the reaction was carried out with K2CO3 and DMAP under reflux in DCM for 72 h (entry 13) [44]. Finally, control experiment revealed that base and catalyst are essential for this reaction (entries 14 and 15).
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | Catalyst | Solvent | Base (equiv.) | Yield (%)b |
| 1 | Cu(MeCN)4PF6 | DMF | Cs2CO3 (2) | 83 |
| 2 | CuBr | DMF | Cs2CO3 (2) | 53 |
| 3 | Cu(OTf)2 | DMF | Cs2CO3 (2) | 45 |
| 4 | CuBr2 | DMF | Cs2CO3 (2) | 41 |
| 5 | Fe(OTf)2 | DMF | Cs2CO3 (2) | 68 |
| 6 | ZnCl2 | DMF | Cs2CO3 (2) | Trace |
| 7 | Cu(MeCN)4PF6 | DMF | DBU (2) | 34 |
| 8 | Cu(MeCN)4PF6 | DMF | t-BuOK (2) | 30 |
| 9 | Cu(MeCN)4PF6 | DMF | K2CO3 (2) | 37 |
| 10 | Cu(MeCN)4PF6 | DMA | Cs2CO3 (2) | 70 |
| 11 | Cu(MeCN)4PF6 | DMSO | Cs2CO3 (2) | 12 |
| 12 | Cu(MeCN)4PF6 | Toluene | Cs2CO3 (2) | 28 |
| 13 | Cu(MeCN)4PF6 | DMF | Cs2CO3 (3) | 94 (trace)c |
| 14 | Cu(MeCN)4PF6 | DMF | – | n.r. |
| 15 | – | DMF | Cs2CO3 (3) | Trace |
| a Reaction conditions: 1a (0.3 mmol, 1.5 equiv.), 2a (0.2 mmol, 1.0 equiv.), catalyst (10 mol%), base (x equiv.), solvent (2 mL), r.t., for 24 h, under N2. b Yield of isolated product. c K2CO3 (4 equiv.), DMAP (20 mol%), DCM, reflux, 72 h. |
||||
With the optimized conditions established, we firstly investigated the scope of indazoles 2 using ethyl 1-(bromomethyl)-2-oxocyclopentane-1-carboxylate 1a as model substrate (Scheme 2). Among them, 4–bromo-1H-indazole and 6–bromo-1H-indazole afforded the products 3b and 3c in 53% and 73% yields, respectively. Substrates with an electron-donating group on heterocyclic ring gave better yields than those with an electron-withdrawing group (3d vs. 3e, 3f). Notably, in addition to 3e, the dechlorination product 3a was also isolated in 35% yield when 3–chloro-1H-indazole was used as substrate. Subsequently, we explored the scope of α-haloalkyl cycloketone derivatives 1 using indazole 2a as a nucleophile under the optimized conditions (Scheme 2). The desired product 3a was obtained from both the analogous chloride and iodide in 54% and 75% yields, respectively. A range of bromides with five- to eight-membered rings also afforded the corresponding products 4a-4d in moderate to excellent yields. The 1-indanone-derived substrate also gave the product 4e in 53% yield. With the 1-tetralone-derived substrate, both the desired product 4f and a by-product 4f' were isolated. In addition, the 4f' could not transfer to 4f. Notably, substrate 1 with an acetyl group instead of the ester group also worked well, giving the target product 4 g in 70% yield. Substrate with a cyano group was also compatible with the reaction system, giving the desired product 4h in 56% yield. However, replacing the ester with a phenyl or methyl group resulted in no formation of the desired products, with 81% and 78% of 2a recovered, respectively. Unfortunately, 2-(bromomethyl)cyclopentan-1-one and substrates with two to four-carbon side chain did not undergo this ring-opening reaction.
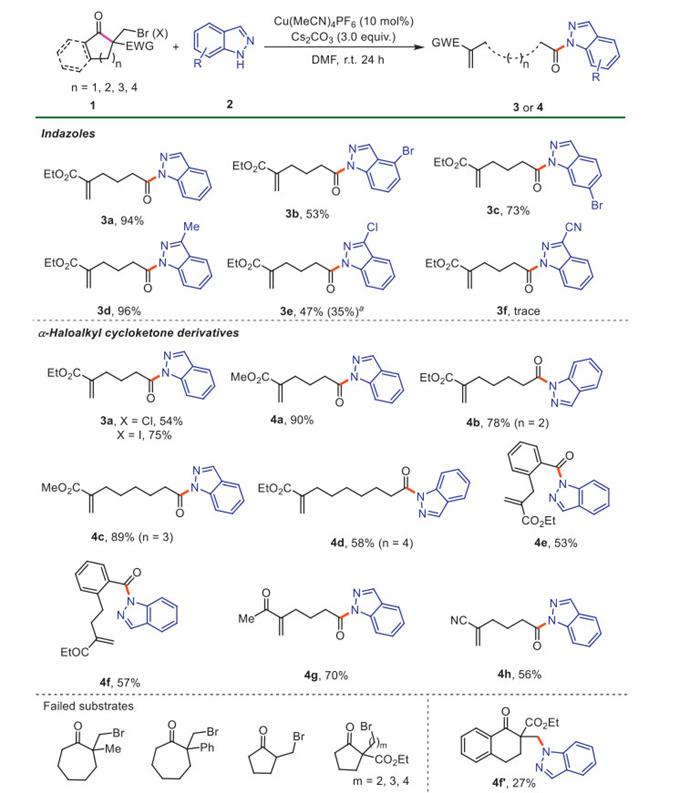
Next, we continued to evaluate the scope of amines 2 using bromide 1a as model substrate (Scheme 3). Both aromatic and aliphatic amines reacted well with 1a to afford the chain-unsaturated amides 5 in moderate to excellent yields. The structure of product 5m was further verified by X-ray crystal diffraction. 5-Methoxyindole furnished the target product 5a in 51% yield. Remarkably, the tryptamine was also amenable, furnishing the diacylated product 5b in 83% yield when 2 equiv. of 1a was used. The dipeptide Gly-Trp afforded product 5c in 60% yield. The natural product melatonin afforded the desired product 5d in acceptable yield. In addition, it was found that both electronic and steric effects on the aromatic ring had an obvious influence on the reaction efficiency. Aromatic amines with electron-donating groups at the para position of benzene ring gave better yields than those with an electron-withdrawing group (5f-5j vs. 5k-5n). Arylamines with o-methyl and o-phenyl groups gave the products 5u and 5v in relatively lower yields. Notably, a variety of functional groups, including ester (5o), sulfonyl (5p), Bpin (5q) and alkynyl (5r) were well preserved under this copper catalyst system. 3,5-Dimethylaniline and 2-naphthylamine were also converted into the corresponding products 5w and 5x in excellent yields. Heteroaryl amines, such as 5-benzothiazolamine and 2-aminopyridine, also participated well in this reaction, giving the products 5y and 5z in moderate yields. When indoline and tetrahydroquinoline were employed, 5aa and 5ab were obtained in good yields. The alkyl amines were then evaluated. Satisfactorily, both primary and secondary alkyl amines were engaged well in this reaction, giving the expected products 5ac-5ak in good to excellent yields. Drug molecules and fragments, such as lurasidone and amoxapine, were successfully acylated by the ring-opening of 1a to give the desired products in satisfactory yields (5al-5am). In addition, benzamide also furnished desired product 5an in 42% yield with t-BuOK as base. In particular, in this study we successfully converted various amines, including aromatic (hetero) amines and aliphatic amines (such as 5a, 5e, 5ae, 5ai, 5aj), which were inaccessible with a previous K2CO3/DMAP-catalyzed method. These results suggest that the main reaction pathway differ from Grob fragmentation [44].
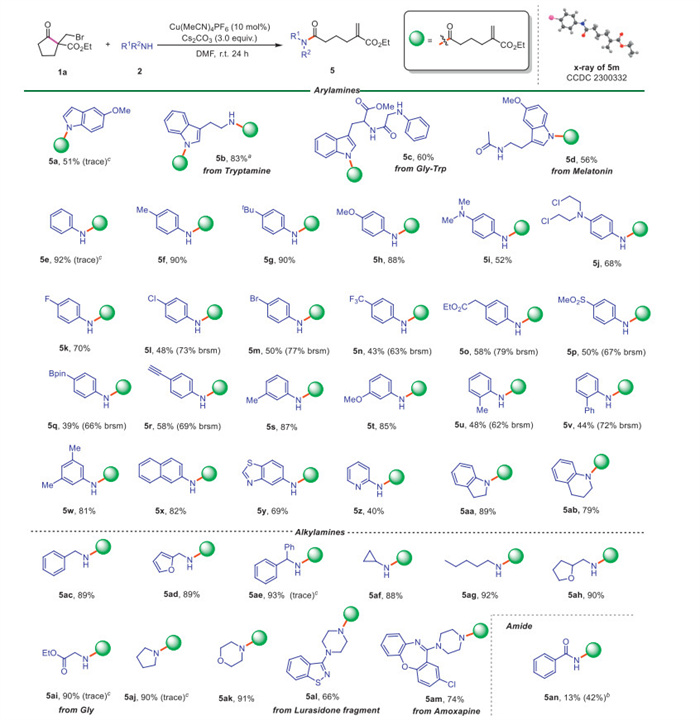
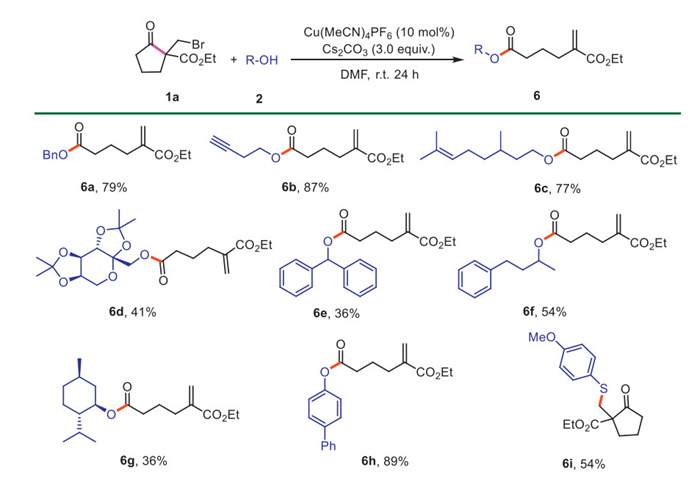
Encouraged by the above results, we explored a structurally diverse range of alcohols as nucleophiles for this reaction (Scheme 4). Benzyl alcohol reacted well to afford the expected ester 6a in 79% yield. Alkenyl and alkynyl alcohols also successfully produced the products 6b and 6c in 87% and 77% yields, respectively. Diacetonefructose was also applicable and gave the product 6d in 41% yield. Additionally, secondary alcohols, including diphenylmetanol (6e), 4-phenyl-2-butanol (6f) and menthol (6 g), provided the target products in moderate yields. Reasonably, sterically bulky tertiary alcohol, such as butanol, failed to yield the expected product (not shown). Notably, p-phenylphenol was a viable substrate, furnishing the desired product 6h in excellent yield. Unfortunately, thiophenol was not compatible with this reaction, leading to the formation of thioether 6i as a byproduct.
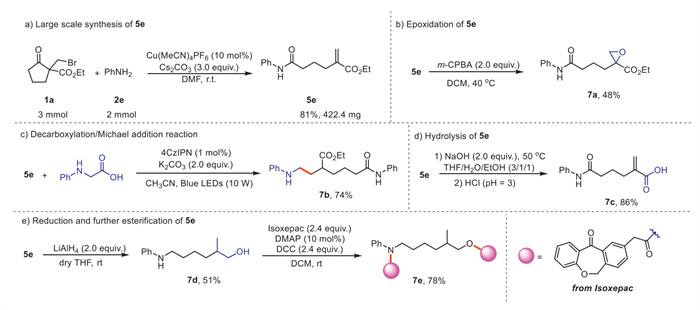
To demonstrate the application of this protocol, a 2.0 mmol scale model reaction was conducted, yielding 5e in 81% yield after extending the reaction time (Scheme 5a). Subsequently, various derivations of product 5e were explored. The epoxidation of 5e with m-CPBA resulted in the product 7a in a 48% yield (Scheme 5b). A visible light-mediated decarboxylative Michael addition of 5e with N-phenylglycine also proceeded efficiently to provide product 7b in good yield (Scheme 5c). Product 5e can be hydrolyzed chemoselectively to afford the carboxylic acid 7c in 86% yield (Scheme 5d). Notably, treatment of 5e with LiAlH4 provided the alcohol 7d in moderate yield, which could be further esterified with the drug molecule isoxepac to give the aminyl-containing ester 7e in 78% isolated yield (Scheme 2e). In addition, the effect of product 5j on the cell viability of HeLa cells was examined, MTT assay shown 5j has potential in vitro anticancer activity against HeLa cells and significant reduction in MTT activity with increasing concentrations of 5j (Fig. S1 in Supporting information).
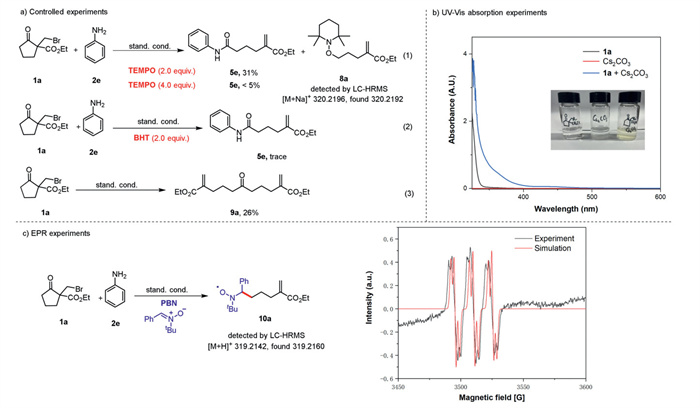
To elucidate the mechanism of this reaction, several control experiments were performed (Scheme 6). Addition of different amounts of TEMPO led to obviously decreased yields of 5e and a decarbonylative TEMPO-adduct 8a was detected by LC—HMRS (Scheme 6a, Eq. 1). The addition of BHT (2.0 equiv.) also resulted in a significant inhibition of this reaction (Scheme 6a, Eq. 2). These results suggest that a radical pathway may be involved in this transformation. Furthermore, in the absence of 2e, substrate 1a decomposed to the compound 9a under standard conditions (Scheme 6a, Eq. 3), and the release of CO could be detected by GC (for details, see Supporting information). These findings imply that an acyl radical might be generated in this transformation. To clarify the role of the base, NMR titration experiments of 1a were performed with different amounts of Cs2CO3 (Scheme 6b). It was observed that the signals of α-H or α-C of bromide 1a shifted downfield in the 1H NMR and 13C NMR spectra, suggesting the possible formation of a halogen-bonding complex [40,41]. Additionally, UV–vis analysis indicated the formation of a complex between bromide 1a and Cs2CO3 (Scheme 6c). Notably, the EPR experiment using the spin trapping agent N–tert–butyl–α-phenylnitrone (PBN) confirmed the formation of a radical intermediate in this transformation (Scheme 6d). Finally, the Cu(Ⅲ) complexes were successfully detected by LC—HMRS (for details, see Supporting information).
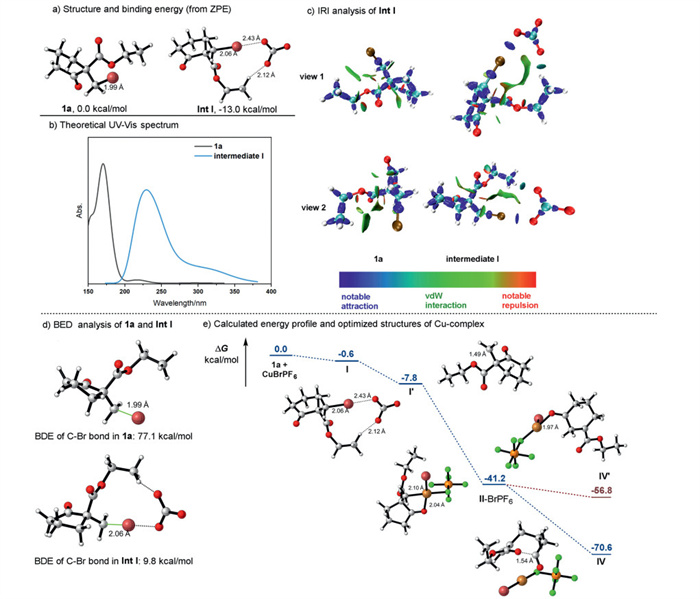
To gain a deeper understanding of the reaction mechanism, we employed density functional theory (DFT) for a comprehensive theoretical study of the intermediate Ⅰ through halogen bond that formed by 1a and CO32− negative ion, was carried out, and the resulting structure and binding energy are presented in Scheme 7. The C‒Br bond in intermediate Ⅰ was found to be elongated by 0.07 Å (from 1.99 Å to 2.06 Å) compared to 1a, suggesting a possible weakening of the C‒Br bond. The distance between O atom and Br atom is 2.43 Å, and the distance between O atom and H atom is 2.12 Å, indicating a potential halogen bond between O atom and Br atom, as well as a non-covalent interaction between O atom and H atom. The favorable binding energy of intermediate Ⅰ, measured at −13.0 kcal/mol, indicates its energetically favorable formation (Fig. 7a). Subsequently, we analyzed the interactions within intermediate Ⅰ using the interaction region indicator (IRI) analysis, as shown in Scheme 7c. The analysis also revealed a robust halogen bond between the O atom and the Br atom, depicted in blue, and an additional interaction between the O atom and the H atom, with a strength comparable to that of a van der Waals interaction. Furthermore, time-dependent density functional theory (TD-DFT) was used to calculate the theoretical UV–vis spectra of 1a and intermediate Ⅰ, as illustrated in Scheme 7b. The UV–vis absorption of intermediate Ⅰ exhibited an obvious redshift compared to 1a, aligning with the experimental observations. Then, the FMOs analysis of 1a and intermediate Ⅰ were also performed and shown in Fig. S9a (Supporting information). We found that the energy gap of intermediate Ⅰ was significantly lower than 1a. Finally, we calculate the BDE of the C‒Br bond and find it decreases significantly after combining the CO32− (Scheme 7d), which may provide an explanation for the activation of substrate 1a by the halogen bond.
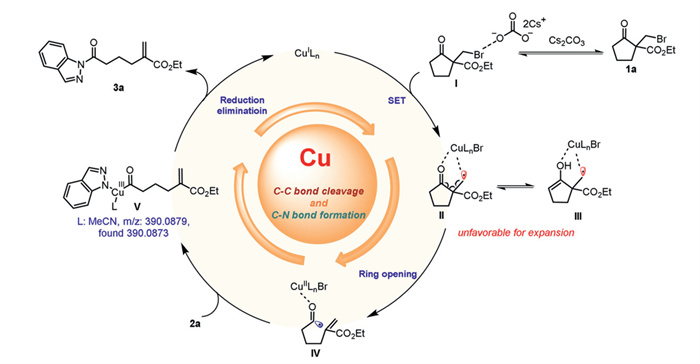
Based on the above results and literature [44-52], a probable mechanism for the ring-opening/amination of the α-haloalkyl cycloketone derivatives is proposed (Scheme 8). Initially, substrate 1a is activated by Cs2CO3, forming a halogen-bonding complex Ⅰ. Then, a single electron transfer (SET) event occurs between Cu(Ⅰ) and complex Ⅰ to give the Cu-complexed radical intermediate Ⅱ or the enolized complex Ⅲ. Subsequently, intermediate Ⅱ undergoes a homolytic C‒C cleavage, likely driven by the formation of stable α,β-unsaturated fragments, to give the acyl radical intermediate Ⅳ. This intermediate rapidly recombines with Cu(Ⅱ) in the presence of indazole 2a to give the Cu(Ⅲ) complex Ⅴ. Finally, reductive elimination of Cu(Ⅲ) complex Ⅴ produces the desired product 3a and regenerates the Cu(Ⅰ) complex. For this unusual ring-opening transformation, we hypothesize that the formation of bicyclic Cu-complex intermediate Ⅱ or Ⅲ may effectively inhibit the ring expansion reaction. To gain more insight into these intermediates, we conducted the structure and energy analysis using DFT calculations. The findings of this study demonstrate that the carbon-centered radical intermediates produced by the SET process have the capacity to form stable complexes Ⅱ-BrPF6 with CuBrPF6. Furthermore, the energy of the corresponding ring-opening product Ⅳ is significantly reduced to −70.6 kcal/mol, indicating that the ring-opening process of Ⅱ-BrPF6 is energetically favorable. In contrast, the energy of the Dowd-Beckwith ring expansion product Ⅳ' is −56.8 kcal/mol, which is significantly higher than that of Ⅳ. From the perspective of product stability, the Dowd-Beckwith ring expansion is not a favorable pathway (Scheme 7e). IRI analysis reveals the presence of notable attractive interactions between Cu—O and Cu—•CH2, suggesting the potential for coordination between the Cu—O and the possibility of covalent bonding between Cu—C cannot be ruled out (Fig. S9b in Supporting information). Spin density analysis was performed to study the spin delocalization of Cu complex Ⅱ-BrPF6, the unpaired electrons on the Cu atoms are dispersed to the ketocarbonyl oxygen (Fig. S9c in Supporting information). In addition, the charge analysis also shown that the charge of the carbonyl carbon increased from 0.433 to 0.518 (Fig. S9d in Supporting information). These results revealed the pre-cleavage carbon-carbon bond was activated after formation Cu complex Ⅱ-BrPF6. Consequently, the generation of the stabilized Cu complex may be the pivotal factors in the ring-opening reaction.
In conclusion, we have developed a novel carbon-centered radical-mediated ring-opening/amination or esterification cascade of α-haloalkyl cycloketone derivatives under copper catalysis. A range of α-haloalkyl cycloketone derivatives underwent a unique C‒C bond cleavage model, producing the chain-unsaturated amides and esters that are traditionally challenging to synthesize. This protocol is characterized by mild conditions, operational simplicity, broad substrate scope and excellent functional group compatibility. Mechanistic studies and DFT calculations indicate that the halogen-bonding interaction between halide and base is crucial for the activation of the C‒Br bond. Furthermore, the formation of a bicyclic Cu-complex, including O–Cu–•CH2, favors the ring-opening reaction over the typical ring expansion. This work would bring new vitality for the radical-mediated C‒C bond cleavage.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Hong Xin: Writing – original draft, Methodology, Data curation, Conceptualization. Shutao Wang: Formal analysis, Data curation. Li-Na Guo: Writing – original draft. Cheng Guan: Methodology. Jinyi Liao: Methodology. Zi-Hang Yuan: Data curation. Zhengze Zhang: Software. Xin-Hua Duan: Project administration, Conceptualization.
This work was financial support from the National Natural Science Foundation of China (No. 22171220); Fundamental Research Funds for the Central Universities (Nos. xtr072022003, 11913224000014), Shaanxi Postdoctoral Science Foundation (No. 31271000000006) are greatly appreciated. We also thank Mr. Zhang and Miss Bai at the Instrument Analysis Center of Xi'an Jiao tong University for their assistance with NMR and HRMS analysis.
Supplementary material associated with this article can be found, in the online version, at doi:
F. Song, T. Gou, B.Q. Wang, Z.J. Shi, Chem. Soc. Rev. 47 (2018) 7078–7115. doi: 10.1039/c8cs00253c
X.X. Wu, C. Zhu, Chin. J. Chem. 37 (2019) 171–182. doi: 10.1002/cjoc.201800455
S.P. Morcillo, Angew. Chem. Int. Ed. 58 (2019) 14044–14054. doi: 10.1002/anie.201905218
P. Sivaguru, Z.K. Wang, G. Zanoni, X.H. Bi, Chem. Soc. Rev. 48 (2019) 2615–2656. doi: 10.1039/c8cs00386f
Y.F. Liang, M. Bilal, L.Y. Tang, et al., Chem. Rev. 123 (2023) 12313–12370. doi: 10.1021/acs.chemrev.3c00219
J.B. Roque, Y. Kuroda, L.T. Göttemann, R. Sarpong, Science 361 (2018) 171–174. doi: 10.1126/science.aat6365
J.B. Roque, Y. Kuroda, L.T. Göttemann, R. Sarpong, Nature 564 (2018) 244–248. doi: 10.1038/s41586-018-0700-3
A. Riahi, J. Muzart, M. Abe, N. Hoffmann, New J. Chem. 37 (2013) 2245–2249. doi: 10.1039/c3nj00457k
A.S.K. Tsang, A. Kapat, F. Schoenebeck, J. Am. Chem. Soc. 138 (2016) 518–526. doi: 10.1021/jacs.5b08347
H. Xin, X.H. Duan, L. Liu, L.N. Guo, Chem. Eur. J. 26 (2020) 11690–11694. doi: 10.1002/chem.202001032
H. Xin, X.H. Duan, M. Yang, Y. Zhang, L.N. Guo, J. Org. Chem. 86 (2021) 8263–8273. doi: 10.1021/acs.joc.1c00708
L. Deng, G. Dong, Trends Chem. 2 (2020) 183–198. doi: 10.1016/j.trechm.2019.12.002
A. Matsumoto, K. Maruoka, Bull. Chem. Soc. Jpn. 94 (2021) 513–524. doi: 10.1246/bcsj.20200321
H. Xin, Z.H. Yuan, M. Yang, et al., Green Chem. 23 (2021), 9549–9553. doi: 10.1039/d1gc03230e
S. Liu, P. Ma, L. Zhang, et al., Chem. Sci. 14 (2023) 5220–5225. doi: 10.1039/d2sc06157k
J.J. Guo, A. Hu, Z. Zuo, Tetrahedron Lett. 59 (2018) 2103–2111. doi: 10.1016/j.tetlet.2018.04.060
L. Chang, Q. An, L. Duan, K. Feng, Z. Zuo, Chem. Rev. 122 (2022) 2429–2486. doi: 10.1021/acs.chemrev.1c00256
P.R. Murray, J.H. Cox, N.D. Chiappini, et al., Chem. Rev. 122 (2022) 2017–2291. doi: 10.1021/acs.chemrev.1c00374
X. Fan, H. Zhao, J. Yu, X. Bao, C. Zhu, Org. Chem. Front. 3 (2016) 227–232. doi: 10.1039/C5QO00368G
D. Wang, J. Mao, C. Zhu, Chem. Sci. 9 (2018) 5805–5809. doi: 10.1039/c8sc01763h
H. Yan, G.S. Smith, F.E. Chen, Green Synth. Catal. 3 (2022) 219–226.
R.N. Yi, W.M. He, Chin. Chem. Lett. 36 (2025) 110787. doi: 10.1016/j.cclet.2024.110787
X. Wu, C. Zhu, Chem. Rec. 18 (2018) 587–598. doi: 10.1002/tcr.201700090
M. Wang, M. Li, S. Yang, et al., Nat. Commun. 11 (2020) 672–679. doi: 10.1038/s41467-020-14435-5
P. Dowd, S.C. Choi, J. Am. Chem. Soc. 109 (1987) 3493–3494. doi: 10.1021/ja00245a069
P. Dowd, S.C. Choi, J. Am. Chem. Soc. 109 (1987) 6548–6549. doi: 10.1021/ja00255a071
A.L.J. Beckwith, D.M. O'Shea, S. Gerba, S.W. Westwood, J. Chem. Soc., Chem. Commun. 9 (1987) 666–667.
A.L.J. Beckwith, D.M. O'Shea, S.W. Westwood, J. Am. Chem. Soc. 110 (1988) 2565–2575. doi: 10.1021/ja00216a033
K. Nishikawa, T. Ando, K. Maeda, T. Morita, Y. Yoshimi, Org. Lett. 15 (2013) 636–638. doi: 10.1021/ol303460u
E. Hasegawa, M. Tateyama, T. Hoshi, et al., Tetrahedron 70 (2014) 2776–2783. doi: 10.1016/j.tet.2014.02.078
M. Deguchi, A. Fujiya, E. Yamaguchi, et al., RSC Adv. 8 (2018) 15825–15830. doi: 10.1039/c8ra02383b
L. Chen, L.N. Guo, S. Liu, L. Liu, X.H. Duan, Chem. Sci. 12 (2021) 1791–1795. doi: 10.1039/d0sc04399k
H. Xin, L.N. Guo, M. Yang, et al., Org. Chem. Front. 10 (2023) 1147–1152. doi: 10.1039/d2qo02001g
T. Songha, G.A. Kadam, D.P. Hari, Chem. Sci. 14 (2023) 6930–6935. doi: 10.1039/D3SC01908J
Q. Zhang, M.F. Chiou, C. Ye, et al., Chem. Sci. 13 (2022) 6836–6841. doi: 10.1039/d2sc00902a
T. Hashimoto, Y. Kawamata, K. Maruoka, Nat. Chem. 6 (2014) 702–705. doi: 10.1038/nchem.1998
Z.Q. Zhang, X.Y. Meng, J. Sheng, Q. Lan, X.S. Wang, Org. Lett. 21 (2019) 8256–8260. doi: 10.1021/acs.orglett.9b03012
F.F. Feng, J.A. Ma, D. Cahard, J. Org. Chem. 86 (2021) 13808–13816. doi: 10.1021/acs.joc.1c01886
Y. Liu, J.L. Sui, W.Q. Yu, et al., J. Org. Chem. 88 (2023) 8563–8575. doi: 10.1021/acs.joc.3c00486
Y. Zhang, C. Zhao, C. Ma, et al., Angew. Chem. Int. Ed. 62 (2023) e202300166. doi: 10.1002/anie.202300166
C. Zhao, W. Ma, K. Liu, et al., Org. Chem. Front. 11 (2024) 4663–4670. doi: 10.1039/d4qo00996g
P.R. Khoury, J.D. Goddard, W. Tam, Tetrahedron 60 (2004) 8103–8112. doi: 10.1016/j.tet.2004.06.100
J.W. Zhang, Y.R. Wang, J.H. Pan, et al., Angew. Chem. Int. Ed. 59 (2020) 3900–3904. doi: 10.1002/anie.201914623
J. Hierold, T. Hsia, D.W. Lupton, Org. Biomol. Chem. 9 (2011) 783–792. doi: 10.1039/C0OB00632G
A. Elhage, P. Costa, A. Nasim, A.E. Lanterna, J.C. Scaiano, J. Phys. Chem. A 123 (2019) 10224–10229. doi: 10.1021/acs.jpca.9b06716
H.F. Piedra, M. Plaza, Chem. Sci. 14 (2023) 650–657. doi: 10.1039/d2sc05556b
P. Dowd, W. Zhang, Chem. Rev. 93 (1993) 2091–2115. doi: 10.1021/cr00022a007
A.J. Clark, Chem. Soc. Rev. 31 (2002) 1–11.
K.W. Shimkin, D.A. Waton, Beilstein J. Org. Chem. 11 (2015) 2278–2288. doi: 10.3762/bjoc.11.248
Q.M. Kainz, C.D. Matier, A. Bar-toszewicz, et al., Science 351 (2016) 681–684. doi: 10.1126/science.aad8313
A. Hossain, A. Bhattacharyya, O. Reiser, Science 364 (2019) eaav9713. doi: 10.1126/science.aav9713
Y. Luo, Y. Li, J. Wu, et al., Science 381 (2023) 1072–1079. doi: 10.1126/science.adg9232

Scheme 2 Scope of indazoles and α-haloalkyl cycloketone derivatives. Reaction conditions: 1 (0.3 mmol, 1.5 equiv.), 2 (0.2 mmol, 1.0 equiv.), 10 mol% of Cu(MeCN)4PF6, Cs2CO3 (0.6 mmol, 3.0 equiv.), DMF (2.0 mL), r.t., for 24 h, under N2. Isolated yields. a Yield of 3a.

Scheme 3 Scope of structurally diverse amines. Reaction conditions: 1a (0.3 mmol, 1.5 equiv.), 2 (0.2 mmol, 1.0 equiv.), 10 mol% of Cu(MeCN)4PF6, Cs2CO3 (0.6 mmol, 3.0 equiv.), DMF (2.0 mL), r.t., for 24 h, under N2. Isolated yields. a 1a (0.4 mmol, 2.0 equiv.) was used. b t-BuOK (3.0 equiv.) instead of Cs2CO3. c K2CO3 (4 equiv.), DMAP (20 mol%), DCM, reflux, 72 h.

Scheme 4 Scope of structurally diverse alcohols. Reaction conditions: 1a (0.3 mmol, 1.5 equiv.), 2 (0.2 mmol, 1.0 equiv.), 10 mol% of Cu(MeCN)4PF6, Cs2CO3 (0.6 mmol, 3.0 equiv.), DMF (2.0 mL), r.t., for 24 h, under N2. Isolated yields.
Table 1. Optimization of the reaction conditions.a
|
||||
| Entry | Catalyst | Solvent | Base (equiv.) | Yield (%)b |
| 1 | Cu(MeCN)4PF6 | DMF | Cs2CO3 (2) | 83 |
| 2 | CuBr | DMF | Cs2CO3 (2) | 53 |
| 3 | Cu(OTf)2 | DMF | Cs2CO3 (2) | 45 |
| 4 | CuBr2 | DMF | Cs2CO3 (2) | 41 |
| 5 | Fe(OTf)2 | DMF | Cs2CO3 (2) | 68 |
| 6 | ZnCl2 | DMF | Cs2CO3 (2) | Trace |
| 7 | Cu(MeCN)4PF6 | DMF | DBU (2) | 34 |
| 8 | Cu(MeCN)4PF6 | DMF | t-BuOK (2) | 30 |
| 9 | Cu(MeCN)4PF6 | DMF | K2CO3 (2) | 37 |
| 10 | Cu(MeCN)4PF6 | DMA | Cs2CO3 (2) | 70 |
| 11 | Cu(MeCN)4PF6 | DMSO | Cs2CO3 (2) | 12 |
| 12 | Cu(MeCN)4PF6 | Toluene | Cs2CO3 (2) | 28 |
| 13 | Cu(MeCN)4PF6 | DMF | Cs2CO3 (3) | 94 (trace)c |
| 14 | Cu(MeCN)4PF6 | DMF | – | n.r. |
| 15 | – | DMF | Cs2CO3 (3) | Trace |
| a Reaction conditions: 1a (0.3 mmol, 1.5 equiv.), 2a (0.2 mmol, 1.0 equiv.), catalyst (10 mol%), base (x equiv.), solvent (2 mL), r.t., for 24 h, under N2. b Yield of isolated product. c K2CO3 (4 equiv.), DMAP (20 mol%), DCM, reflux, 72 h. |
||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们





 下载:
下载: