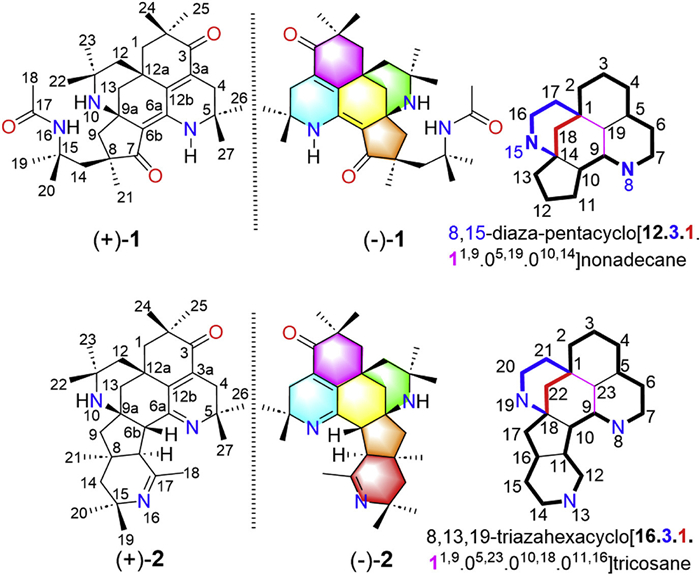
Figure 1.
Chemical structures of pyrethalkalines A (1) and B (2) and the nomenclature of two unprecedented ring systems.
Discovery of pyrethalkalines A and B as potent analgesics with unprecedented chemical skeletons dual targeting TRPM8 and Kv1.2 ion channels
Hui Chen , Xieraili Tuerxun , Amina Abula , Yenan Sun , Hanqi Zhang , Guangmin Yao , Haji Akber Aisa

It is estimated that nearly 600 million people worldwide are afflicted by neuropathic pain and cancer-related pain [1,2]. Currently, opioid drugs remain indispensable in managing these conditions, particularly advanced cancer pain [3-5]. However, their addictive nature, respiratory depression, cold hyperalgesia, and public health risks necessitate urgent attention [6,7]. Consequently, developing non-addictive, highly effective non-opioid analgesics is critical [8]. Currently, three major strategies have been reported to develop non-opioid analgesic drugs, such as developing spinal inhibitory synapse enhancers such as γ-aminobutyric acid (GABA) modulators [9,10], implementing neuroimmune axis blockers such as C-C chemokine receptor 2 (CCR2) and CCR5 antagonists [11,12], exploring new peripheral analgesic targets such as transient receptor potential melastatin 8 (TRPM8) [13-15] and Nav1.8 [8] ion channels.
TRPM8 is a type of TRPM subtype ion channel that has been shown to mediate cold perception and pain sensation [16]. The distribution of this channel is extensive, being found in sensory neurons such as dorsal root ganglia, trigeminal ganglia, and prostate tissues [17,18]. Under normal physiological conditions, TRPM8 activation by temperatures below 28 ℃ or menthol mediates Ca2+ influx to convey cold sensation; conversely, in pathological states such as nerve injury and chemotherapy-induced neuropathy, its aberrant hyperactivity induces cold hyperalgesia, characterized by severe pain evoked by mild cold stimuli [17-19]. TRPM8 antagonists selectively block ion channels to inhibit Ca2+ influx, directly suppressing abnormal neuronal discharges [17,20]. Furthermore, they downregulate spinal mGluR-mediated central sensitization and reverse cold hyperalgesia, thereby interrupting pathological pain signaling in neuropathic, cancer, and inflammatory pain [14,18,19]. Despite clinical studies of pain-treatment antagonists such as VBJ103 have begun, their clinical application may be limited due to hypothermia side effects and low bioavailability [19]. Meanwhile, Kv1.2 regulates cold sensation thresholds through molecular interactions with TRPM8, serving a core regulatory function in cold stimulus-triggered action potential conduction [20,21]. Dual targeting of TRPM8 and Kv1.2 channels concurrently inhibits calcium influx and potassium efflux, significantly reducing neuronal excitability and peripheral nociceptive signaling [22]. Consequently, this approach potently suppresses pain transmission, elicits non-opioid analgesia, and mitigates adverse effects associated with selective TRPM8 inhibition, such as hypothermia. This emphasizes the necessity for the development of novel, non-opioid dual-target potent analgesics capable of targeting both TRPM8 and Kv1.2.
The roots of Anacyclus pyrethrum were traditionally employed in systems such as Ethnomedicine from China (Xinjiang region), and Ayurvedic medicine from India to treat neuralgias, including migraine, trigeminal neuralgia, and toothache [13,23]. Previously phytochemical studies on the roots of A. pyrethrum revealed the presence of structurally novel and potent non-opioid analgesic alkaloids by targeting TRPM8, transient receptor potential canonical 6 (TRPC6), Kv1.2, Kv1.3, and Cav2.1 ion channels [13,24,25].
To search for more potent non-opioid analgesics, the roots of A. pyrethrum were re-investigated, leading to the isolation of two pairs of alkaloid enantiomers with unprecedented chemical skeletons, named pyrethalkalines A (1) and B (2) (Fig. 1). Pyrethalkaline A (1) is an unprecedented 6/6/6/6/5-fused pentacyclic triamino alkaloid featuring a unique 8,15-diaza-pentacyclo[12.3.1.11,9.05,19.010,14]nonadecane core, and pyrethalkaline B (2) is a novel 6/6/6/6/5/6-fused hexacyclic triamino alkaloid possessing an unprecedented 8,13,19-triazahexacyclo[16.3.1.11,9.05,23.010,18.011,16]tricosane motif, respectively. All the isolates exhibited significant analgesic activities, even at lower doses of 1, 0.2, and 0.04 mg/kg, (±)-1, (−)-1, and (+)-2 still showed potent analgesic activities comparable to the positive control morphine. Herein, the isolation, structural elucidation, plausible biosynthetic pathways, analgesic activity, and analgesic targets evaluation of pyrethalkalines A (1) and B (2) were described.
Pyrethalkaline A (1) was obtained as yellow crystals. The (+)-high resolution electrospray ionization mass spectroscopy (HRESIMS) ion at m/z 496.3532 [M + H]+ (calcd. for C30H46N3O3, 496.3534) and 13C nuclear magnetic resonance (NMR) data assigned the molecular formula of 1 to be C30H45N3O3, indicating ten indices of hydrogen deficiency. Its 1H NMR data (Table S1 in Supporting information) displayed characteristic resonances for ten singlet methyl groups at δH 0.98 (H3-23), 1.06 (H3-22), 1.08 (H3-25), 1.16 (H3-21), 1.16 (H3-26), 1.27 (H3-27), 1.30 (H3-24), 1.33 (H3-19), 1.42 (H3-20), and 1.84 (H3-18), six methylenes at δH 1.39 (d, J = 11.8 Hz, H-13β)/1.52 (d, J = 11.8 Hz, H-13α), 1.44 (d, J = 12.4 Hz, H-12α)/1.94 (d, J = 12.4 Hz, H-12β), 1.60 (d, J = 13.6 Hz, H-9β)/2.13 (d, J = 13.6 Hz, H-9α), 1.68 (d, J = 13.9 Hz, H-1α)/1.73 (d, J = 13.9 Hz, H-1β), 1.80 (d, J = 15.3 Hz, H-14α)/2.10 (d, J = 15.3 Hz, H-14β), 2.36 (d, J = 17.3 Hz, H-4)/2.51 (d, J = 17.3 Hz, H-4), and two singlet NH at δH 7.17 (16-NH) and 8.83 (6-NH). The 13C NMR (Table S1) spectroscopic data of 1 revealed the presence of 30 carbon resonances, assignable to be three carbonyls (δC 207.4, C-7; 204.1, C-3; 169.5, C-17), four olefinic carbons (δC 148.5, C-6a; 146.6, C-12b; 132.8, C-3a; 108.5 C-6b), seven sp3 quaternary carbons (δC 55.8, C-9a; 54.7, C-15; 50.3, C-11; 49.6, C-5; 48.0, C-8; 41.6, C-2; 35.6, C-12a), six sp3 methylenes, and ten sp3 methyls, respectively, by HSQC and DEPT-135 ° data. Two double bonds and three ketone carbonyls accounted for five indices of hydrogen deficiency, the remaining five indices of hydrogen deficiency indicated a pentacyclic ring system in 1.
The 1H-1H correlation spectroscopy (COSY) spectrum of 1 only exhibited correlations of geminal coupling of methylenes as deduced from the coupling constants and peak multiplets, and no useful information to construct the partial structures. Heteronuclear multiple bond correlation (HMBC) data were used to determine the planar structure of 1. The HMBC correlations (Fig. 2A) of H3-22/H3-23 to C-11/C-12, H2-12/H2-13 to C-12a/C-12b, H2-13 to C-9a/C-6b, and 6-NH to C-6b, as well as the chemical shifts of C-11 (δC 50.3) and C-9a (δC 55.8) established the rings A and B, forming the 2-azabicyclo[3.3.1]nonane substructure. HMBC correlations of H3-26/H3-27 to C-4/C-5, H2-4 to C-3/C-3a/C-12b, H3-24/H3-25 to C-1/C-2/C-3, H2-1 to C-12a/C-12b, and 6-NH to C-4/C-5/C-12b constructed the rings C and D, forming an octahydroisoquinolinone substructure, which was connected to the 2-azabicyclo[3.3.1]nonane unit via C-12a/C-12b/C-6a. The cyclopentanone ring E fused to 2-azabicyclo[3.3.1]nonane unit by C-6b/C-9a was deduced from the HMBC correlations of H2-9 to C-6b/C-9a, H3-21 to C-7/C-8/C-9. Additionally, the HMBC correlations of H2-14 to C-7, H3-19/H3-20 to C-14/C-15, 16-NH to C-14/C-15/C-17, and H3-18 to C-17 indicated the presence of a CH3(C-18)–C(C-17)–NH(N-16)–C(CH3)2(C-15 or C-19 or C-20)–CH2(C-14) fragment, which was connected to the ring E through the C-8, as supported by the HMBC correlations of H2-9/H3-21 to C-14. Accordingly, the planar structure of 1 was determined to be a 6/6/6/6/5-fused pentacyclic triamino alkaloid featuring an unprecedented 8,15-diaza-pentacyclo[12.3.1.11,9.05,19.010,14]nonadecane core, which follows the nomenclature of International Union of Pure and Applied Chemistry (IUPAC) [26].
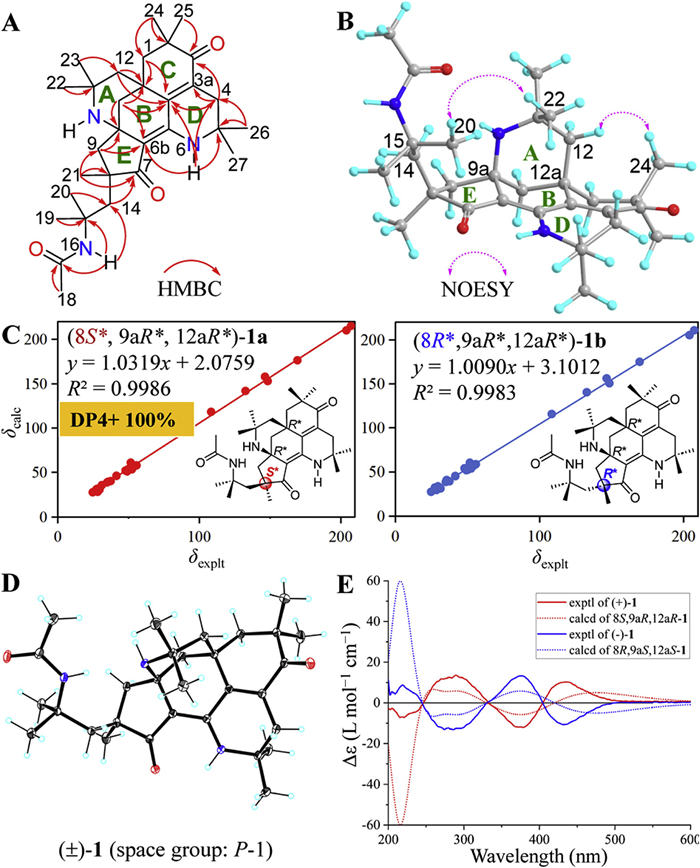
The relative configuration of 1 was defined by the two-dimensional nuclear Overhauser effect spectroscopy (NOESY) data and DP4+ probability analysis [27-32]. The NOESY (Fig. 2B) correlation of H3-20 and H3-22 indicated that the chain of 14-CH2–15-C(CH3)2–16-NH–17-C=O–18-CH3 had the same orientation as the bridge of 10-NH–11-C(CH3)2–12-CH2 between C-9a and C-12a. To further verify the relative configurations of 1, the NMR data of two potential isomers, (8S*,9aR*,12aR*)-1a and (8R*,9aR*,12aR*)-1b, were calculated using the gauge-independent atomic orbital (GIAO) method at the mPW1PW91/6-311G(d, p) level using the Gaussian 09 software [33-35]. Results (Fig. 2C) showed that (8S*,9aR*,12aR*)-1a had a higher coefficient of determination (R2 = 0.9986) of the linear correlation between the calculated and experimental 13C NMR data than (8R*,9aR*,12aR*)-1b (R2 = 0.9983). The relative configuration of 1 is further determined to be 8S*,9aR*,12aR* with a higher probability of 100% based on the DP4+ probability calculation (Table S2 in Supporting information).
Finally, the structure of 1 was confirmed by single-crystal X-ray diffraction analysis (Fig. 2D). However, the symmetric space group (P-1) revealed the racemic nature of 1. Two optically pure enantiomers, (+)-1 and (−)-1 in a mass ratio of 1:1, were then successfully separated by a chiral HPLC (Fig. S1 in Supporting information).
To establish their absolute configurations, (8S,9aR,10aR)-1 and its enantiomer (8R,9aS,10aS)-1 were subjected to time-dependent density functional theory electronic circular dichroism (TDDFT ECD) calculation at PBE0-SCRF/TZVP (methanol, IEFPCM solvent model) level of theory [36-39]. The calculated ECD data of (8S,9aR,10aR)-1 matches well with the experimental ECD curve of (+)-1 (Fig. 2E), while is opposite to that of (−)-1. Accordingly, the absolute configurations of (+)-1 and (−)-1 were established as 8S,9aR,12aR and 8R,9aS,12aS, respectively.
Pyrethalkaline B (2) was obtained as a yellow powder. Its molecular formula was determined to be C30H45N3O by (+)-HRESIMS ion at m/z 464.3636 [M + H]+ (calcd. for C30H46N3O, 464.3635) and 13C NMR data, indicating ten degrees of unsaturation. The NMR data (Table S3 in Supporting information) analysis of 2 suggested the presence of ten sp3 methyl groups, six sp3 methylenes, two sp3 methines, one carbonyl (δC 204.2, C-3), four non-hydrogen olefinic carbons (δC 143.1, C-12b; 131.9, C-3a; 164.8, C-6a; 165.0, C-17), and seven sp3 quaternary carbons, which showed structural similarities to pyrethalkaline A (1). The larger chemical shifts of C-6a (δC 164.8) and C-17 (δC 165.0) suggested the presence of two imines in 2. One carbonyl, one double bond, and two imines occupied four degrees of unsaturation, the remaining six degrees of unsaturation revealed the hexacyclic system in 2.
As shown in Fig. S2 (Supporting information), the 1H–1H COSY spectrum of 2 (Fig. S2A) mainly exhibited correlations of geminal coupling of methylenes, except for the H-6b and H-7 correlation. Similar to 1, the chemical shifts and the HMBC correlations (Fig. S2A) of H3-22/H3-23 to C-11/C-12, H2-1/H2-12/H2-13 to C-12a/C-12b, H-6b to C-6a/C-12b, H3-24/H3-25 to C-1/C-2, H2-4 to C-3/C-3a, H3-26/H3-27 to C-4/C-5, H2-9 to C-6b/C-9a, and H3-21 to C-7/C-8/C-9 constructed the substructure of rings A–E in 2, which possesses the unprecedented 8,15-diaza-pentacyclo[12.3.1.11,9.05,19.010,14]nonadecane motif as in 1. In consideration of the remaining degree of unsaturation and the chemical shifts of C-15 (δC 55.4) and C-17 (δC 165.0), the HMBC correlations of H3-19/H3-20 to C-14/C-15, H3-21 to C-14, and H3-18 to C-7/C-17 constructed the ring F, a tetrahydropyridine moiety in 2. Thus, the planar structure of 2 was identified to be a novel 6/6/6/6/5/6-fused hexacyclic triamino alkaloid possessing an unprecedented 8,13,19-triazahexacyclo[16.3.1.11,9.05,23.010,18.011,16]tricosane motif, based on the nomenclature of IUPAC [26].
The relative configuration of 2 was determined by coupling constants analysis, NOESY data, and NMR calculations. The orientation of H-6b was randomly assigned to be β-orientation. The larger coupling constant JH-6b/H-7 = 12.4 Hz of H-7 and H-6bβ defined the α-orientation of H-7. The α-orientation of 21-CH3 was deduced from the NOESY correlation (Fig. S2B) of H3-21 and H-7α. The NOESY correlations of H-6bβ to H3-19 and H-14β further supported the above conclusion. The weak NOESY correlation between H-6bβ and H3-22 suggested the β-orientation of the bridge of 10-NH–11-C(CH3)2–12-CH2 between C-9a and C-12a. To further prove the relative configurations of 2, the 13C NMR data and DP4+ probability analysis of two possible isomers of 2 were calculated by the gauge-independent atomic orbital (GIAO) method at the mPW1PW91/6-311G(d, p) level using Gaussian 09 software [33,34]. As shown in Fig. S2C, (6bR*,7R*,8R*,9aS*,12aR*)-2a had a superior coefficient of determination (R2 = 0.9989) for the linear correlation between the experimental and calculated 13C NMR data than (6bR*,7R*,8R*,9aR*,12aS*)-2b. The higher DP4+ probability of 100% of (6bR*,7R*,8R*,9aS*,12aR*)-2a (Fig. S2D) defined the relative configuration of 2 to be 6bR*,7R*,8R*,9aS*,12aR*. Same as 1, 2 is a racemic mixture, and two optically pure enantiomers (+)-2 and (−)-2 were successfully resolved by a chiral HPLC (Fig. S2E). To determine their absolute configurations, the ECD spectra of (6bR,7R,8R,9aS,12aR)-2 and its enantiomer were calculated at the LC-wPBE/6-311G(d, p) level using Gaussian 09 software [40,41]. The absolute configurations of (+)-2 and (−)-2 were determined to be 6bR,7R,8R,9aS,12aRand6bS,7S,8S,9aR,12aS, respectively, by comparison of the calculated and experimental ECD data (Fig. S2F).
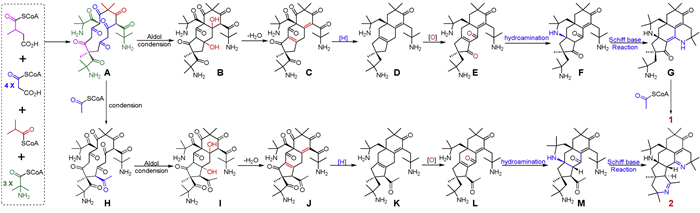
Pyrethalkaline A (1) is a highly conjugated 6/6/6/6/5-fused pentacyclic triamino alkaloid featuring an unprecedented 8,15-diaza-pentacyclo[12.3.1.1,9.05,19.010,14]nonadecane motif, while pyrethalkaline B (2) is a conjugated 6/6/6/6/5/6-fused hexacyclic triamino alkaloid possessing an unprecedented 8,13,19-triazahexacyclo[16.3.1.11,9.05,23.010,18.011,16]tricosane core. In a view of the structural perspective, 1 and 2 shared the same skeleton containing a unique 1,1-dimethyl ethylamine unit, and 2-aminoisobutyric-CoA may be the key building block [42]. Similar to the previously reported octacyclic tetraazabic alkaloid anacyphrethine A from the roots of A. pyrethrum [13,24,25], the biosynthetic pathways for 1 and 2 are proposed to start from the condensation of the polyketides and 2-aminoisobutyric-CoA (Scheme 1). The condensation of one 2-methylsuccinyl-CoA, four malonyl-CoA, and one isobutyryl-CoA forms a thirteen-membered polyketide, which further condenses with three 2-aminoisobutyryl-CoA to produce the nitrogen-containing thirteen-membered polyketide A. An intramolecular aldol reaction in A forms the nitrogen-containing tricyclic polyketide B. After dehydration, reduction, and oxidation, the key 6/6/6/5-fused tetracyclic alkaloid F is formed by the key hydroamination reaction in E. The 6/6/6/6/5-fused pentacyclic alkaloid G is derived from F via a key Schiff base reaction. Finally, pyrethalkaline A (1) is obtained by the condensation of G with an acetyl-CoA. Similarly, the condensation of A with an acetyl-CoA yields the nitrogen-containing polyketide H. In the same way, pyrethalkaline B (2) is obtained from H by the successive intramolecular aldol reaction, dehydration, reduction, oxidation, hydroamination reaction, and final Schiff base formation.
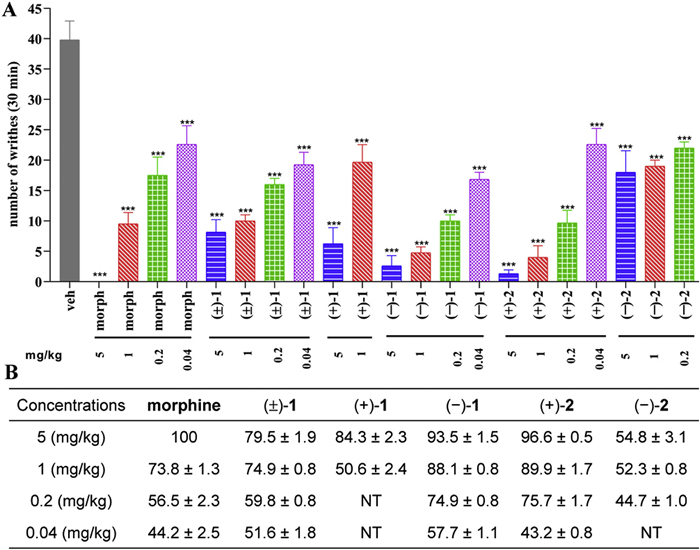
Since the roots of A. pyrethrum have been used as a traditional Chinese and Uyghur medicine to treat headaches, migraines, and cold toothaches, the isolates (±)-1, (+)-1, (−)-1, (+)-2, and (−)-2 were evaluated for their analgesic activity using an acetic acid-induced writhing model in mice, with morphine as the positive control [27]. As shown in Fig. 3, the intraperitoneal administration of all the test isolates at a dose of 5 mg/kg exhibited significant analgesic effects with percentage inhibitions ranging from 54.8% to 96.6%. At a lower dose of 1 mg/kg, (±)-1, (−)-1, and (+)-2 demonstrated more potent analgesic effects with percentage inhibitions of 74.9% ± 0.8%, 88.1% ± 0.8%, and 89.9% ± 1.7%, respectively, than morphine with a percentage inhibition of 73.8% ± 1.3%. Remarkably, at the lower dose of 0.2 mg/kg, the dosage of morphine in clinical practice, (±)-1 (percentage inhibition 59.8% ± 0.8%), (−)-1 (percentage inhibition 74.9% ± 0.8%), and (+)-2 (percentage inhibition 75.7% ± 1.7%) maintained superior analgesic activity than morphine (percentage inhibition 56.5% ± 2.3%). Even at the lowest dose of 0.04 mg/kg, (±)-1 and (−)-1 still showed more potent analgesic effects with percentage inhibitions of 51.6% ± 1.8% and 57.7% ± 1.1%), respectively, than morphine (44.2% ± 2.5%). It is noteworthy that both 1 and 2 exhibit chiral specificity. In all the test doses, the analgesic activity of the levorotatory isomer (−)-1 was found to be stronger than that of the dextrorotatory isomer (+)-1 and the racemic mixture (±)-1. While, the dextrorotatory isomer (+)-2 demonstrates more potent analgesic activity than the levorotatory isomer (−)-2 at two test doses of 5 and 1 mg/kg. Due to the more potent analgesic activity of 1 than morphine at lower doses of 0.2 and 0.04 mg/kg, (+)-1 and (−)-1 were selected for further investigations.
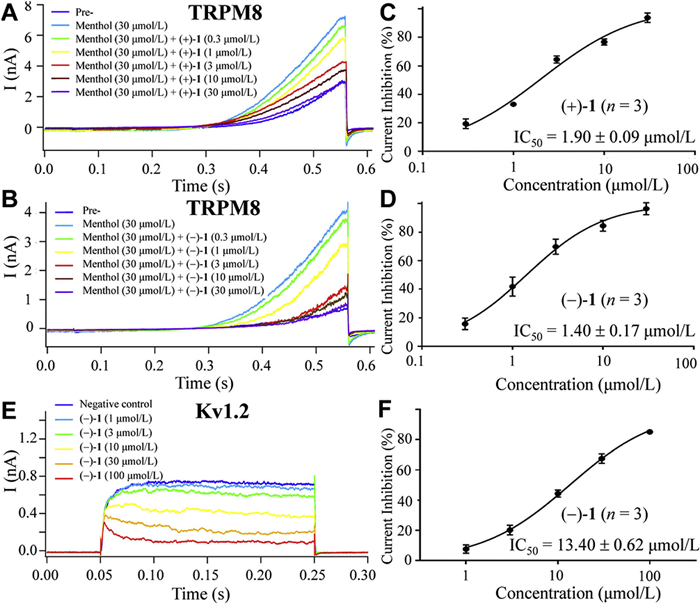
Since anacyphrethine A, an octacyclic tetraazabic alkaloid from the roots of A. pyrethrum, had been reported to be a non-opioid, multitargets potent analgesic targeting TRPM8, TRPC6, Kv1.2, Kv1.3, and CaV2.1 ion channels rather than opioid receptors [13,24,25]. Inspired by this, to identify the analgesic target of the most potent analgesic 1, (+)-1 and (−)-1 were tested for their inhibitory effects against TRPM8, TRPC6, Kv1.2, Kv1.3, and CaV2.1 ion channels using the whole-cell patch-clamp technique [43]. As shown in Table S4 (Supporting information), at a concentration of 10 µmol/L, (+)-1 showed significant inhibitory effects against the peak current in TRPM8-expressing HEK 293T cells with a percentage inhibition of 68.76% ± 0.50%, while moderate inhibitory effects against Kv1.2 ion channel with a percentage inhibition of 30.95% ± 1.17% (Table S4). In contrast, (−)-1 exhibited significant inhibitory effects against TRPM8 and Kv1.2 ion channels with percentage inhibitions of 68.37% ± 5.88% and 46.20% ± 1.19%, respectively (Table S4). However, both (+)-1 and (−)-1 did not exhibit significant inhibitory effects against the TRPC6, Kv1.3 and Cav2.1 ion channels. (+)-1 and (−)-1 dose-dependently inhibited TRPM8 peak currents (Figs. 4A and B) with half-maximal inhibitory concentration (IC50) values of 1.90 ± 0.09 µmol/L (Fig. 4C) and 1.40 ± 0.17 µmol/L (Fig. 4D), respectively. (−)-1 also dose-dependently blocked Kv1.2 peak currents (Fig. 4E) with an IC50 value of 14.40 ± 0.62 µmol/L (Fig. 4F). Notably, (−)-1 demonstrated significantly stronger inhibitory activity against TRPM8 and Kv1.2 channels than its enantiomer (+)-1, indicating a chirality dependent efficacy difference. These in vitro results could be used to explain why (−)-1 exhibited superior in vivo analgesic efficacy than (+)-1.
TRPM8, a human cold receptor predominantly expressed in trigeminal and dorsal root ganglia, has been shown to mediate sensitised pain signalling [44]. Kv1.2 has been demonstrated to regulate the cold sensitivity threshold of TRPM8 through direct interaction, thereby governing low-temperature-triggered action potential conduction [20,21]. Dual targeting TRPM8 and Kv1.2 ion channels concurrently inhibits calcium influx and reduces potassium efflux, significantly diminishing neuronal excitability and peripheral nociceptive signalling, thereby suppressing pain transmission [22]. Interestingly, TRPM8 inhibitors have been reported to antagonise morphine-induced cold hyperalgesia, thus providing an enhanced efficacy and reduced toxicity therapeutic strategy for morphine co-administration [45,46]. However, the TRPM8 inhibitor-based analgesic VBJ103, currently in clinical research, is limited by low oral bioavailability and cold hypersensitivity adverse reactions[19]. Heretofore, there is no analgesics targeting both TRPM8 and Kv1.2 in clinic or clinical trials. Therefore, (+)-1 and (−)-1 showed the potential for the effective treatment of neuropathic pain by dual regulating TRPM8 and Kv1.2 ion channels in the central nervous system.
To further investigate their modes of action, molecular docking [27] and molecular dynamics (MD) [47] simulations were carried out to study the binding modes of (+)-1 and (−)-1 to TRPM8 and Kv1.2 (Table S5 and Figs. S3–S7 in Supporting information).
Molecular docking reveals that (+)-1 and (−)-1 occupy almost the same position and share almost the same binding mode within the TRPM8 pocket (Fig. S3). Both of them have hydrophobic interactions with Phe846, Ile746, Leu 842, Tyr835, Thr839, Ala879, and Val878 residues, and π-alkyl interactions with Leu749, Leu750, Ala874, Phe871, and Val875 residues (Fig. S4). However, there is no significant chiral difference, and the major difference between them is the orientation of the acetamide C-17 carbonyl. The C-17 carbonyl in (−)-1 extends into a hydrophobic region, forming a conventional hydrogen bond with Tyr907 residue and a hydrophobic interaction with Met877 residue (Figs. S4A–C). Whereas, no such interactions are found in (+)-1 (Figs. S4D–F). This difference could be used to rationally explain the enhanced TRPM8 inhibition of (−)-1.
Similar to those in TRPM8, the activity pocket and binding modes of (+)-1 and (−)-1 to Kv1.2 are almost same (Fig. S5), and both of them have hydrophobic interactions with Val89, Tyr90, Trp121, and Trp243 residues, π-alkyl interactions with Trp57 and Trp272 residues, and conventional hydrogen bonds with Arg189 and Lys276 residues (Fig. S6). There is no significant chiral difference, and the primary conformational difference between them originates from the slight spatial orientation of the C-17 carbonyl group. In (+)-1, the C-17 carbonyl forms only one conventional hydrogen bond with Arg189 residue (Figs. S6A–C). While, the C-17 carbonyl of compound (−)-1 adopts an extended conformation, and forms two conventional hydrogen bonds with Arg189 residue (Figs. S6D–F). This slight difference may explain the stronger inhibitory effects of (−)-1 than (+)-1 against Kv1.2 ion channel.
To further confirm the binding modes, all-atom molecular dynamics simulations were performed on the (−)-1/TRPM8 complex system. The results (Fig. S7) demonstrate that the conformation of (−)-1 within the TRPM8 binding site exhibits high stability, with no significant conformational drift observed. Notably, the root-mean-square deviation (RMSD) (Fig. S7A) of the (−)-1/TRPM8 complex (blue line) increases rapidly in the early stages of the simulation, reaching equilibrium after approximately 20 ns. It then stabilizes within the range of 0.7−1.0 nm. Residue-specific root-mean-square fluctuation (RMSF) analysis (Fig. S7B) shows that most residues have RMSF values less than 1.2 nm, indicating that these regions of the protein structure are relatively stable and constitute the stable core. The lowest free energy region (in blue) in the Gibbs energy landscape (GEL) is located near PC1 at approximately 0.90 and PC2 at approximately 4.9 kJ/mol (Fig. S7C), thereby substantiating the dynamic stability of the complex structure.
In summary, two pairs of potent analgesic alkaloids enantiomers with unprecedented chemical architectures, named pyrethalkalines A (1) and B (2), were isolated from the roots of A. pyrethrum. Pyrethalkaline A (1) is a highly conjugated 6/6/6/6/5-fused pentacyclic triamino alkaloid featuring an unprecedented 8,15-diaza-pentacyclo[12.3.1.1,9.05,19.010,14]nonadecane core, and pyrethalkaline B (2) is 6/6/6/6/5/6-fused hexacyclic triamino alkaloid possessing an unprecedented 8,13,19-triazahexacyclo[16.3.1.11,9.05,23.010,18.011,16]tricosane motif. (±)-1, (+)-1, (−)-1, (+)-2, and (−)-2 exhibited significant analgesic effects at a dose of 5 mg/kg. At lower doses of 1, 0.2, and 0.02 mg/kg, (±)-1 and (−)-1 demonstrated more potent analgesic effects than morphine. While, (+)-2 showed more potent analgesic effects than morphine at a dose of 1 mg/kg. Further investigation revealed that (+)-1 and (−)-1 were TRPM8 and Kv1.2 dual inhibitors, conferring synergistic potential for neuropathic pains. Molecular dockings provide a novel structural template for developing potent analgesics dual targeting TRPM8 and Kv1.2 ion channels. These results would attract the interests from communities of synthetic chemistry, molecular pharmacology, and neuroscience.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Hui Chen: Writing – original draft, Software, Methodology, Investigation, Data curation. Xieraili Tuerxun: Methodology, Investigation. Amina Abula: Software, Data curation. Yenan Sun: Software, Data curation. Hanqi Zhang: Methodology, Investigation, Data curation. Guangmin Yao: Writing – review & editing, Validation, Supervision, Project administration, Funding acquisition, Formal analysis, Conceptualization. Haji Akber Aisa: Validation, Supervision, Resources, Project administration, Funding acquisition, Conceptualization.
This work was supported by the National Key R & D Program of China (No. 2020YFE0205600), Tian-Shan Talent Program (No. 2022TSYCLJ0064), National Natural Science Foundation of China (Nos. 22277034 and 22477034), Interdisciplinary Research Program of Huazhong University of Science and Technology (No. 2023JCYJ037), and International Cooperation Project of Hubei Provincial Key R & D Plan (No. 2023EHA040), and Xinjiang Key Laboratory of Natural Medicines Active Components and Drug Release Technology (No. 2025XJTRZ07). We are grateful to the Analytical and Testing Center at Huazhong University of Science and Technology for IR, ECD, and single crystal X-ray diffraction data collection, Medical Subcenter at Huazhong University of Science and Technology for NMR data acquisition. The computation is completed in the HPC Platform of Huazhong University of Science and Technology. We are grateful to the Institutional Center for Shared Technologies and Facilities of SIMM for X-ray diffraction data collection.
Supplementary material associated with this article can be found, in the online version, at doi:
W.F. Vieira, D.R.A. Coelho, S.T. Litwiler, et al., Neurosci. Biobehav. Rev. 161 (2024) 105673. doi: 10.1016/j.neubiorev.2024.105673
A.M. Filho, M. Laversanne, J. Ferlay, et al., Int. J. Cancer 156 (2024) 1336–1346.
S. Mercadante, G. Sapienza, A.L. Cascio, et al., Pain. Ther. 14 (2025) 999–1006. doi: 10.1007/s40122-025-00728-4
M. Jamshidi, C.M.P. Jones, A.V. Langford, et al., CNS Drugs 39 (2025) 345–360. doi: 10.1007/s40263-025-01165-9
S. Yennurajalingam, J. Arthur, S. Reddy, et al., JAMA Oncol. 7 (2021) 404. doi: 10.1001/jamaoncol.2020.6789
N.D. Volkow, C. Blanco, Mol. Psychiatry 26 (2021) 218–233. doi: 10.1038/s41380-020-0661-4
D. Dowell, T.M. Haegerich, R. Chou, J. Am. Med. Assoc. 315 (2016) 1624–1645. doi: 10.1001/jama.2016.1464
J.D. Osteen, S. Immani, T.L. Tapley, et al., Pain. Ther. 14 (2025) 655–674. doi: 10.1007/s40122-024-00697-0
M. Pandey, J.H. Zhang, P.R. Adikaram, et al., JCI Insight 8 (2023) e134685. doi: 10.1172/jci.insight.134685
X. Qian, X. Zhao, L. Yu, et al., Biomed. Pharmacother. 168 (2023) 115800. doi: 10.1016/j.biopha.2023.115800
É. Midavaine, R.L. Brouillette, E. Théberge, et al., Pharmacol. Res. 205 (2024) 107242. doi: 10.1016/j.phrs.2024.107242
K. Kwiatkowski, K. Pawlik, K. Ciapała, et al., Front. Immunol. 11 (2020) 615327. doi: 10.3389/fimmu.2020.615327
H. Chen, H. Zhang, C. Niu, et al., Acta Pharm. Sin. B 15 (2025) 3725–3737. doi: 10.1016/j.apsb.2025.04.032
A. Bertamino, N. Iraci, C. Ostacolo, et al., J. Med. Chem. 61 (2018) 6140–6152. doi: 10.1021/acs.jmedchem.8b00545
V. Chubanov, M. Köttgen, R.M. Touyz, et al., Nat. Rev. Nephrol. 20 (2023) 175–187.
S. Wang, D. Zhang, J. Hu, et al., EMBO Mol. Med. 9 (2017) 802–815. doi: 10.15252/emmm.201607300
Y. Yin, M. Wu, L. Zubcevic, et al., Science 359 (2018) 237–241. doi: 10.1126/science.aan4325
Y. Yin, C.G. Park, F. Zhang, et al., Sci. Adv. 10 (2024) eadp2211. doi: 10.1126/sciadv.adp2211
M.S. Gold, J.B. Pineda-Farias, D. Close, et al., Br. J. Pharmacol. 181 (2024) 3527–3543. doi: 10.1111/bph.16429
A.I. Basbaum, D.M. Bautista, G. Scherrer, et al., Cell 139 (2009) 267–284. doi: 10.1016/j.cell.2009.09.028
R.W. Teichert, T. Memon, J.W. Aman, et al., Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 2319–2324. doi: 10.1073/pnas.1324019111
A. González, G. Ugarte, C. Restrepo, et al., J. Neurosci. 37 (2017) 3109–3126. doi: 10.1523/JNEUROSCI.3553-16.2017
H. Manouze, O. Bouchatta, A.C. Gadhi, et al., Front. Pharmacol. 8 (2017) 598. doi: 10.3389/fphar.2017.00598
H. Chen, H.A. Aisa, Phytochemistry 229 (2025) 114255. doi: 10.1016/j.phytochem.2024.114255
Q.B. Chen, J. Gao, G.A. Zou, et al., J. Nat. Prod. 81 (2018) 1474–1482. doi: 10.1021/acs.jnatprod.8b00239
H.A. Favre, W.H. Powell, Nomenclature of Organic Chemistry-IUPAC Recommendations and Preferred Names 2013, The Royal Society of Chemistry2013, 3th. ed., Royal Society of Chemistry, Cambridge, 2014.
G. Zheng, K. Lv, H. Wang, et al., J. Am. Chem. Soc. 145 (2023) 3196–3203. doi: 10.1021/jacs.2c12963
Y. Feng, S. Zha, H. Zhang, et al., Chin. Chem. Lett. 34 (2023) 107742. doi: 10.1016/j.cclet.2022.107742
N. Grimblat, M.M. Zanardi, A.M. Sarotti, J. Org. Chem. 80 (2015) 12526–12534. doi: 10.1021/acs.joc.5b02396
H. Zhang, B. Gao, Y. Feng, et al., Chin. Chem. Lett. 36 (2025) 111234. doi: 10.1016/j.cclet.2025.111234
W.Y. Zhou, Z.H. Xi, N.N. Du, et al., Chin. Chem. Lett. 35 (2024) 109030. doi: 10.1016/j.cclet.2023.109030
J. Zhang, F. Liu, Q. Jin, et al., Chin. Chem. Lett. 35 (2024) 108881. doi: 10.1016/j.cclet.2023.108881
G. Zheng, L. Huang, Y. Feng, et al., Bioorg. Chem. 142 (2024) 106928. doi: 10.1016/j.bioorg.2023.106928
Y. Feng, H. Zhang, B. Gao, et al., Bioorg. Chem. 132 (2023) 106374. doi: 10.1016/j.bioorg.2023.106374
M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Revision D. 01, Gaussian, Inc., Wallingford CT, 2013.
Y. Feng, S. Zha, B. Gao, et al., Chin. J. Chem. 40 (2022) 1019–1027. doi: 10.1002/cjoc.202100764
L. Huang, G. Zheng, Y. Feng, et al., Chin. J. Chem. 40 (2022) 2285–2295. doi: 10.1002/cjoc.202200348
Y.F. Liu, H.F. Du, Y.H. Zhang, et al., Chin. Chem. Lett. 36 (2025) 109858. doi: 10.1016/j.cclet.2024.109858
W.Y. Liu, X.X. Lei, W.J. Wang, et al., Chin. Chem. Lett. 36 (2025) 110478. doi: 10.1016/j.cclet.2024.110478
P. Jin, X. Yuan, X. Ma, et al., Chin. J. Chem. 39 (2021) 1997–2008. doi: 10.1002/cjoc.202100077
P. Jin, G. Zheng, X. Yuan, et al., Bioorg. Chem. 111 (2021) 104870. doi: 10.1016/j.bioorg.2021.104870
J. Li, W.X. Wang, H.P. Chen, et al., Org. Lett. 21 (2019) 1511–1514. doi: 10.1021/acs.orglett.9b00312
Y. Gong, R. Liu, H. Zha, et al., Angew. Chem. Int. Ed. 63 (2023) e202313461.
Y. Yin, S.Y. Lee, Trends. Biochem. Sci. 45 (2020) 806–819. doi: 10.1016/j.tibs.2020.05.008
M. Iftinca, L. Basso, R. Flynn, et al., Mol. Brain 13 (2020) 61. doi: 10.1186/s13041-020-00599-0
K. Gong, L. Jasmin, J. Pain 18 (2017) 212–221. doi: 10.1016/j.jpain.2016.10.015
P.W. Hildebrand, A.S. Rose, J.K.S. Tiemann, Trends. Biochem. Sci. 44 (2019) 902–913. doi: 10.1016/j.tibs.2019.06.004

Figure 1 Chemical structures of pyrethalkalines A (1) and B (2) and the nomenclature of two unprecedented ring systems.

Figure 2 Structural elucidation of pyrethalkaline A (1). Key HMBC (A) and NOESY (B) correlations, linear correlation plots between the experimental and calculated 13C NMR data for two isomers (C), ORTEP drawing of the crystal structure with the ellipsoid contour at 20% probability level (D), and experimental and calculated ECD spectra of (+)-1 and its enantiomer (E).

Figure 3 Analgesic activities of 1 and 2 at a dose of 5.0, 1.0, 0.2, and 0.04 mg/kg with morphine (morph) as a positive control. ***P < 0.001, **P < 0.01 statistically significant differences between 1 and 2 and vehicle (veh) or morph and veh. (A) Numbers of writhes in 30 min. (B) Analgesic percentage inhibition. NT: not test.

Figure 4 Inhibitory effects of (+)-1 and (-)-1 against TRPM8 and Kv1.2 in HEK 293T cells. Inhibition of TRPM8 peak currents (A, B) and dose-response relationship and IC50 data suppressed by different concentrations of (+)-1 (C) and (−)-1 (D); Inhibition of Kv1.2 peak currents (E) and dose-response relationship and IC50 data (F) suppressed by different concentrations of (−)-1.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: