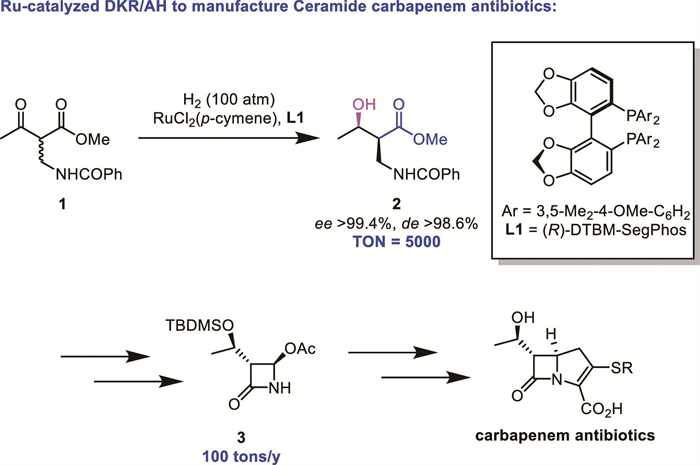
Scheme 1.
A landmark industrial application of DKR-based reduction.

Asymmetric synthesis plays a pivotal role in modern chemistry, particularly in the development of enantiomerically pure compounds for applications in pharmaceuticals, agrochemicals, and materials science. Among the various strategies for achieving enantioselectivity, DKR has emerged as a powerful method for asymmetric transformations [1-3]. DKR combines the principles of kinetic resolution (KR) with in situ racemization of the less reactive enantiomer, allowing for the full conversion of racemic mixtures into a single enantiomer with ideally 100% yield. This dual process overcomes the 50% yield limitation of traditional KR while maintaining excellent stereocontrol, making it an indispensable tool in modern asymmetric synthesis.
On the other hand, asymmetric hydrogenation (AH) has evolved into a robust and practical method to access chiral molecules [4-9]. The integration of DKR with asymmetric (transfer) hydrogenation has garnered significant attention due to its ability to selectively reduce racemic substrates to enantiomerically enriched products in high yields. This strategy is particularly valuable for the synthesis of enantiomerically pure diastereomers of chiral alcohols, amines, and other functionalized molecules bearing multiple consecutive stereogenic centers, which serve as key intermediates in the manufacture of bioactive compounds. In industrial settings, DKR-based reductions are particularly valuable for synthesizing chiral alcohols with high atom economy and operational efficiency. A landmark industrial application is the synthesis of a carbapenem antibiotic key intermediate 2 via AH of an α-substituted-β-ketoester under DKR conditions, producing >100 tons annually (Scheme 1) [10].
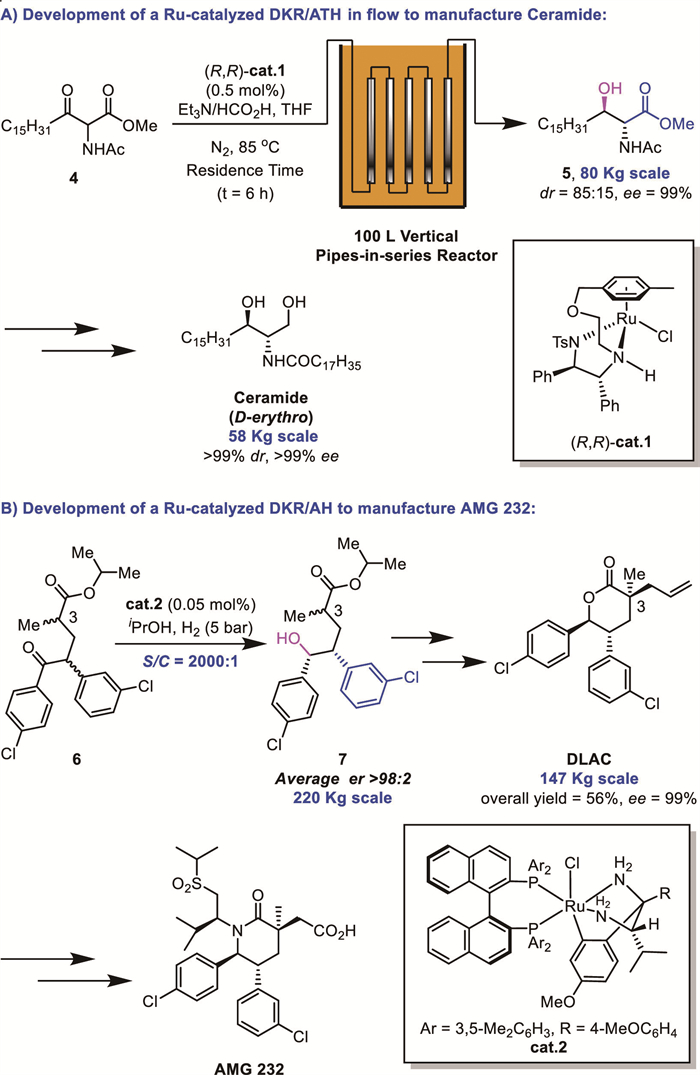
In 2018, scientists from Takasago reported a large-scale asymmetric transfer hydrogenation (ATH) reaction in a pipes-in-series flow reactor. Using (R,R)-Ts-DENEB-ligated ruthenium cat.1 as catalyst (0.5 mol%) and a HCOOH/Et3N azeotrope as hydrogen donor, α-amino-β-ketoester 4 underwent DKR-based reduction to afford anti-β–hydroxy α-aminoester 5 with excellent enantioselectivity (99% ee) and moderate diastereoselectivity (85:15 dr) (Scheme 2A). Subsequent transformations yielded D–erythro-Ceramide on a 58 kg scale with >99% ee and >99:1 dr after recrystallization [11]. In 2020, Smith and co-workers developed a ruthenium-catalyzed DKR/AH process of 6 for the synthesis of DLAC (a δ-lactone precursor to AMG 232) (Scheme 2B). Employing the ruthenabicyclic complex cat.2 at a substrate-to-catalyst ratio (S/C) of 2000:1, product 7 was achieved with >98:2 er. This optimized process delivered 147 kg of DLAC in 56% overall yield with 99% ee [12].
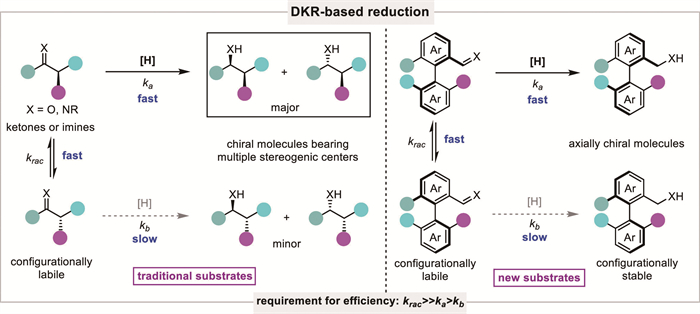
The success of DKR-based reductions relies heavily on the interplay between two catalytic systems: one to facilitate racemization (acid, base or catalyst, etc.) and the other to enable enantioselective reduction. To obtain high efficiency, the racemization rate (krac) of the starting substrate must be much faster than the rate of the asymmetric reduction (ka and kb) and the matched enantiomer must react faster than the mismatched one (ka > kb) (Scheme 3). The careful design and optimization of these catalytic systems are critical for achieving high efficiency, selectivity, and compatibility under mild reaction conditions. In addition, the design of new substrates incorporating labile stereocenters and diverse functional groups is also important, as this can lead to useful chiral products possessing vicinal or non-vicinal chiral centers. So far, the majority of successful substrates are enolizable ketones or activated imines, while other types of substrates are relatively less explored. However, benefiting from the development of new catalytic systems and innovative concepts, some challenging structures, such as racemic α-chiral esters, amides, aromatic N-heterocycles, and even axially chiral compounds, have been conquered recently, which further broadens the applicability of this strategy (Fig. 1).
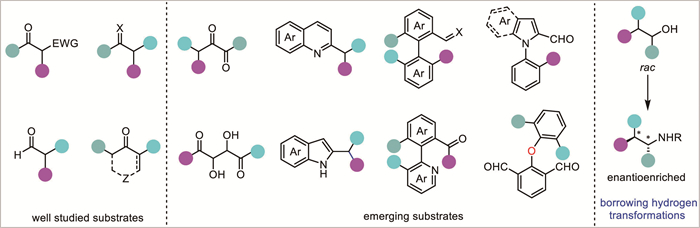
Although the transition-metal-catalyzed AH of ketones and imines through DKR have previously been respectively reviewed by Ratovelomanana-Vidal’s group [13,14] and Zhou’s group [15], the rapid development of this field, including emerging conceptually new reaction designs and their applications in the synthesis of challenging axially chiral compounds or novel N-heterocycles which have not been summarized before, deserves a timely summary. In addition, the emerging asymmetric amination of β-branched alcohols through borrowing hydrogen catalysis [16] has achieved promising advances, and the enantioselectivity-determining step involves a DKR-based asymmetric reduction. Therefore, the key advances in this topic are also included in this review.
This review aims to provide a comprehensive overview of the important advances in DKR-based reduction mainly since 2020, highlighting the latest developments in catalytic systems, mechanistic insights, and synthetic applications. However, a few significant works before that have also been included to better understand the context. By examining the progress in this field, we seek to identify the latest developments, current limitations and outline future directions for research, with a focus on expanding the utility of DKR in asymmetric synthesis. The research progress is summarized according to the substrate types, which is accompanied by significant synthetic applications to underscore the importance of this strategy. The current limitations and prospects of this field are also discussed in the end. To be noted, examples on DKR-based reduction via biocatalysis is not covered.
Due to the facile keto-enol tautomerization, α-substituted β-keto esters 8 are among the most popular substrates for the evaluation of the ability of new catalytic systems on DKR-based hydrogenation. Since the pioneering work of Noyori's [17] and Genêt’s group [18] on the stereoselective hydrogenation of β-keto esters using a RuⅡ-BINAP or RuⅡ–CHIRALPHOS catalyst, various catalytic systems were designed to fit this reaction.
Recently, several new catalytic systems, including the combinations of nickel/bisphosphine, iridium/P,N,N-tridentate ligands, iridium/P,N,O-tridentate ligands, and ruthenium/DIPSkewphos have been developed and tested in the DKR hydrogenation of α-substituted β-keto esters (Scheme 4) [19-23]. The α-substituent can be a simple alkyl group, an amino group, an allyl group, and others. Generally, excellent enantiocontrol and diastereocontrol have been achieved.
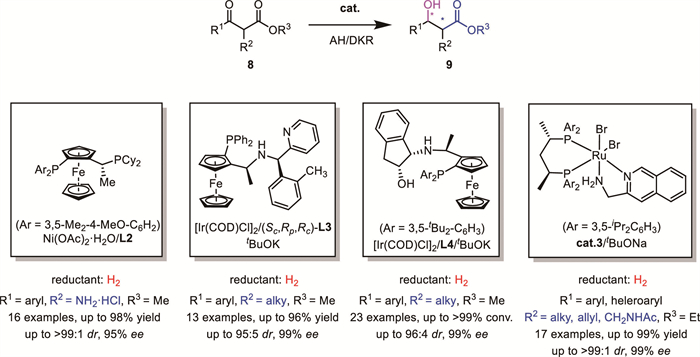
In 2020, Cheung's group developed an innovative protocol for the synthesis of monobactam antibiotic LYS228, discovered by Novartis, using a DKR-based asymmetric hydrogenation strategy to efficiently construct two stereocenters in a single step (Scheme 5) [24]. This method significantly reduced the synthetic steps and cost of LYS228, highlighting its practical applicability in the synthesis of monobactams.
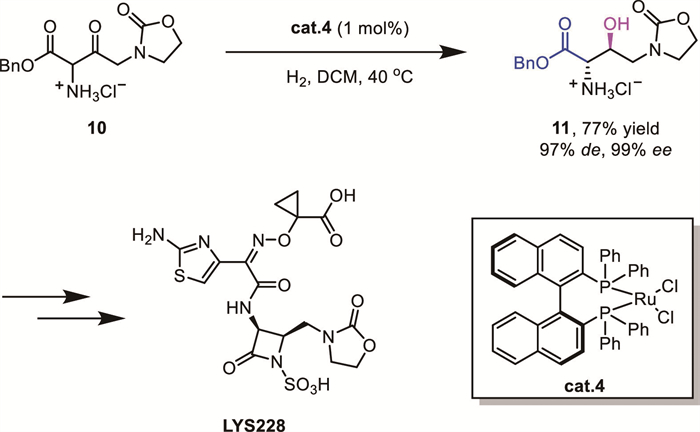
Meanwhile, α-substituted β-ketoamides 12 or β‐keto lactams 14 had also been widely investigated with different metal systems using either H2 or other hydride surrogates, and syn-β–hydroxy amides are typically obtained as the major diastereomer with high enantioselectivity. For instance, Tan’s group reported a Ru-catalyzed DKR-based transfer hydrogenation of α-substituted β-ketoamides 12 (Scheme 6A) [25]. While Xie and Zhou’s group reported an Ir/PNS complex-catalyzed asymmetric hydrogenation of β‐keto lactams 14 (Scheme 6B) [26]. These methodologies are extensively employed in asymmetric catalytic systems for the stereoselective construction of key chiral intermediates in complex molecular architectures.
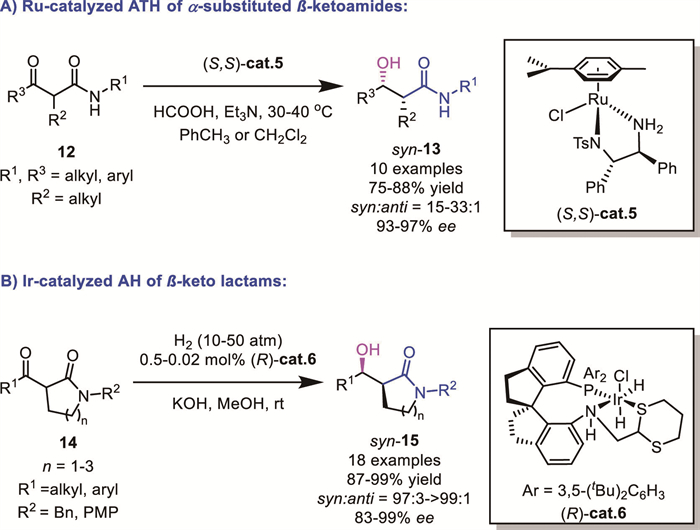
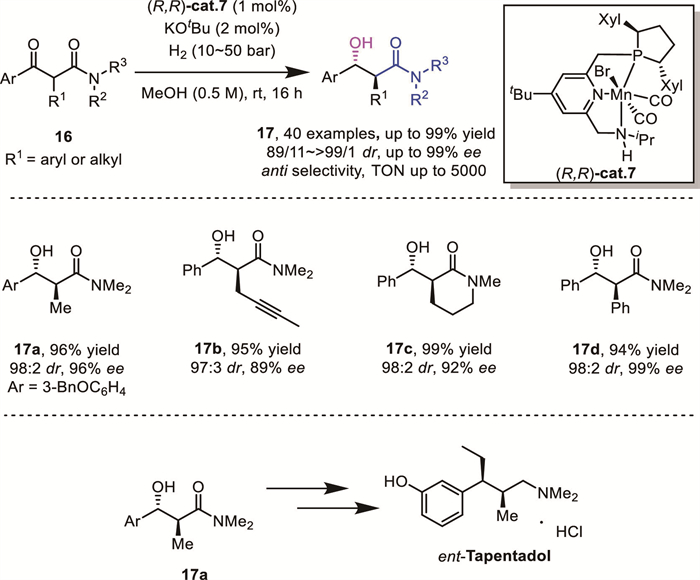
While noble metals still dominate this field, recent development on AH using earth-abundant metals brings new advances in the regulation of the reactivity and selectivity. For instance, in 2020, Ding's group successfully achieved a diastereo- and enantioselective AH of α-substituted β-ketoamides 16 by using well-defined MnⅠ complexes containing lutidine-based P,N,N ligands (Scheme 7), directly affording a broad range of unique anti-α-substituted β–hydroxy amides, which are useful building blocks for the synthesis of bioactive molecules and chiral drugs [27]. A comparative study showed that a cheap Mn catalyst outperformed the noble-metal Ru catalyst bearing the same P,N,N ligand. Importantly, the key intermediate for the asymmetric synthesis of the chiral analgesic drug tapentadol was prepared through the hydrogenation of 16a. Remarkably, the turnover number (TON) could achieve up to 5000, which represented the highest level in the transition-metal-catalyzed asymmetric hydrogenation of α-substituted β-ketoamides. These results demonstrated the practicality of the Mn catalyst system. DFT calculations disclosed that the origin of the unique anti-stereoselectivity lay in an attractive π···π stacking interaction between the phenyl ring of the substrate and the pyridyl unit of the ligand in the favored transition state.
Direct asymmetric reduction of easily accessed α-branched ketones bearing various substituents 18 provides a rapid way to access chiral secondary alcohols with vicinal chirality, which are prevalent in natural products and biorelevant agents. Related works have been continuously reported [13-15]. Noteworthily, from 2020 onward, a variety of novel catalytic systems have been developed to achieve asymmetric reduction of various α-branched ketones via DKR (Scheme 8). With these elegant catalysts, α-branched ketones bearing functional groups such as halogens, sulfonyl, sulfonamide, phosphonate, and secondary amino groups have been successfully reduced with generally excellent diastereo- and enantiocontrol [28-33].
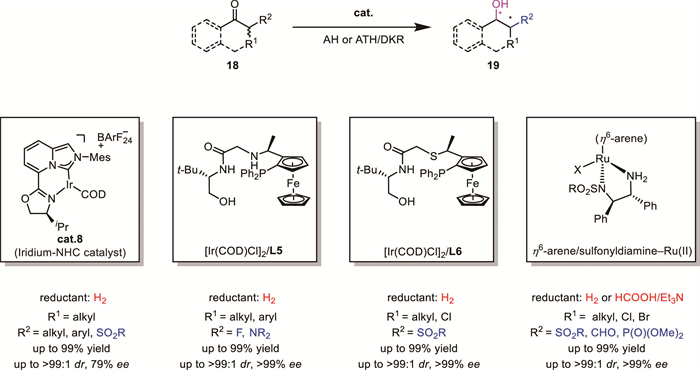
In 2020, Wills’ group reported that the sulfone group served as a versatile and removable directing group for the ATH of ketones [34]. This strategy utilizes the sulfone group to control the reduction selectivity, followed by its removal, enabling access to challenging dialkyl-substituted secondary alcohols with high stereoselectivity. After systematic optimization, a series of α-sulfonyl ketones 20 were reduced with high yields (up to 96% yield) and excellent ee and dr (up to 99% ee, >99:1 dr) (Scheme 9). X-ray crystallographic analysis of the products confirmed that the reduction followed the same stereochemical model as previously reported, wherein the sulfone group preferentially occupies a position distal to the η6-arene of the catalyst in the transition state, while aryloxy and alkoxy groups adopt positions adjacent to the η6-arene of the catalyst. Notably, the authors further demonstrated that the sulfone group could be reductively cleaved, furnishing enantioenriched dialkyl-substituted secondary alcohols 22, a class of compounds that are otherwise difficult to access via conventional methods.
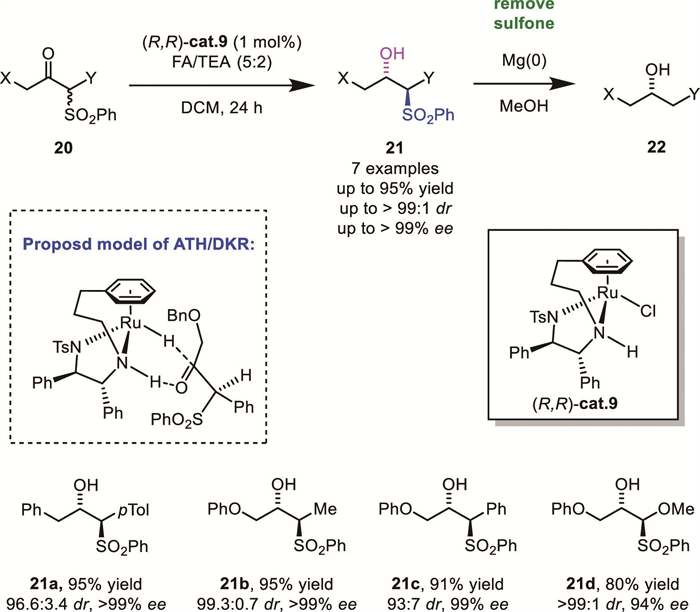
Chiral β–hydroxy carbonitriles are valuable building blocks in organic synthesis because the -CN group can be transformed to amino, carbonyl, and amide groups; therefore, the efficient synthesis of enantiopure β–hydroxy carbonitriles has been a long-standing goal. The direct DKR reduction of α-substituted-β-keto carbonitriles is among the most straightforward methods for this goal. However, due to the metal inhibitory effect derived from the strong coordination property of the cyano group, very limited progress had been achieved using metal catalysis until recently.
In 2021, Zhang and Chen's group successfully developed an efficient strategy for the synthesis of chiral α-substituted-β–hydroxy carbonitriles 24 through ATH of α-substituted-β-keto carbonitriles 23. Their optimal reaction conditions employed the tethered RhⅢ complex (R,R)-cat.10 as catalyst and HCOOH/Et3N azeotrope as the hydrogen donor (Scheme 10A) [35]. Under these conditions, a series of acyclic α-substituted-β-keto carbonitriles 23 were efficiently reduced, affording the corresponding α-substituted-β–hydroxy carbonitriles 24 with high enantio– and diastereoselectivities (up to >99% ee and >99:1 dr). Computational studies revealed that the transition state leading to the anti-product was more stable than that of the syn-product, rationalizing the preferential formation of the anti-isomer as the major product. Furthermore, the synthetic utility of this method was demonstrated through the gram-scale synthesis of key intermediates for Ipenoxazone and Tapentadol.
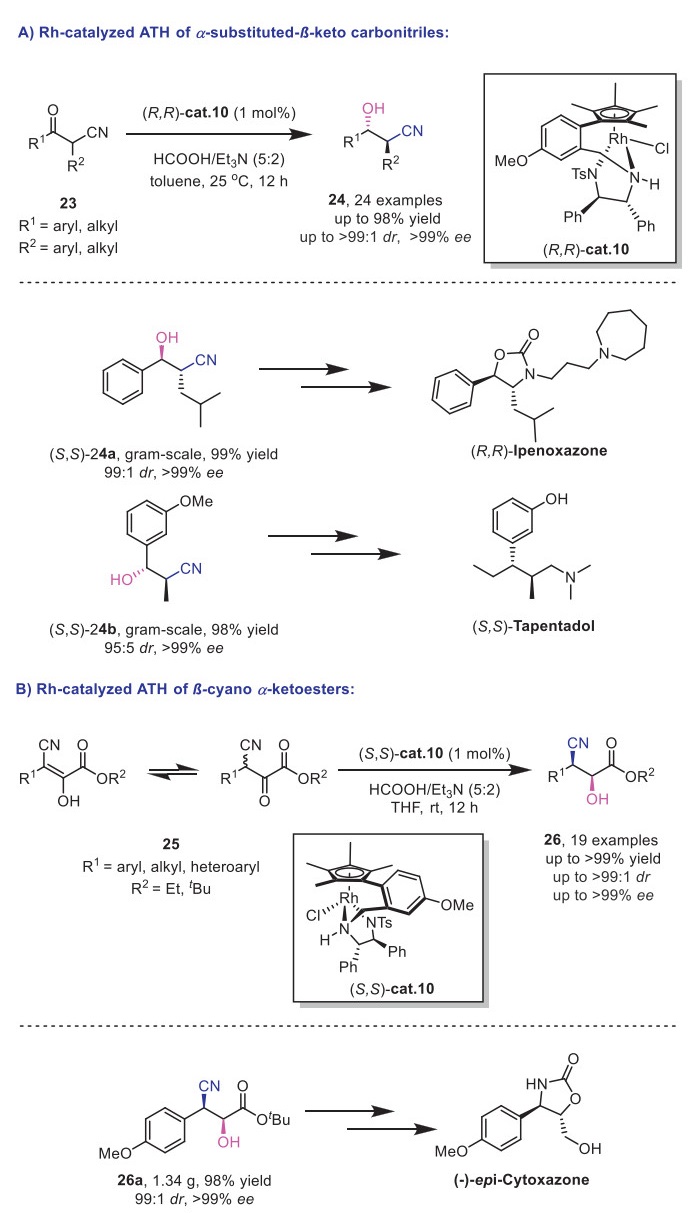
In 2024, the same group further expanded their work by applying a similar ATH/DKR strategy to β-cyano-α-ketoesters 25 (Scheme 10B) [36], providing access to a diverse range of chiral β-cyano-α–hydroxy esters with excellent enantio– and diastereoselectivities (up to >99% ee, 99:1 dr). Additionally, the synthetic potential of this transformation was highlighted by its application in the preparation of a key precursor 26a for (−)–epi–cytoxazone.
Indanols or indanones possessing multiple contiguous stereocenters are widely found in lots of natural compounds or bioactive molecules. DKR-based hydrogenation of α-substituted benzocyclic ketones offers a convenient route to attain these valuable skeletons. Recently, Mohar's group developed an ATH of rac-2-substituted 1-indanones 27 to afford chiral 2,3-disubstituted-1-indanols 28 through a DKR process [37]. A tethered RuⅡ complex was identified as the optimal catalyst, which afforded the desired products with up to 99% ee and >99:1 dr (Scheme 11). With this methodology, they were able to develop a practical protocol toward the synthesis of a drug analog 29 which contains four contiguous stereogenic centers.
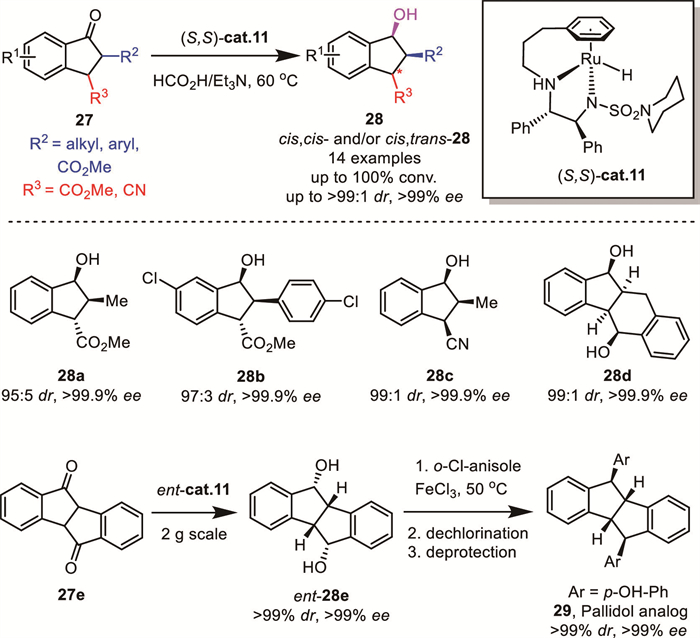
Chiral cyclic β-amino alcohols with contiguous stereocenters are important building blocks for the synthesis of many biologically active molecules and chiral catalysts. DKR-based enantioselective reduction of easily available α-amino benzocyclic ketones represents one of the most effective methods to achieve these useful structures [38,39].
In 2019, Mohar’s group reported a highly stereoselective reduction of α-amino benzocyclic ketones 30 using a home-made chiral RuⅡ complex cat.12, yielding trans-β-amino alcohols 31 with excellent diastereoselectivity and high enantioselectivity [40]. The authors speculated that trans-diastereoselectivity was attributed to the attractive ligand-substrate interaction. Specifically, the H-bond between O=S=O of the ligand and H–NAc of the substrate, as well as a CH/π interaction with T-shaped geometry between the ligand and substrate, contributed to the overall trans-selectivity (Scheme 12).
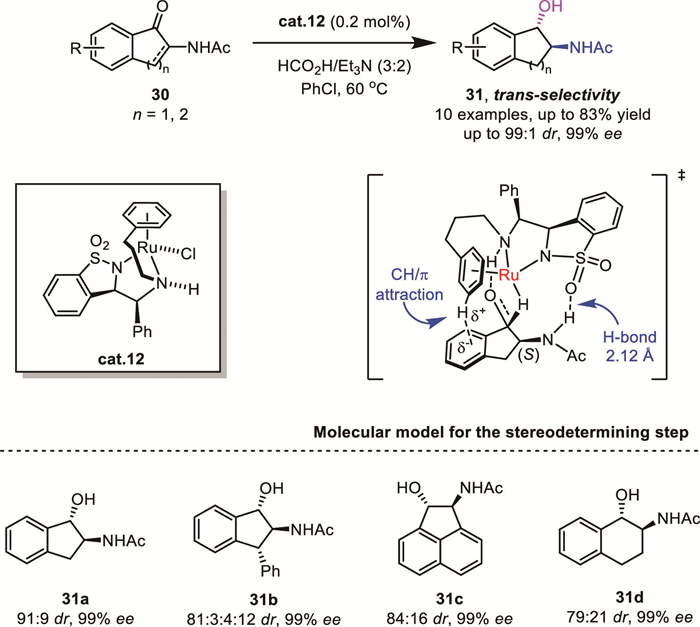
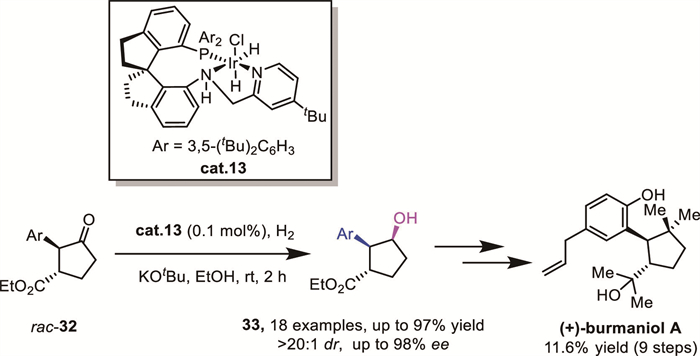
In 2021, Xie and Zhou's group achieved a significant breakthrough in AH of racemic α-aryl-β-ethoxycarbonyl cyclopentanones 32 by utilizing a chiral Ir-SpiroPAP catalyst cat.13 (Scheme 13) [41]. This transformation afforded a range of ester-functionalized chiral 2-arylcyclopentanols 33 with three contiguous stereocenters in high yields with excellent enantio– and diastereoselectivities. Notably, this methodology enabled the first total synthesis of the phenylpropanoid (+)-burmaniol A, showcasing the synthetic utility of their approach.
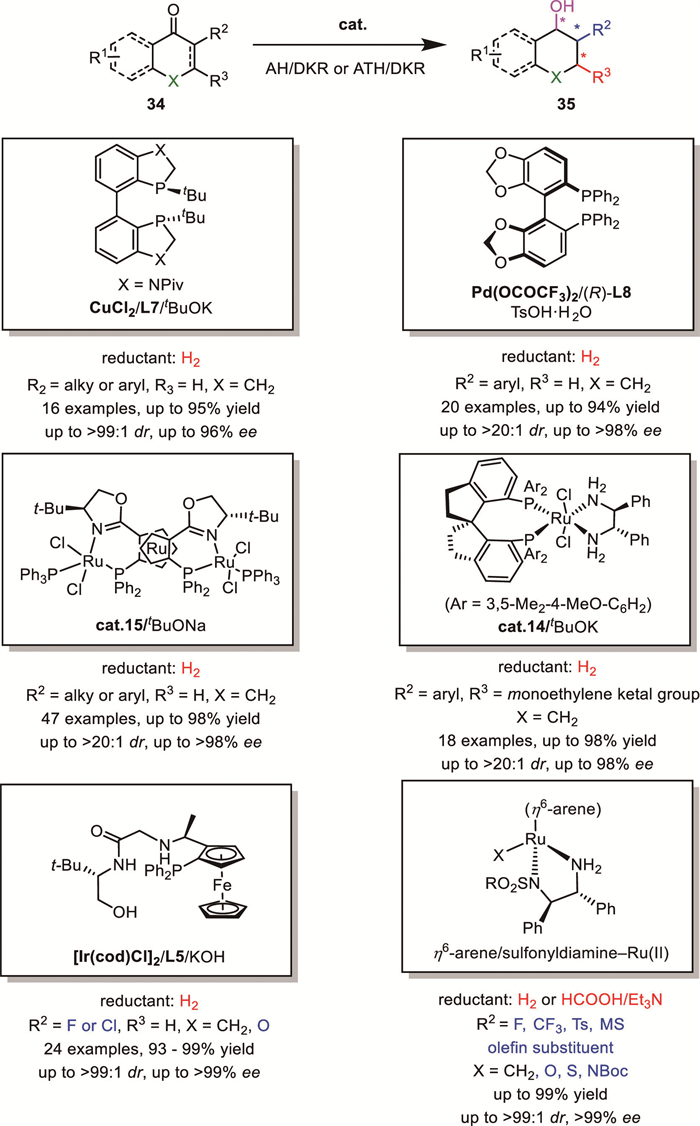
Chiral β-substituted (benzofused) cyclohexanol 35 is also an important unit presented in numerous naturally occurring compounds or biologically active molecules. α-Substituted cyclohexanone or conjugated enones 34 are optional substrates to synthesize them upon DKR-based reduction. Lots of catalytic systems have been developed to realize this important reaction [42-48]. For example, in 2018, Senanayake’s group reported the first Cu-catalyzed hydrogenation of 2-substituted-1-tetralones and related heteroaryl ketones via dynamic kinetic resolution [49]. Subsequently, Zhou’s group accomplished a palladium-catalyzed AH of 2-aryl cyclic ketones [50]. In 2021, Xie and Zhou's group developed an AH approach using a chiral spiroruthenium catalyst to access functionalized β-aryl cyclohexanols [51]. In 2022, Zhang's group successfully achieved the RuPHOX–Ru catalyzed AH of α-substituted tetralones via DKR [52]. Most recently, Zhang’s group further advanced the field by successfully applying Ir/f-phamidol to the AH/DKR of similar substrates (Scheme 14) [53]. In addition to hydrogenation, transfer hydrogenation using DPEN-ligated or tethered Ru or Rh catalysts has also been widely applied in DKR reduction of cycloketones or benzofused cycloketones, and quite a lot of related works have previously been summarized and are therefore not included in this report [13-15, 54-59].
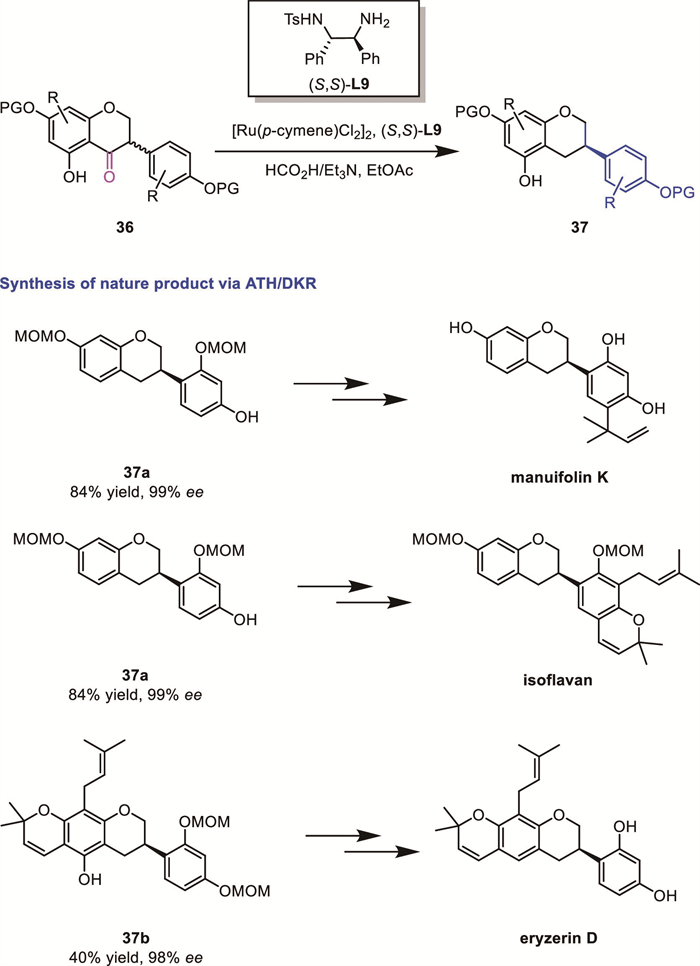
In 2018, Metz's group reported an interesting ruthenium-catalyzed DKR-based ATH/deoxygenation cascade reaction for the synthesis of structurally diverse isoflavones from a single chromone precursor 36 (Scheme 15) [60]. This innovative strategy enabled enantioselective synthesis of several biologically relevant natural products, including isoflavan, manuifolin K and eryzin D. Key to this strategy is a DKR of isoflavanones using a domino ATH/deoxygenation reaction.
In 2020, the same group successfully adapted a consecutive reduction strategy of isoflavones 38 via DKR to enable the first catalytic asymmetric synthesis of (−)-glyceollin Ⅰ and (−)-glyceollin Ⅱ (Scheme 16) [61]. Moreover, their approach enabled the efficient synthesis of several other naturally occurring phytoalexins, a class of plant defense compounds, with high enantiomeric purity, including (−)-3,9-dihydroxypterocarpan, (−)-homopterocarpin, and (−)-variabilin.
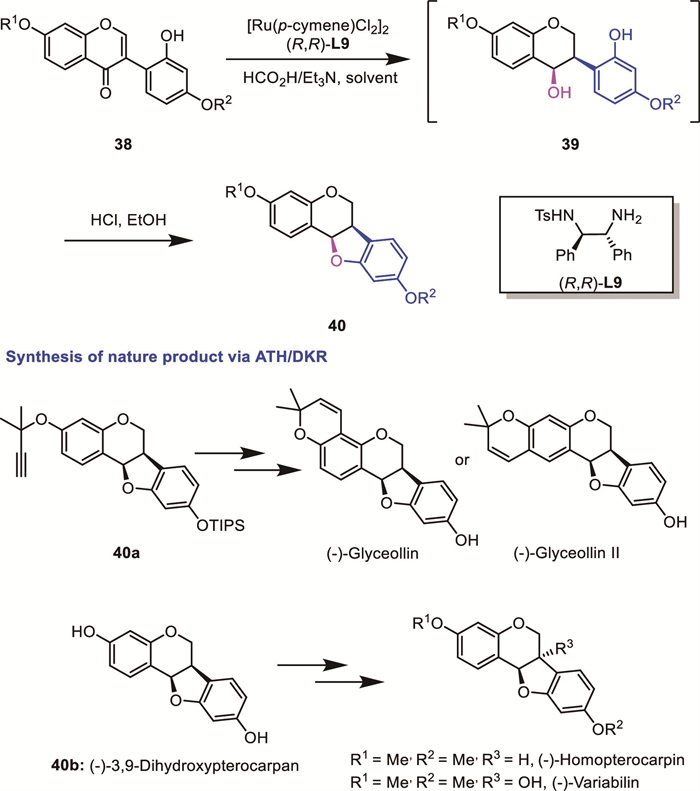
As a kind of chromanols, chiral 3-amino-4-chromanols, due to their high frequency in biological molecules, have attracted chemists’ attention. Following their work on Rh-catalyzed AH of five-membered cyclic α-dehydroamino ketones [62], Zhang's group recently reported a Rh-catalyzed consecutive AH of 3-amino chromones 41 for the synthesis of enantiomerically enriched 3-amino-4-chromanols 43 via an unusual DKR process (Scheme 17) [63]. Among the evaluated reaction parameters, the biaryl diphosphines BridgePhos L10 demonstrated superior performance. This was attributed to the adjustable dihedral angles of BridgePhos/Rh complexes. Control experiments revealed that C10-BridgePhos/Rh complex firstly catalyzed the asymmetric hydrogenation of the C═C double bond in substrate 41, which afforded the intermediate 42 with ~80% ee. The subsequent hydrogenation of the C=O group of the intermediate 42 proceeded via a stereomutation pathway from an undesired enantiomer to the desired enantiomer, instead of proceeding via a traditional racemization process (e.g., keto–enol tautomerism) of the undesired enantiomer. Through derivatization of the products, they successfully accessed a series of bioactive compounds encompassing chiral drugs and natural products, thereby unequivocally establishing the synthetic versatility of this protocol.
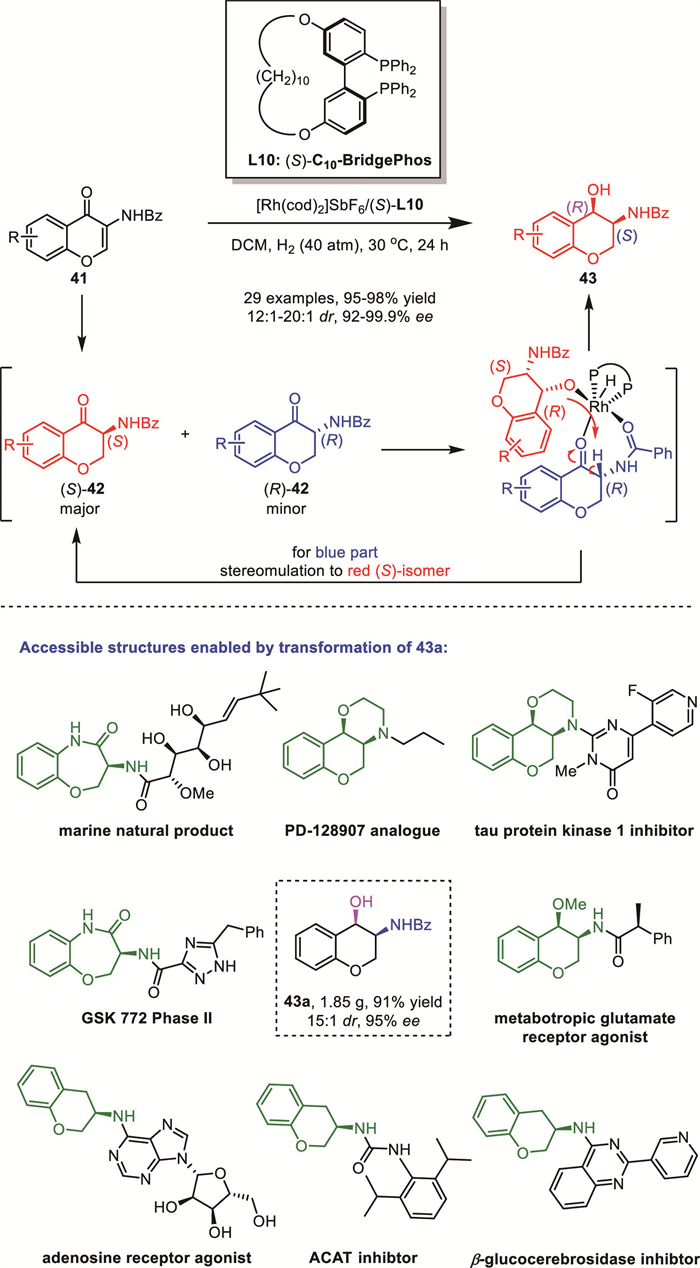
In 2019, Kayaki's group reported a cascade lactone formation via DKR driven by the ATH of keto acids with oxo-tethered ruthenium catalysts [64]. Similar to the seminal work from Johnson’s group [65], the oxo-tethered Ru catalysts successfully enabled multiple stereoinductions and allowed the sequential ring-closing transformation of the in situ generated alcohol intermediates 47. In addition to simply substituted α-tetralone and β-tetralone derivatives, substrates bearing a tethered carboxyl group at α-position 44 or 45 were also compatible with this ATH–lactonization protocol, leading to the formation of the corresponding γ-lactones in high yields with excellent diastereoselectivity and enantioselectivity (up to >99% ee, up to >99:1 dr) (Scheme 18).
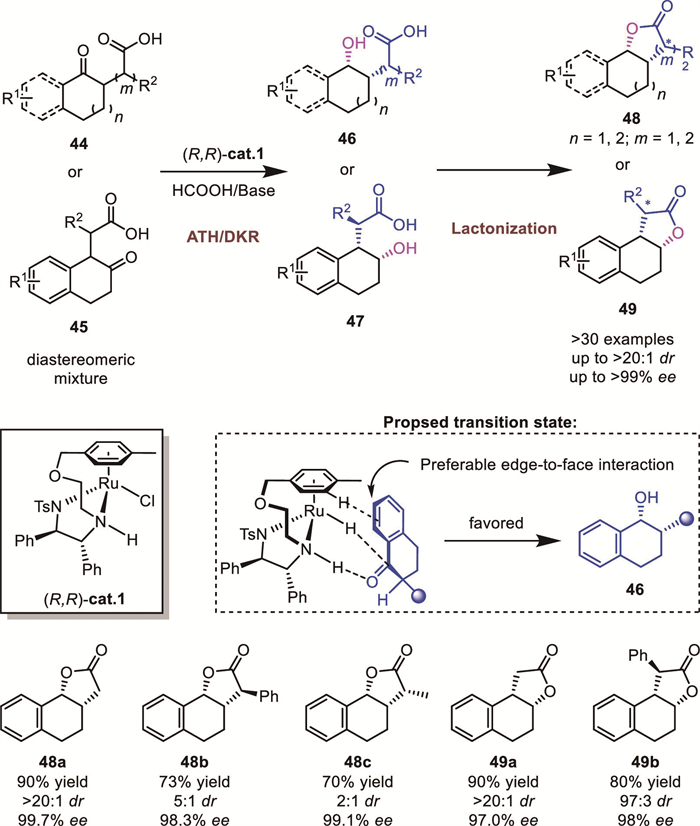
The proposed transition-state model for (R,R)-cat.1-catalyzed DKR-ATH of 2-substituted α-tetralone derivatives is illustrated in Scheme 18. A well-defined edge-to-face interaction between the η6-arene ligand of the catalyst and the substrate's aryl group stabilizes the favored transition state, accounting for the observed syn-diastereoselectivity in the alcohol product. Compared to β-tetralone derivatives 45, where the substituent on the side chain is closer to the fused arene, the lactonization of α-tetralones 44 exhibited lower diastereocontrol. The substituent adjacent to the carboxylic acid significantly influenced the diastereoselectivity. When aryl groups are introduced at positions adjacent to the carboxylic group in both classes of compounds, a moderate decrease in diastereoselectivity was observed. Notably, replacing these aryl groups with methyl substituents resulted in a more pronounced reduction in diastereoselectivity.
Later, Lv and Zhang’s group reported a related DKR-ATH protocol employing an arene-tethered Wills’ catalyst cat.9 [66]. The synthetic utility of this methodology was demonstrated through the concise synthesis of (+)-GR24 and (+)–epi–GR24, which are bioactive strigolactone analogues (Scheme 19).
The asymmetric hydrogenation of ortho-substituted phenols has been a long-standing problem in homogeneous catalysis. In 2022, Fan’s group reported a highly efficient ruthenium-catalyzed enantioselective hydrogenation of 9-phenanthrols under mild conditions. Notably, their Ru(diamine)/base catalytic system was extended to 9,10-disubstituted phenanthren-9-ols 52, affording 9,10-dihydrophenanthrenes 53 with up to 99% ee and >99:1 dr via a DKR process (Scheme 20) [67]. Control experiments supported a proposed mechanism involving base-promoted tautomerization of the substrate, followed by ruthenium hydride trapping of the transient keto intermediate. In the favored transition state, hydride transfers from ruthenium to the Si-face of the C=O moiety to yield the (R)-configured product.
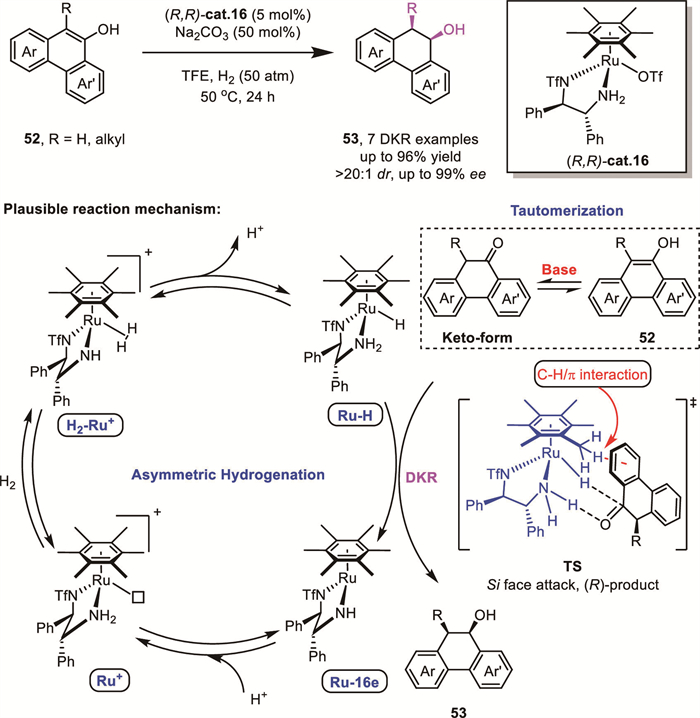
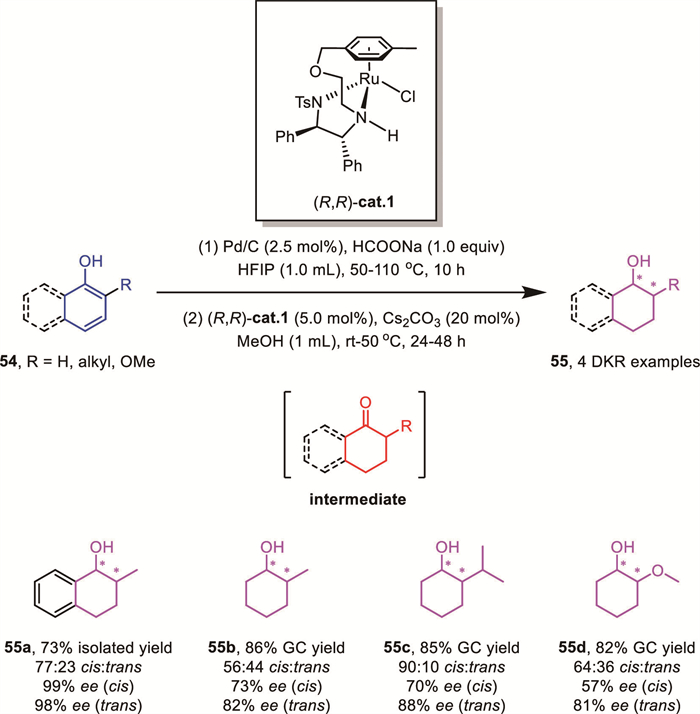
Compared to 9-phenanthrol, naphthols and phenols possess enhanced aromatic stability, which typically necessitates robust heterogeneous catalysts for their disruption. In 2023, the same group reported a conceptually innovative strategy for the reduction of naphthol and phenol derivatives via cooperative bimetallic heterogeneous-homogeneous catalysis. This approach involves initial selective partial hydrogenation of the arene ring using Pd/C to generate achiral ketone intermediates. These intermediates subsequently undergo homogeneous asymmetric reduction catalyzed by an arene-tethered TsDPEN Ru-complex, either sequentially or in a tandem fashion, affording the final chiral saturated cyclic compounds [68].
Notably, this strategy was successfully extended to ortho-substituted phenols and 1-naphthols 54 via a one-pot, two-step procedure. By leveraging a DKR process, the reaction delivered target products 55 bearing contiguous stereocenters with moderate diastereoselectivity and excellent enantioselectivity (Scheme 21). This work establishes a novel paradigm for asymmetric reduction of all-carbon arenes through a synergistic catalytic system.
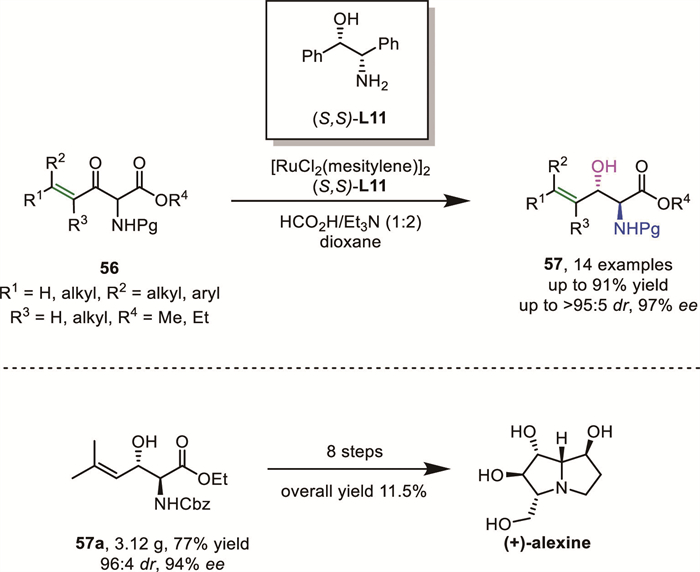
In 2019, Somfai's group expanded the scaffold of labile α-amido-β-keto ester by the incorporation of a vinyl moiety in the α-position of the ketone, and successfully achieved ATH of racemic α-amino-γ,δ-unsaturated β-ketoesters 56 via DKR. After extensive evaluation of various reaction parameters, the combination of [RuCl2(mesitylene)]2 and the chiral amino alcohol ligand (S,S)-DPAE L11 was identified as the optimal catalyst and HCOOH/Et3N as the hydrogen sources, which afforded excellent asymmetric control (up to > 95:5 dr, 97% ee) and minimized by-product formation (Scheme 22) [69]. The versatility of this methodology was demonstrated in the asymmetric synthesis of polyhydroxylated pyrrolizidine alkaloid (+)-alexine within 8 steps with an overall yield of 11.5%.
In the same year, Xie and Zhou's group reported an enantioselective total synthesis of (−)-goniomitine using a PNN/Ir complex-catalyzed hydrogenation of an exocyclic enone ester 58 as the key step [70]. Racemic 5-membered exocyclic enone esters 58 served as applicable substrates for a DKR-based reduction, which afforded the critical chiral allylic alcohol 59 that could be converted to the final natural product with a reported route. Remarkably, the hydrogenation step could be amplified to a 32 g scale without erosion of the asymmetric control (Scheme 23A).
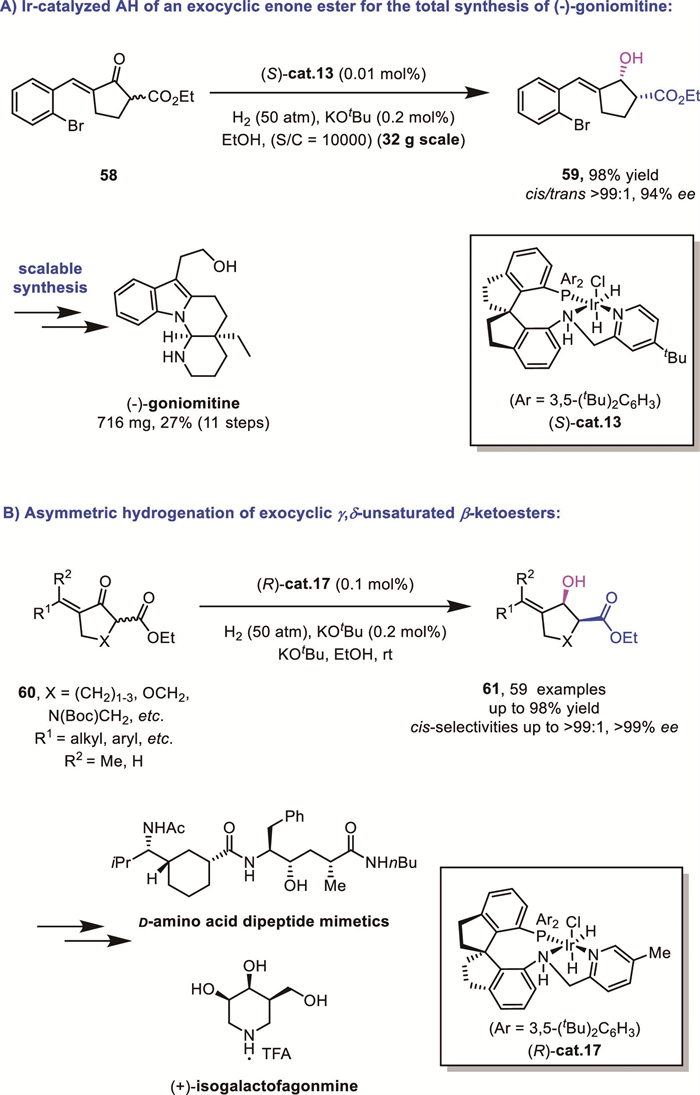
Building on this highly efficient asymmetric hydrogenation, the same group applied a similar strategy in 2021 in the DKR-based AH of exocyclic enone esters 60 (Scheme 23B) [71], leading to concise and efficient routes to chiral carbocyclic δ-amino esters 61 and other biologically active molecules.
Chiral 3-substituted or 3,4-disubstituted succinimide substructures are the core scaffolds of lots of natural products and drugs. However, few methodologies have been developed concerning the synthesis of 3,4-disubstituted succinimides compared with well-established synthetic methods for 3-substituted succinimides, due a large part to the challenging control of the stereochemistry of both chiral centers of 3,4-disubstituted succinimides.
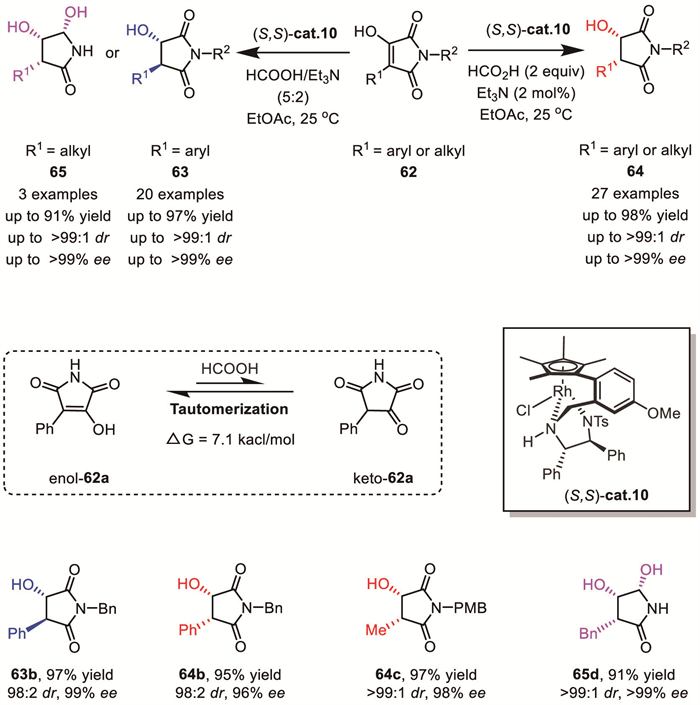
In 2022, Yu, Chen, and Zhang's group reported an ATH reaction of 3–hydroxy-4-substituted-maleimide derivatives 62 (Scheme 24) [72]. Their optimal reaction conditions included TsDPEN-derived Rh complexes cat.10 as the catalyst and azotropic mixture of HCOOH/Et3N as the hydrogen donor. Notably, by modulating the ratio of HCOOH and Et3N, anti-3–hydroxy-4-substituted-succinimides 63 (up to >99% ee, up to >99:1 dr) or syn-3–hydroxy-4-substituted-succinimides 64 could be selectively obtained. A stereodivergent approach to all 4 stereoisomers of succinimides was thus demonstrated. Furthermore, for the N-unprotected substrates, both the enol and the imide group could be reduced by prolonging the reaction time and enhancing the catalyst loading. Overall, this work was very efficient with high asymmetric control (up to >99% ee, up to >99:1 dr). Mechanistic studies confirmed the keto-enol equilibrium was existed and the reaction probably proceeded via reduction of the keto form.
Chiral α–hydroxy-β-lactams are the key fragment of many pharmaceutically active molecules such as glycosides, anti-microbial drugs and anti-cancer reagents. The asymmetric hydrogenation of α-keto-β-lactams is one of the most straightforward approaches to access them.
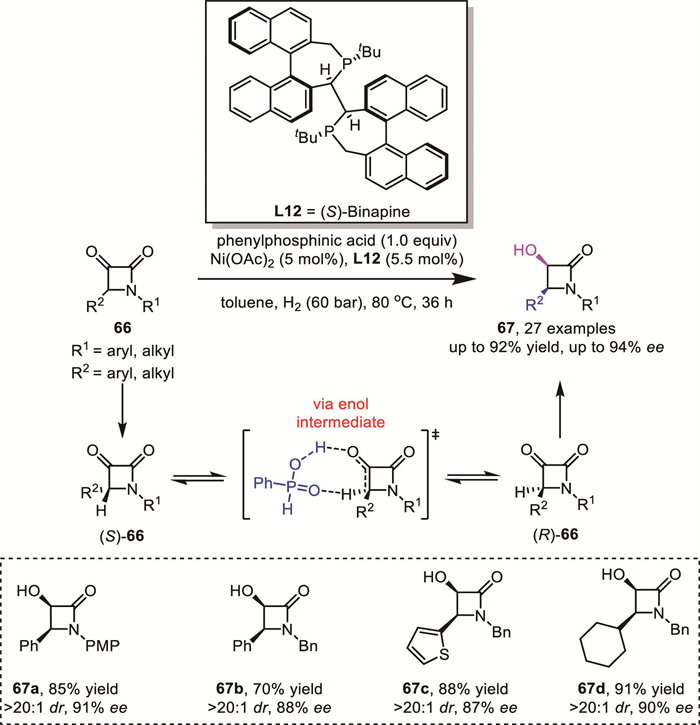
In 2020, Zhang's group reported a DKR-based hydrogenation of α-keto-β-lactams 66 using an earth-abundant nickel catalyst ligated with a chiral bisphosphine (Scheme 25) [73]. The corresponding products 67 were obtained in high yields with excellent enantioselectivity and diastereoselectivity (up to 92% yield, > 20:1 dr, up to 94% ee). Control experiments revealed that phenylphosphonic acid played a pivotal role in the enolization of the substrate through a novel proton-shuttling strategy, which enabled an efficient DKR process.
Chiral 1,2-diols with vicinal chirality are highly significant in biochemistry and pharmaceutical science due to their unique biological activity. Besides, they can serve as precursors for chiral ligands, auxiliaries or protecting groups in organic synthesis. Direct double reduction of 1,2-diketones offers a rapid way to them. However, the control of stereochemistry poses a challenge.
Tang’s group developed a Ru-catalyzed DKR-based ATH of benzils 68, affording syn-(S,S)-hydrobenzoins 72 in excellent yields with high diastereoselectivity and enantioselectivity (up to 97.1%, syn/anti up to 30/1, up to 99% ee) [74]. The rapid keto-enol tautomerism of the in situ formed α–hydroxy ketone intermediates is critical to achieve high DKR efficiency (Scheme 26A).
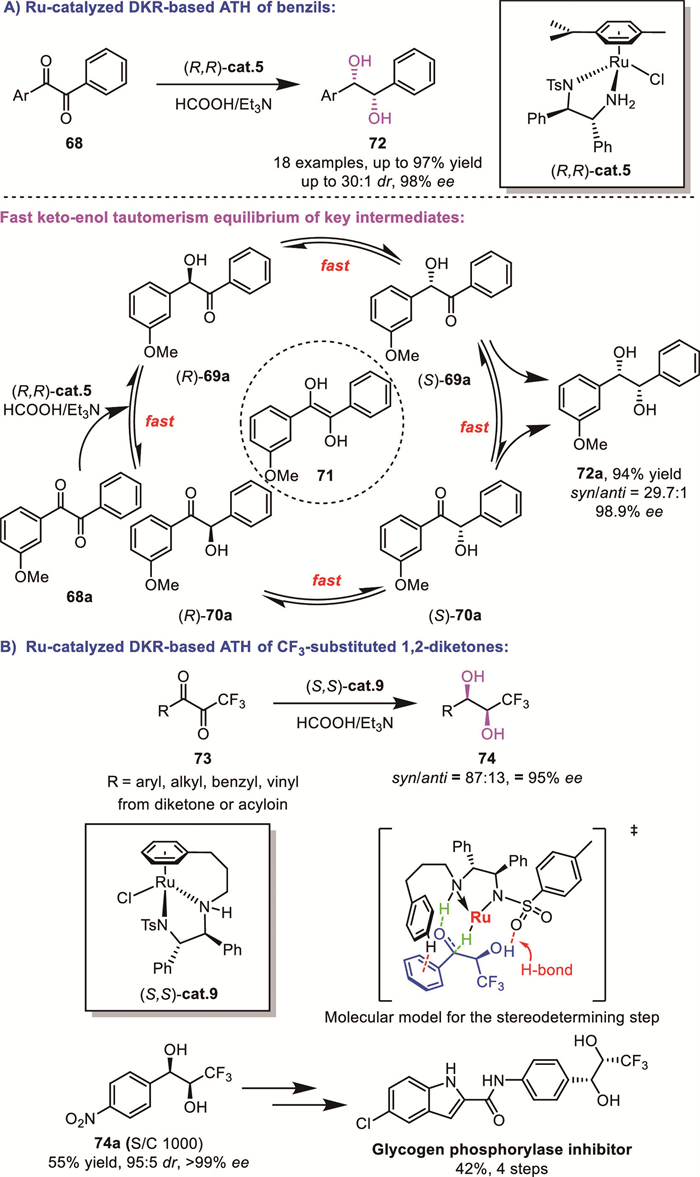
In 2023, Cotman’s group extended this strategy to the synthesis of stereopure CF3-substituted syn-1,2-diols 74 via the reductive DKR of the in situ formed racemic α-hydroxyketones using HCO2H/Et3N as a hydride source (Scheme 26B) [75]. Their work not only enabled enantioselective access to these sterically demanding diols but also demonstrated their potential as precursors to pharmacologically relevant molecules. Based on DFT calculations, the origin of stereoselectivity was ascribed to the H-bonding between the hydroxyl group of the in situ formed alcohol intermediate and the catalyst’s SO2 moiety. The synthetic utility of this methodology was demonstrated through an efficient asymmetric synthesis of a glycogen phosphorylase inhibitor, achieved in two steps.
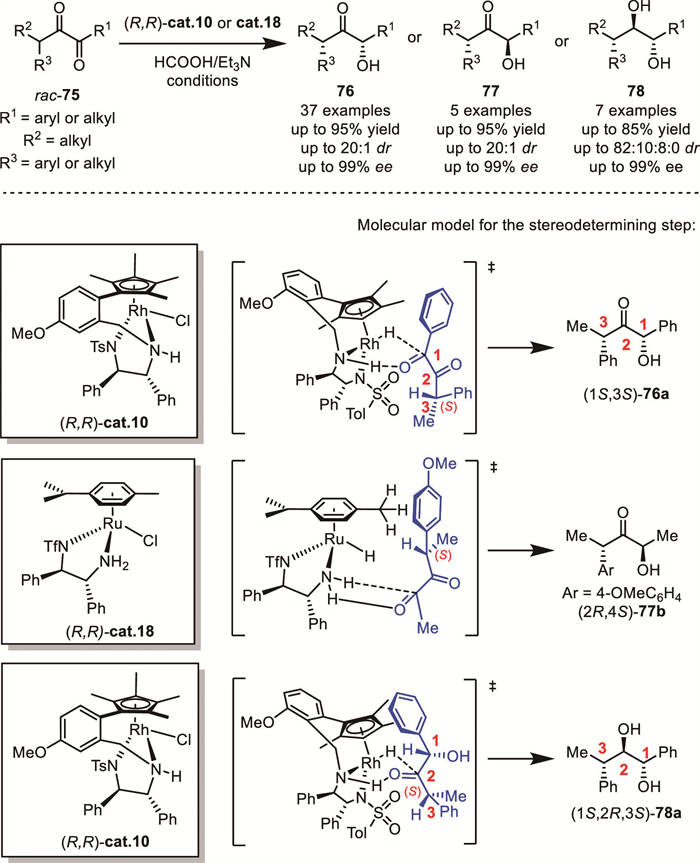
In 2023, Song and Fang's group reported a unique regio- and stereoselective semi-transfer hydrogenation of α-branched diketones 75 via an interesting DKR mode (Scheme 27). By using Noyori-Ikariya-type ruthenium or tethered rhodium catalysts, the external carbonyl group of β-substituted α-diketones was reduced, while the internal carbonyl group adjacent to the labile stereocenter remained intact [76]. A series of α–hydroxy-α'-chiral ketones 76 or 77 with different R1 and R2 groups were obtained in high enantio– and diastereoselectivity (up to >99% ee and up to >99:1 dr). Notably, by altering the reaction conditions, some substrates could be further reduced to the corresponding diols 78, and all diols with three stereogenic centers were efficiently generated with up to 99% ee. Based on the stereochemical outcomes arising from catalyst-substrate interactions, two distinct transfer hydrogenation pathways governing the formation of products 76 and 77 were proposed, along with a hydrogenation model accounting for the stereoselective transformation of the trans-perhydrogenated substrate 78.
In 2024, Zhang and Fang’s group systematically developed the aryloxy group-assisted ATH of 1,2-diketones 79 using Noyori-Ikariya-type ruthenium catalysts with HCOOH/tetramethylethylenediamine (TMEDA) as the hydrogen source (Scheme 28A) [77]. This protocol achieved excellent regio-, diastereo-, and enantioselectivity (up to 94% yield, up to >20/1 dr, up to 99% ee), notably enabling the synthesis of all eight stereoisomers of diaryl triols 81. Through combined experimental and computational studies, the authors elucidated the crucial role of the para-methoxyphenyl (OPMP) group in controlling the reaction selectivity. Transition state analysis revealed that the OPMP group: (1) adopts a conformation distal to the η6-arene ligand to minimize steric hindrance distortions, while (2) forms enhanced Ru-H···C and N–H···O interactions and hydrogen bonds between N–H2 and the O atom in OPMP. A plausible reaction mechanism was proposed: under the reaction conditions, the 3-position of rac-79a undergoes facile deprotonation to form enol-79a, which serves as the key intermediate enabling interconversion between (S)-79a and (R)-79a through an enolization process. Subsequent fast ATH of (S)-79a preferentially generates the (2S,3S)-80a product, while the ATH pathway for (R)-79a remains energetically unfavorable. This DKR process drives the continuous conversion of (R)-79a to (S)-79a, which subsequently proceeds to form the desired product (Scheme 28B).
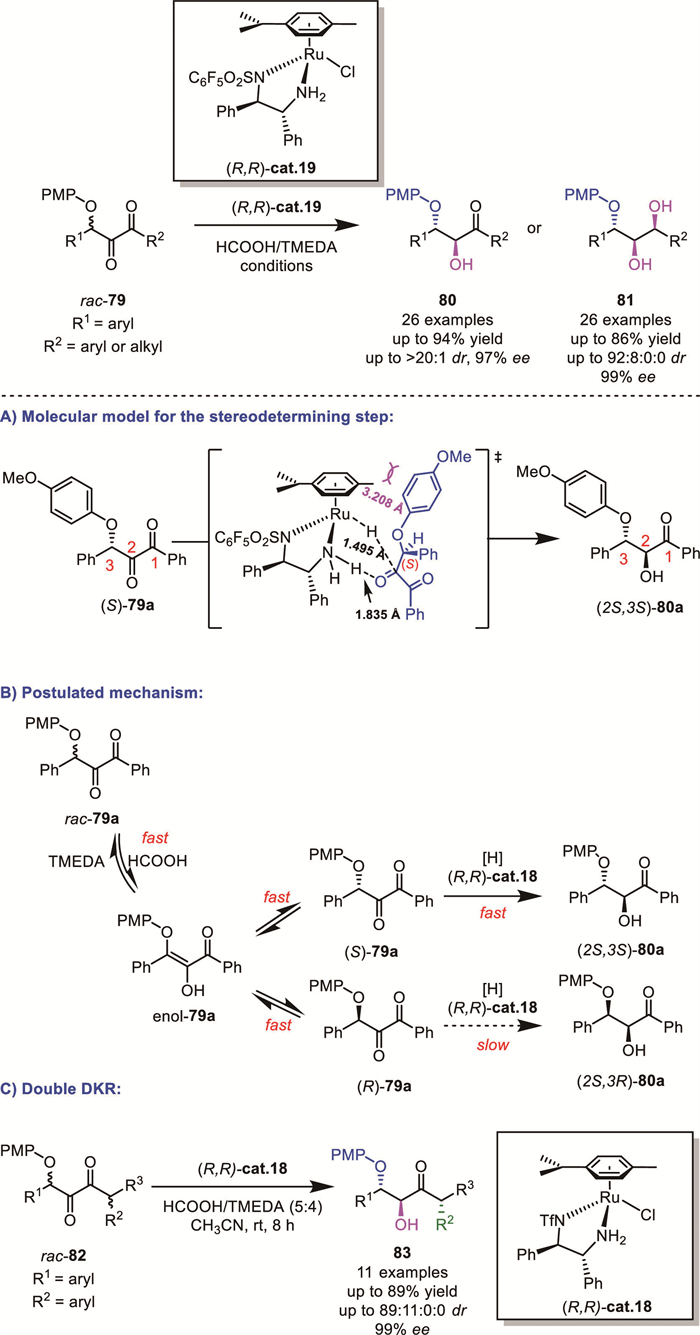
They also realized an elegant double DKR transformation of more challenging racemic diketones 82 bearing two stereocenters, which led to the formation of a single major product 83 with three stereocenters, demonstrating the synthetic potential of ATH in the synthesis of complicated chiral alcohols with consecutive or non-consecutive stereogenic centers (Scheme 28C).
The asymmetric reduction of α-substituted 1,3-diketones provides a straightforward access to valuable 1,3-diols or β–hydroxy ketones with vicinal chirality. However, for unsymmetric 1,3-diketones, the control of chemo- and stereoselectivity is very challenging. So far, only limited progress has been achieved.
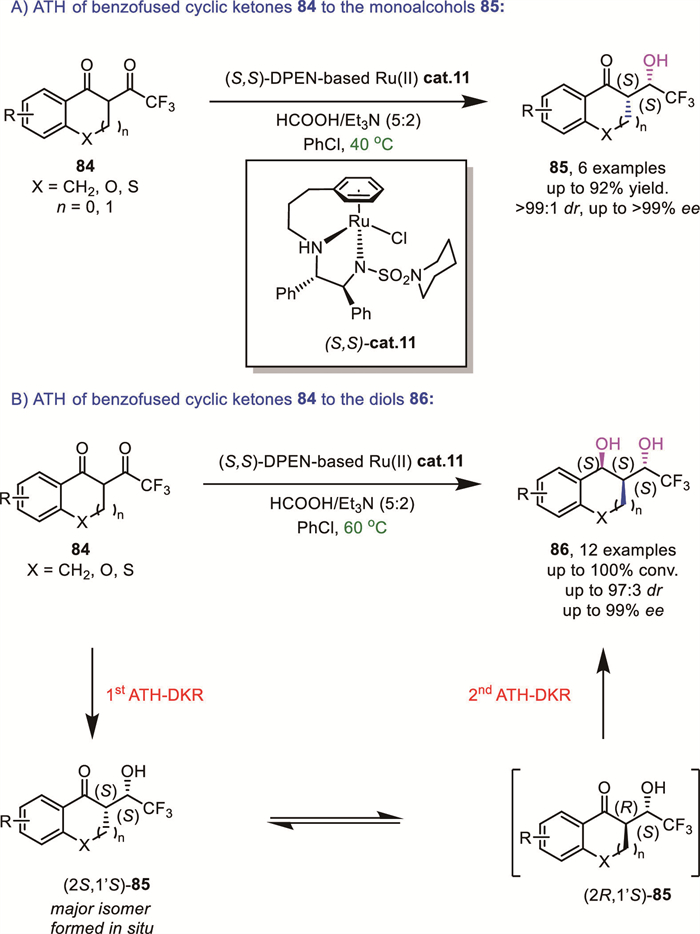
Mohar's group developed an ATH reaction of rigid α-CF3C(O)-substituted benzofused five- to seven-membered cyclic ketones 84 using a homemade ansa-ruthenium(Ⅱ) complex cat.11 and formic acid and triethylamine azeotrope as the hydrogen source. This method afforded either stereodefined CF3-substituted β–hydroxy ketones 85 or 1,3-diols 86 in high yields, diastereo- and enantioselectivities (mostly 100% conv., >20:1 dr and up to 99% ee), depending on the reaction conditions [78]. The newly designed catalyst (cat.11), incorporating piperidino-SO2DPEN-(CH2)3(η6-Ph) conjugate ligands, exhibited higher levels of activity and increased longevity compared to previous systems (Scheme 29A). Racemic reactants 84 were reduced to the corresponding monoalcohol 85 at 40 ℃, validating the occurrence of a DKR process. More interestingly, complete reduction from intermediate 85 to 1,3-diols 86 can be achieved at 60 ℃ with increased catalyst loading via the second DKR-ATH (Scheme 29B).
1,2,3,4-Tetraols (C4 sugar alcohol), with four contiguous stereogenic oxidized carbons, have been considered a kind of “privileged scaffold” due to their potential to act as sugar mimics and oxygen-functionalized chiral synthons. Traditional synthesis to assemble 1,2,3,4-tetraols frameworks is often laborious and time-consuming, involving multi-step processes that require complex protection-deprotection strategies and precise stereochemical control. These limitations have spurred significant interest in developing more efficient and stereoselective methodologies to access these important motifs.
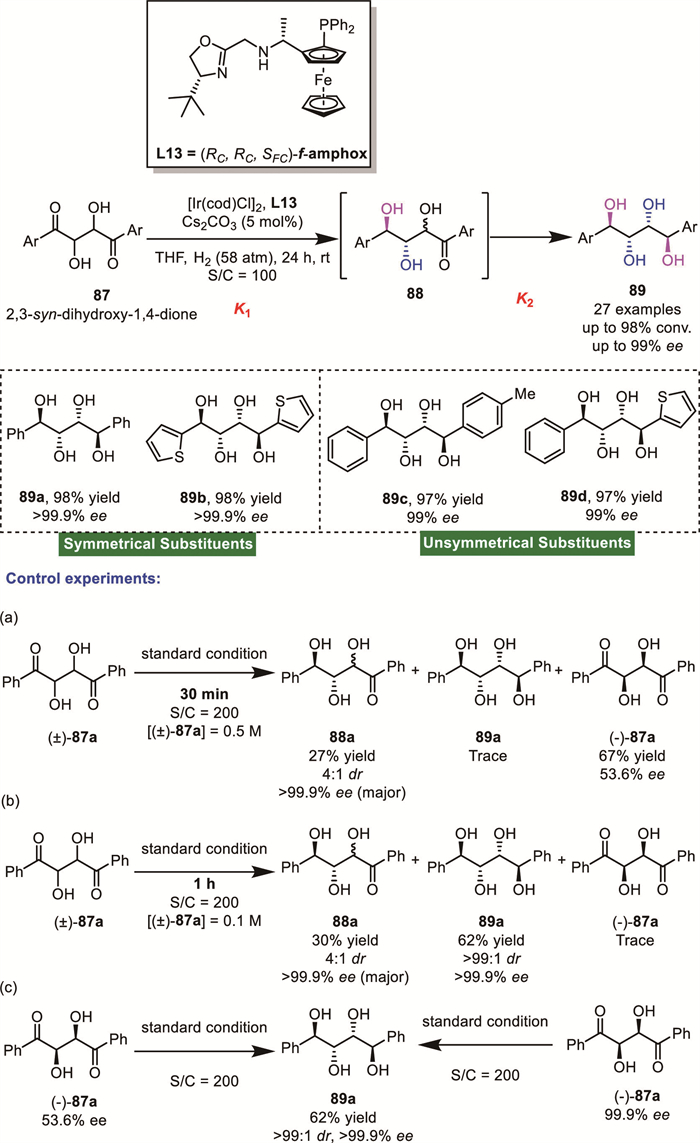
In 2020, Zhang's group reported an innovative Ir-catalyzed AH of racemic 2,3-syn-dihydroxy-1,4-diones 87 [79]. This protocol established an efficient and straightforward route to sugar alcohols containing four contiguous stereocenters 89, delivering products with excellent stereocontrol (typically >99:1 dr and up to 99% ee) across a broad substrate scope (Scheme 30). To enhance comprehension of the mechanism, a series of controlled experiments were conducted. Initially, the reaction kinetics were monitored by in situ Fourier-transform infrared spectroscopy (FTIR), revealing that the mono-reduced 88 was formed at a significantly faster rate than the further hydrogenation to the double-reduced tetraol 89 (k1 >> k2). Furthermore, in a series of quenching experiments, it was observed that only (+)-87 undergoes conversion initially; once (+)-87 is consumed, the racemization of (-)-87 begins. It is noteworthy that under the standard conditions, the isolated mono-reduced product 88, whether optically pure or not, could be reduced to the desired 1,2,3,4-tetraol 85 via a DKR reduction.
DKR-based asymmetric reduction of racemic α-chiral ketones is extensively studied; in contrast, there are far fewer reports of DKR reduction of racemic aldehydes, esters, and amides due to either reactivity or stereoselectivity problems.
For asymmetric reduction of prochiral ketones, at least one new stereogenic center is formed. However, no new stereogenic center is produced in the reduction of α-chiral aldehydes, making the stereocontrol extremely challenging. Compared to enzymatic catalysis that can stereoselectively reduce the aldehydes under mild conditions, chemocatalysis has lagged behind.
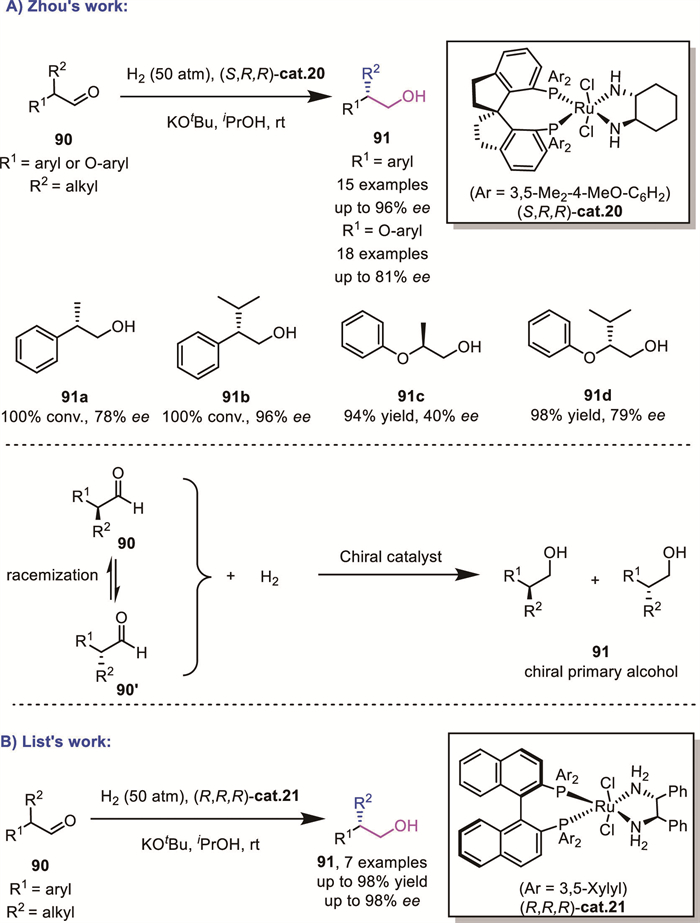
In a pioneering work, Zhou's group reported the AH of two different types of α-branched aldehyde substrates 90 using [RuCl2(SDP)(diamine)] complexes cat.20 via a DKR process (Scheme 31A). The Ru catalyst could selectively hydrogenate one enantiomer of the racemic aldehyde, while the other enantiomer underwent rapid base-mediated racemization [[80], [81]]. As a result, both enantiomers are ultimately converted into the primary alcohols 91 with high enantioselectivity. This elegant approach leverages the DKR strategy to overcome the inherent challenges of enantiocontrol in α-branched aldehyde hydrogenation. During the same period, List's group also achieved a similar transformation using a [Ru(diphosphine)(diamine)] complex cat.21 (Scheme 31B) [82].
Traditional methods for the preparation of β-substituted alcohols from α-substituted esters generally require the prior synthesis of optically pure α-substituted esters, along with careful control to avoid racemization during the reduction process. These methods, such as the use of chiral auxiliaries or enzymatic resolutions, have been extensively developed and are well-documented in the literature. However, direct hydrogenation of racemic α-substituted esters to enantioenriched β-substituted alcohols through DKR has rarely been reported.
In 2011, Ikariya's group reported the first application of a well-defined Cp*Ru catalyst, in conjunction with an inorganic base, for the enantioselective hydrogenation of racemic lactones 92 via DKR [83]. Although the resulting chiral diol 93 exhibited limited enantioselectivity (only 32% ee), the substoichiometric amount of inorganic base likely induced reversible deprotonation of substrates with acidic C-H bonds, leading to racemization of chiral nonracemic substrates bearing a tertiary stereogenic center at the α-carbon. This mechanistic insight provided a valuable conceptual framework for subsequent studies in asymmetric catalysis, inspiring further exploration of DKR strategies for challenging substrates (Scheme 32A).
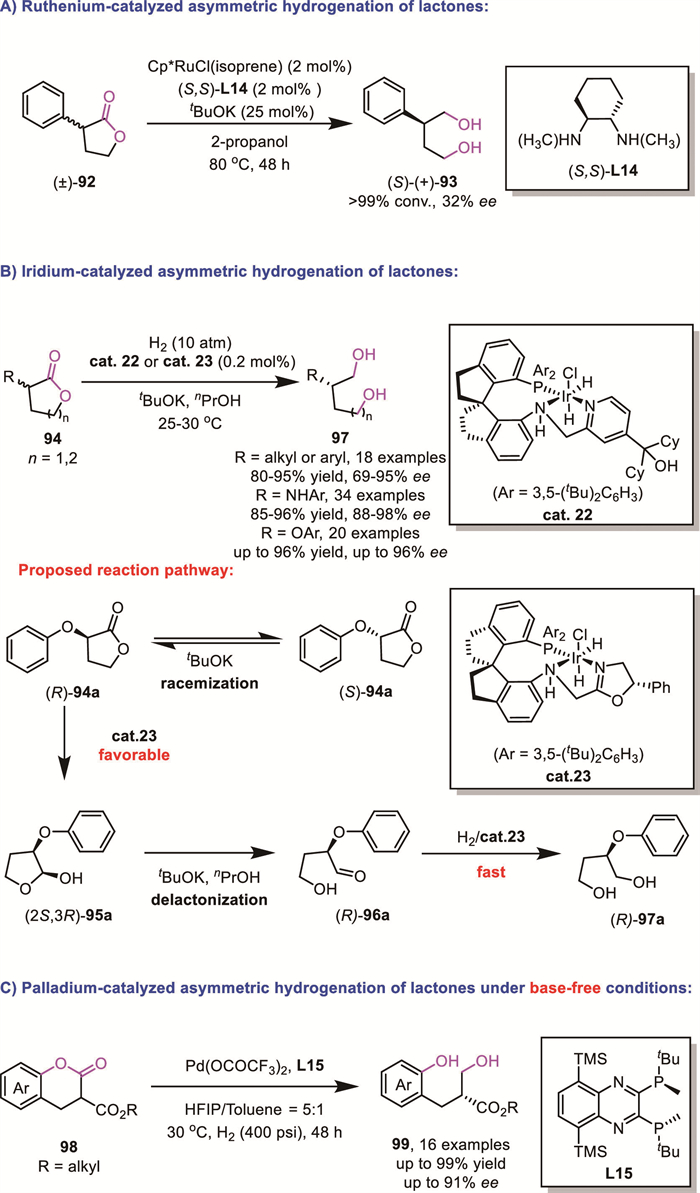
Later, Zhou's group improved the asymmetric control to a useful level using homemade Ir-SpiroPAP catalysts cat.22 or cat.23 [84,85]. The high reactivity of these catalysts enabled the reactions to be successfully conducted at room temperature, which was helpful to the enantiocontrol. These transformations consistently afforded the corresponding chiral diols 97 in high yields and with excellent enantioselectivity (up to 96% yield, up to 98% ee). Based on a metal-ligand bifunctional mechanism, density functional theory studies were conducted to investigate the hydrogenation of rac-α-aryloxy lactones, leading to the proposed DKR pathway as follows: the catalyst (S,S)-cat.23 preferentially recognizes and hydrogenates (R)-94a to generate the hemiacetal (2S,3R)-95a, while the remaining opposite (S)-94a undergoes racemization to (R)-94a via enolization under strong basic conditions. Subsequently, the hemiacetal (2S,3R)-95a undergoes delactonization to form the aldehyde (R)-96a, which is further hydrogenated by the iridium catalyst cat.23 to produce the final product (R)-97a. Since the hydrogenation of aldehydes is generally more facile than that of esters, the enantioselectivity of the hydrogenation of rac-94a is primarily determined by the first step involving the hydrogenation of the lactone rac-94a to the hemiacetal 95a. Consequently, the hydrogenation product would be dominated by (R)-97a, consistent with experimental observations (Scheme 32B).
In 2024, Zhou’s group developed a palladium-catalyzed asymmetric hydrogenation of more reactive benzofused lactones 98 under base-free conditions through a DKR process [86]. This method delivered products 99 with high enantioselectivity and reactivity while maintaining broad functional group compatibility (Scheme 32C).
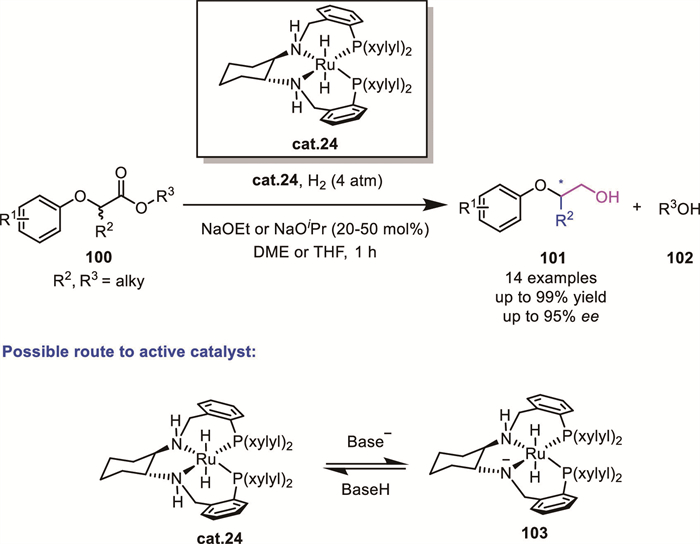
Generally, the asymmetric reduction of α-branched esters presents a significant challenge due to their inherently lower reactivity compared to lactones. In 2019, Bergens' group addressed this challenge by developing a DKR process for the hydrogenation of racemic α-functionalized esters 100 to β-chiral primary alcohols 101 [87]. Using highly reactive chiral ruthenium hydride complexes, the reaction achieved excellent yields (up to 99%) and enantioselectivities (up to 95% ee) (Scheme 33). Mechanistic studies disclosed the complexity of the reaction process, involving the initial reduction of the ester to an aldehyde intermediate or equivalent oxidation state, which remained associated with the Ru catalyst, and the following reduction to the alcohol products. The authors proposed that the role of the large excess strong base was to activate the putative dihydride cat.24 by deprotonation of the NH group of DPEN.
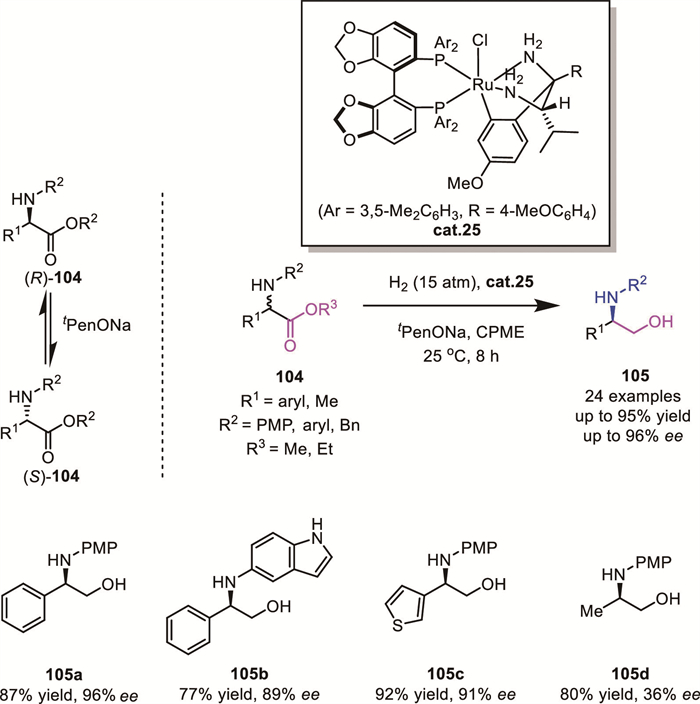
In 2023, Ohkuma’s group reported an elegant ruthenium-catalyzed AH of racemic α-amino esters 104, affording enantioenriched β-amino alcohols 105 in high yields with excellent enantioselectivity (up to 95% yield, 96% ee) [88]. Deuteration experiments revealed a non-conventional mechanism involving dehydrogenation of the amino group to generate an imino intermediate, rationalizing the essential role of N-aryl substituents in facilitating this pathway. Furthermore, base-mediated reversible epimerization at the α-stereocenter enabled an efficient DKR process under catalytic conditions (Scheme 34). However, alkyl-substituted α-amino esters displayed poor asymmetric control.
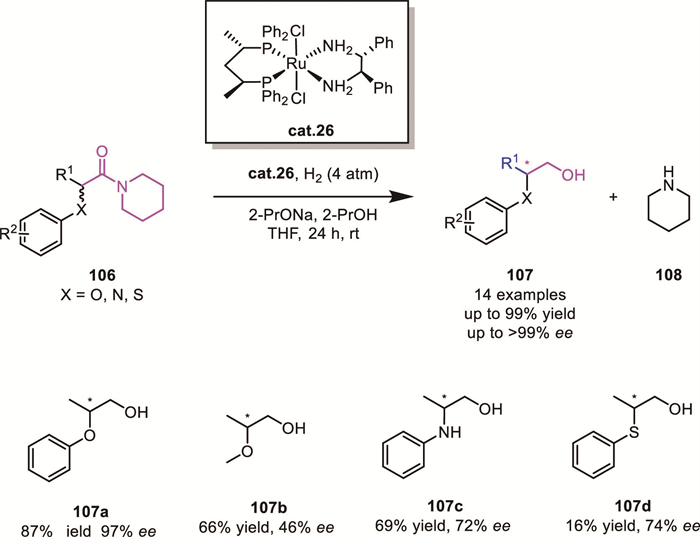
Amides rank among the least reactive carboxylic acid derivatives, typically requiring a stoichiometric amount of metal reductants. This inherent inert reactivity poses significant challenges for developing catalytic asymmetric hydrogenation methods for amides. To date, only one efficient catalytic system exhibiting both high reactivity and excellent stereocontrol has been reported.
In 2017, Bergens' group developed a catalytic system comprising trans-RuCl2((S,S)-Skewphos)((R,R)-DPEN) cat.26, sodium isopropoxide, and isopropanol [89]. This system successfully achieved the hydrogenation of α-phenoxy amides 106 under mild conditions, affording β-chiral primary alcohols 107 with high yields (up to 99% yield) and enantioselectivity (up to 99% ee). This approach leverages base-promoted rapid substrate tautomerization to enable DKR, converting both amide enantiomers into a single alcohol enantiomer (Scheme 35). However, substrate scope limitations were observed, with methoxyl, anilinyl, and thiophenoxyl substituents exhibiting diminished reactivity.
Chiral amines featuring vicinal stereocenters constitute highly prevalent structural motifs in natural products and bioactive pharmaceuticals, driving significant interest in medicinal chemistry. Although asymmetric reductive amination (ARA) has emerged as a powerful method for constructing chiral amines, its application to racemic ketones via a DKR process remains underdeveloped [90-94]. This stagnation stems from inherent challenges: (1) competitive ketone reduction pathways complicating reaction outcomes, and (2) the formidable task of controlling multiple stereogenic centers with high fidelity.
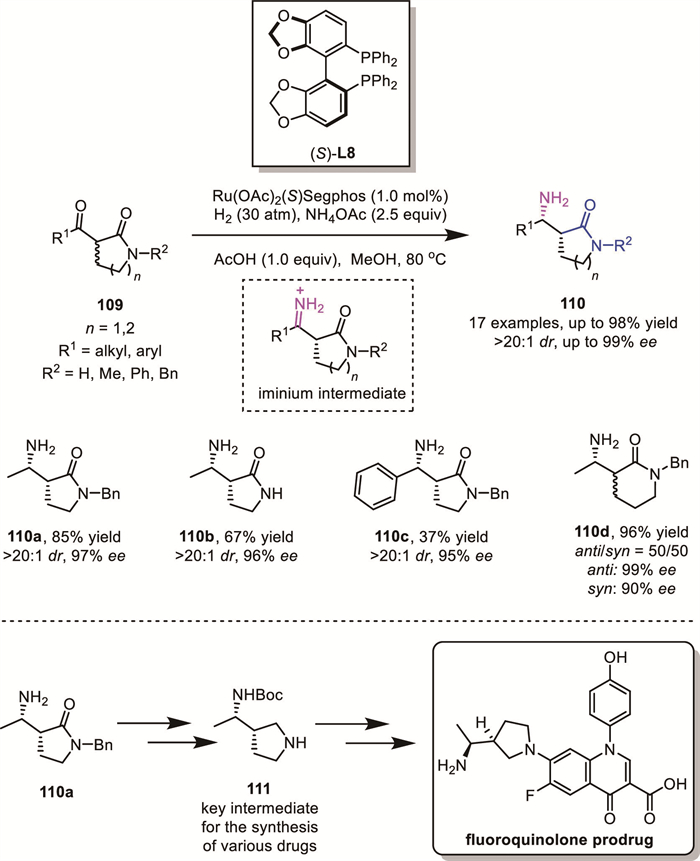
In 2018, Yin and Zhang's group achieved a highly efficient ruthenium-catalyzed ARA of racemic β-keto lactams 109 using molecular hydrogen (H2) and ammonium salts (Scheme 36) [95]. This strategy enabled the synthesis of a range of syn-primary β-amino lactams 110 in high yields with excellent chemo-, enantio–, and diastereoselectivity (up to 98% yield, 99% ee, >20:1 dr). Notably, six-membered lactam substrates displayed much lower diastereocontrol compared to that of five-membered lactams, which was ascribed to the different ring conformation. Mechanistic studies, supported by control experiments and prior work, suggested hydrogenation of an iminium intermediate as the key step. Simultaneously, they demonstrated the utility of their method through gram-scale synthesis and the preparation of key intermediates 111, which serve as crucial building blocks for the synthesis of pharmaceutical products containing a chiral γ-aminopyrrolidine moiety, such as fluoroquinolone prodrugs.
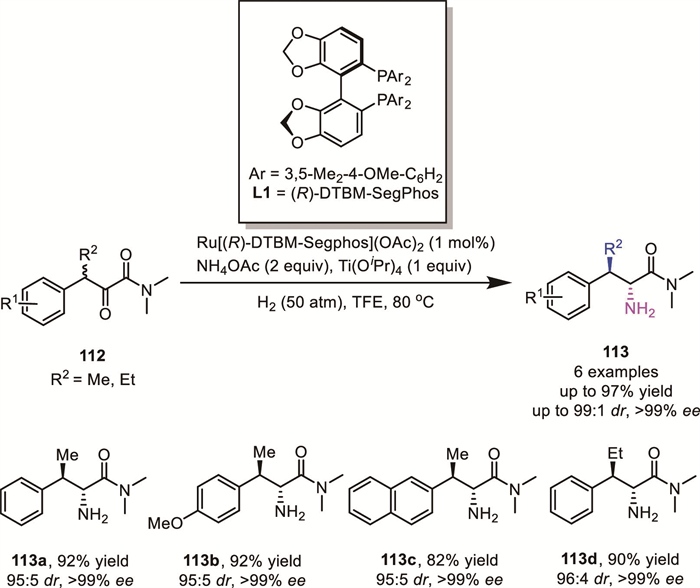
Enantiopure β-branched α-amino carboxylic derivatives serve as crucial scaffolds for the synthesis of modified peptides and proteins with improved biological properties. In 2022, Yin and Zhang's group further reported a ruthenium-catalyzed ARA of α-keto amides using ammonium salts [96]. In addition to simple α-keto amides, their strategy was successfully applied to α-branched keto amides 112 via a DKR process (Scheme 37). This approach delivered a series of β-branched α-amino amides bearing two contiguous stereocenters 113 with high yields (up to 97% yield), excellent diastereoselectivity (up to 99:1 dr), and enantioselectivity (up to >99% ee).
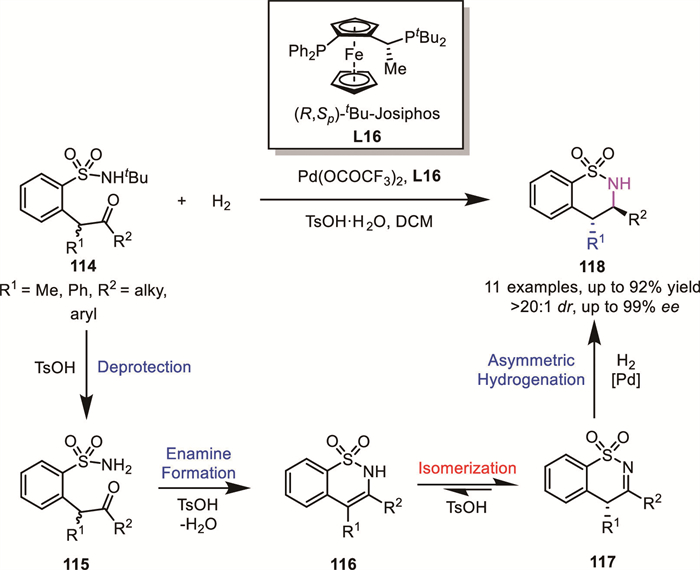
Zhou's group successfully developed a palladium-catalyzed intramolecular ARA of racemic α-branched ketones 114 bearing a tethered sulfonamide group via a DKR process (Scheme 38) [97]. This approach delivered chiral δ-sultams 118 containing two contiguous stereogenic centers with high chemo-, enantio–, and diastereoselectivity (up to 92% yield, 99% ee, >20:1 dr). Mechanistic studies suggested that, under the assistance of Brønsted acid, the starting material 114 is initially deprotected to generate intermediate 115, followed by cyclodehydration to form intermediate 116. Intermediate 116 then undergoes acid-mediated tautomerization between the enamine and imine forms. Finally, one enantiomer of intermediate 117 undergoes stereoselective reduction to afford the anti-configured δ-sultams.
In 2010, List's group reported an organocatalytic one-pot ARA and lactamization of α-branched ketones 119 using para-anisidine 120 as the amine source and Hantzsch ester as the reductant, yielding syn-bicyclic lactams 121 with high enantioselectivity (94% ee) and moderate diastereoselectivity (5:1 dr) (Scheme 39A) [98].
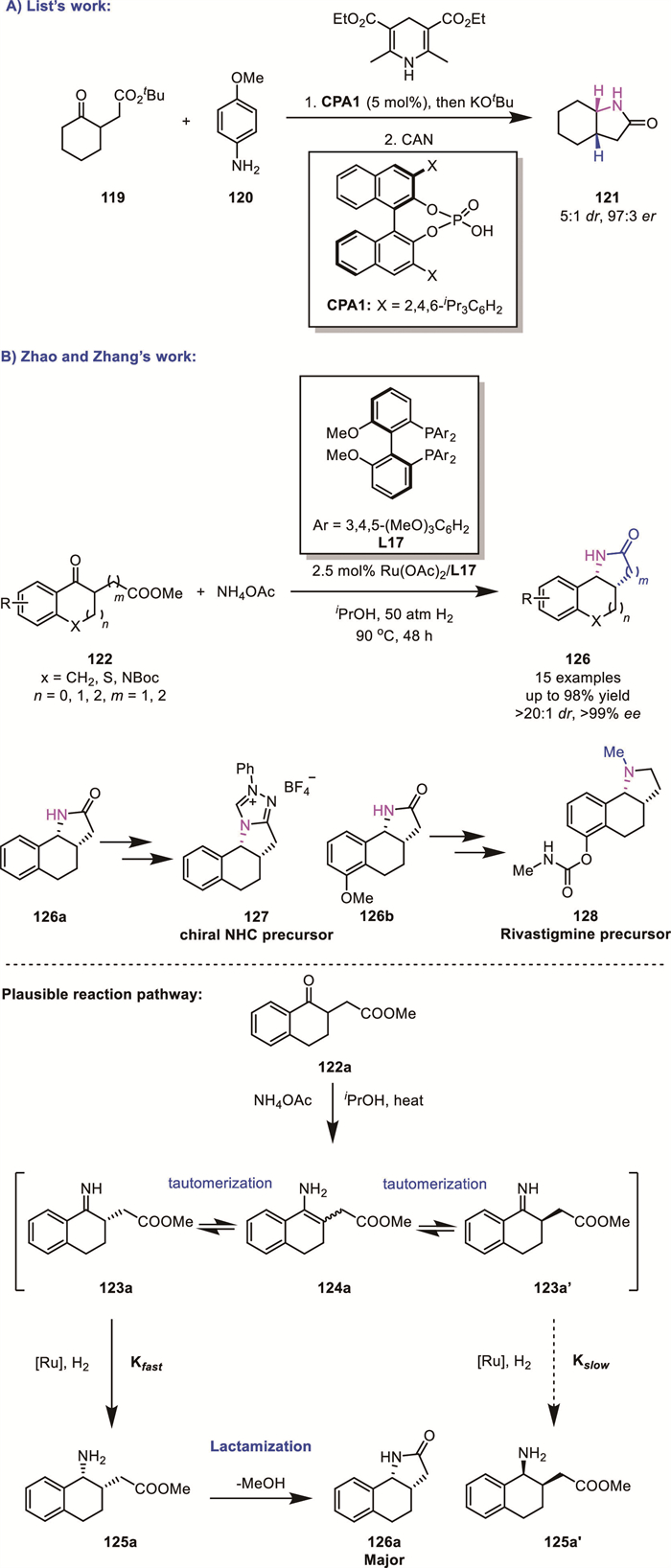
Inspired by this pioneering work, in 2023, Zhao and Zhang’s groups recently developed an efficient synthesis of chiral fused tricyclic lactams 126 via a ruthenium-catalyzed tandem DKR-ARA/lactamization (Scheme 39B) [99]. In their protocol, ammonium salts served as the nitrogen source, enabling the efficient transformation of α- or β-tetralone derivatives 122 into syn-lactam products 126 in high yields with excellent diastereoselectivity and enantioselectivity (up to >99% ee, >20:1 dr, and 98% yield). Based on control experiments, they proposed a plausible reaction mechanism as follows: Initially, the starting material 122a condenses with the amine source to generate imine intermediates 123a and 123a'. These substrates undergo rapid racemization via the achiral enamine intermediate 124a. The selective reduction of 123a instead of 123a' occurs with the chiral Ru−H species, resulting in the formation of chiral amine 125a (instead of 125a’). Subsequent lactamization delivers 126a. Notably, the utility of this strategy was demonstrated by the syntheses of a chiral N-heterocyclic carbene precursor and a drug analogue.
Chiral N-heterocycles are crucial structural motifs in medicinal chemistry, due to their widespread occurrence in biologically active molecules (Scheme 40).
Among the methods toward N-heterocycles, the AH of easily available N-heteroarenes has been recognized as an efficient and direct method [100-103]. Various catalytic systems based on precious metals Ru, Pd, Rh, and Ir, or earth-abundant metals Mn, Co have been developed. In addition to hydrogenation with molecular H2, transfer hydrogenation with diverse hydride sources has also been reported. Both metal and organocatalysts have displayed high reactivity in transfer hydrogenation, which can meet the requirements of various purposes. Quinolines and indoles are among the most explored substrates due to their high reactivity and the importance of the resulting chiral N-heterocycle skeletons after reduction.
Chiral indolines are prevalent structures existing in many natural products and bioactive molecules. Asymmetric hydrogenation of substituted indoles represents one of the most direct methods toward them. In addition to mono-substituted indoles, C2,C3-disubstituted indoles have also been examined, affording chiral indolines with consecutive endocyclic chirality.
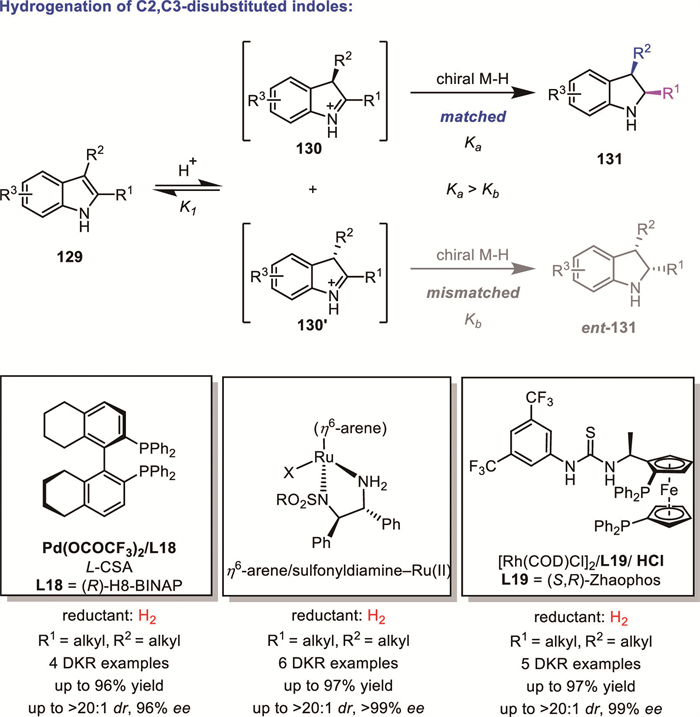
The seminal contributions of Zhou and Zhang's group marked a milestone with the development of the first efficient DKR-AH of unprotected 2,3-disubstituted indoles 129 using a Pd(OCOCF3)2/(R)-H8-BINAP catalytic system in combination with a Brønsted acid as an activator [104]. Subsequently, Arai's group and Fan's group independently reported chiral ruthenium diamine complexes-catalyzed hydrogenation of unprotected 2,3-disubstituted indoles 129 under relatively mild conditions [105–106]. In 2018, Chung and Zhang’s group reported an attractive Rh/ZhaoPhos catalytic system for the AH of N-unprotected indoles [107]. It is worth noting that all of these catalytic systems rely on the assistance of external Brønsted acids or acids generated in situ from the catalyst-mediated cleavage of hydrogen to facilitate the protonation of the C=C double bond of indoles, as well as the subsequent isomerization to iminium salt intermediates and their hydrogenation. According to mechanistic studies, these reductions, in fact, proceed via a DKR process (Scheme 41). The protonation of an indole molecule will yield a pair of enantiomers of the chiral indolium intermediate (130 and 130′), which can be converted to each other rapidly through rearomatization.
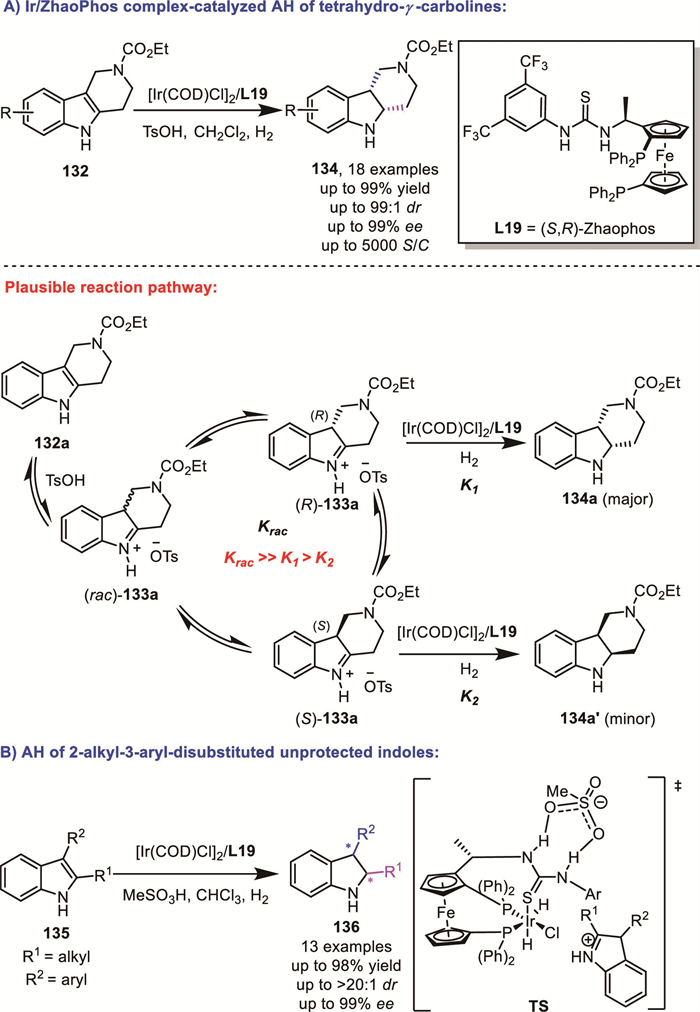
In 2022, Chen and Zhang’s group reported a novel route for the construction of chiral cis-hexahydro-γ-carboline derivatives 134 through an Ir/ZhaoPhos complex-catalyzed AH of the corresponding tetrahydro-γ-carbolines 132. This transformation achieved high yields (up to 99% yield), excellent diastereoselectivities (up to >99:1 dr) and enantioselectivities (up to 99% ee), as well as high substrate-to-catalyst ratios (up to 5000) (Scheme 42A) [108]. Based on previous studies on the mechanism of the AH of 2,3-disubstituted indolines, they proposed a plausible reaction pathway involving a DKR process. They also proposed an outer-sphere mechanism on the hydride-transfer step. Later, Zhang and Dong's group successfully reported the AH of challenging 2-alkyl-3-aryl-disubstituted unprotected indoles 135, catalyzed also by the Ir/ZhaoPhos complex (Scheme 42B) [109].
Despite significant progress in previous studies enabling the construction of various 2,3-disubstituted indolines, there has been no example of directly accessing chiral indolines bearing exocyclic vicinal stereogenic centers through asymmetric hydrogenation until recently. Remarkably, these chiral skeletons exist broadly in natural products (Scheme 43).
Yin's group recently introduced a configurationally labile substituent at the 2-position of unprotected indoles, thus extending the DKR process from the endocyclic to the exocyclic domain (Scheme 44) [110]. By leveraging Zhou’s palladium catalytic system, they successfully achieved a novel DKR-based hydrogenation of racemic α-substituted indole-2-acetates 137 to afford chiral indolines bearing both endocyclic and exocyclic vicinal stereogenic centers 141 in high yields with excellent diastereoselectivity and enantioselectivity (up to >99% ee, >20:1 dr, and 94% yield). Based on control experiments, they proposed a plausible reaction pathway involving a unique DKR model as follows: First, in the presence of a strong acid, racemic 137 undergoes rapid protonation to provide a pair of indolium intermediates (R)-138 and (S)-138, which can isomerize to each other through enamine intermediate 139. With a chiral Pd–H species 143 formed in situ, only (R)-138 is selectively reduced to yield 141 while (S)-138 is inert. To explain the diastereoselectivity control, they proposed that H-bonding exists between the indolium ion and the tethered ester group, affording a rigid six-membered-ring structure that is beneficial to diastereoselective differentiation at the reduction step. The Pd–H species then reduces (R)-138 from the less hindered side (H vs. R1) to yield the enantioenriched indoline product. Importantly, the practical utility of their strategy has been demonstrated by transforming the products into non-natural β-amino acids and amino alcohols.
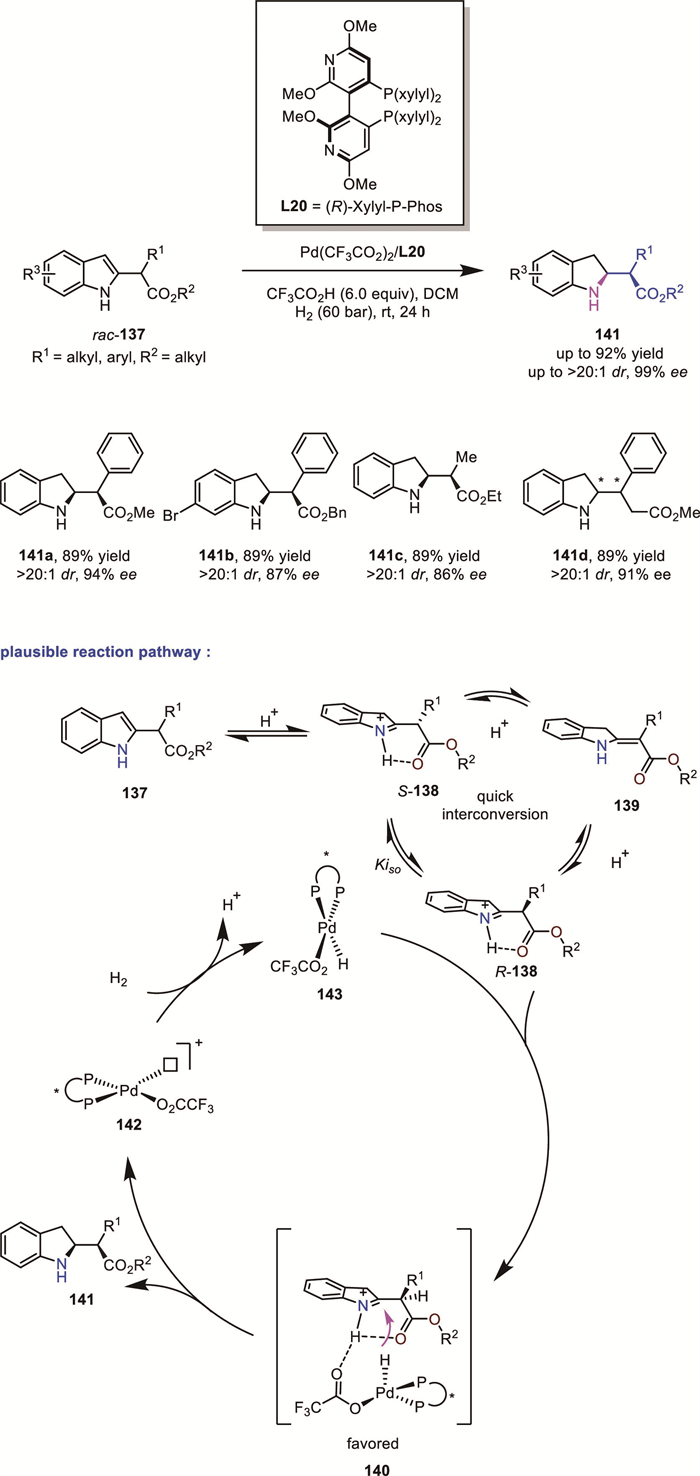
1,2,3,4-Tetrahydroquinolines (THQs) represent a significant class of chiral N-heterocycles existing in a number of natural alkaloids and medicinal agents. Therefore, their synthesis attracts great attention. The direct hydrogenation of easily accessible quinolines offers convenient and practical access to them [111-113]. In addition to widely used quinolines bearing only one substituent at the pyridine ring, quinolines bearing more than one substituent have also been evaluated as feasible substrate types for asymmetric hydrogenation, which affords 1,2,3,4-tetrahydroquinolines with multiple contiguous stereocenters. These reactions normally proceed via a DKR process.
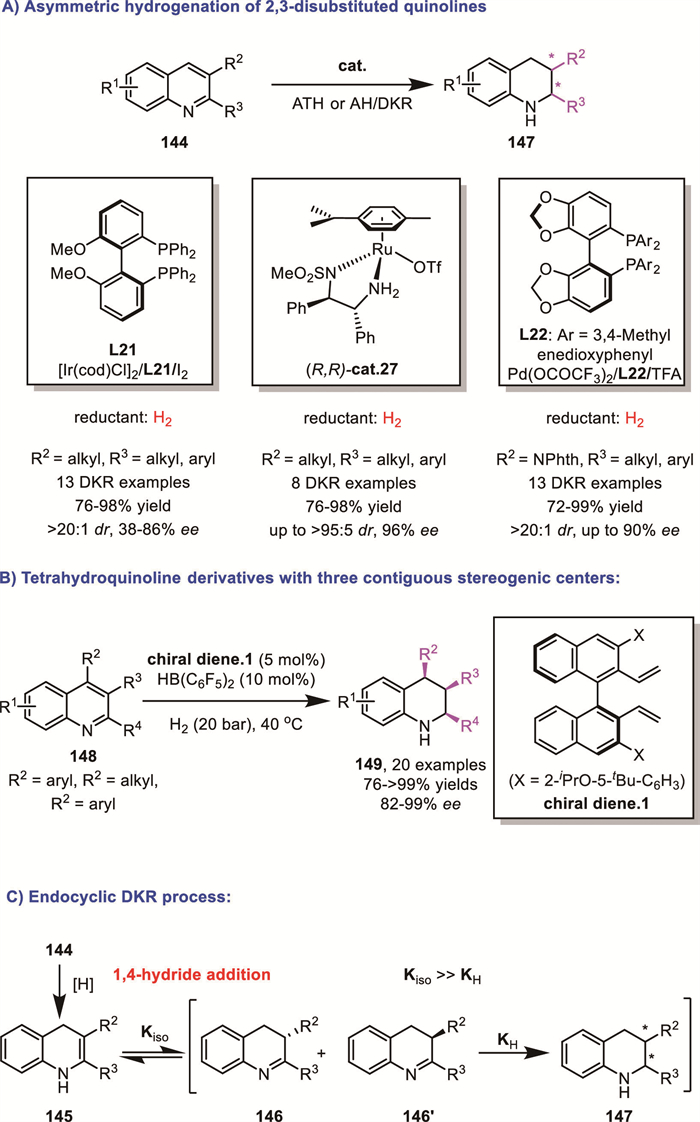
In 2009, Zhou and Li's group extended the application of the [Ir(COD)Cl]2/chiral bisphosphine ligands/I2 catalytic system from the hydrogenation of 2-substituted quinolines to the asymmetric hydrogenation of 2,3-disubstituted quinolines 144 [114]. In 2011, Fan and Yu's group reported the asymmetric hydrogenation of quinolines by using chiral cationic η6-arene–N-tosylethylenediamine–Ru(Ⅱ) complexes cat.27 [115]. In 2014, Zhou’s group developed a homogeneous Pd-catalyzed asymmetric hydrogenation of 3-phthalimido-substituted quinolones (Scheme 45A) [116]. In 2015, Du’s group achieved a groundbreaking advancement in asymmetric catalysis by employing chiral borane catalysts generated in situ from chiral dienes. This innovative approach enabled the first highly enantioselective cis-hydrogenation of 2,3,4-trisubstituted quinolines 148 (Scheme 45B) [117]. A reasonable description of the DKR process in these examples is that, following the 1,4-hydride addition, the resulting enamine intermediate undergoes rapid isomerization to the corresponding imine intermediates. Subsequently, only one specific enantiomer of the imine intermediates is selectively recognized and hydrogenated by the catalyst (Scheme 45C).
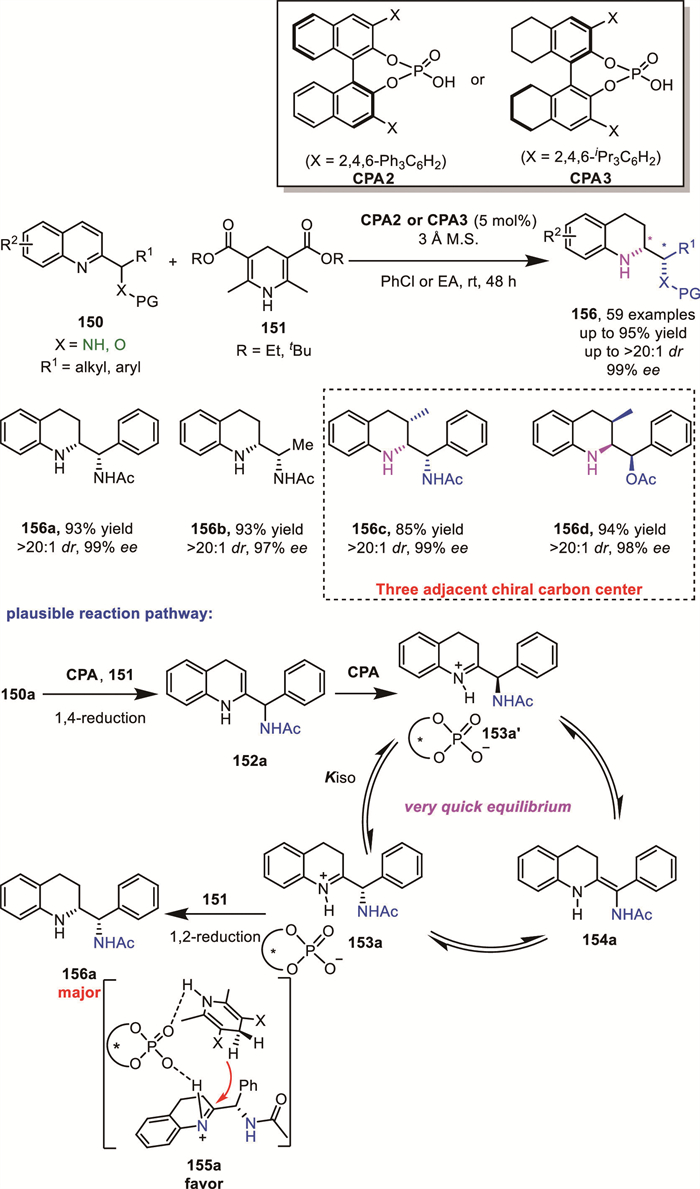
Inspired by Zhou’s work [118] and their own work [110], Yin’s group tried to expand the scope of quinolines by introducing a chiral substituent at the C2 position of quinolines, which could offer more diverse substituent patterns. In 2025, they successfully reported a DKR-based ATH of quinolines containing a chiral aminomethyl or hydroxymethyl substituent 150 at the C2 position using chiral phosphoric acid (CPA) catalysis, affording structurally novel chiral THQs with consecutive chiral centers 156 (up to three consecutive stereocenters) [119]. This method provides excellent yields, diastereoselectivity and enantioselectivities for a broad range of substrates (up to 95% yield, up to > 20:1 dr, 99% ee). Based on control experiments and previous studies, they proposed a plausible reaction pathway involving a DKR model as shown below: In the presence of a CPA, the racemic substrate 150a undergoes a 1,4-reduction with Hantzsch ester (HE), generating the key endocyclic enamine intermediate 152a. The enamine intermediate 152a can be further transformed under the influence of the CPA catalyst to yield the corresponding imine intermediates 153a or 153a’. The two configurations of the imine intermediates rapidly interconvert through an exocyclic enamine intermediate 154a via tautomerization. Subsequently, only one enantiomer of the endocyclic imine intermediate is selectively reduced by the catalyst (which is also the stereoselectivity-determining step). This process ultimately leads to the formation of the products 156a with excellent diastereo- and enantiocontrol (Scheme 46). This method puts forward a novel idea on the asymmetric synthesis of unknown N-heterocycles bearing exocyclic chirality. With this method, a series of THQ-derived diamines and amino esters have been obtained.
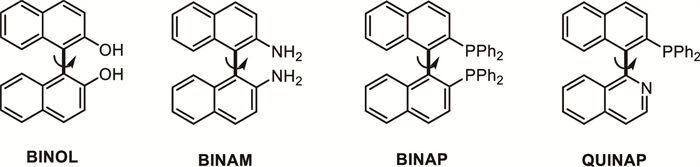
Axially chiral biaryls represent a highly valuable class of organic compounds characterized by restricted rotation around a single bond, leading to stable axial chirality. These compounds are of critical importance in asymmetric catalysis, medicinal chemistry, and materials science due to their distinct structural properties and broad functional utility. Notable examples include BINOL, BINAM, BINAP, QUINAP, and their derivatives (Scheme 47), which have been widely employed as chiral ligands and catalysts in a variety of enantioselective transformations. The synthesis of axially chiral biaryls with high enantiopurity, however, poses a considerable synthetic challenge, particularly when dealing with racemic or prochiral starting materials. In this context, DKR has also been applied as a useful strategy for the asymmetric synthesis of axially chiral compounds. The key point is the utilization of the lability of the axial, either caused by the inert property or internal interaction of various functional groups.
As a pioneering work, the DKR-based reduction of configurationally labile “bridged biaryl lactones”, first developed by Bringmann’s group, represents a unique and highly efficient strategy for the synthesis of axially chiral compounds. The first catalytic example utilized a stoichiometric amount of chiral hydride-transfer reagents derived from borane [120–121], subsequently followed by the development of strategies employing Co-catalysts with NaBH₄ as the reductant [122].
In 2018, Yin and Zhang’s group successfully synthesized the O-SPINOL-derived ligand O-SpiroPAP L23 and applied it to the iridium-catalyzed DKR-based AH of Bringmann lactones 157 (Scheme 48A) [123]. Their strategy enabled the synthesis of valuable chiral biaryl molecules 160 in high yields with excellent enantioselectivity (up to 99% yield, >99% ee). Based on control experiments and literature precedents, they proposed a plausible reaction pathway. Bringmann lactones 157 are first reduced to lactol 158, which undergoes rapid racemization and tautomerizes into its aldehyde form 159. Subsequently, the enantiopure intermediate 159A is selectively reduced to yield the final product 160 with high enantioselectivity while the other enantiomer is mismatched.
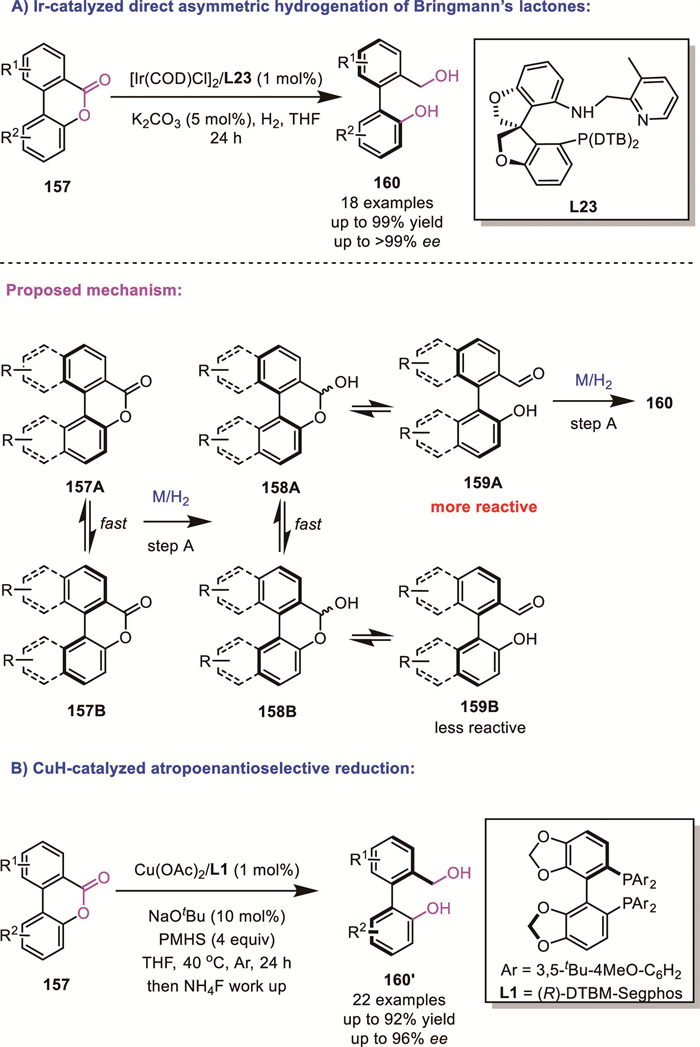
In 2019, the same group reported a similar atropoenantioselective reduction of Bringmann’s lactones using an inexpensive copper-based catalyst and silane as hydride source (Scheme 48B) [124].
In 2018, Lassaletta's group first reported a novel zinc-catalyzed hydrosilylation of configurationally labile heterobiaryl ketones 161, affording secondary alcohols bearing both axial and central chirality 162 [125]. Although most of the products exhibit only moderate dr values, this strategy demonstrates its potential through the stereospecific transformation of the products into N,N-ligands 163 or attractive axially chiral bifunctional thiourea organocatalysts 164. This DKR process relies on the transient Lewis acid-base interaction (LABI) between the nitrogen atom in the pyridine and the carbonyl group, which facilitates the racemization of the stereogenic axis in the heterobiaryl ketones (Scheme 49).
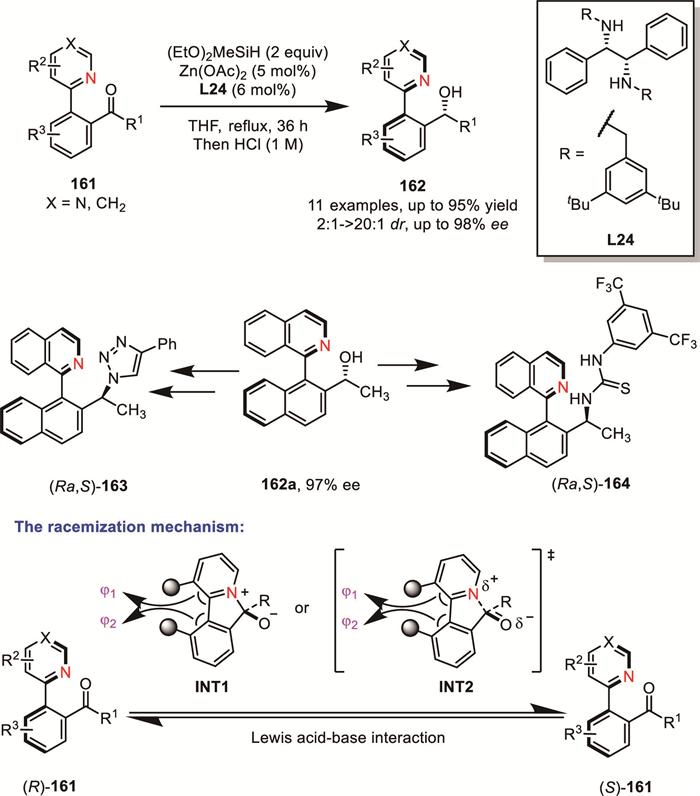
In 2024, Hu’s group reported a manganese-catalyzed AH of configurationally labile heterobiaryl ketone N-oxides 165, employing a structurally fine-tuned chiral P,N,N-ligand (Rc,Sp)-L25 [126]. Under the optimal conditions, a wide range of biaryl ketone N-oxides, including isoquinoline, pyridine, and quinoline derivatives, were efficiently hydrogenated. This transformation provided access to chiral heterobiaryl secondary alcohol N-oxides 167 bearing both central and axial chirality in high yields (up to 99% yield) with excellent diastereo- and enantioselectivity (up to >20:1 dr and >99% ee).
Based on literature precedents, they proposed that the success of this methodology relies on the rapid racemization of heterobiaryl ketone N-oxides via a six-membered cyclic O,O-ketal 166 closing/opening event between the oxygen atom of N-oxide and the ketone carbonyl group. Notably, the Mitsunobu reaction of the product 167a enables the rapid construction of diastereomers of these chiral heterobiaryl secondary alcohol N-oxides (Scheme 50). These novel compounds, featuring both central and axial chirality, can serve as highly efficient catalysts for the asymmetric allylation of benzaldehyde with allyltrichlorosilane, further highlighting their synthetic utility and potential in asymmetric catalysis.
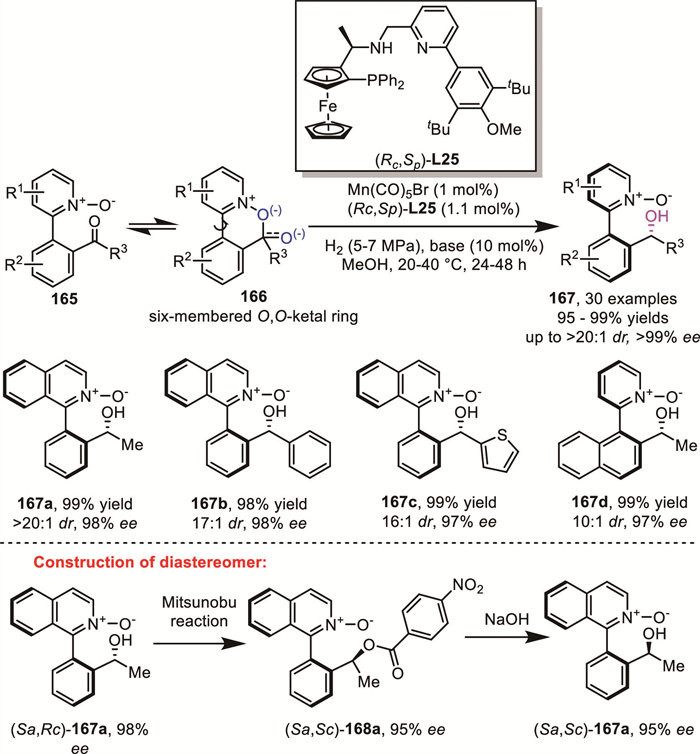
By integrating the DKR approach with reductive amination, a series of versatile and efficient methodologies have been developed for the synthesis of functionally diverse axially chiral biaryls.
In 2016, Akiyama's group reported a CPA-catalyzed approach for the synthesis of chiral biaryls 172 via ARA of 169 (Scheme 51) [127]. Notably, the atroposelectivity of the product can be completely controlled by the choice of the hydroxyaniline derivative. Based on their control experiments, they proposed a mechanism in which the in situ formed biaryl N,O-acetal 170 and biaryl imine 171 undergo rapid racemization through a ring-opening/ring-closing tautomerization. This dynamic interconversion enables the continuous equilibration of enantiomers. Furthermore, the different configurations of biaryl imine 171 are hydrogenated at distinct rates due to their different reactivity with the catalyst, leading to the selective formation of chiral biaryls (S)- or (R)-enantiomers of 172 with excellent enantioselectivities (up to 99% ee).
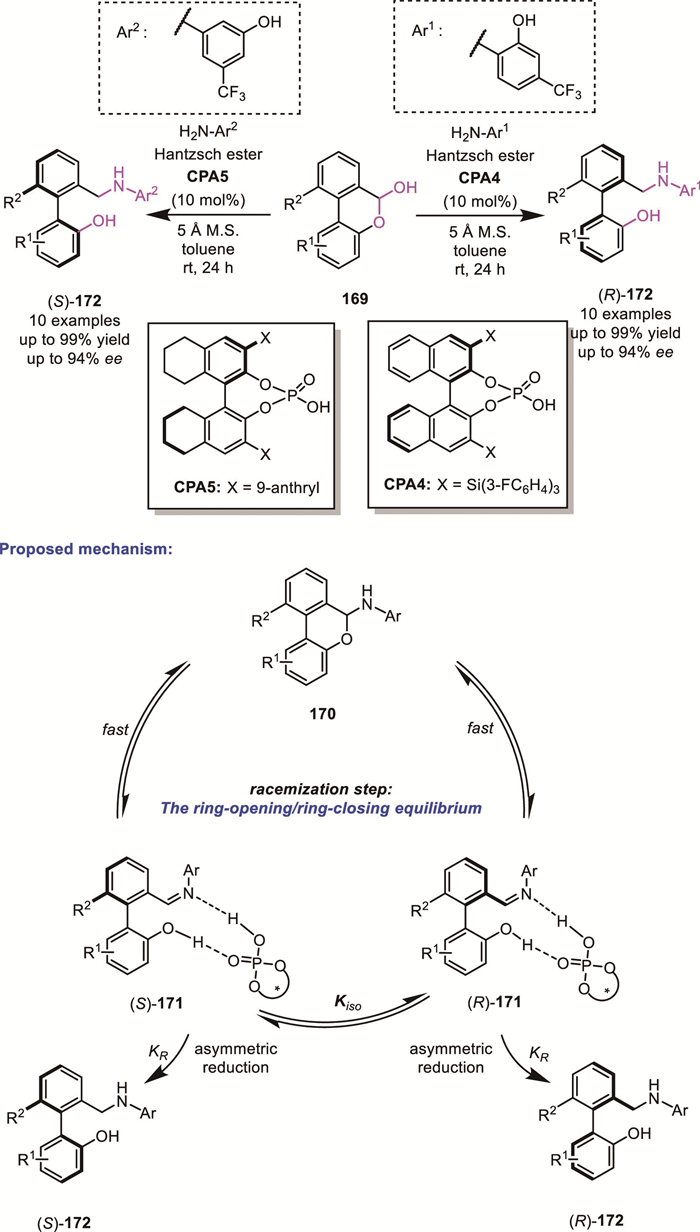
In 2021, Hronillos' group developed a ruthenium-catalyzed ARA of biaryl aminals 173, affording axially chiral diamines 176 in high yields with excellent enantioselectivities (up to 91% yield, up to 99% ee) [128]. The ring-opening–ring-closing equilibrium between imine 174 and biaryl aminals 175, as supported by DFT calculations, enables a DKR process through Ru-catalyzed ATH. Their strategy allows for the synthesis of a series of axially chiral analogues of BINAM under relatively mild conditions (Scheme 52).
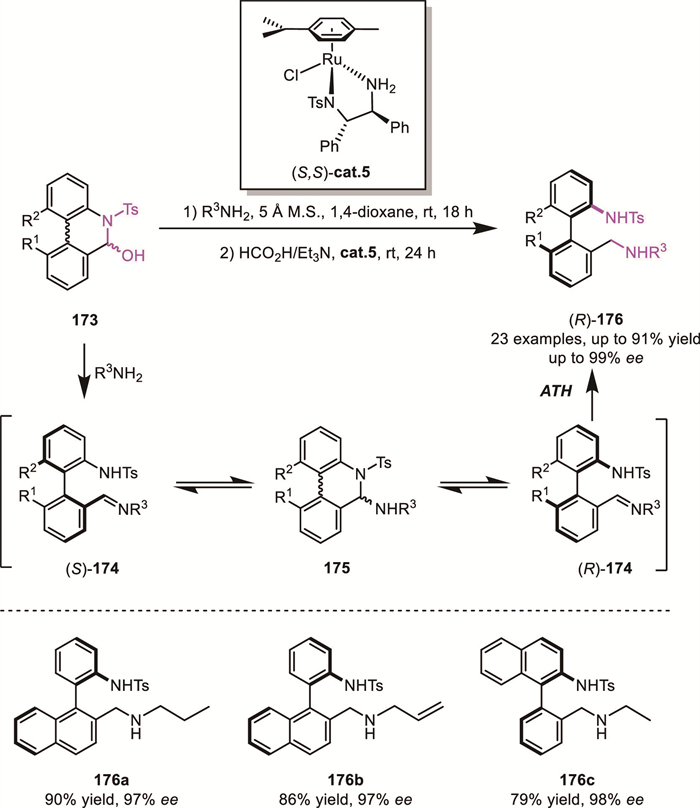
On the other hand, Wang's group successfully reported the ruthenium-catalyzed atropoenantioselective transfer hydrogenation reductive amination of biaryl aldehydes 177 using alkylamines as the amine source, significantly expanding the scope of accessible products (Scheme 53A) [129]. In 2022, Cheng's group further extended this strategy to the CPA-catalyzed enantioselective synthesis of axially chiral styrene-type allylamines 183 (Scheme 53B) [130]. This innovative approach facilitated the synthesis of novel axially chiral aryl–alkene architectures, further underscoring the value and broad applicability of the DKR-based reductive amination strategy.
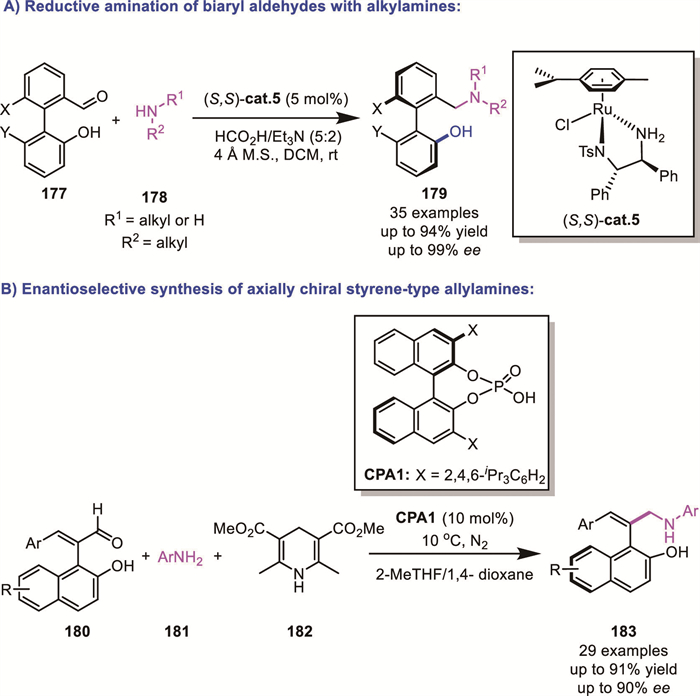
Due to the inherent structural properties of the C–N axis, which often lead to decreased stability, the asymmetric synthesis of C–N axially chiral compounds has been less explored compared to their C–C axially chiral biaryl counterparts. As previously discussed [125], relatively weak Lewis acid-base interactions (LABI) between strategically located functionalities in (hetero)biaryl systems substantially reduce the barrier to rotation, facilitating the racemization of relatively hindered systems and thus enabling DKR processes.
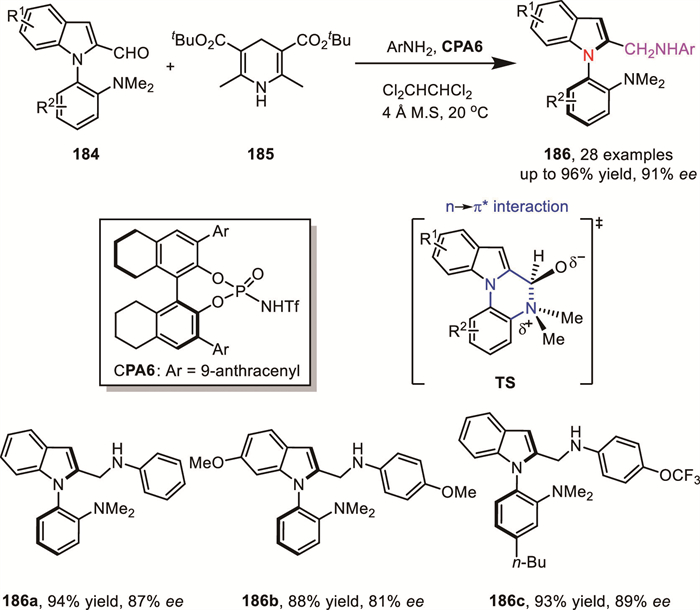
In 2024, Cheng’s group reported a chiral N-triflyl phosphoramide-catalyzed reductive amination of configurationally labile N-arylindoles 184, using Hantzsch esters as the hydrogen source [131]. This method provided access to a series of N-arylindole atropisomers 186 in high yields with excellent enantioselectivity (up to 96% yield, up to 91% ee). Notably, under the influence of the chiral N-triflyl phosphoramide catalyst, the noncovalent n→π* interaction within the cyclic transition state plays a crucial role in ensuring the successful DKR process (Scheme 54).
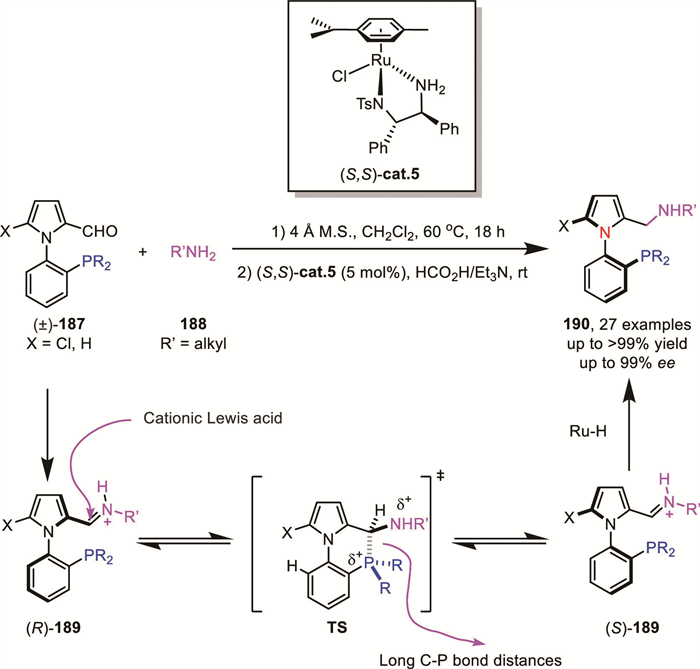
In 2024, Hornillos and Lassaletta's group proposed the use of phosphine groups as Lewis bases to achieve LABI between functional groups, successfully achieving a highly enantioselective DKR through the reductive amination of heterobiaryl phosphino-aldehydes 187 (Scheme 55) [132]. By employing ruthenium-catalyzed ATH of in situ generated imines, a series of N-aryl pyrrole aminophosphines 190 were synthesized with up to 99% yield and 99% ee. This methodology provided access to a diverse range of P,N derivatives, demonstrating again the versatility and efficiency of the LABI and DKR strategy in constructing C–N axially chiral compounds with high stereocontrol.
Within the broad family of axially chiral molecules, substituted biaryl derivatives based on C–C bond linkage are the most widely studied and utilized. Unlike biaryl derivatives linked by C–C bonds, the atroposelective synthesis of axially chiral diaryl ethers remains comparatively unexplored and thus appealing.
In 2023, Zhong and Zeng's group developed a DKR approach to axially chiral diary ethers via chiral phosphoric acid-catalyzed atroposelective reductive amination reaction of dicarbaldehydes 191 with anilines (Scheme 56) [133]. The desired diaryl ethers 195 could be obtained in moderate to good yields (up to 79%) and excellent enantioselectivities (up to 95% ee). Based on control experiments and literature precedents, they proposed a plausible reaction pathway. The in situ generated imine 194, formed from dicarbaldehyde 191 and aniline 192, cannot undergo racemization spontaneously. Instead, rapid racemization is achieved through hydrolysis back to dicarbaldehyde 191. In the presence of a chiral phosphoric acid, imine (R)-194 undergoes transfer hydrogenation at a faster rate compared to (S)-194, leading to the formation of the corresponding axially chiral diaryl ethers 195 with excellent enantioselectivity.
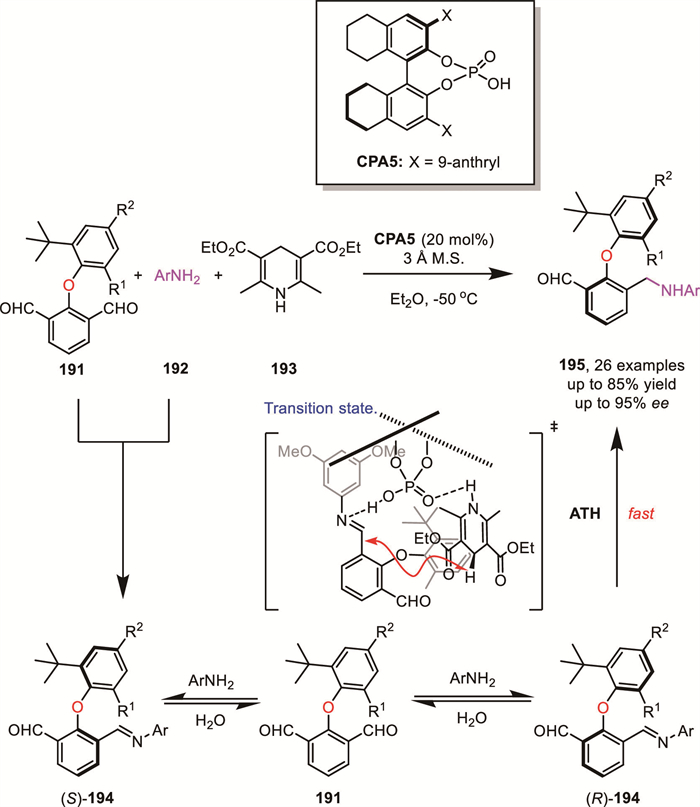
In the last decades, various enantioenriched oxaheterocycles have been accessed via a chirality transfer strategy utilizing enantioenriched alcohols bearing a terminal hydroxyl group or Williamson ether formation with enantioenriched alcohols containing a chloride at the proper position. However, the catalytic synthesis of enantioenriched aliphatic cyclic ethers has rarely been reported.
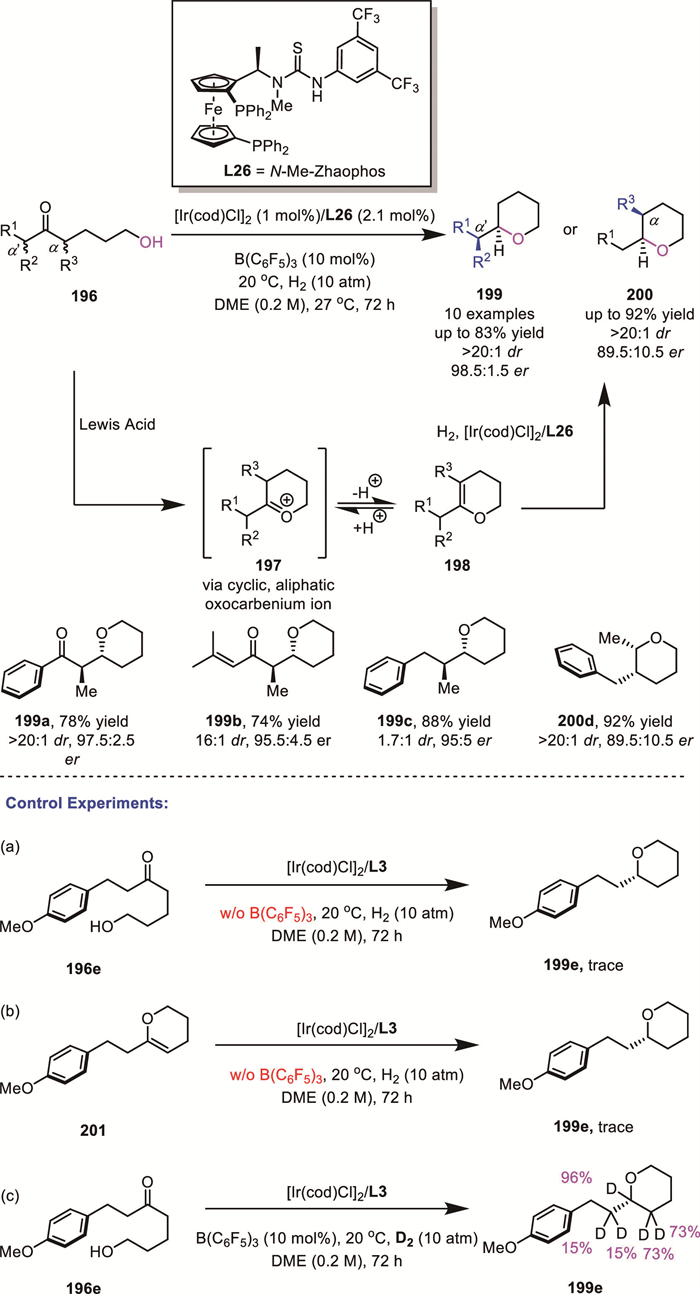
In 2024, Chen and Zhang's group successfully achieved an efficient Ir-catalyzed reductive cycloetherification of γ- or δ–hydroxy ketones 196 for the synthesis of chiral THFs and THPs 199 and 200 via asymmetric hydrogenation of cyclic, aliphatic oxocarbenium ions (Scheme 57) [134]. Several α- or α’-substituted hydroxy ketones were tolerated. To elucidate the reaction mechanism, they conducted a series of control experiments. Only a trace amount of target product 199e could be produced in the absence of B(C6F5)3 from 196e or enol ether 201, which indicated the critical role of the Lewis acid for this transformation. Meanwhile, the reaction of 196e with D2 under standard conditions resulted in deuterium incorporation of 73% and 15% for the adjacent endocyclic and exocyclic prochiral carbon atoms, respectively, suggesting the existence of an equilibrium between the oxocarbenium ion and enol ether. Moreover, the significant discrepancy in deuteration ratios suggested the ease of formation of an endocyclic enol compared with the exocyclic one. The interconversion was faster than the hydrogenation. The key to the success of DKR relies on the rapid isomerization between intermediates.
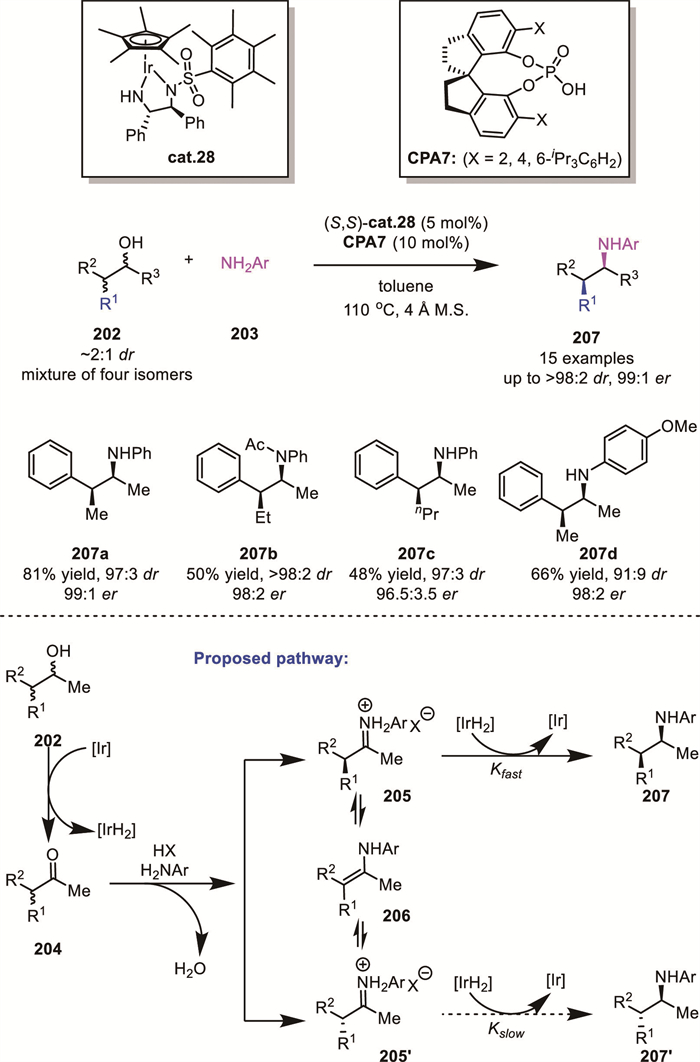
In 2015, Zhao's group reported an elegant example of dynamic kinetic asymmetric amination of racemic α-branched alcohols 202, which was catalyzed by an Ir-complex in combination with a chiral phosphoric acid (Scheme 58) [135]. This transformation converted all four stereoisomers of the substrate alcohol into acyclic chiral amines 207 with high stereoselectivity (up to >98:2 dr, 99:1 er). According to their proposed mechanism, the Ir catalyst captures the hydrogen from the substrate and oxidizes 202 to form ketone 204. Subsequently, the two iminium intermediates (205 and 205′) undergo tautomerization via enamine 206, enabling rapid racemization. Concurrently, diastereoselective reduction of the matched intermediate 205 (over 205′) by the Ir-hydride species delivers the target amine product 207.
Inspired by previous reports from Oe's studies [136], Zhao's group further investigated the monoamination of diols 208. By introducing an achiral Brønsted acid in the presence of a ruthenium/Josiphos co-catalytic system, a series of amino alcohols 213 could be obtained with high yields and enantioselectivity (up to 96% yield, up to 94% ee) [137]. Based on a series of control experiments, they proposed that this enantioselective process was realized via a dynamic kinetic asymmetric reduction of in situ formed iminium ions 212 (Scheme 59A).
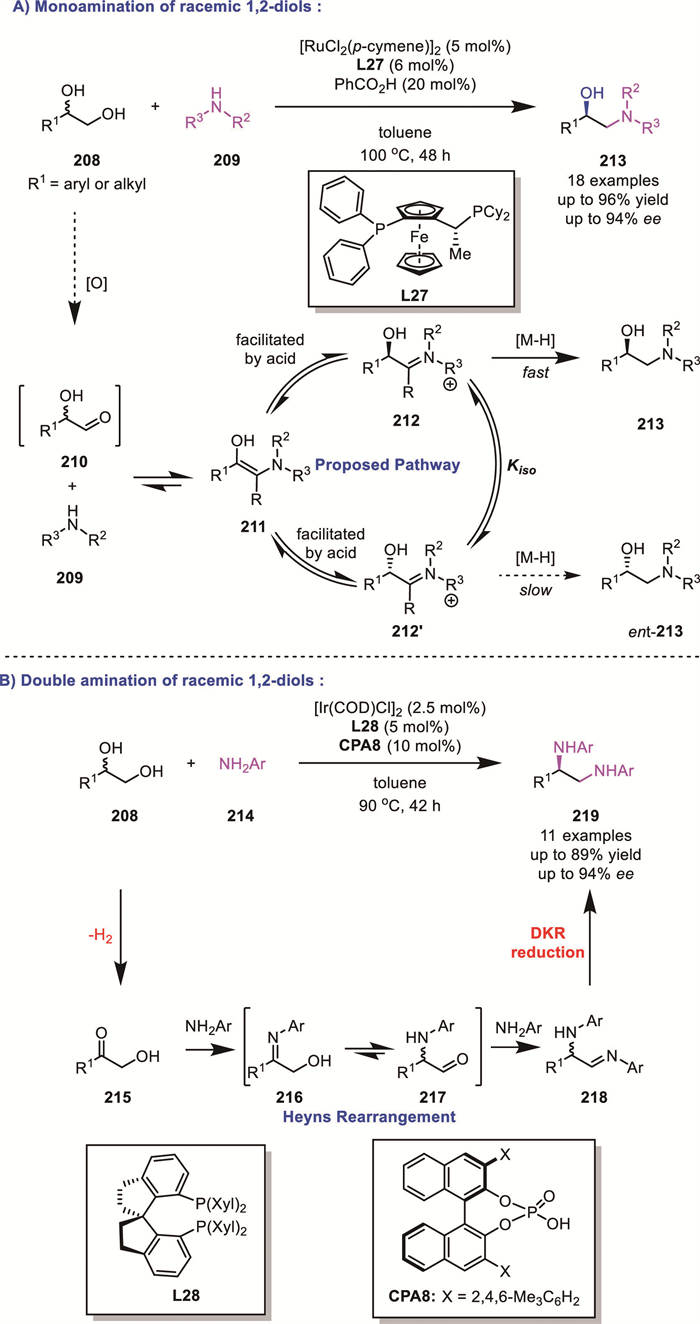
In 2021, they overcame the longstanding limitation in previous catalytic systems, where diols could only undergo monoamination, by developing a novel iridium/phosphoric acid co-catalyzed strategy (Scheme 59B) [138]. In this case, they achieved the enantioconvergent double amination of racemic 1,2-diols 208 for the first time, providing practical access to highly valuable enantioenriched vicinal diamines 219 (up to 89% yield, up to 94% ee). Their control experiments demonstrated that the double amination does not proceed via two individual aminations by borrowing hydrogen. Instead, mono-oxidation of diol followed by imine condensation would yield a hydroxy imine intermediate 216. Such structures would undergo a facile Heyns Rearrangement to generate α-amino carbonyl 217. The intermediate then undergoes a second imine condensation followed by the well-established DKR process, to afford the diamines 219.
In 2025, the same group reported a DKR for achieving enantioconvergent synthesis of C2-substituted benzomorpholines bearing a challenging-to-establish stereocenter 224 from readily available racemic amino primary alcohols 220 [139]. It was recognized that effective imine-enamine tautomerization is imperative for this DKR process, which could be influenced by an acid cocatalyst. So they examined the effect of various acid cocatalysts. In contrast to previous studies, CPA was ineffective in this case. Instead, the addition of Fe(OTf)3 was found to significantly improve both yield and enantioselectivity. This improvement is attributed to the Lewis acid-promoted rapid racemization of the imine intermediates 222 and 222′ through the achiral enamine 223 before reduction, a step that is essential for an effective DKR process (Scheme 60).
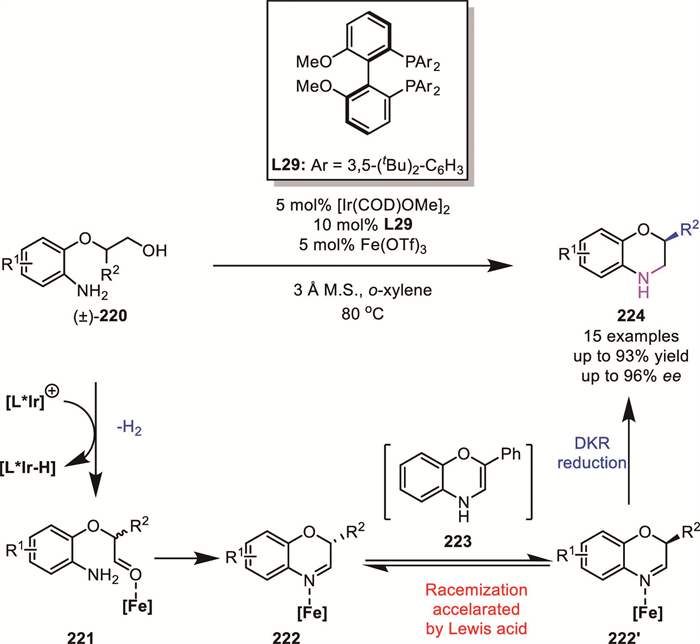
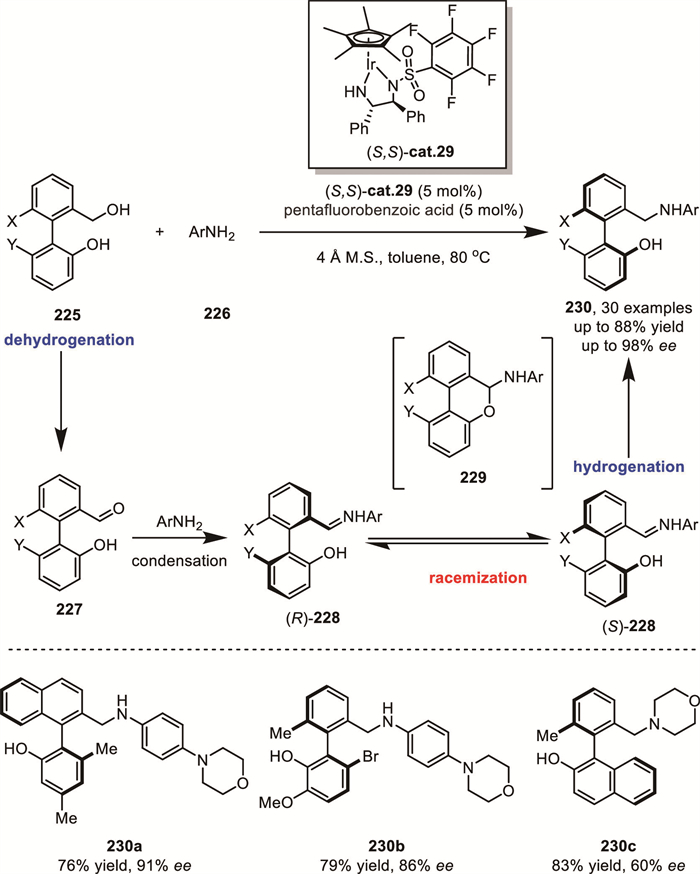
Building upon the previously introduced DKR of “bridged biaryl lactones”, the synthesis of axially chiral biaryl molecules 230 from racemic biaryl phenol-benzyl alcohol 225 typically requires at least two steps (oxidation followed by asymmetric reduction). However, Wang's group reported a novel iridium-catalyzed atropoenantioselective redox-neutral amination reaction (Scheme 61) [140], providing a new synthetic route to assemble axially chiral phenol-benzylamine compounds in high yields with excellent enantioselectivity (up to 88% yield, up to 98% ee). In this process, racemic biaryl phenol-benzyl alcohol 225 undergoes dehydrogenation mediated by the activated iridium catalyst, generating a biaryl phenol imine intermediate 229. This intermediate then participates in a well-established ring-opening/ring-closing equilibrium (racemization), followed by DKR-based asymmetric reduction to afford the target product.
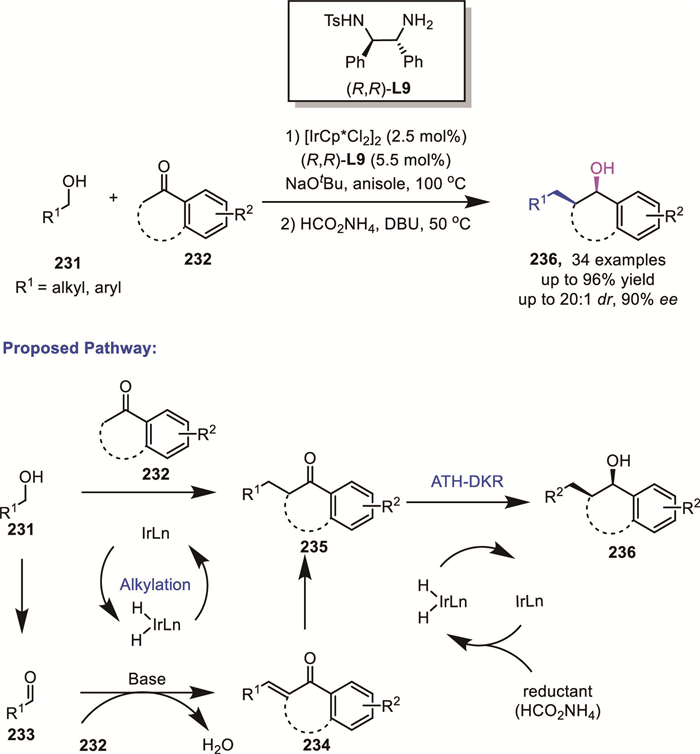
Conventional approaches to the synthesis of enantioenriched 1,2,3,4-tetrahydronaphthalen-1-ols typically relied on α,β-unsaturated or saturated tetralones as starting materials, requiring additional synthetic steps for their preparation. In 2024, Sarjeant's group developed an asymmetric synthesis of enantioenriched tetrahydronaphthalen-1-ol products bearing two contiguous stereogenic centers 236 directly from commercially available primary alcohols 231 and tetralones 232 via a hydrogen-borrowing/ATH-DKR sequence (Scheme 62) [141]. This iridium-catalyzed one-pot process afforded high yields with excellent enantio– and diastereoselectivity (up to 96% yield, up to 95:5 er, 20:1 dr). By employing a single iridium catalyst, this strategy enables direct access to the target products, bypassing the need for pre-synthesized α,β-unsaturated or saturated tetralones. They further proposed that an analogous DKR-ATH process, similar to that described earlier, occurs following α-alkylation.
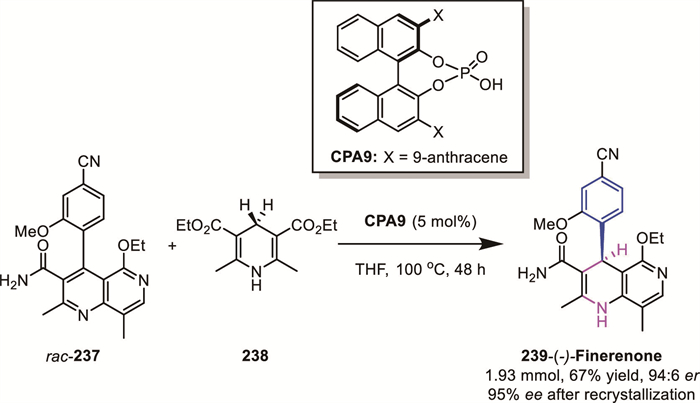
In 2020, Grayson and Aggarwal’s group reported a concise 6-step enantioselective synthesis of the nonsteroidal mineralocorticoid receptor antagonist (−)-finerenone 239 [142]. A key step in establishing stereochemistry was the CPA-catalyzed ATH of a naphthyridine derivative 237 under DKR conditions (Scheme 63). Mechanistic studies revealed that the naphthyridine substrate adopts two atropisomeric configurations that react at distinct rates and with differing enantioselectivity, enabling initial kinetic resolution. Elevated temperature facilitated efficient substrate racemization, converting the process into a DKR. This transformation delivered (−)-finerenone in 67% yield with 94:6 er.
Tetraphenylene, bearing four benzene rings located alternatively above and below its eight-membered ring, is comprised of a rigid saddle-shaped geometry. Owing to the high inversion barrier, the scaffold possessing unsymmetrical substituents will exhibit inherent chirality. In 2024, Yang's group reported a CPA-catalyzed ATH of imine-type eight-membered azaheterocycles 240, using Hantzsch ester 241 as the hydrogen donor and operating through a DKR mechanism (Scheme 64) [143]. Control experiments revealed that during the dehydrogenation of enantioenriched C10-unsubstituted 9,10-dihydrotribenzoazocine (-)-242a (95% ee), the corresponding imine-type eight-membered azaheterocycle 240a was obtained in a fully racemic form. This observation provided evidence that the aldimine imine-type eight-membered azaheterocycle 240a undergoes facile ring-opening and ring-closing reactions, enabling rapid racemization. In a manner analogous to the common DKR, the configuration matched with CPA could be reduced to responding product at a faster rate.
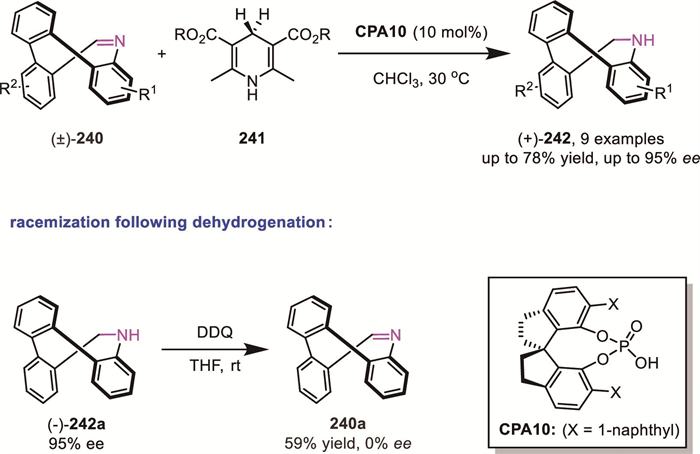
In 2023, Zhao’s group reported a CPA-catalyzed asymmetric reductive amination of cyclophane-derived aldehydes 243, providing planar-chiral cyclophanes 247 in high yields (up to 98%) and excellent enantioselectivities (up to >99% ee) [144]. Their studies demonstrated that the variable conformational flexibility of the ansa bridge dictates the reaction pathway: substrates with higher rotational barriers underwent kinetic resolution, while those possessing a > 15-membered ansa bridge underwent efficient racemization, enabling a DKR process. Control experiments supported a racemization mechanism involving hydrolysis of the imine intermediate. Consequently, substrates with the larger bridge reacted smoothly with amines via the DKR pathway (Scheme 65).
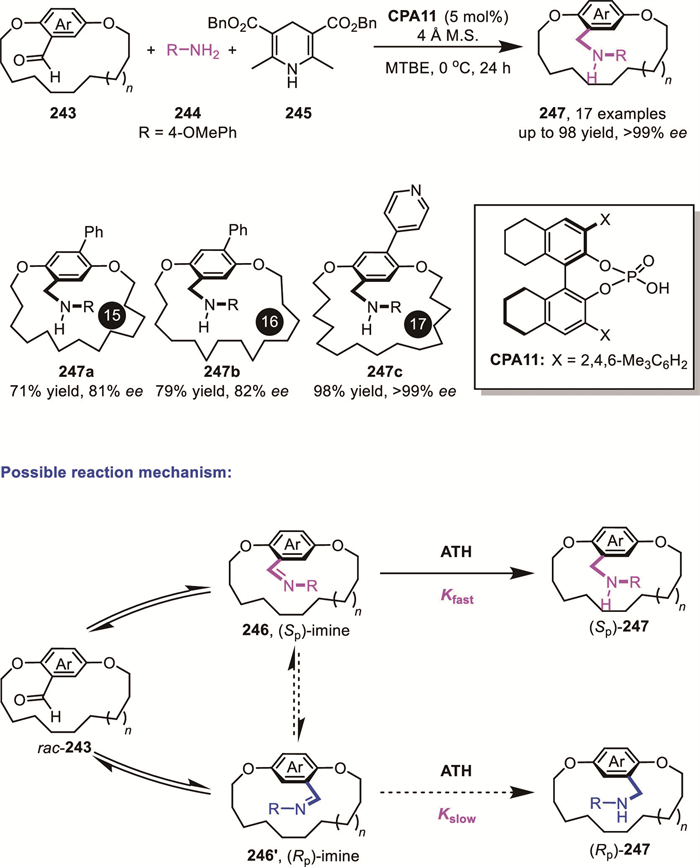
Although reduction-based DKR strategies have predominantly been applied to carbonyl or imine substrates, efforts to expand the substrate scope have been actively pursued.
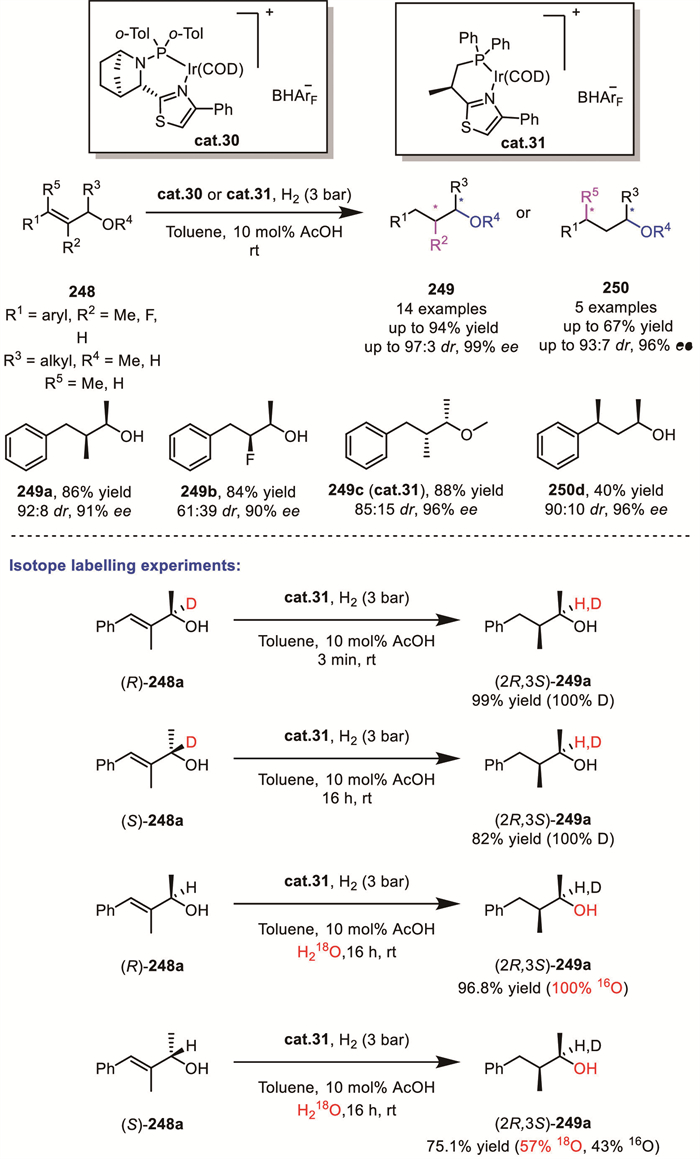
Recently, Andersson’s group reported an elegant example of Ir/N,P-ligand catalyzed asymmetric hydrogenation of racemic allylic alcohols 248 through a DKR process (Scheme 66) [145]. Interestingly, in addition to products bearing vicinal chirality, products with 1,3-chirality can also be achieved, albeit with moderate yield. Isotopic labeling with deuterium and 18O demonstrated that this DKR process does not involve tautomerism analogous to the enol-keto tautomerism, where a C=O double bond and a C–H single bond interconvert, but rather proceeds through cleavage and reforming of the C–O bond. This novel DKR mechanism reveals a unique paradigm that differs from traditional tautomerism-based DKR processes and underscores the need for further exploration to fully understand this important transformation.
In the past several years, considerable progress has been made in the field of DKR-based asymmetric reduction, enabling it as a powerful and sustainable method to address challenges in the asymmetric synthesis of chiral alcohols, amines, and N-heterocycles bearing multiple stereogenic centers. Advances in transition-metal catalysis, organocatalysis, and synergistic approaches have expanded the scope of substrates. Furthermore, the application of innovative catalytic systems, including chiral ligands and complexes, has enhanced the robustness and versatility of DKR methodologies. In addition, through rational design, the racemic stereo-labile axially chiral compounds can be transformed into enantioenriched axially chiral molecules, which have found extensive utility in organic synthesis, broadening the scope and synthetic potential of this method.
Despite these advancements, several challenges remain. Substrate generality, scalability, and compatibility with complex functional groups continue to be areas requiring further optimization. Additionally, achieving high efficiency under milder and greener reaction conditions is critical for advancing the sustainability of these processes. The integration of computational tools, such as machine learning and quantum chemical calculations, holds great promise for accelerating the rational design of catalysts and reaction pathways. Moreover, deeper insights into mechanistic aspects, including transition states and the interplay of kinetic and thermodynamic factors, will be essential for refining existing methods and discovering new ones.
Looking ahead, future research will likely focus on extending the utility of DKR to a broader range of challenging substrates, including those with multiple stereocenters and medicinally relevant scaffolds. The combination of DKR-based reduction with emerging technologies such as flow chemistry may unlock new avenues for synthetic applications. Synthetic chemists also expect to explore more sustainable catalytic systems, including bio-inspired and earth-abundant metal catalysts as well as customized enzymes to align with green chemistry principles. Ultimately, continued efforts in this field will not only deepen our understanding of stereochemical control in asymmetric reductions but also contribute to the development of innovative tools for drug discovery, materials science, and fine chemical synthesis.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ao Zhou: Writing – review & editing, Writing – original draft, Conceptualization. Mostafa M.K. Amer: Writing – review & editing, Writing – original draft. Qin Yin: Writing – review & editing, Supervision, Conceptualization.
This work was supported by the National Natural Science Foundation of China (Nos. 22522109, 22271307), the Shenzhen Science and Technology Innovation Program (No. JCYJ20240813161112017). Q.Y. is indebted to Shenzhen University of Advanced Technology and Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, for providing a starting grant.
H. Pellissier, Chirality from Dynamic Kinetic Resolution, RSC Publishing, 2011.
V. Bhat, E.R. Welin, X. Guo, et al., Chem. Rev. 117 (2017) 4528–4561. doi: 10.1021/acs.chemrev.6b00731
S.L. Bartlett, J.S. Johnson, Acc. Chem. Res. 50 (2017) 2284–2296. doi: 10.1021/acs.accounts.7b00263
D.J. Ager, A.H.M. de Vries, J.G. de Vries, Chem. Soc. Rev. 41 (2012) 3340–3380. doi: 10.1039/c2cs15312b
Z. Zhang, N.A. Butt, W. Zhang, Chem. Rev. 116 (2016) 14769–14827. doi: 10.1021/acs.chemrev.6b00564
V. Ratovelomanana-Vidal, P. Phansavath, Asymmetric Hydrogenation and Transfer Hydrogenation, Wiley-VCH, 2021.
F. Zhao, W. Ye, K. Wang, Chin. J. Org. Chem. 41 (2021) 2650–2665. doi: 10.6023/cjoc202011018
Y. Tian, L. Hu, Y.Z. Wang, et al., Org. Chem. Front. 8 (2021) 2328–2342. doi: 10.1039/D1QO00300C
A. Cabré, X. Verdaguer, A. Riera, Chem. Rev. 122 (2022) 269–339. doi: 10.1021/acs.chemrev.1c00496
H. Shimizu, I. Nagasaki, K. Mastsumura, et al., Acc. Chem. Res. 40 (2007) 1385–1393. doi: 10.1021/ar700101x
T. Touge, M. Kuwana, Y. Komatsuki, et al., Org. Process Res. Dev. 23 (2019) 452–461. doi: 10.1021/acs.oprd.8b00338
A.G. Smith, M.M. Bio, J.T. Colyer, et al., Org. Process Res. Dev. 24 (2020) 1164–1174. doi: 10.1021/acs.oprd.9b00427
R.M. Betancourt, P.G. Echeverria, T. Ayad, et al., Synthesis 53 (2021) 30–50. doi: 10.1017/9781108868532.002
M. Kosiuha, A. Karapetyan, O. Charron, et al., Synthesis 57 (2025) 2909–2934. doi: 10.1055/a-2646-6085
J.H. Xie, Q.L. Zhou, Dynamic Kinetic Resolution in Asymmetric Hydrogenation and Transfer hydrogenation, Science of Synthesis: Dynamic Kinetic Resolution (DKR) and Dynamic Kinetic Asymmetric Transformations (DYKAT), Thieme, 2022, p. 371.
Y. Gao, G. Hong, B.M. Yang, et al., Chem. Soc. Rev. 52 (2023) 5541–5562. doi: 10.1039/d3cs00424d
R. Noyori, T. Ikeda, T. Ohkuma, et al., J. Am. Chem. Soc. 111 (1989) 9134–9135. doi: 10.1021/ja00207a038
S. Jugé, J.P. Genêt, S. Mallard, Patent, EP 0489071A1, 1992.
Y. Hamada, Y. Koseki, T. Fujii, et al., Chem. Commun. (2008) 6206–6208. doi: 10.1039/b816524f
C.J. Hou, X.P. Hu, Org. Lett. 18 (2016) 5592–5595. doi: 10.1021/acs.orglett.6b02828
G. Gu, J. Lu, O. Yu, et al., Org. Lett. 20 (2018) 1888–1892. doi: 10.1021/acs.orglett.8b00433
Y.Z. Wang, B. Lu, G.Q. Chen, et al., Chem. Commun. 61 (2025) 1148–1151. doi: 10.1039/D4CC05644B
T. Yurino, R. Nishihara, T. Yasuda, et al., Org. Lett. 26 (2024) 2872–2876. doi: 10.1021/acs.orglett.3c04036
Z. Fei, Q. Wu, L. Li, et al., J. Org. Chem. 85 (2020) 6854–6861. doi: 10.1021/acs.joc.9b01916
J. Limanto, S.W. Krska, B.T. Dorner, et al., Org. Lett. 12 (2010) 512–515. doi: 10.1021/ol902715d
D.H. Bao, X.S. Gu, J.H. Xie, et al., Org. Lett. 19 (2017) 118–121. doi: 10.1021/acs.orglett.6b03397
L. Zhang, Z. Wang, Z. Han, et al., Angew. Chem. Int. Ed. 59 (2020) 15565–15569. doi: 10.1002/anie.202006383
C. Ayya Swamy P, A. Varenikov, G. de Ruiter, Chem. Eur. J. 26 (2020) 2333–2337. doi: 10.1002/chem.201904911
B. He, J. Song, C. Yin, et al., Org. Chem. Front. 9 (2022) 6247–6251. doi: 10.1039/d2qo01121b
X. Tan, W. Zeng, J. Wen, et al., Org. Lett. 22 (2020) 7230–7233. doi: 10.1021/acs.orglett.0c02565
Y.Z. Wang, Y.N. Duan, G.Q. Chen, et al., Org. Lett. 26 (2024) 8594–8598. doi: 10.1021/acs.orglett.4c03264
S.K. Gediya, G.J. Clarkson, M. Wills, J. Org. Chem. 85 (2020) 11309–11330. doi: 10.1021/acs.joc.0c01438
D.P. Lapa, L.H.S. Araújo, S.R. Melo, et al., Molecules 29 (2024) 3420. doi: 10.3390/molecules29143420
V.K. Vyas, G.J. Clarkon, M. Wills, Angew. Chem. Int. Ed. 59 (2020) 14265. doi: 10.1002/anie.202004658
F. Wang, T. Yang, T. Wu, et al., J. Am. Chem. Soc. 143 (2021) 2477–2483. doi: 10.1021/jacs.0c13273
R. Hu, F. Wang, F. Pan, et al., Org. Lett. 26 (2024) 7457–7462. doi: 10.1021/acs.orglett.4c02844
A.E. Cotman, B. Modec, B. Mohar, Org. Lett. 20 (2018) 2921–2924. doi: 10.1021/acs.orglett.8b00980
V.K. Vyas, B.M. Bhanage, Org. Lett. 18 (2016) 6436–6439. doi: 10.1021/acs.orglett.6b03334
V.K. Vyas, B.M. Bhanage, Asian J. Org. Chem. 7 (2018) 346–349. doi: 10.1002/ajoc.201700688
A.E. Cotman, M. Lozinšek, B. Wang, et al., Org. Lett. 21 (2019) 3644–3648. doi: 10.1021/acs.orglett.9b01069
Y. Xiong, H. Lin, C.L. Zhu, et al., Org. Lett. 23 (2021) 8883–8887. doi: 10.1021/acs.orglett.1c03384
D. Zhao, B. Beiring, F. Glorius, Angew. Chem. Int. Ed. 52 (2013) 8454–8458. doi: 10.1002/anie.201302573
B. He, P. Phansavath, V. Ratovelomanana-Vidal, Org. Lett. 21 (2019) 3276–3280. doi: 10.1021/acs.orglett.9b01002
Y.M. Zhang, Q.Y. Zhang, D.C. Wang, et al., Org. Lett. 21 (2019) 2998–3002. doi: 10.1021/acs.orglett.9b00451
M. Heo, B. Lee, K. Sishtla, et al., J. Org. Chem. 84 (2019) 9995–10011. doi: 10.1021/acs.joc.9b01134
Z. Luo, Z. Wang, G. Sun, et al., Org. Lett. 22 (2020) 4322–4326. doi: 10.1021/acs.orglett.0c01361
T. Touge, H. Nara, M. Kida, et al., Org. Lett. 23 (2021) 3070–3075. doi: 10.1021/acs.orglett.1c00739
K. Wang, S. Niu, X. Guo, et al., J. Org. Chem. 87 (2022) 3804–3809. doi: 10.1021/acs.joc.1c02916
O.V. Zatolochnaya, S. Rodríguez, Y. Zhang, et al., Chem. Sci. 9 (2018) 4505–4510. doi: 10.1039/c8sc00434j
X. Li, Z.B. Zhao, M.W. Chen, et al., Chem. Commun. 56 (2020) 5815–5818. doi: 10.1039/d0cc00480d
D. Yang, A.J. Yang, Y. Chen, et al., Org. Lett. 23 (2021) 1616–1620. doi: 10.1021/acs.orglett.1c00044
J. Li, J. Ye, J. Zhou, et al., Chem. Commun. 58 (2022) 4905–4908. doi: 10.1039/d2cc01193j
B. He, G.Q. Chen, X. Zhang, Chem. Commun. 60 (2024) 9785–9788. doi: 10.1039/d4cc02529f
E. Pierre-Georges, T. Ayad, P. Phansavath, et al., Synthesis 48 (2016) 2523–2539. doi: 10.1055/s-0035-1561648
R.M. Betancourt, L. Bacheley, A. Karapetyan, et al., ChemCatChem 14 (2022) e202200595. doi: 10.1002/cctc.202200595
R.M. Betancourt, P. Phansavath, V. Ratovelomanana-Vidal, Molecules 27 (2022) 995. doi: 10.3390/molecules27030995
R.M. Betancourt, P. Phansavath, V. Ratovelomanana-Vidal, J. Org. Chem. 86 (2021) 12054–12063. doi: 10.1021/acs.joc.1c01415
G.S. Caleffi, J.O.C. Brum, A.T. Costa, et al., J. Org. Chem. 86 (2021) 4849–4858. doi: 10.1021/acs.joc.0c02981
R.M. Betancourt, P. Phansavath, V. Ratovelomanana-Vidal, Org. Lett. 23 (2021) 1621–1625. doi: 10.1021/acs.orglett.1c00047
A. Keßberg, T. Lübken, P. Metz, Org. Lett. 20 (2018) 3006–3009. doi: 10.1021/acs.orglett.8b01034
P. Ciesielski, P. Metz, Nat. Commun. 11 (2020) 3091. doi: 10.1038/s41467-020-16933-y
Q. Hu, J. Chen, Z. Zhang, et al., Org. Lett. 18 (2016) 1290–1293. doi: 10.1021/acs.orglett.6b00212
Y. Xu, Y. Luo, J. Ye, et al., J. Am. Chem. Soc. 144 (2022) 20078–20089. doi: 10.1021/jacs.2c09266
T. Touge, K. Sakaguchi, N. Tamaki, et al., J. Am. Chem. Soc. 141 (2019) 16354–16361. doi: 10.1021/jacs.9b07297
K.M. Steward, M.T. Corbett, C.G. Goodman, et al., J. Am. Chem. Soc. 134 (2012) 20197–20206. doi: 10.1021/ja3102709
Z. Xiong, J. Tian, P. Xue, et al., Org. Chem. Front. 7 (2020) 104–108. doi: 10.1039/c9qo01047e
S.X. Zhang, C. Xu, N. Yi, et al., Angew. Chem. Int. Ed. 61 (2022) e202205739. doi: 10.1002/anie.202205739
X. Li, W. Hao, N. Yi, Y.M. He, Q.H. Fan, CCS Chem. 5 (2023) 2277–2289. doi: 10.31635/ccschem.023.202303007
L. Yu, P. Somfai, RSC Adv. 9 (2019) 2799–2802. doi: 10.1039/c9ra00173e
H.Y. Bin, K. Wang, D. Yang, et al., Angew. Chem. Int. Ed. 58 (2019) 1174. doi: 10.1002/anie.201812822
H.Y. Bin, L. Cheng, X. Wu, et al., Chem. Sci. 12 (2021) 7793–7799. doi: 10.1039/d1sc02044g
F. Wang, Z. Zhang, Y. Chen, et al., Nat. Commun. 13 (2022) 7794. doi: 10.1038/s41467-022-35124-5
F. Wang, X. Tan, T. Wu, et al., Chem. Commun. 56 (2020) 15557–15560. doi: 10.1039/d0cc05599a
H. Zhang, D. Feng, H. Sheng, et al., RSC Adv. 4 (2014) 6417–6423. doi: 10.1039/c3ra47129b
M. Sterle, M. Huš, M. Lozinšek, et al., ACS Catal. 13 (2023) 6242–6248. doi: 10.1021/acscatal.3c00980
T. Chen, W. Liu, W. Gu, et al., J. Am. Chem. Soc. 145 (2023) 585–599. doi: 10.1021/jacs.2c11149
W. Liu, C. Ren, L. Zhou, et al., J. Am. Chem. Soc. 146 (2024) 20092–20106. doi: 10.1021/jacs.4c04171
A.E. Cotman, D. Cahard, B. Mohar, Angew. Chem. Int. Ed. 55 (2016) 5294–5298. doi: 10.1002/anie.201600812
J. Wang, P.L. Shao, X. Lin, et al., Angew. Chem. Int. Ed. 59 (2020) 18166–18171. doi: 10.1002/anie.202006661
J.H. Xie, Z.T. Zhou, W.L. Kong, et al., J. Am. Chem. Soc. 129 (2007) 1868–1869. doi: 10.1021/ja0680109
Z.T. Zhou, J.H. Xie, Q.L. Zhou, Adv. Synth. Catal. 351 (2009) 363–366. doi: 10.1002/adsc.200800634
X. Li, B. List, Chem. Commun. 41 (2007) 1739–1741. doi: 10.1039/b703977h
M. Ito, T. Ootsuka, R. Watari, et al., J. Am. Chem. Soc. 133 (2011) 4240–4242. doi: 10.1021/ja1117254
X.H. Yang, H.T. Yue, N. Yu, et al., Chem. Sci. 8 (2017) 1811–1814. doi: 10.1039/C6SC04609F
X.S. Gu, N. Yu, X.H. Yang, et al., Org. Lett. 21 (2019) 4111–4115. doi: 10.1021/acs.orglett.9b01290
H. Wang, S.S. Xun, C.B. Yu, et al., Chem. Sci. 15 (2024) 11038–11042. doi: 10.1039/d4sc01890g
R.T. Endean, L. Rasu, S.H. Bergens, ACS Catal. 9 (2019) 6111–6117. doi: 10.1021/acscatal.9b01037
H. Ishikawa, T. Yurino, R. Komatsu, et al., Org. Lett. 25 (2023) 2355–2360. doi: 10.1021/acs.orglett.3c00740
L. Rasu, J.M. John, E. Stephenson, et al., J. Am. Chem. Soc. 139 (2017) 3065–3071. doi: 10.1021/jacs.6b12254
S. Hoffmann, M. Nicoletti, B. List, J. Am. Chem. Soc. 128 (2006) 13074–13075. doi: 10.1021/ja065404r
Q. Yin, Y. Shi, J. Wang, et al., Chem. Soc. Rev. 49 (2020) 6141–6153. doi: 10.1039/c9cs00921c
Y. Tian, L. Hu, Y.Z. Wang, et al., Org. Chem. Front. 8 (2021) 2328–2342. doi: 10.1039/D1QO00300C
N.U.D. Reshi, V.B. Saptal, M. Beller, et al., ACS Catal. 11 (2021) 13809–13837. doi: 10.1021/acscatal.1c04208
Z. Dai, X.M. Zhang, Q. Yin, Chin. J. Org. Chem. 42 (2022) 2261–2274. doi: 10.6023/cjoc202203058
Y. Lou, Y. Hu, J. Lu, et al., Angew. Chem. Int. Ed. 57 (2018) 14193–14197. doi: 10.1002/anie.201809719
L. Hu, Y.Z. Wang, L. Xu, et al., Angew. Chem. Int. Ed. 61 (2022) e202202552. doi: 10.1002/anie.202202552
B. Song, M.W. Chen, Y.G. Zhou, Org. Chem. Front. 5 (2018) 1113–1117. doi: 10.1039/c7qo01098b
V.N. Wakchaure, J. Zhou, S. Hoffmann, et al., Angew. Chem. Int. Ed. 49 (2010) 4612–4614. doi: 10.1002/anie.201001715
J. Wang, Y. Shi, F. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202303868. doi: 10.1002/anie.202303868
Y.G. Zhou, Acc. Chem. Res. 40 (2007) 1357–1366. doi: 10.1021/ar700094b
B.R. Shao, L. Shi, Y.G. Zhou, et al., Chem. Commun. 57 (2021) 12741–12753. doi: 10.1039/d1cc04722a
R. Gunasekar, R.L. Goodyear, I.P. Silvestri, et al., Org. Biomol. Chem. 20 (2022) 1794–1827. doi: 10.1039/d1ob02331d
L. Lückemeier, M. Pierau, F. Glorius, Chem. Soc. Rev. 52 (2023) 4996–5012. doi: 10.1039/d3cs00329a
D.S. Wang, Q.A. Chen, W. Li, et al., J. Am. Chem. Soc. 132 (2010) 8909–8911. doi: 10.1021/ja103668q
T. Touge, T. Arai, J. Am. Chem. Soc. 138 (2016) 11299–11305. doi: 10.1021/jacs.6b06295
Z. Yang, F. Chen, Y. He, et al., Angew. Chem. Int. Ed. 55 (2016) 13863–13866. doi: 10.1002/anie.201607890
J. Wen, X. Fan, R. Tan, et al., Org. Lett. 20 (2018) 2143–2147. doi: 10.1021/acs.orglett.8b00312
L.S. Zheng, C. Yin, F. Wang, et al., Chem. Commun. 58 (2022) 3286–3289. doi: 10.1039/d1cc06888a
C. Liu, L. Zheng, K. Tian, et al., CCS Chem. 5 (2023) 1398–1410. doi: 10.31635/ccschem.022.202101643
N.X. Rong, A. Zhou, M. Liang, et al., J. Am. Chem. Soc. 146 (2024) 5081–5087 8. doi: 10.1021/jacs.4c00298
W.B. Wang, S.M. Lu, P.Y. Yang, et al., J. Am. Chem. Soc. 125 (2003) 10536–10537. doi: 10.1021/ja0353762
S.M. Liu, X.W. Han, Y.G. Zhou, Adv. Synth. Catal. 346 (2004) 909–912. doi: 10.1002/adsc.200404017
S.M. Lu, Y.Q. Wang, X.W. Han, et al., Angew. Chem. Int. Ed. 45 (2006) 2260–2263. doi: 10.1002/anie.200503073
D.W. Wang, X.B. Wang, D.S. Wang, et al., J. Org. Chem. 74 (2009) 2780–2787. doi: 10.1021/jo900073z
T. Wang, L.G. Zhuo, Z. Li, et al., J. Am. Chem. Soc. 133 (2011) 9878–9891. doi: 10.1021/ja2023042
X.F. Cai, W.X. Huang, Z.P. Chen, et al., Chem. Commun. 50 (2014) 9588–9590. doi: 10.1039/C4CC04386C
Z. Zhang, H. Du, Org. Lett. 17 (2015) 2816–2819. doi: 10.1021/acs.orglett.5b01240
M.W. Chen, X.F. Cai, Z.P. Chen, et al., Chem. Commun. 50 (2014) 12526–12529. doi: 10.1039/C4CC06060A
M.R. Liang, X. Du, J. Lin, et al., J. Am. Chem. Soc. 147 (2025) 4239–4248. doi: 10.1021/jacs.4c14200
G. Bringmann, D.C.T. Hartung, Angew. Chem. Int. Ed. 31 (1992) 761–762. doi: 10.1002/anie.199207611
G. Bringmann, M. Breuning, P. Henschel, J. Hinrichs, Org. Synth. 79 (2002) 72, doi: 10.15227/orgsyn.079.0072.
T. Ashizawa, S. Tanaka, T. Yamada, Org. Lett. 10 (2008) 2521–2524. doi: 10.1021/ol800802c
G.Q. Chen, B.J. Lin, J.M. Huang, et al., J. Am. Chem. Soc. 140 (2018) 8064–8068. doi: 10.1021/jacs.8b03642
L. Hu, Y. Zhang, G.Q. Chen, et al., Org. Lett. 21 (2019) 5575–5580. doi: 10.1021/acs.orglett.9b01907
V. Hornillos, J.A. Carmona, A. Ros, et al., Angew. Chem. Int. Ed. 57 (2018) 3777–3781. doi: 10.1002/anie.201713200
Y.B. Wan, X.P. Hu, ACS Catal. 14 (2024) 17633–17641. doi: 10.1021/acscatal.4c04979
K. Mori, T. Itakura, T. Akiyama, Angew. Chem. Int. Ed. 55 (2016) 11642–11646. doi: 10.1002/anie.201606063
J.A. Carmona, C. Rodríguez-Franco, J. López-Serrano, et al., ACS Catal. 11 (2021) 4117–4124. doi: 10.1021/acscatal.1c00571
D. Guo, J. Zhang, B. Zhang, J. Wang, Org. Lett. 20 (2018) 6284–6288. doi: 10.1021/acs.orglett.8b02785
Y.D. Shao, J.S. Feng, D.D. Han, et al., Org. Chem. Front. 9 (2022) 764–770. doi: 10.1039/d1qo01672e
Y.D. Shao, D.D. Han, H.X. Jiang, et al., Org. Chem. Front. 11 (2024) 3894–3899. doi: 10.1039/d4qo00616j
C. Rodríguez-Franco, E. Roldán-Molina, A. Aguirre-Medina, et al., Angew. Chem. Int. Ed. 63 (2024) e202409524. doi: 10.1002/anie.202409524
L. Dai, Y. Liu, Q. Xu, et al., Angew. Chem. Int. Ed. 62 (2023) e202216534. doi: 10.1002/anie.202216534
Y. Zong, X. Zou, H. Tao, et al., J. Am. Chem. Soc. 146 (2024) 34107–34117. doi: 10.1021/jacs.4c13709
Z.Q. Rong, Y. Zhang, R.H.B. Chua, et al., J. Am. Chem. Soc. 137 (2015) 4944–4947. doi: 10.1021/jacs.5b02212
A.E. Putra, Y. Oe, T. Ohta, Eur. J. Org. Chem. 2013 (2013) 6146–6151. doi: 10.1002/ejoc.201300692
L.C. Yang, Y.N. Wang, Y. Zhang, et al., ACS Catal. 7 (2017) 93–97. doi: 10.1021/acscatal.6b02959
H.J. Pan, Y. Lin, T. Gao, et al., Angew. Chem. Int. Ed. 60 (2021) 18599–18604. doi: 10.1002/anie.202101517
Y. Gao, G. Hong, L. Zhang, et al., CCS Chem. 7 (2025) 80–90. doi: 10.31635/ccschem.024.202404559
J. Zhang, J. Wang, Angew. Chem. Int. Ed. 57 (2018) 465–469. doi: 10.1002/anie.201711126
S.W. Kim, E.A. Foker, W.J. Wolf, et al., Org. Lett. 26 (2024) 3103–3108. doi: 10.1021/acs.orglett.4c00718
A. Lerchen, N. Gandhamsetty, E.H.E. Farrar, et al., Angew. Chem. Int. Ed. 59 (2020) 23107–23111. doi: 10.1002/anie.202011256
D. Zhang, J. Zhou, T. Qin, et al., Chem. Catal. 4 (2024) 100827.
J. Li, C. Zhao, ACS Catal. 13 (2023) 14155–14162. doi: 10.1021/acscatal.3c03718
J. Liu, S. Krajangsri, J. Yang, et al., Nat. Catal. 1 (2018) 438–443. doi: 10.1038/s41929-018-0070-0

Scheme 10 Rh-catalyzed ATH of α-substituted β-keto carbonitriles and β-cyano α-keto esters.

Scheme 13 Enantioselective total synthesis of the phenylpropanoid (+)-burmaniol A via AH/DKR.

Scheme 19 Enantioselective synthesis of chiral multicyclic γ-lactones and its application.

Scheme 53 Enantioselective DKR-based reductive amination for novel axially chiral architectures.

Scheme 55 Ru-catalyzed atroposelective reductive amination of heterobiaryl phosphino-aldehydes.

Scheme 56 CPA-catalyzed atroposelective reductive amination reaction of dicarbaldehydes.

Scheme 61 Ir-catalyzed amination of biaryl phenol-benzyl alcohol through borrowing hydrogen.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:



























































 下载:
下载: