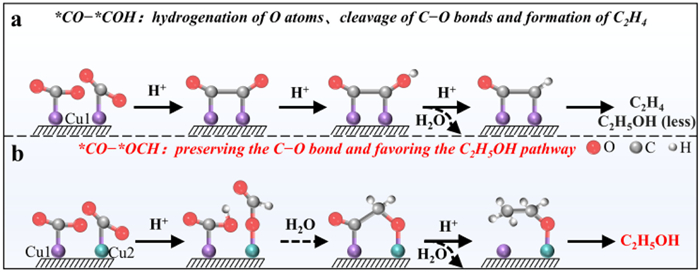
Scheme 1.
Poroposed reaction paths for yielding C2H4 vs. C2H5OH on the surface of (a) symmetric Cu1-Cu1 sites and (b) asymmetric Cu1-Cu2 sites in this work.
Efficient large-current conversion of CO2 to C2H5OH via a *CO-*OCH2 coupling pathway on alkanethiol-modified Cu2O array electrode
Min Zhang , Weimin Wang , Jun Li , Xun Zhu , Qian Fu

Zero-gap membrane electrode assembly electrolyzer significantly reduces the inter-electrode distance and markedly improve energy efficiency, which is conducive to promote industrial electro-synthesis [1-5]. The electrocatalytic reduction of CO2 (CO2RR) to valuable fuels and feedstocks using renewable electricity presents a carbon-neutral pathway for sustainable fuel generation [6]. Among various hydrocarbons and oxygenates, ethanol (C2H5OH) stands out as an important liquid product due to its widespread applications as an organic solvent, disinfectant, and fuel additive [7,8]. Thus, the efficient electrosynthesis of C2H5OH from CO2 holds considerable economic and environmental significance.
Recently, numerous copper-based catalytic sites, including homo-Cu sites and multi-Cu sites structures, have been designed to enhance the CO2RR performance toward C2H5OH [9-11]. However, in most cases, the Faradaic efficiency for C2H5OH (FEC2H5OH) remains lower than that for ethylene (C2H4), another major C2 product. This discrepancy primarily stems from the higher energy barrier associated with C2H5OH formation [12,13]. Specifically, during CO2RR, both C2H5OH and C2H4 share a common key intermediate, *CO–*COH (where * denotes an adsorption site) [14]. It is crucial to note that for C2H5OH to be produced, one of the two C–O bonds in the *CO–*COH intermediate must be preserved [15].
However, during the CO2RR process, the key intermediate *CO–*COH becomes coordinated to two Cu sites via each of its C atoms. This bidentate coordination facilitates the hydrogenation of O atoms, promoting facile cleavage of the C–O bond and leading preferentially to C2H4 formation (Scheme 1a) [16]. Moreover, the adsorbed *CO−*COH species may undergo structural rearrangement, transforming into an enol-like configuration where one of the C atoms originally bound to Cu is replaced by an O atom. Nevertheless, even in this enol-form intermediate, C–O bond rupture remains favorable, resulting again in C2H4 as the dominant product [17]. Consequently, the FEC2H5OH is typically two to three times lower than that for FEC2H4, highlighting the significant challenge in achieving high selectivity of C2H5OH by Cu-based catalysts with homo-sites and multi-sites. Therefore, developing more efficient catalyst architectures and gaining deeper mechanistic insights into the pathways are essential for advancing CO2RR toward practical C2H5OH synthesis.
A promising strategy to suppress C2H4 formation and promote ethanol selectivity involves steering the CO2RR pathway toward distinct mechanistic routes for C2H5OH production [18,19]. This alternative mechanism proceeds through unique C1 and C2 intermediates and involves an unconventional C–C coupling step. By enabling one of the C1 intermediates to coordinate to the metal center via its O atom, rather than the C atom, premature hydrogenation of O can be circumvented, thereby preserving the C–O bond and favoring C2H5OH formation [20]. Theoretical studies have identified several such O-bound intermediates, including *OC, *OCHO, *OCH2OH, and *OCH2 [21,22]. Furthermore, an asymmetric C–C coupling between *OCH2 and another C1 species, *CO, can generate the critical intermediate CO–OCH2 (Scheme 1b), which exhibits a higher propensity toward C2H5OH than C2H4 [23-25]. Consequently, it is imperative that adjacent copper atoms in the catalytic site possess asymmetric electronic or geometric properties to facilitate this distinct reaction pathway.
Cu-based catalysts with strong affinity for O atoms are conducive to the formation of *OCH2 during CO2RR process. In particular, cuprous oxide (Cu2O) has been reported to preferentially stabilize *OCHO with respect to *CO species [26,27], which accounts for its widespread application in the electrosynthesis of HCOOH. Recent studies show that introducing Cu+ vacancies into Cu2O via surface modification can selectively enhance the adsorption and fixation of *CO [28]. These findings indicate that adjacent active sites within copper catalysts, featuring distinct affinities for carbon and oxygen, respectively, can simultaneously promote the generation of *CO and *OCH2 species. This asymmetric environment facilitates the critical C–C coupling between *CO and *OCH2, forming the *CO–*OCH2 intermediate and ultimately promoting the selective production of C2H5OH from CO2, enriching the local reaction concentration at the catalytic surface.
To verify the above hypothesis, the alkanethiols molecule-modified Cu2O nanowire array (OTT-Cu2O) with unprecedented asymmetric Cu sites (namely, a pair of Cu–O and Cu–S sites) was designed to overcome aforementioned limitation, which exhibits an impressive selectivity of CO2RR to C2H5OH reaches up to 45% at 300 mA/cm2, representing a more than two-fold improvement compared to the Cu2O electrode. Mechanistic studies indicate that, in contrast to the Cu–O site which exhibits higher oxygen affinity, the Cu–S site facilitates unique carbon bonding, playing a critical role in stabilizing key intermediates such as *OCH2 and *CO. Consequently, the asymmetric S–Cu–O sites are shown to be thermodynamically more favorable for the asymmetric C–C coupling between *CO and *OCH2, leading to the formation of the critical CO–OCH2 intermediate, compared to symmetric O–Cu–O sites that mainly produce HCOOH. This configuration significantly promotes the selective formation of C2H5OH. This work not only establishes a promising strategy for achieving high selectivity in CO2-to-C2H5OH conversion through the rational design of multi-active-site catalysts, but also provides deeper mechanistic insights into the pathway of CO2 reduction toward C2H5OH.
Copper mesh (CM, 200 mesh) was purchased from anping hangying silkscreen Co., Ltd. Potassium bicarbonate (KHCO3, > 99.5%), potassium hydroxide (KOH, > 90%), hydrochloric acid (HCl, 36%−38%) and anhydrous ethanol (C2H5OH, > 99.7%) were purchased from Chengdu Kelong Chemical Co., Ltd. Phosphoric acid (H3PO4, > 85%) was obtained from Chongqing Chuandong Chemical Co., Ltd. Dimethylsulfoxide (DMSO, 99.8%), deuterium oxide (D2O, 99.9%), and 1-octadecanethiol (C18H38S, 97%) was acquired from Aladdin Co., Ltd. All reagents were used as received without further purification. Deionized water (18.2 MΩ/cm) was generated from an ultra-pure purified water system.
Pretreatment of Cu mesh: The purchased Cu mesh cleaned in a variety of ways. Firstly, Cu mesh was washed with HCl to remove surface impurities. Whereafter, surface electropolish was conducted in a two-electrode system by using 85% H3PO4 solution as electrolyte, Cu mesh as working electrode (2 × 2 cm2), and a platinum plate as counter electrode, respectively. A constant voltage of 4 V was applied to the Cu surface for 75 s. After polishing, the electrode was washed with deionized water several times.
Preparation of Cu2O nanowires (Cu2O electrode): The dense Cu2O nanoneedles layers can synthesize by electrochemical oxidation coupled with heating dehydration strategy. Firstly, the cleaned Cu mesh as anode electrode was immersed in 3 mol/L KOH electrolyte, a platinum plate as counter electrode, and Ag/AgCl as reference electrode, respectively. A constant current of 11 mA/cm2 was applied for 350 s to obtain Cu(OH)2 nanoneedles. Afterward, Cu(OH)2 nanoneedles were dehydrated at 180 ℃ for 1 h, and the Cu2O electrode was prepared.
Preparation of OTT modified Cu2O nanowires (Cu2O-OTT electrode): The hydrophilic Cu2O was immersed in an N2-saturated ethanol solution containing 10 mmol/L of 1-octadecanethiol at 60 ℃ in continuous N2 flow for natural drying, and the Cu2O@OTT electrode was prepared. Subsequently, the Cu2O@OTT precursor was electrochemically converted to OTT-Cu2O by applying a potential of −1.4 V vs. Ag/AgCl for 30 min in a CO2-saturated 0.5 mol/L KHCO3 electrolyte.
X-ray diffraction (XRD) measurements were performed on D8 ADVANCE X-ray diffractometer from Bruker AG at 40 kV and 40 mA using Cu Kα radiation. X-ray photoemission spectroscopy (XPS) data were collected on a ESCALAB250Xi spectrometer equipped with an Al-Kα radiation source (1486.6 eV) using C 1s (284.8 eV) as the reference. Fourier transform infrared spectroscopy (FTIR) measurements were recorded on Nicolet IS 10. Scanning electron microscopy (SEM) images were acquired on GeminiSEM 300 with an accelerating voltage of 3 kV. Transmission electron microscope (TEM) images and EDX mapping images were taken on JEM1200EX with an operated voltage at 200 kV. X-ray absorption fine structure (XAFS) measurement was collected on an Easy XAFS300+ system, and the acquired data were processed via the standard procedure using the ATHENA module of Demeter software packages. The contact angle was measured using a contact angle measuring instrument XC-CAM. The gas-phase product was detected by gas chromatography (GC) from Agilent 8890. The liquid phase product was tested by nuclear magnetic resonance (NMR) from Bruker AVANCE Ⅲ. 600 M. In-situ ATR-SEIRAS spectra were collected by an FT-IR spectrometer (IRTracer100+CHI 660E) equipped with an MCT-A detector at different applied potentials. In-situ Raman spectra obtained by the LabRAM HR Evolution at different applied potentials.
Electrochemical characterization of the prepared electrodes was performed on an electrochemical workstation (VMP3, Biologic) in a two-electrode MEA system (Wuhan Zhisheng New Energy Co., Ltd.), which is comprised of the Cu2O@OTT electrode cathode, an nickel foam anode, and a piece of Nafion 115 membrane. During electrolysis, the CO2-saturated 1 mol/L KHCO3 was directly fed to cathodic compartment at a rate of 100 mL/min, and 1 mol/L KOH was forced to continuously circulate through the anode compartment with a rate of 40 mL/min. Chronopotentiometry measurements were performed in the current density of 50 mA/cm2 to 600 mA/cm2, and full-cell voltages reported were non-iR corrected full-cell potentials.
In general, the thiol end group are among the most stable, suggesting that, even under electrochemical reducing conditions, the ligand could remain chemisorbed to the catalyst surface. To address the simultaneous the requirements, we engineered a hierarchical Cu2O nanowires array with a thiol-modified surface (Fig. 1). Initially, copper hydroxide (Cu(OH)2) nanowires on Cu foam (Cu(OH)2 NWs) were prepared via a straightforward oxidizing process. After annealing under argon condition, these were transformed into Cu2O NWs, subsequently converted into OTT-Cu2O NWs array by simple wet-chemical adsorption combined with electrochemical activation strategies. As shown in Fig. S1 (Supporting information), for Cu2O NWs, SEM images feature its one-dimensional wire with a smooth surface, and the fine wire average diameter circa of 100 nm. Afterward, the resulting Cu2O NWs were immersed in an C2H5OH solution containing 1-octadecanethiol (OTT) for the synthesis of Cu2O@OTT, and then subjected to an electrochemical activation to remove excess weak molecular layer, leading to a strongly bonded OTT molecular surface on the one-dimensional wire (Cu2O-OTT), which feature asymmetric Cu–S–Cu–O sites for significantly enhancing CO2-to-C2H5OH conversion.

The powder X-ray diffraction (XRD) pattern of as-prepared Cu2O electrode exhibited a characteristic peak corresponding to the (111) plane of Cu2O (Fig. 2a), which is more favorable to bond *OCHO species. After OTT modification, no additional diffraction peaks emerged in the XRD pattern, indicating that the crystalline structure of Cu2O remained unaltered. To confirm the existence of the OTT layer, contact angle measurements were conducted and results shown in Fig. 2b, the electrode contact angle to increase dramatically from 6.1° to 130.1° after OTT modification. Moreover, the strong Fourier transform infrared spectroscopy (FTIR) absorption bands at 785,1473 and 2067 cm−1 (Fig. 2c) further suggested that OTT covered the surface of Cu2O electrode. The successful functionalization with the OTT was further corroborated through X-ray photoelectron spectroscopy (XPS) spectroscopy. As shown in Fig. S3 (Supporting information), C, S, O, and Cu elements in Cu2O@OTT electrode. Prior to treatment, Cu2O exhibited characteristic peaks consistent with Cu2O at 932.5 and 952.4 eV, while corresponding to Cu2+ environments at 942.7 and 962.9 eV. After thiolation of the OTT layer, the Cu2+ environment on the surface was removed, leaving Cu-S bonds and forming a Cu+ surface (Fig. S4 in Supporting information). Additionally, a new S 2p peak appeared at 163.0 eV, indicating the presence of Cu-S bonds (Fig. 2d).
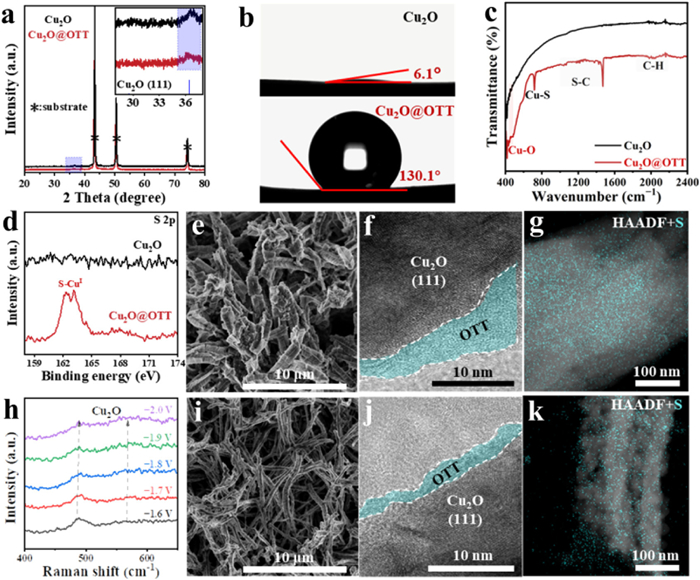
SEM of the Cu2O NWs after treatment confirmed that the nanostructure remained intact (Fig. 2e and Fig. S2 in Supporting information) and was coated with a monolayer between 4 nm and 5 nm in thickness (Fig. 2f). Through HAADF-STEM element-specific signal analysis, S elemental mapping analysis visually demonstrates uniform coverage of OTT on the Cu2O surface (Fig. 2g). After maintaining a reduction potential in CO2-saturated 0.5 mol/L KHCO3 electrolyte for over 30 min, the activation of the Cu2O@OTT electrode was converted into a Cu2O-OTT electrode. Meanwhile, the presence of Cu2O species are confirmed by in-situ Raman spectra during electrolysis (Fig. 2h and Fig. S6 in Supporting information). SEM characterization confirmed that the activated Cu2O-OTT electrode retained its integrity (Fig. 2i and Fig. S5 in Supporting information), the Cu2O (111) crystal plane still presented, and the Cu2O surface still retains an approximately 2 nm-thick OTT layer (Fig. 2j), consisting with a surface of 1-octadecanethiol molecules bound upright (the chain length is 2.3 nm between the surface-bound S and terminal C). HAADF-STEM elemental analysis revealed that although the OTT coverage decreased after electrochemical activation, it maintained uniform distribution (Fig. 2k), which indicates selective peeling of partial OTT layers during potential application. Preliminary electrochemical characterization demonstrates that the ECSA decreases after OTT coating, the active area of Cu2O-OTT electrodes (1.98 mF/cm2) is smaller than that of Cu2O electrodes (6.26 mF/cm2) (Fig. S7c in Supporting information), which stems from reduced electrolyte contact area due to hydrophobic surface properties.
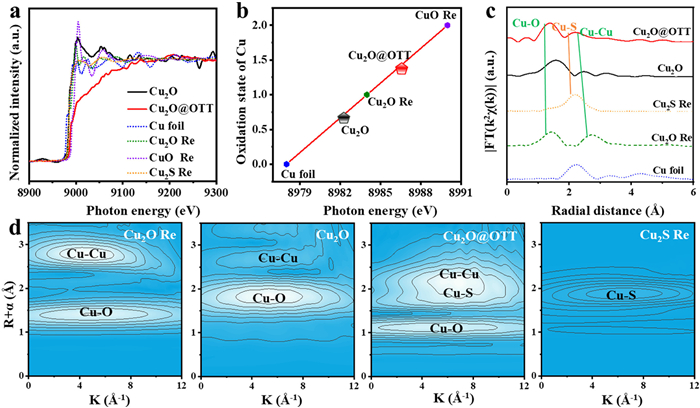
X-ray absorption spectroscopy (XAS) was employed to characterize the chemical states and local fine structures of the Cu in Cu2O before and after OTT modification. The X-ray absorption near-edge structure (XANES) spectra in Fig. 3a reveal that the absorption edge positions of Cu2O-OTT shifting to the higher energy side with respect to Cu2O, which indicates that Cu2O-OTT had a different coordination environment from that of Cu2O. As shown in Fig. 3b, the valence of Cu2O lies between the characteristic values of Cu2O Re and Cu Re, whereas the Cu2O-OTT is sized of a valence between Cu2O Re and standard CuO Re, implying that the oxidation state of Cu in Cu2O increase after the modification of OTT coating layers. The Fourier transform extended k-edge X-ray absorption fine structure (EXAFS) spectra in Fig. 3c reveals that Cu2O-OTT consisted of Cu–O, Cu–S, and Cu–Cu bonds with simulated coordination numbers of 1.13, 1.85, and 2.43 Å, respectively. While the fitting results of Cu2O show that coordination numbers of Cu-O and Cu-Cu bonds are 1.75 and 2.61 Å, suggesting the formation of many unsaturated sites in Cu2O after OTT modification. Wavelet transform extended XAFS (WT-EXAFS) spectra was conducted to further highlight the local structural changes in Cu2O (Fig. 3d). The reduced backscattering amplitude at Cu–O in Cu2O-OTT compared with Cu2O, indicating the lower coordination state of Cu2O-OTT. As a result of OTT modification, unprecedented asymmetric Cu sites (namely, a pair of Cu–O and Cu–S sites) was successfully created for the further selective reduction of CO2.
To evaluate the CO2RR activity of the OTT-modified Cu2O electrode, the electrocatalytic CO2RR performance was measured in a zero-distance electrolyzer with a CO2-saturated 0.5 mol/L KHCO3 electrolyte at applied current density from 50 mA/cm2 to 500 mA/cm2 (Figs. S8 and S9 in Supporting information). The products were characterized by nuclear magnetic resonance (NMR) and gas chromatography (GC), including CO, HCOO-, CH4, and C2+ products (C2H4, C2H5OH, CH3COOH and C3H7OH) (Fig. S10 in Supporting information). Fig. 4a shows the LSV of the hydrophobic Cu2O@OTT electrode and equivalent wettable Cu2O electrode in a CO2-saturated KHCO3 electrolyte (0.5 mol/L, pH 6.8). To achieve a current density of −300 mA/cm2, the Cu2O electrode required a voltage of 2.78 V, whereas the Cu2O-OTT electrode required a significantly higher voltage of 3.15 V. This reduction in current density at a given voltage is partially attributable to the lower ECSA of the hydrophobic electrode.
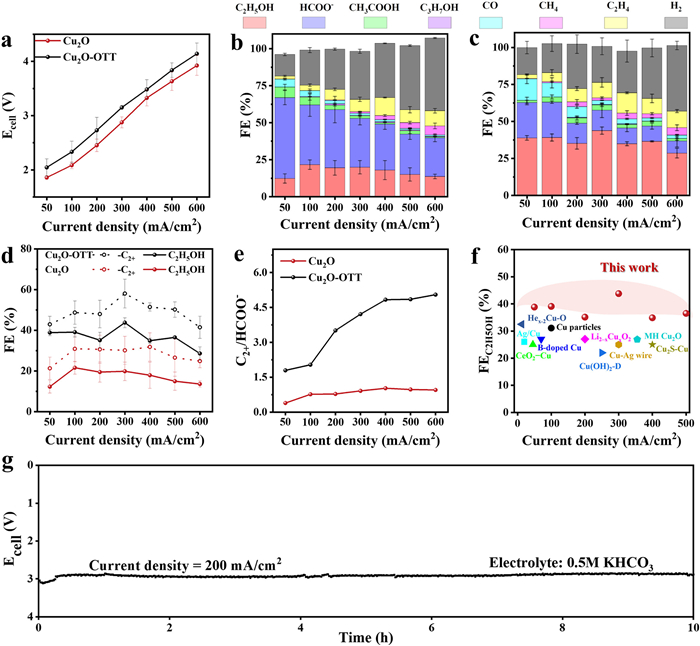
The distribution of gases and liquid products generated are presented in Fig. 4 and c. For Cu2O electrode, HCOO− is the primary product along with little C2H5OH. As the current density becomes large, C2H4 and CH4 products begin to appear, while the maximum FEC2H5OH reached only 18% at 100 mA/cm2 (Fig. 4b). However, C2H5OH is the primary product for Cu2O-OTT electrode, reaching a peak Faradaic efficiency of 45% at 300 mA/cm2 (Fig. 4c). It is worth noting that the selectivity of H2 over Cu2O electrode does not change significantly after OTT modification. To visually represent the influence of OTT on the reduction products, the selectivity data is re-plotted in Fig. 4d. The FEC2+ of the CuO-OTT electrode is 60%, contrasting with 30% for the Cu2O electrode, with C2H5OH comprising the majority of C2+ products, showcasing an enhancement of more than 2-fold. Besides, the ratio of C2+/HCOO− is 4.8 for CuO-OTT at 600 mA/cm2, which is much higher than that of Cu2O (Fig. 4e). Furthermore, energy efficiency of C2H5OH for Cu2O-OTT is also significantly higher with respect to Cu2O (Fig. S11 in Supporting information). Notably, to the best of our knowledge, the outstanding CO2RR performance of Cu2O-OTT for C2H5OH production is record high in comparison with the previously reported (Fig. 4f and Table S1 in Supporting information).
As shown in Fig. 4g, the Cu2O-OTT electrode exhibited a relatively stable bias under a constant current density of 200 mA/cm2, with a negligible performance degradation within 10 h, evidencing the excellent stability of Cu2O-OTT electrode under CO2RR operating conditions. In addition, SEM image illustrates that the nanowire structure is maintained, XRD analysis confirmed retention of the Cu2O (111) crystal plane in the Cu2O-OTT electrode, while XPS detected persistent S species. Contact angle measurements further demonstrated maintained hydrophobicity, collectively illustrating excellent stability and activity (Fig. S12 in Supporting information).
Typical Cu-based electrocatalysts cannot achieve high FEC2H5OH due to the competitive thermodynamics of the products of C2H4. To further investigate the possible reaction pathways and intermediates of the CO2RR to C2H5OH on the Cu2O-OTT surface, in situ spectroscopy are performed in CO2-saturated 0.5 mol/L KHCO3 electrolyte (Fig. 5a, Figs. S13 and S14 in Supporting information). In our observations, we noted five distinct infrared signals at 1120, 1226, 1473, 1583, and 1646 cm−1 for Cu2O-OTT during continuous operation (Fig. 5b). The first peak signal at 1120 cm−1 corresponds to the key intermediate *OC2H5 involved in the conversion of CO2 to C2H5OH [29]. The simultaneous observation of C–C (1226 cm−1) and C–O (1253 cm−1) vibrational peak signals further verified the formation of the C2H5OH [30]. It is worth noting that the characteristic peak signal at 1583 cm−1 can be attributed to the C2H5OH key coupling *CO−*OCH2 intermediate over the asymmetric S-Cu-O sites, which is generated by C–C coupling through *OCH and *CO [31]. By contrast, the main characteristic peak signals of the Cu2O appear at the *OCHO intermediate displayed at 1450 cm−1 and the *H2O around 1650 cm−1 (Fig. 5c).

In Raman spectra (Fig. 5d), two regions appearing at about 283 and 374 cm−1 for Cu2O-OTT are associated with the surface-absorbed *CO, belonging to the Cu–CO frustrated rotation and Cu–CO stretch, respectively [10,13,15]. At the same time, two extra bands appearing at 1435 and 1580 cm−1 for the Cu2O-OTT were also noticed at all testing potentials. The former is tentatively assigned to the symmetric stretching vibration of the carboxylate group (namely, v–O–(CH)–O), corresponding to the surface-adsorbed *OCHO intermediates [32]. While the band at 1580 cm−1 is assigned to the C═O stretching mode of surface-adsorbed *CHxCO species [33], a pivotal intermediate responsible for the yielding of C2H5OH, which maybe originate from the asymmetric C–C coupling between the *CO and *OCHO species on Cu2O-OTT with Cu–O and Cu–S pairs. In comparison, only a peak at 1435 cm−1 corresponding to *OCHO emerged in the Raman spectra of Cu2O (Fig. 5e).
Additionally, there appear two peaks at 1923 and 2044 cm−1 for Cu2O and Cu2O-OTT, associating with the bridge–adsorbed *CO (*CObridge) and atop–adsorbed *CO (*COatop), respectively. In general, *CObridge protonation to *OCH2 is proved to be more energetically favorable than that of *COatop [34-36], and the ratio *CObridge/*COatop for Cu2O@OTT is significantly greater than that for Cu2O over the potential range (Fig. 5f), which could be in favor of C2H5OH generated from CO2 (Fig. S16 in Supporting information).
Based on the above results, we proposed a comprehensive reaction mechanism, as outlined in Fig. 5g. The Cu2O primarily follows the pathway of CO2-to-HCOOH to generate HCOO− due to its inherent crystal characteristics (Fig. S15 in Supporting information). OTT layer not only creates a favorable microenvironment for CO2/CO enrichment, but also allow the formation of S–Cu–O asymmetric site in Cu2O-OTT electrode, which induces more *CO absorbed on the catalyst surface. Afterward, the coupling of *CO and *OCHO intermediates via asymmetric active sites of Cu–S and Cu–O enables significant enhancement of C2H5OH selectivity through synergistic dual-sites pathway. Additionally, the OTT layer enriches CO, allowing C–C coupling between different adsorbed CO states to produce C2H5OH. These combined mechanisms achieve the switch of products from HCOO− to C2H5OH, and surface modification engineering strategy provides a novel perspective for designing efficient CO2RR electrocatalysts.
In summary, this work demonstrates the rational design of an alkanethiol-modified Cu2O nanowire array electrocatalyst featuring asymmetric Cu–O/Cu–S dual sites for significantly enhancing CO2-to-C2H5OH conversion. The catalyst exhibits a notable C2H5OH Faradaic efficiency of 45% at 300 mA/cm2, representing a more than two-fold improvement over bare Cu2O. Mechanistic studies reveal that the O–Cu–S site facilitates critical carbon-bonding interactions that stabilize key *CO and *OCH2 intermediates, complementing the oxygen-affinitive nature of the Cu–O site. This asymmetric configuration enables thermodynamically favored C–C coupling between *CO and *OCH2 via the asymmetric S–Cu–O motif, leading to the formation of the CO–OCH2 intermediate and selectively promoting C2H5OH production, in contrast to symmetric O–Cu–O sites that favor HCOOH formation. Our findings provide both a promising catalyst design strategy for multi-site synergistic CO2RR and fundamental insights into the mechanism of asymmetric C–C coupling toward C2H5OH.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Min Zhang: Writing – review & editing, Writing – original draft, Visualization, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Weimin Wang: Visualization, Data curation. Jun Li: Supervision, Resources. Xun Zhu: Supervision, Resources, Funding acquisition. Qian Fu: Writing – review & editing, Supervision, Resources, Funding acquisition.
The authors are grateful for the financial supports of the National Natural Science Foundation of China (NSFC, Nos. 52394202, 52476056, and 52301232) and the Natural Science Foundation of Chongqing Province (No. 2024NSCQ-MSX1109).
Supplementary material associated with this article can be found, in the online version, at doi:
M. Zhang, A. Cao, Y. Xiang, et al., Nano-Micro Lett. 16 (2024) 50. doi: 10.1007/s40820-023-01264-6
M. Zhang, W. Wei, S. Zhou, et al., Energy Environ. Sci. 14 (2021) 4998–5008. doi: 10.1039/D1EE01495A
M. Zhang, X. Wang, J. Ding, et al., Nanoscale 16 (2024) 7076–7084. doi: 10.1039/D4NR00361F
M. Zhang, S. Zhou, W. Wei, et al., Chem. Catal. 2 (2022) 3528–3545.
M. Zhang, Z. Tan, M. Xing, et al., J. Energy Chem. 112 (2026) 90–96. doi: 10.1016/j.jechem.2025.08.050
X. Wang, Z. Wang, F.P. García de Arquer, et al., Nat. Energy 5 (2020) 478–486. doi: 10.1038/s41560-020-0607-8
Z.H. Zhao, J.R. Huang, P.Q. Liao, et al., J. Am. Chem. Soc. 145 (2023) 26783–26790. doi: 10.1021/jacs.3c08974
J. Bi, P. Li, J. Liu, et al., Nat. Commun. 14 (2023) 2823. doi: 10.1038/s41467-023-38524-3
Z. Chen, Z. Ma, G. Fan, et al., ACS Appl. Mater. Interfaces 16 (2024) 35143–35154. doi: 10.1021/acsami.4c05973
G. Feng, S. Wang, S. Li, et al., Angew. Chem. Inter. Ed. 62 (2023) e202218664. doi: 10.1002/anie.202218664
A. Herzog, M.L. Luna, H.S. Jeon, et al., Nat. Commun. 15 (2024) 3986. doi: 10.1038/s41467-024-48052-3
Y. Lin, T. Wang, L. Zhang, et al., Nat. Commun. 14 (2023) 3575. doi: 10.1038/s41467-023-39351-2
P. Luan, X. Dong, L. Liu, et al., ACS Catal. 14 (2024) 8776–8785. doi: 10.1021/acscatal.4c01579
C. Peng, J. Ma, G. Luo, et al., Angew. Chem. Int. Ed. 63 (2024) e202316907. doi: 10.1002/anie.202316907
D. Wakerley, S. Lamaison, F. Ozanam, et al., Nat. Mater. 18 (2019) 1222–1227. doi: 10.1038/s41563-019-0445-x
J. Wang, H. Yang, Q. Liu, et al., ACS Energy Lett. 6 (2021) 437–444. doi: 10.1021/acsenergylett.0c02364
P. Wang, H. Yang, C. Tang, et al., Nat. Commun. 13 (2022) 3754. doi: 10.1038/s41467-022-31427-9
X. Wang, Z. Jiang, P. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202313646. doi: 10.1002/anie.202313646
A. Xu, S.F. Hung, A. Cao, et al., Nat. Catal. 5 (2022) 1081–1088. doi: 10.1038/s41929-022-00880-6
F. Xu, B. Feng, Z. Shen, et al., J. Am. Chem. Soc. 146 (2024) 9365–9374. doi: 10.1021/jacs.4c01610
H. Zhang, Y. Sun, J. Wang, et al., ACS Appl. Energy Mater. 6 (2023) 11448–11457. doi: 10.1021/acsaem.3c01463
T. Zhang, S. Xu, D.L. Chen, et al., Angew. Chem. Int. Ed. 63 (2024) e202407748. doi: 10.1002/anie.202407748
Y. Zhang, Y. Chen, X. Wang, et al., Nat. Commun. 15 (2024) 5172. doi: 10.1038/s41467-024-49247-4
J. Zhou, Q. Liang, P. Huang, et al., Nanoscale 16 (2024) 13011–13018. doi: 10.1039/D4NR01173B
J. Li, R. Cai, H. Mu, et al., ACS Catal. 14 (2024) 3266–3277. doi: 10.1021/acscatal.3c05857
X. Ma, Y. Zhang, T. Fan, et al., Adv. Funct. Mater. 33 (2023) 2213145. doi: 10.1002/adfm.202213145
D. Tan, B. Wulan, J. Ma, et al., Nano Lett. 22 (2022) 6298–6305. doi: 10.1021/acs.nanolett.2c01942
S. Li, H. Duan, J. Yu, et al., ACS Catal. 12 (2022) 9074–9082. doi: 10.1021/acscatal.2c01750
D. Wakerley, S. Lamaison, J. Wicks, et al., Nat. Energy 7 (2022) 130–143. doi: 10.1038/s41560-021-00973-9
X. Kong, J. Zhao, J. Ke, et al., Nano Lett. 22 (2022) 3801–3808. doi: 10.1021/acs.nanolett.2c00945
Y.C. Li, Z. Wang, T. Yuan, et al., J. Am. Chem. Soc. 141 (2019) 8584–8591. doi: 10.1021/jacs.9b02945
H. Li, P. Wei, D. Gao, et al., Curr. Opin. Green Sustain. Chem. 34 (2022) 100589. doi: 10.1016/j.cogsc.2022.100589
F. Li, Z. Men, S. Li, et al., Spectrochim. Acta A 189 (2018) 621–624. doi: 10.1016/j.saa.2017.08.077
S. Jiang, K. Klingan, C. Pasquini, et al., J. Chem. Phys. 150 (2019) 041718. doi: 10.1063/1.5054109
H.H. Heenen, H. Shin, G. Kastlunger, et al., Energy Environ. Sci. 15 (2022) 3978–3990. doi: 10.1039/D2EE01485H
D. Karapinar, C.E. Creissen, J.G. Rivera de la Cruz, et al., ACS Energy Lett. 6 (2021) 694–706. doi: 10.1021/acsenergylett.0c02610

Scheme 1 Poroposed reaction paths for yielding C2H4 vs. C2H5OH on the surface of (a) symmetric Cu1-Cu1 sites and (b) asymmetric Cu1-Cu2 sites in this work.

Figure 2 Composition and structure characterizations: (a) XRD pattern, (b) the contact angle, (c) FTIR spectra, (d) S 2p XPS spectra of Cu2O and Cu2O@OTT. (e) SEM image, (f) TEM image, (g) HAADF-STEM image of Cu2O@OTT. (h) In-situ Raman spectra of Cu2O@OTT at electrolysis from −1.6 V to −2.0 V vs. Ag/AgCl in CO2-saturated 0.5 mol/L KHCO3. (i) SEM image, (j) TEM image, (k) HAADF-STEM image of Cu2O-OTT. Equivalent images from i-to-k after electrolysis at –1.4 V vs. Ag/AgCl in CO2-saturated 0.5 mol/L KHCO3 for 30 min.

Figure 3 XAS characterizations: (a) Normalized Cu K-edge XANES spectra of Cu2O and Cu2O@OTT with the references. (b) the mean chemical valence analysis of Cu species. (c) FT-EXAFS spectra of Cu2O and Cu2O@OTT with the references. (d) wavelet transform for the Cu K-edge EXAFS spectra of Cu2O Re, Cu2O, Cu2O@OTT and Cu2S Re.

Figure 4 CO2RR performance in a MEA electrolyzer: (a) I-V curves of Cu2O and Cu2O-OTT; FE of products of the (b) Cu2O and (c) Cu2O-OTT. (d) FE of C2+ and C2H5OH. (e) Ratios of C2+ to-HCOO- for Cu2O and Cu2O-OTT. (f) Performance comparison of Cu-based catalysts for C2H5OH production by CO2RR pathway. (g) Stability test of Cu2O-OTT in 0.5 mol/L KHCO3 electrolyte.

Figure 5 Mechanistic investigation: (a) The schematic illustration of in-situ FTIR and Raman electrolytic cell. (b, c) In-situ FTIR spectra at different potentials vs. Ag/AgCl. (d, e) In-situ Raman spectra at different potentials vs. Ag/AgCl. (f) The ratio of *CObridge to *COatop obtained from Raman spectra. (g) Schematic illustration for the catalytic mechanism for CO2RR to C2H5OH on Cu2O-OTT.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: