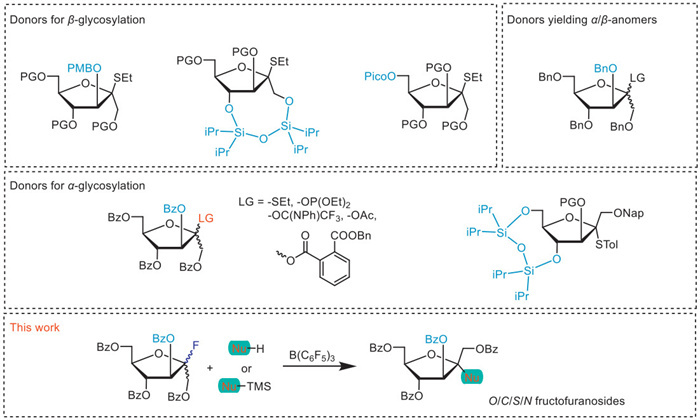
Scheme 1.
Glycosyl donors developed in previous studies and the present study for constructing fructofuranosidic bonds.
Synthesis of α-d-fructofuranosides and difructose dianhydride Ⅳ using glycosyl fluorides as donors
Yidian Mo , Ao Sun , Hang Dong , Zhongtang Li , Zhongjun Li

Fructofuranosides are widely distributed in nature and they play crucial roles in biological processes. These compounds are found in plants and bacteria, as constituents of sucrose, β-(2→1)-linked inulins, β-(2→6)-linked levans, and α- or β-fructofuranosides [1-3]. In addition to their biological functions, such as energy storage and structural roles in cells, fructofuranosides also exhibit pharmacological properties, including antiviral, antioxidant, antitumor, and anti-inflammatory activities [4,5]. Compared with enzymatic synthesis, chemical glycosylation methods offer easier product isolation, broader substrate compatibility, and greater suitability for synthesizing structurally defined fructosides and derivatives. Despite their importance, the chemical synthesis of fructofuranosides, especially with high stereoselectivity and yield, remains a significant challenge in carbohydrate chemistry.
One major obstacle to synthesizing fructofuranosides is the difficulty in controlling the stereochemistry during the glycosylation process. Employing donors with nonparticipating groups like benzyl often results in a mixture of α- and β-anomers, complicating their separation. Several methods have been developed for synthesizing β-d-fructofuranosides. The internal aglycone delivery (IAD) method developed by Oscarson and colleagues involves the use of thiofructoside donors and is notable for its β-stereoselectivity (Scheme 1). However, the intermediate acetal formed by the protocol is unstable, and this strategy fails in the synthesis of complex bacterial oligosaccharides [6,7]. The other strategy uses conformationally restricted donors that possess a siloxane spanning O1 and O4 in fructofuranose rings to block α-face attack and promote the formation of β-d-fructofuranosides. However, the drawback of this method is the high cost and complexity involved in preparing the fructosyl donors, limiting its broader application in synthetic chemistry [8-10]. Our previous work developed an elegant hydrogen-bond-mediated aglycone delivery (HAD) method for constructing β-d-fructofuranosidic linkages with 6-O-picoloyl-protected fructofuranosyl thioglycoside as the glycosyl donor [11]. On the other hand, α-selective fructosylation could be achieved through neighboring group participation by an acyl group at C3. Thio-fructofuranosides [6], fructofuranosyl phosphites [12], acetates [13], and trifluoroacetimidates [14] have been employed as donors in the synthesis of α-d-fructofuranosides. Currently, there are limited options for leaving groups available for fructose glycosylation, especially for application in orthogonal glycosylation strategies. Moreover, we were surprised to find that fructosyl fluorides have not been explored, despite their ease of synthesis and relative stability.
Glycosyl fluorides are widely and effectively utilized in both O- and C-glycosylation reactions. One key advantage of glycosyl fluorides as glycosyl donors is their remarkably high thermal and chemical stability. With these favorable synthetic properties, glycosyl fluorides were first developed in practical glycosylation by Mukaiyama and co-workers in 1981 with a fluorophilic activator, SnCl2-AgClO4 [15]. Since their significant development, numerous effective chemical methods for O-glycosylations and C-glycosylations using glycosyl fluorides have been established. Different reagents and cooperative systems for the activation of glycosyl fluorides as glycosyl donors including tin-based activation systems (SnCl2 or SnCl4 in combination with AgOTf or AgClO4 or TrClO4 or AgB(C6F5)3, SnF4) [15-20], group IVB metal-based activators (TiF4, Cp2MCl2 in combination with AgOTf or AgClO4, M = Ti or Zr or Hf) [21,22] and other Lewis acids (SiF4, TMSOTf, BF3·OEt2, TrB(C6F5)4, HB(C6F5)4, B(C6F5)3, LiClO4, Tf2O, Ca(OTf)2, Cu(OTf)2, Yb(OTf)3, La(ClO4)3·nH2O) have been developed [23-36], providing a diverse and versatile synthetic toolbox for oligosaccharide synthesis, particularly for the development of orthogonal glycosylation strategies. Furthermore, glycosyl fluorides exhibit versatile synthetic accessibility as they can be readily synthesized from diverse precursors including anomeric acetates, glycosyl hemiacetals, and thioglycosides, and are efficiently interconvertible with thioglycosides. Notably, this reactivity has enabled the development of a novel orthogonal activation strategy in oligosaccharide synthesis based on the independent activation of glycosyl fluoride donors and thioglycoside acceptors under distinct reaction conditions [37]. Since no applications of glycosyl fluoride donors in the synthesis of fructofuranosides have been reported, in pursuit of advancing glycosylation methodologies, herein we synthesized a fructofuranosyl fluoride donor and investigated its reactivity in corresponding glycosylation processes. Additionally, this work explored the application potential of fructofuranosyl fluoride in the orthogonal synthesis of fructooligosaccharides, offering a novel approach for the synthesis of complex oligosaccharides.
The fructofuranosyl fluoride donor 2 was prepared by direct fluorination of fructofuranosyl hydroxy sugar 1 in a satisfactory yield of 98% (Scheme S1 in Supporting information), which maintained stability at room temperature. Separately, the armed donor 3 was prepared from thio-fructofuranosides by treatment with NIS/DAST in 88% yield (Scheme S2 in Supporting information).
Under different conditions for the activation of glycosyl fluoride, the glycosylation of donor 2 (1.2 equiv.) was conducted with 2-thiopheneethanol 4a (1.0 equiv.) in the presence of 4 Å molecular sieves (MS) at room temperature in anhydrous CH2Cl2 (Table 1). Systematic screening of Lewis acids revealed reactivity trends. Strikingly, Ca(OTf)2, Cu(OTf)2, and camphorsulfonic acid (CSA) failed to activate the fluoride donor (Table 1, entry 1–3). BF3·OEt2 exhibited partial activation to yield glycoside products, along with significant leftover of donor and acceptor (entry 5). The stronger acid BBr3 caused donor degradation without glycoside formation (entry 4). In comparison, B(C6F5)3, with intermediate acidity, enabled efficient donor activation and quantitative coupling with the acceptor. Additionally, activation systems including SnCl4, Cp2ZrCl2 in combination with AgOTf, and SnCl2 in combination with AgOTf have also demonstrated remarkable efficiency and stereospecificity in α-glycoside formation (entries 6–10). In contrast, the reaction of armed donor 3 resulted in the formation of inseparable α/β-anomeric glycosides, which were identified by NMR analysis to have an α/β ratio of 1.5/1. This outcome suggests that the α-stereoselectivity in donor 2 was likely controlled by the neighboring group participation of the C-3 benzoyl group.
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | Donor | Activator (equiv.) | Yield (%)b | Ratio of α/βc |
| 1 | 2 | Cu(OTf)2 (1.2) | NR | – |
| 2 | 2 | Ca(OTf)2 (1.2) | NR | – |
| 3 | 2 | CSA (1.2) | NR | – |
| 4 | 2 | BBr3 (1.2) | 0 | – |
| 5 | 2 | BF3·OEt2 (1.2) | 27 | α only |
| 6 | 2 | SnCl4 (1.2) | 97 | α only |
| 7 | 2 | B(C6F5)3 (0.4) | 95 | α only |
| 8 | 2 | Cp2ZrCl2/AgOTf (0.6/1.2) | 94 | α only |
| 9 | 2 | SnCl2/AgOTf (0.6/0.6) | 93 | α only |
| 10 | 3 | B(C6F5)3 (0.4) | 93 | 1.5/1 |
| a Glycosylations were conducted with fructofuranosyl fluoride donor 2 or 3 (1.2 equiv.) in anhydrous CH2Cl2 (0.025 mol/L, calculated based on donor) in the presence of 4 Å MS (80 mg/mL, calculated based on solvent) at room temperature for 3 h. b Isolated yield. NR = No reaction. c The α/β ratio was determined by NMR analysis. |
||||
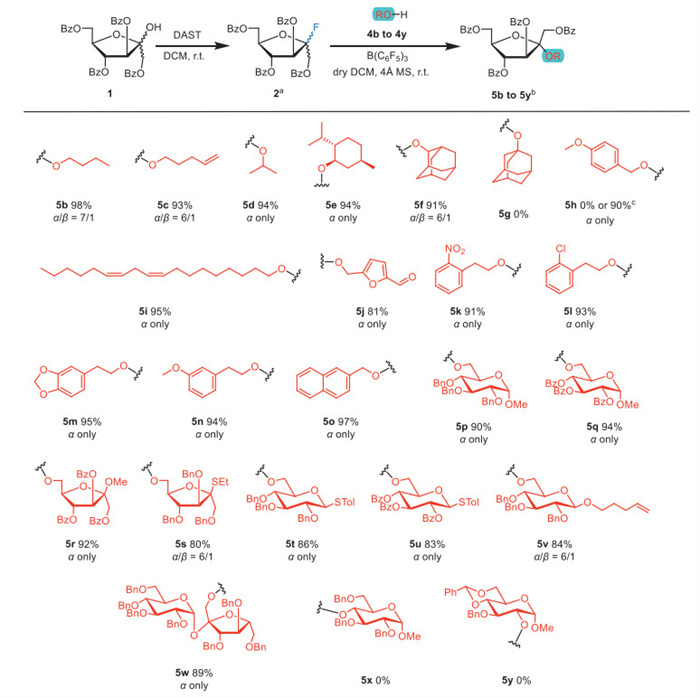
Subsequently, we investigated the O-glycosylation scope of donor 2 (1.2 equiv.) with various acceptors (4b–4y) under the optimal conditions (Scheme 2, donor: 1.2 equiv., acceptor: 1.0 equiv., CH2Cl2: 0.025 mol/L calculated based on donor, 4 Å MS: 80 mg/mL calculated based on solvent, r.t.). Except for the acid-sensitive PMB group, which would be cleaved under SnCl4 or B(C6F5)3 conditions and required switching to milder Cp2ZrCl2/AgOTf activation, all three activation systems afforded α-fructofuranosides with good stereoselectivity (α/β ratio ≥ 6/1) and high yields (80%–98%) in glycosylation of donor 2 with primary alcohols. For the reactions with thioglycoside acceptors, the glycosylation process between donor 2 and acceptor 4s–4u produced α-fructofuranosyl disaccharides 5s–5u with yields ranging from 80% to 86%, demonstrating the orthogonal reactivity of thioglycosides and fluorides and their potential utility in complex oligosaccharide synthesis. Notably, glycosylation reactions involving secondary hydroxyl–containing glycosyl acceptors or tertiary hydroxyl acceptors failed to yield detectable products, an outcome that may be attributed to steric hindrance effects at these nucleophilic oxygen centers, consistent with prior findings [11].
The biosynthesis of amino sugars proceeds through the hexosamine biosynthetic pathway (HBP), with the rate-limiting step being the conversion of fructose-6-phosphate to glucosamine-6-phosphate. Therefore, fructose derivatives hold promise as molecular probes for investigating glycobiological processes and developing novel enzymatic inhibitors. Furthermore, C-, S-, and N-glycosides exhibit unique characteristics compared to O-glycosides, demonstrating resistance to enzymatic hydrolysis [38]. However, as a crucial metabolic molecule, fructose has rarely been reported in the synthesis of its C- and N-glycosides. Thus, we attempted to broaden the structural diversity of fructose derivatives by coupling fructofuranosyl fluoride donor 2 with common nucleophilic reagents 7a–7g (Scheme 3).
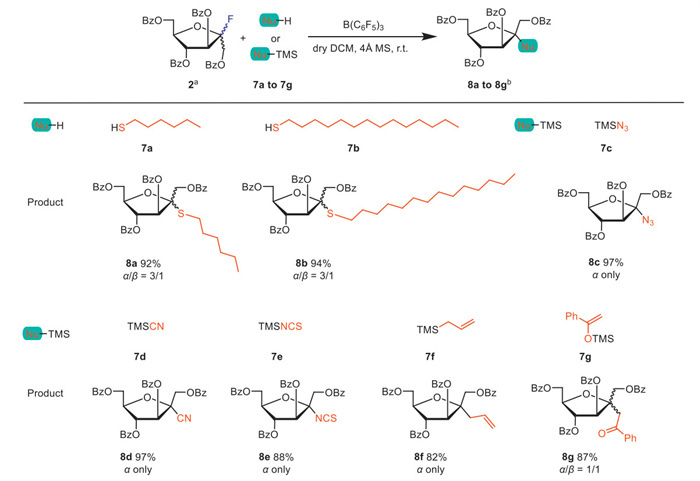
Under standard glycosylation conditions, donor 2 was coupled with thiol 7a or 7b, resulting in over 90% yield of thio-fructorufanoside 8a or 8b with a ratio of α/β at 3/1. Donor 2 reacted with nitro or carbo nucleophilic reagents 7c–7g to afford the corresponding glycoside derivatives 8c–8g in high yields (82%–97%), with 8c–8f exhibiting complete α-stereoselectivity. The results demonstrate the significant potential of fructosyl fluoride in the synthesis of fructose derivatives.
Di-d-fructose-2,6′:6,2′-dianhydride, also known as difructose anhydride Ⅳ (DFA Ⅳ), is a centrally symmetric cyclic disaccharide composed of two fructose units linked by β-glycosidic bonds. Similar to cyclodextrins and inulin-type cyclofructans [39,40], DFA Ⅳ is derived from the conversion of levan by levan fructotransferase (LFTase) [41] and is rarely found in nature, making it a promising low-calorie sugar substitute. Several beneficial effects associated with the consumption of DFA Ⅳ have been reported [42,43]. To date, the preparation of DFA Ⅳ has been achieved solely through enzymatic conversion using LFTase [44-47], whereas there are no known reports of its chemical synthesis, which may be attributed to the lack of orthogonal glycosylation methods for fructose. To better serve human health, basic research and the practical applications of DFA Ⅳ deserve attention.
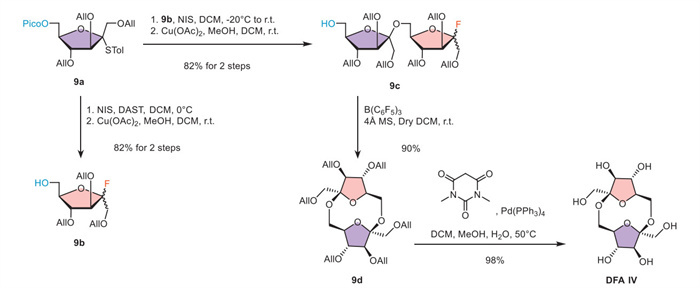
Given the intriguing structural properties of DFA Ⅳ, its chemical synthesis inherently demands the implementation of two orthogonal glycosylation strategies, exemplified by thioglycoside and glycosyl fluoride methodologies, to address the unique challenges posed by its molecular architecture. The HAD methodology enables the stereoselective construction of β-glycosidic bonds, while the facile interconversion and orthogonal reactivity between thioglycosides and glycosyl fluorides strategically define the reactive sites for subsequent cycloglycosylation. As shown in Scheme 4, thioglycoside donor 9a was first converted to glycosyl fluoride using DAST, followed by deprotection of the picoloyl group via Cu(OAc)2 to afford glycosyl acceptor 9b. The initial β-glycosidic linkage was constructed via the HAD method employing thioglycoside donor 9a. Subsequently, glycosyl fluoride 9c was subjected to cycloglycosylation under high dilution conditions (2 × 10–3 mol/L), consistent with the concentrations required in other reports on cyclic sugar synthesis [48,49]. The reaction afforded DFA Ⅳ as allylated derivative 9d in 90% yield with complete β-selectivity, presumably due to the symmetric nature of the intermediate. Finally, deprotection of the allyl group via Pd(PPh3)4 afforded the target molecule DFA Ⅳ in 98% yield.
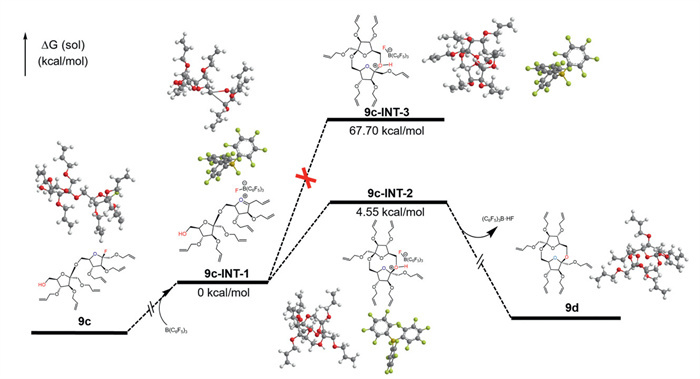
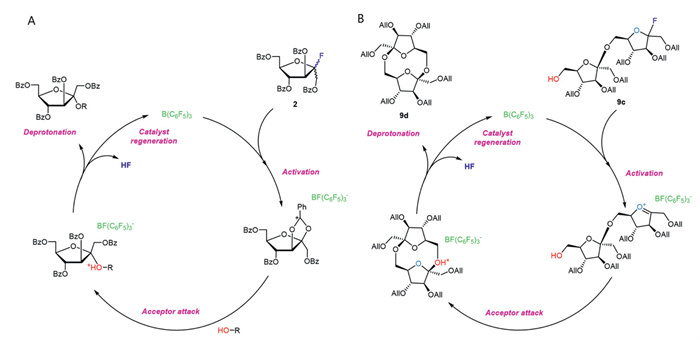
We further explored the reaction mechanism of cycloglycosylation by density functional theory (DFT) calculations and evaluated the configurational stereoselectivity of the reaction. As shown in Fig. 1, the donor 9c undergoes C–F bond cleavage under the activation of B(C6F5)3, forming an oxonium ion intermediate 9c-INT-1, with its ΔG set to 0 kcal/mol. Subsequently, the hydroxyl group of the acceptor attacks the anomeric carbon of the donor from the α-side to form intermediate 9c-INT-2, or from the β-side to form intermediate 9c-INT-3. However, due to the higher ΔG required for the formation of 9c-INT-3 (67.70 kcal/mol) compared to 9c-INT-2 (4.55 kcal/mol), this intermediate is difficult to generate. As a result, only the symmetric β,β-product 9d is formed via 9c-INT-2, and the computational results align with experimental observations. Scheme 5 shows the proposed reaction mechanism of cycloglycosylation with 9c. Tris(pentafluorophenyl)borane activates the fluoride donor, breaking the C–F bond and forming an oxonium ion intermediate. The hydroxyl group of the acceptor attacks from the α-side because of the lower energy barrier for forming the symmetric β,β-cyclodisaccharide. After proton dissociation, DFA Ⅳ is formed with complete β-stereoselectivity.
In summary, we developed fructofuranosyl fluorides as donors for synthesizing α-d-fructofuranosides. Promoted by tris(pentafluorophenyl)borane, this method proved highly effective, yielding the glycosylation products in almost quantitative yields with primary alcohols, thiols and N- and C-nucleophiles. These fructofuranosyl derivatives represent new structural types for bioactive molecules or molecular probes for investigating HBP processes. Furthermore, the fluoride donor can undergo facile interconversion with thioglycoside and enable orthogonal glycosylation, based on which we achieved the first chemical synthesis of DFA Ⅳ through orthogonal glycosylation reactions involving thioglycosides and glycosyl fluorides.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yidian Mo: Writing – review & editing, Writing – original draft, Visualization, Validation, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Ao Sun: Writing – review & editing, Methodology, Investigation. Hang Dong: Writing – review & editing, Investigation. Zhongtang Li: Writing – review & editing, Methodology, Investigation, Funding acquisition, Formal analysis. Zhongjun Li: Writing – review & editing, Project administration, Funding acquisition.
This work was supported by the National Key R&D Program of China (Nos. 2022YFC2303700, 2022YFF1203005), the Ningbo Science and Technology Plan Project of China (No. 2024Z189), the National Natural Science Foundation of China (Nos. 92478133, 21977005), Beijing Natural Science Foundation (No. 7254478), China Postdoctoral Science Foundation (No. 2024M760155) and Postdoctoral Fellowship Program of CPSF (No. GZB20250832).
Supplementary material associated with this article can be found, in the online version, at doi:
B. Lindberg, Adv. Carbohydr. Chem. Biochem. 48 (1990) 279–318.
Z. Feng, Z. Zhan, Y. Yang, J. Jiang, P. Zhang, Bioorg. Chem. 74 (2017) 10–14. doi: 10.1016/j.bioorg.2017.07.004
L.V. Romashov, F.A. Kucherov, K.S. Kozlov, V.P. Ananikov, Int. J. Mol. Sci. 24 (2023) 3997. doi: 10.3390/ijms24043997
D. Chen, X. Yang, J. Yang, et al., Front. Aging Neurosci. 9 (2017) 403. doi: 10.3389/fnagi.2017.00403
D. Ni, W. Xu, Y. Zhu, et al., Biotechnol. Adv. 37 (2019) 306–318. doi: 10.1016/j.biotechadv.2019.01.002
C. Krog-Jensen, S. Oscarson, J. Org. Chem. 61 (1996) 4512–4513. doi: 10.1021/jo960776b
C. Krog-Jensen, S. Oscarson, J. Org. Chem. 63 (1998) 1780–1784. doi: 10.1021/jo970983r
S. Oscarson, F.W. Sehgelmeble, J. Am. Chem. Soc. 122 (2000) 8869–8872. doi: 10.1021/ja001439u
S. Oscarson, F.W. Sehgelmeble, J. Org. Chem. 67 (2002) 8457–8462. doi: 10.1021/jo020341q
S. Oscarson, F.W. Sehgelmeble, Tetrahedron Asymmetry 16 (2005) 121–125. doi: 10.1016/j.tetasy.2004.11.051
P. Wang, Y. Mo, X. Cui, et al., Org. Lett. 22 (2020) 2967–2971. doi: 10.1021/acs.orglett.0c00702
T. Müller, R. Schneider, R.R. Schmidt, Tetrahedron Lett. 35 (1994) 4763–4766. doi: 10.1016/S0040-4039(00)76961-9
T. Yamanoi, N. Misawa, M. Watanabe, Tetrahedron Lett. 48 (2007) 6458–6462. doi: 10.1016/j.tetlet.2007.07.092
G. Lian, Q. Gao, F. Lin, Carbohydr. Res. 343 (2008) 2992–2996. doi: 10.1016/j.carres.2008.09.001
T. Mukaiyama, Y. Murai, S. Shoda, Chem. Lett. 10 (1981) 431–432. doi: 10.1246/cl.1981.431
Y. Takahashi, T. Ogawa, Carbohydr. Res. 164 (1987) 277–296. doi: 10.1016/0008-6215(87)80136-2
T. Mukaiyama, Y. Hashimoto, S. Shoda, Chem. Lett. 12 (1983) 935–938. doi: 10.1246/cl.1983.935
H. Jona, H. Maeshima, T. Mukaiyama, Chem. Lett. 30 (2001) 726–727. doi: 10.1246/cl.2001.726
K.C. Nicolaou, A. Chucholowski, R.E. Dolle, J.L. Randall, J. Chem. Soc., Chem. Commun. 17 (1984) 1155–1156.
M. Kreuzer, J. Thiem, Carbohydr. Res. 149 (1986) 347–361. doi: 10.1016/S0008-6215(00)90057-0
T. Matsumoto, M. Katsuki, K. Suzuki, Tetrahedron Lett. 29 (1988) 6935–6938. doi: 10.1016/S0040-4039(00)88479-8
T. Matsumoto, M. Katsuki, K. Suzuki, Tetrahedron Lett. 30 (1989) 833–836. doi: 10.1016/S0040-4039(01)80628-6
S. Hashimoto, M. Hayashi, R. Noyori, Tetrahedron Lett. 25 (1984) 1379–1382. doi: 10.1016/S0040-4039(01)80163-5
H.P. Wessel, Tetrahedron Lett. 31 (1990) 6863–6866. doi: 10.1016/S0040-4039(00)97191-0
G. Böhm, H. Waldmann, Tetrahedron Lett. 36 (1995) 3843–3846. doi: 10.1016/0040-4039(95)00637-R
S. Hosono, W.S. Kim, H. Sasai, M. Shibasaki, J. Org. Chem. 60 (1995) 4–5. doi: 10.1021/jo00106a002
T. Mukaiyama, M. Yanagisawa, D. Iida, I. Hachiya, Chem. Lett. 29 (2000) 606–607. doi: 10.1246/cl.2000.606
H. Jona, H. Mandai, T. Mukaiyama, Chem. Lett. 30 (2001) 426–427. doi: 10.1246/cl.2001.426
H. Yamada, T. Hayashi, Carbohydr. Res. 337 (2002) 581–585. doi: 10.1016/S0008-6215(02)00029-0
G. Pelletier, A. Zwicker, C.L. Allen, A. Schepartz, S.J. Miller, J. Am. Chem. Soc. 138 (2016) 3175–3182. doi: 10.1021/jacs.5b13384
G.C. Sati, J.L. Martin, Y. Xu, et al., J. Am. Chem. Soc. 142 (2020) 7235–7242. doi: 10.1021/jacs.0c03165
A. Sun, Z. Li, S. Li, et al., Chin. Chem. Lett. 36 (2025) 109972. doi: 10.1016/j.cclet.2024.109972
X. Zhou, G. Liu, M. Yang, et al., Chin. Chem. Lett. 36 (2025) 110734. doi: 10.1016/j.cclet.2024.110734
Q. Long, J. Gao, N. Yan, P. Wang, M. Li, Org. Chem. Front. 8 (2021) 3332–3341. doi: 10.1039/d1qo00211b
Y. Manabe, T. Matsumoto, Y. Ikinaga, et al., Org. Lett. 24 (2022) 6–10. doi: 10.1021/acs.orglett.1c03233
Q. Zhu, Y. Tang, B. Yu, Org. Lett. 24 (2022) 3626–3630. doi: 10.1021/acs.orglett.2c01146
Y. Singh, S.A. Geringer, A.V. Demchenko, Chem. Rev. 122 (2022) 11701–11758. doi: 10.1021/acs.chemrev.2c00029
Y. Yang, B. Yu, Chem. Rev. 117 (2017) 12281–12356. doi: 10.1021/acs.chemrev.7b00234
J. Szejtli, Chem. Rev. 98 (1998) 1743–1754. doi: 10.1021/cr970022c
Y. Wu, C. Tang, J.T. Lee, et al., J. Am. Chem. Soc. 146 (2024) 9801–9810. doi: 10.1021/jacs.3c14427
K. Tanaka, T. Uchiyama, K. Yamauchi, Y. Suzuki, S. Hashiguchi, Carbohydr. Res. 99 (1982) 197–204. doi: 10.1016/S0008-6215(00)81911-4
K. Saito, T. Hira, T. Suzuki, et al., Biosci. Biotechnol. Biochem. 63 (1999) 655–661. doi: 10.1271/bbb.63.655
H. Mineo, H. Hara, N. Shigematsu, Y. Okuhara, F. Tomita, J. Nutr. 132 (2002) 3394–3399. doi: 10.1093/jn/132.11.3394
K. Saito, H. Goto, A. Yokota, F. Tomita, Biosci. Biotechnol. Biochem. 61 (1997) 1705–1709. doi: 10.1271/bbb.61.1705
X. Wang, S. Yu, T. Zhang, B. Jiang, W. Mu, Appl. Microbiol. Biotechnol. 99 (2015) 175–188. doi: 10.1007/s00253-014-6238-x
H. Hang, Appl. Microbiol. Biotechnol. 101 (2017) 7477–7486. doi: 10.1007/s00253-017-8500-5
H. Ko, J.H. Bae, B.H. Sung, et al., Sci. Rep. 9 (2019) 15980. doi: 10.1038/s41598-019-52373-5
G. Gattuso, S.A. Nepogodiev, J.F. Stoddart, Chem. Rev. 98 (1998) 1919–1958. doi: 10.1021/cr960133w
X. Li, C. Di Carluccio, H. Miao, et al., Angew. Chem. Int. Ed. 62 (2023) e202307851. doi: 10.1002/anie.202307851

Scheme 1 Glycosyl donors developed in previous studies and the present study for constructing fructofuranosidic bonds.

Scheme 2 Substrate scope of the fructofuranosyl O-glycosylation. a For 5b–5y, glycosylations were conducted with fructofuranosyl fluoride donor 2 (1.2 equiv.) and acceptor 4b–4y (1.0 equiv.) under the activation of B(C6F5)3 (0.4 equiv.) in anhydrous CH2Cl2 (0.025 mol/L, calculated based on donor) for 3 h. b Isolated yield, and the α/β ratio was determined by analysis of NMR for 5b–5y. c The activation system for 5h was replaced with Cp2ZrCl2/AgOTf (0.6 equiv./1.2 equiv.).

Scheme 3 Substrate scope of the fructofuranosyl S-, N-, and C-glycosylation. a For 8a–8g, glycosylations were conducted with fructofuranosyl fluoride donor 2 (1.0 equiv.) and nucleophiles 7a–7g under the activation of B(C6F5)3 in anhydrous CH2Cl2 (0.025 mol/L, calculated based on donor) for 3 h. b Isolated yield, and the α/β ratio was determined by analysis of NMR for 8a–8g. TMS = trimethylsilyl.

Figure 1 The DFT calculation results of the intermediates in cycloglycosylation from 9c to 9d.

Scheme 5 Proposed mechanism of tris(pentafluorophenyl)borane promoted glycosylation with fluoride 2 or 9c.
Table 1. Effect of activator on fructofuranosyl fluoride donor glycosylation.a
|
||||
| Entry | Donor | Activator (equiv.) | Yield (%)b | Ratio of α/βc |
| 1 | 2 | Cu(OTf)2 (1.2) | NR | – |
| 2 | 2 | Ca(OTf)2 (1.2) | NR | – |
| 3 | 2 | CSA (1.2) | NR | – |
| 4 | 2 | BBr3 (1.2) | 0 | – |
| 5 | 2 | BF3·OEt2 (1.2) | 27 | α only |
| 6 | 2 | SnCl4 (1.2) | 97 | α only |
| 7 | 2 | B(C6F5)3 (0.4) | 95 | α only |
| 8 | 2 | Cp2ZrCl2/AgOTf (0.6/1.2) | 94 | α only |
| 9 | 2 | SnCl2/AgOTf (0.6/0.6) | 93 | α only |
| 10 | 3 | B(C6F5)3 (0.4) | 93 | 1.5/1 |
| a Glycosylations were conducted with fructofuranosyl fluoride donor 2 or 3 (1.2 equiv.) in anhydrous CH2Cl2 (0.025 mol/L, calculated based on donor) in the presence of 4 Å MS (80 mg/mL, calculated based on solvent) at room temperature for 3 h. b Isolated yield. NR = No reaction. c The α/β ratio was determined by NMR analysis. |
||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: