Scheme 1.
Pd-catalyzed hydrofunctionalizations of unsaturated bonds.

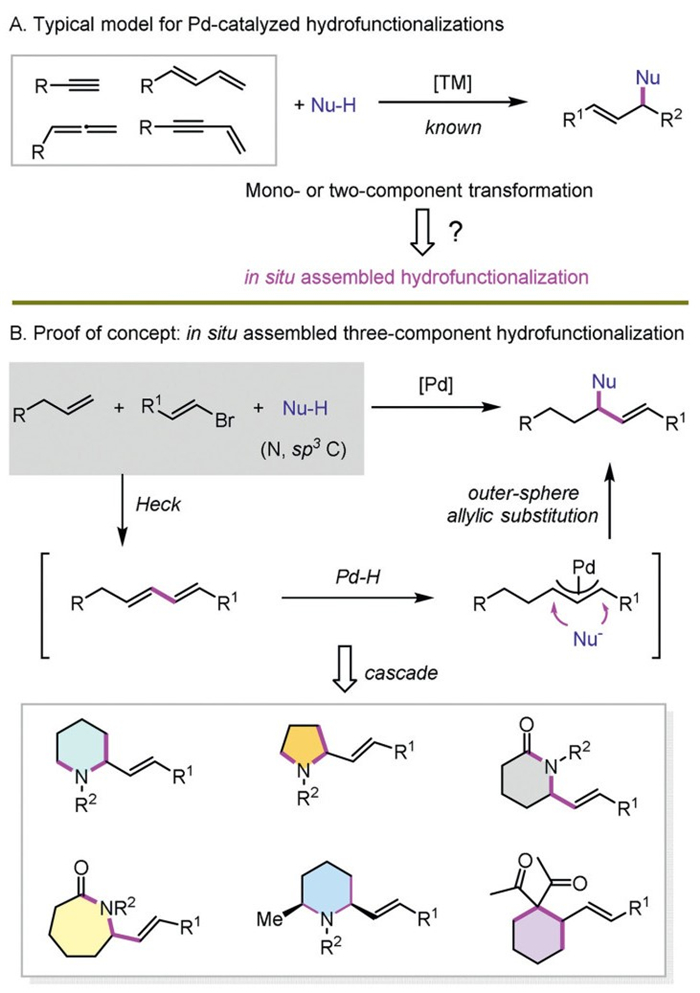
Transition metal-catalyzed hydrofunctionalization of unsaturated hydrocarbons via η3-allyl metal species has emerged as a powerful strategy to construct allyl skeletons [1–12]. A series of typical nucleophiles have been demonstrated suitable for the preparation of allylic C–C, C–N, C–O, C–S and C–P bonds. In contrast to typical Tsuji-Trost reaction, this route usually features the ready availability of both substrates, 100% atom economy and mild reaction conditions. Unsaturated hydrocarbons, including alkynes, dienes, allenes and enynes are most commonly used to react with nucleophiles via an intramolecular or intermolecular route (Scheme 1A) [13–76]. However, different from the well explored mono- and two-component hydrofunctionalization models, a three-component hydrofunctionalization via in situ assembling pathway from simple alkenes is unknown.
Due to the inability to form critical η3-allyl metal species, alkenes as feedstock compounds could not be adopted directly for hydrofunctionalization. It can be imagined that if the alkene could be in situ transformed into a typical unsaturated substrate, such as conjugated diene, then hydrofunctionalization might occur readily. A few elegant studies explored related pathways via Pd- [77–79] or Ni-catalyzed [80] three-component cross couplings of alkenes with cyclic vinyl triflates and aryl boronic acids or B2pin2, all of which, however, focused on inner-sphere reductive elimination coupling route using boron reagents. A different process involving general nucleophiles through outer-sphere allylic substitution pathway is unexplored.
Following our long-standing interest in hydrofunctionalization areas [32–34,39,52,58,63,70–74], we envisioned that the combination of in situ formation of conjugated diene from the reaction of simple alkene and alkenyl bromide via Pd-catalyzed Heck reaction and following hydrofunctionalization with common nucleophiles might provide a potential route for the unprecedented in situ assembled three-component hydrofunctionalization (Scheme 1B). However, this design is not straightforward. The perfect merger of Heck reaction and hydrofunctionalization is challenging, due to competing direct coupling between alkenyl electrophiles and nucleophiles, and the balanced basicity for transmetallation, β-H elimination and allylic substitution. The stereocontrol is another potential issue, especially considering Heck reaction usually requires a high reaction temperature [81–82]. Herein, we report the first stereoselective in situ assembled three-component hydrofunctionalization via Pd catalysis. A series of simple olefins react with alkenyl bromides and amines or carbon nucleophiles smoothly to provide diverse allyl skeletons in moderate to good yields and with high regioselectivities. In addition, the combination of assembled hydrofunctionalization and cascade processes enables convenient access to various intriguing substituted cyclic scaffolds, including piperidines, pyrrolidines, piperidones, azepan-2-ones, and cyclohexanes.
We initiated studies of the assembled hydrofunctionalization by conducting reactions with olefin 1a, alkenyl bromide 2a, and aniline 3a as the substrates, inorganic KHCO3 as the base, and Pd(OAc)2 as the catalyst at 80 ℃ for 12 h (Table 1). A set of solvents were evaluated first. However, low reaction yield of 4a was detected along with most substrates recovered when using both nonpolar and polar solvents (entries 1-5 and see Supporting information for details). Only with DMSO as the solvent, product 4a was observed in 46% yield (entry 6). The evaluation of a couple of palladium sources indicated that Pd2(dba)3 as the catalyst was slightly favorable to the reaction (entries 7-10). In this context, we expected that different ligands might help regulate the reactivity and stereoselectivity. Various ligands were thus examined but all led to the erosion of the yield of 4a (entry 11 and see Supporting information for details), which suggested that DBA (dibenzylideneacetone) was the optimal ligand for Pd metal. Next, weak inorganic bases generally showed comparable reactivity, but strong inorganic bases and organic bases were unfavorable to the transformation (entries 12-15 and see Supporting information for details). Nevertheless, with the adoption of K2CO3 and KHCO3 as mixed bases, 4a was provided in an increased yield (entry 16). Finally, both higher and lower temperatures led to the decrease of reactivity (entry 17), but extended reaction time to 24 h was effective and furnished the optimal reaction yield of 4a in 71% (entry 18). It should be noted that no regioisomers were detected for these trials.
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | [Pd] | Solvent | Base | Yield (%) |
| 1 | Pd(OAc)2 | THF | KHCO3 | 5 |
| 2 | Pd(OAc)2 | DCM | KHCO3 | Trace |
| 3 | Pd(OAc)2 | CH3CN | KHCO3 | Trace |
| 4 | Pd(OAc)2 | DMF | KHCO3 | Trace |
| 5 | Pd(OAc)2 | PhEt | KHCO3 | 3 |
| 6 | Pd(OAc)2 | DMSO | KHCO3 | 46 |
| 7 | Pd(PPh3)4 | DMSO | KHCO3 | 29 |
| 8 | Pd[P(tBu)3]2 | DMSO | KHCO3 | 32 |
| 9 | Pd(PPh3)2Cl2 | DMSO | KHCO3 | 40 |
| 10 | Pd2(dba)3 | DMSO | KHCO3 | 53 |
| 11b | Pd2(dba)3 | DMSO | KHCO3 | <48 |
| 12 | Pd2(dba)3 | DMSO | K2CO3 | 48 |
| 13 | Pd2(dba)3 | DMSO | Na2CO3 | 40 |
| 14 | Pd2(dba)3 | DMSO | K3PO4 | 44 |
| 15 | Pd2(dba)3 | DMSO | Et3N | 10 |
| 16c | Pd2(dba)3 | DMSO | Mixture | 63 |
| 17c,d | Pd2(dba)3 | DMSO | Mixture | <54 |
| 18c,e | Pd2(dba)3 | DMSO | Mixture | 71 |
| a The reaction was carried out in 0.10 mmol scale. The yield was determined by 1H NMR. b A series of ligands were used and see Supporting information for details. c Both K2CO3 (1.5 equiv.) and KHCO3 (1.0 equiv.) were used. d At 60 or 100 ℃. e The reaction time was 24 h and isolated yield was shown. |
||||
Having identified conditions for three-component hydrofunctionalization, the scope of the transformation was examined, and the results are summarized in Scheme 2. First, a series of terminal olefins bearing diverse functional groups showed high compatibility with the reaction. For example, alkenes containing amide, aryl, ketone, hydroxyl, heteroaryl, ester, OTBDPS, NHBoc, and ether units in the alkenes were well tolerated and afforded corresponding products 4a–4m in 51%−71% yield and with excellent regioselectivity (Scheme 2A). In particular, the unaffected functional groups such as ketone, hydroxyl and ester units in the alkenes provided further modification space with the formed nucleophilic secondary amines (4d, 4e, 4k).

Arylamines featuring different electronic and steric characters were also suitable for the in situ assembled hydrofunctionalization (Scheme 2B). Substituents including F, Cl, alkyl, and ether at varying positions in the arylamines exhibited good tolerance in the reaction with alkene 1a and alkenyl bromide 2a, generating allylamines in moderate to good yields (4n–4s). Arylamines containing multiple substituents and large ring system and heteroaryl amines were all good coupling partners in the three-component reaction (4t–4w). In addition to arylamines, alkyl amines were evaluated as well and furnished similarly good reactivity and stereocontrol during the reaction (4x–4y). Finally, a couple of diverse aryl vinyl bromides were tested and gave corresponding products 4z–4ac in 57%–80% yield (Scheme 2C). It should be noted that only one regioisomer was detected for each case. It should be noted that alkyl-substituted alkenyl bromide was not suitable for the protocol at present (see Supporting information for failed trials).
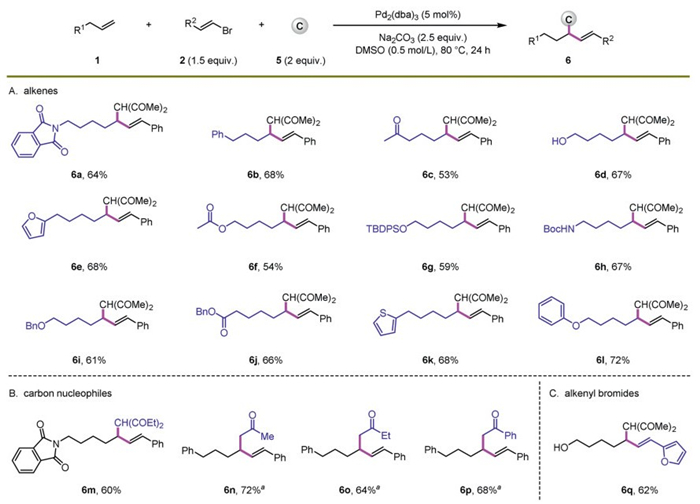
To demonstrate the generality of the assembled hydrofunctionalization concept, hydroalkylation through this in situ assembled process was established simultaneously (Scheme 3). With the use of Na2CO3 as the base instead of K2CO3 and KHCO3 in the conditions for hydroamination, assembled hydroalkylation reaction proceeded smoothly. Alkenes containing various functional groups, such as amide, aryl, ketone, hydroxyl, furyl, ester, OTBDPS, NHBoc, ether, and thienyl units, reacted with alkenyl bromide 2a and acetylacetone 5a well, furnishing assembled allyl skeletons 6a–6l in reasonable yields and with excellent regioselectivities (Scheme 3A). Other carbon nucleophiles were also suitable for the transformation (Scheme 3B, 6m–6p). In particular, with the combination of assembled hydrofunctionalization and decarboxylation, a set of β-alkenyl ketones were prepared readily via a one-pot reaction. In addition, a heteroaryl vinyl bromide underwent the transformation smoothly, furnishing 6q in 62% yield as a single regioisomer (Scheme 3C).
Considering the broad functional group tolerance, we hypothesized that a combination of assembled hydrofunctionalization and various precursor formation or sequential downstream transformations might construct valuable scaffolds conveniently from readily available substrates (Scheme 4). First, with the use of alkene containing a terminal Br atom, alkenyl bromide and amine as substrates, a series of substituted saturated N-heterocycles were achieved facilely (Scheme 4A). For example, piperidines and pyrrolidines bearing different substituents were obtained in good yields through this one-pot process (7a–7h). Similarly, by using ester-derived alkenes, piperidones and azepan-2-ones were readily achieved via sequential assembled hydroamination and amide formation (Scheme 4B, 8a–8d). Furthermore, multiple substituted piperidines 9a–9c could be easily synthesized from the reaction of alkene 1d, phenyl vinyl bromide 2a and arylamine 3 through assembled hydroamination and reductive amination cascade pathway (Scheme 4C). Finally, substituted cyclohexanes 11a–11c were prepared from pent-4-en-1-ol 1e via assembled hydroalkylation and nucleophilic substitution (Scheme 4D). These designed cascade reactions provide new routes to build various cyclic scaffolds, highlighting the value and power of the assembled hydrofunctionalization concept.

To elucidate the mechanistic pathway of the designed assembled hydrofunctionalization, a couple of experiments were carried out (Scheme 5). When deuterated alkene 1a-d was adopted under standard conditions, product 4a-d was observed in 68% yield (Scheme 5A, top). In this case, both allylic and homoallylic positions were observed in high deuteration. This result indicated that a Heck coupling occurred first to generate a diene intermediate int-2 and Pd-D species. The reinsertion of Pd-D with diene species introduced deuterium atom to the homoallylic position of product 4a-d. When deuterated phenylamine was used as nucleophile, no deuteration was observed in the skeleton of prepared product 4a-d (Scheme 5A, bottom). This fact suggested that the formed Pd-D species via β-H elimination was tightly associated with formed diene intermediate. To further support the potential diene formation step, a control experiment with the use of only 0.2 equiv. of 3a as limited nucleophile was conducted (Scheme 5B, top). In this case, product 4a was observed in 4% yield, along with substrate 1a recovered in 67% yield and diene intermediate 12 in 20% yield. Without the use of amine nucleophile 3a, diene 12 was still detected in 34% yield along with 64% yield of 1a. All these results demonstrated that a Heck coupling was involved in the design.
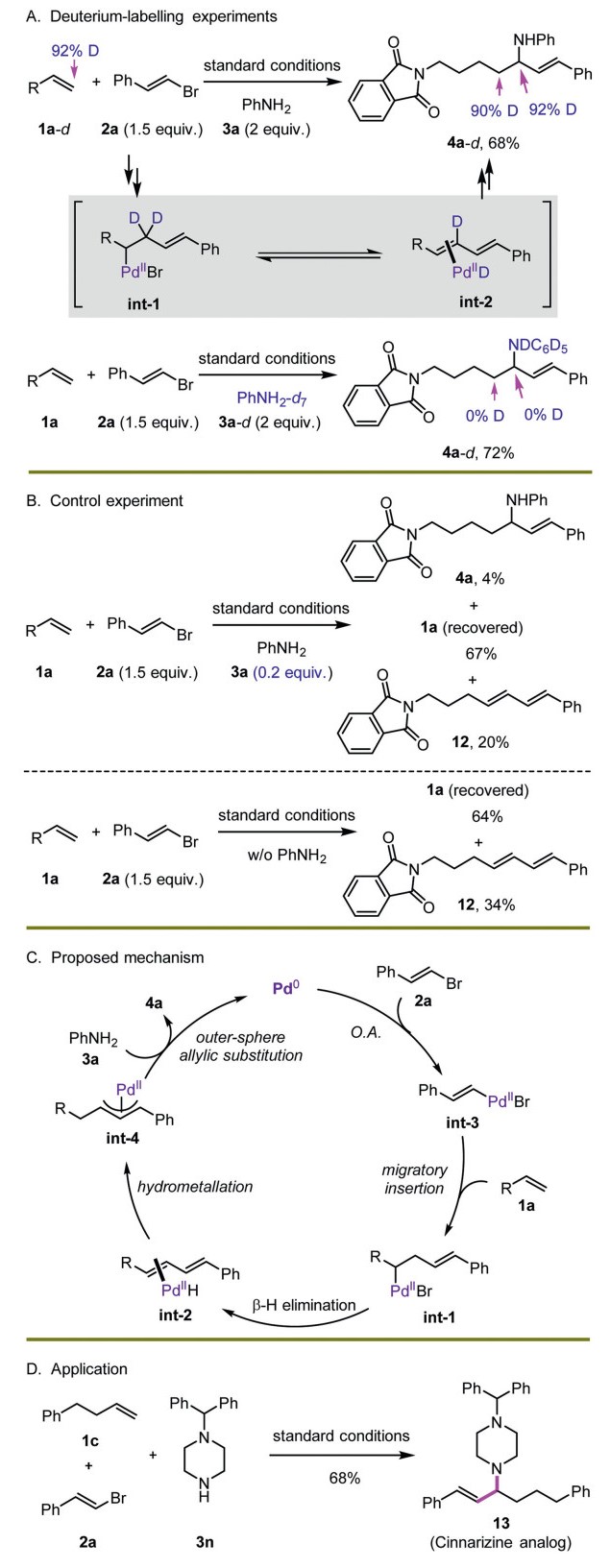
Based on these experiments, a plausible mechanistic pathway was described in Scheme 5C. The low-valent Pd was oxidized by alkenyl bromide first to form alkenyl-Pd species int-3, which reacted with olefin 1a to provide int-1 via migratory insertion. The following β-H elimination from int-1 gave diene intermediate int-2, and then generated electrophilic allyl-Pd species int-4. Finally, an outer-sphere allylic substitution from arylamine 3a provided the product 4a and regenerated Pd catalyst. In addition, the present protocol could be easily applied to the concise synthesis of Cinnarizine analog 13 from the reaction of simple substrates 1c, 2a, and 3n, highlighting the practical value of the methodology (Scheme 5D) [83].
In conclusion, a three-component model for in situ assembled hydrofunctionalizations is established via palladium catalysis, different from conventional mono- and two-component reaction model. Both assembled hydroamination and hydroalkylation are developed through this tandem Heck coupling and hydrofunctionalization pathway. Various allyl skeletons are prepared in moderate to good yields and with high regioselectivities from the reaction of readily available olefins, alkenyl bromides and common nucleophiles. The combination of assembled hydroamination and different downstream transformations provide convenient routes to access a series of valuable substituted cyclic scaffolds, including piperidines, pyrrolidines, piperidones, azepan-2-ones, and cyclohexanes. Preliminary studies support the diene-involved mechanistic pathway.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Li-Hong Xiong: Investigation. Liang Chen: Investigation. Zhi-Tao He: Writing – review & editing, Writing – original draft, Supervision, Project administration, Funding acquisition, Conceptualization.
We acknowledge the National Natural Science Foundation of China (No. 22371292), Ningbo Natural Science Foundation (No. 2023J036), Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB0610000), State Key Laboratory of Organometallic Chemistry, and Shanghai Institute of Organic Chemistry for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
P. Koschker, B. Brei, Acc. Chem. Res. 49 (2016) 1524–1536. doi: 10.1021/acs.accounts.6b00252
Y. Zheng, W. Zi, Tetrahedron Lett. 59 (2018) 2205–2213. doi: 10.1016/j.tetlet.2018.04.057
G. Li, X. Huo, X. Jiang, et al., Chem. Soc. Rev. 49 (2020) 2060–2118. doi: 10.1039/c9cs00400a
N.J. Adamson, S.J. Malcolmson, ACS Catal. 10 (2020) 1060–1076. doi: 10.1021/acscatal.9b04712
R. Blieck, M. Taillefer, F. Monnier, Chem. Rev. 120 (2020) 13545–13598. doi: 10.1021/acs.chemrev.0c00803
A. Flaget, C. Zhang, C. Mazet, ACS Catal. 12 (2022) 15638–15647. doi: 10.1021/acscatal.2c05251
G. Cera, G. Maestri, ChemCatChem 14 (2022) e202200295. doi: 10.1002/cctc.202200295
C. Ma, Y.W. Chen, Z.T. He, Sci. Sin. Chim. 53 (2023) 474–484. doi: 10.1360/ssc-2022-0196
Y.C. Wang, J.B. Liu, Z.T. He, Chin. J. Org. Chem. 43 (2023) 2614–2627. doi: 10.6023/cjoc202302010
F. Panahi, F. Bauer, B. Breit, Acc. Chem. Res. 56 (2023) 3676–3693. doi: 10.1021/acs.accounts.3c00322
L. Li, S. Wang, A. Jakhar, et al., Green Synth. Catal. 4 (2023) 124–134. doi: 10.1117/12.2660370
J.M. Zhang, Z.T. He, Chin. J. Org. Chem. 45 (2025) 592–601. doi: 10.6023/cjoc202406047
K. Takahashi, A. Miyake, G. Hata, Bull. Chem. Soc. Jpn. 45 (1972) 1183–1191. doi: 10.1246/bcsj.45.1183
R.W. Armbruster, M.M. Morgan, J.L. Schmidt, et al., Organometallics 5 (1986) 234–237. doi: 10.1021/om00133a011
P.W. Jolly, N. Kokel, Synthesis 1990 (1990) 771–773. doi: 10.1055/s-1990-27010
B.M. Trost, L. Zhi, Tetrahedron Lett. 33 (1992) 1831–1834. doi: 10.1016/S0040-4039(00)74154-2
O. Löber, M. Kawatsura, J.F. Hartwig, J. Am. Chem. Soc. 123 (2001) 4366–4367. doi: 10.1021/ja005881o
B.M. Trost, C. Jäkel, B. Plietker, J. Am. Chem. Soc. 125 (2003) 4438–4439. doi: 10.1021/ja029190z
L.M. Lutete, I. Kadota, Y. Yamamoto, J. Am. Chem. Soc. 126 (2004) 1622–1623. doi: 10.1021/ja039774g
B.M. Trost, J. Xie, J.D. Sieber, J. Am. Chem. Soc. 133 (2011) 20611–20622. doi: 10.1021/ja209244m
N.J. Adamson, E. Hull, S.J. Malcolmson, J. Am. Chem. Soc. 139 (2017) 7180–7183. doi: 10.1021/jacs.7b03480
N.J. Adamson, K.C.E. Wilbur, S.J. Malcolmson, J. Am. Chem. Soc. 140 (2018) 2761–2764. doi: 10.1021/jacs.7b13300
S.Z. Nie, R.T. Davison, V.M. Dong, J. Am. Chem. Soc. 140 (2018) 16450–16454. doi: 10.1021/jacs.8b11150
Q. Zhang, H. Yu, L. Shen, et al., J. Am. Chem. Soc. 141 (2019) 14554–14559. doi: 10.1021/jacs.9b07600
Z. Zhang, F. Xiao, H.M. Wu, et al., Org. Lett. 22 (2020) 569–574. doi: 10.1021/acs.orglett.9b04341
C.I. Onyeagusi, X. Shao, S.J. Malcolmson, Org. Lett. 22 (2020) 1681–1685. doi: 10.1021/acs.orglett.0c00342
H. Yang, D. Xing, Chem. Commun. 56 (2020) 3721–3724. doi: 10.1039/d0cc00265h
Q. Zhang, D. Dong, W.W. Zi, J. Am. Chem. Soc. 142 (2020) 15860–15869. doi: 10.1021/jacs.0c05976
M.M. Li, L. Cheng, L.J. Xiao, et al., Angew. Chem. Int. Ed. 60 (2021) 2948–2951. doi: 10.1002/anie.202012485
A.Y. Jiu, H.S. Slocumb, C.S. Yeung, et al., Angew. Chem. Int. Ed. 60 (2021) 19660–19664. doi: 10.1002/anie.202105679
Q. Zhang, M. Zhu, W.W. Zi, Chem 8 (2022) 2784–2796. doi: 10.1016/j.chempr.2022.07.014
Y.C. Wang, Z.X. Xiao, M. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202215568. doi: 10.1002/anie.202215568
S.Q. Yang, A.J. Han, Y. Liu, et al., J. Am. Chem. Soc. 145 (2023) 3915–3925. doi: 10.1021/jacs.2c11843
J.M. Zhang, Y.C. Wang, L. Chen, et al., Chem. Eur. J. 30 (2024) e202401350. doi: 10.1002/chem.202401350
L.M. Lutete, I. Kadota, Y. Yamamoto, J. Am. Chem. Soc. 126 (2004) 1622–1623. doi: 10.1021/ja039774g
J.T.D. Lee, Y. Zhao, Chem. Eur. J. 24 (2018) 9520–9524. doi: 10.1002/chem.201802273
M.S. Wu, Z.Y. Han, L.Z. Gong, Org. Lett. 23 (2021) 636–641. doi: 10.1021/acs.orglett.0c03466
J. Zhang, Y.N. Wang, C. You, et al., Org. Lett. 24 (2022) 1186–1189. doi: 10.1021/acs.orglett.1c04334
M.Q. Tang, Z.J. Yang, Z.T. He, Nat. Commun. 14 (2023) 6303. doi: 10.1038/s41467-023-42160-2
Y. Lin, W. Wen, J.H. Liu, et al., Org. Lett. 26 (2024) 7908–7913. doi: 10.1021/acs.orglett.4c02840
Y. Liu, H. Chen, X. Wang, J. Am. Chem. Soc. 146 (2024) 28427–28436. doi: 10.1021/jacs.4c09983
Y. Yamamoto, M. Al-Masum, N. Asao, J. Am. Chem. Soc. 116 (1994) 6019–6020. doi: 10.1021/ja00092a083
B.M. Trost, V.J. Gerusz, J. Am. Chem. Soc. 117 (1995) 5156–5157. doi: 10.1021/ja00123a020
Y. Yamamoto, M. Al-Masum, Synlett 1995 (1995) 969–970. doi: 10.1055/s-1995-5148
M. Li, S. Datta, D.M. Barber, et al., Org. Lett. 14 (2012) 6350–6353. doi: 10.1021/ol303128s
H. Zhou, Y. Wang, L. Zhang, et al., J. Am. Chem. Soc. 139 (2017) 3631–3634. doi: 10.1021/jacs.7b00437
Z. Wu, M. Zhang, Y. Shi, et al., Org. Chem. Front. 7 (2020) 1502–1511. doi: 10.1039/d0qo00174k
M. Zhu, Q. Zhang, W.W. Zi, Angew. Chem. Int. Ed. 60 (2021) 6545–6552. doi: 10.1002/anie.202014510
H.C. Lin, G.J. Knox, C.M. Pearson, et al., Angew. Chem. Int. Ed. 61 (2022) e202201753. doi: 10.1002/anie.202201753
M. Zhu, P. Wang, Q. Zhang, et al., Angew. Chem. Int. Ed. 61 (2022) e202207621. doi: 10.1002/anie.202207621
J.H. Liu, Q. Zhou, Y. Lin, et al., ACS Catal. 13 (2023) 6013–6022. doi: 10.1021/acscatal.3c00790
M.Q. Tang, Z.J. Yang, A.J. Han, et al., Angew. Chem. Int. Ed. 64 (2025) e202413428. doi: 10.1002/anie.202413428
M.M. Salter, V. Gevorgyan, S. Saito, et al., Chem. Commun. 32 (1996) 17–18.
V. Gevorgyan, C. Kadowaki, M.M. Salter, et al., Tetrahedron 53 (1997) 9097–9106. doi: 10.1016/S0040-4020(97)00602-9
U. Radhakrishnan, M. Al-Masum, Y. Yamamoto, Tetrahedron Lett. 39 (1998) 1037–1040. doi: 10.1016/S0040-4039(97)10697-9
N.J. Adamson, H. Jeddi, S.J. Malcolmson, J. Am. Chem. Soc. 141 (2019) 8574–8583. doi: 10.1021/jacs.9b02637
H. Tsukamoto, T. Konno, K. Ito, et al., Org. Lett. 21 (2019) 6811–6814. doi: 10.1021/acs.orglett.9b02439
S.Q. Yang, Y.F. Wang, W.C. Zhao, et al., J. Am. Chem. Soc. 143 (2021) 7285–7291. doi: 10.1021/jacs.1c03157
L. Li, S. Wang, P. Luo, et al., Nat. Commun. 12 (2021) 5667. doi: 10.1038/s41467-021-25981-x
Q. Li, X. Fang, R. Pan, et al., J. Am. Chem. Soc. 144 (2022) 11364–11376. doi: 10.1021/jacs.2c03620
C. You, M. Shi, X. Mi, et al., Nat. Commun. 14 (2023) 2911. doi: 10.1038/s41467-023-38488-4
M. Eaton, Y. Dai, Z. Wang, et al., J. Am. Chem. Soc. 145 (2023) 21638–21645. doi: 10.1021/jacs.3c08151
B.Y. Xie, Z.T. He, ACS Catal. 14 (2024) 9742–9751. doi: 10.1021/acscatal.4c02377
R.C. Larock, Y.D. Lu, A.C. Bain, et al., J. Org. Chem. 56 (1991) 4589–4590. doi: 10.1021/jo00015a002
R.C. Larock, Y. Wang, Y. Lu, et al., J. Org. Chem. 59 (1994) 8107–8114. doi: 10.1021/jo00105a030
Y. Wang, X. Dong, R.C. Larock, J. Org. Chem. 68 (2003) 3090–3098. doi: 10.1021/jo026716p
H. Pang, D. Wu, H. Cong, et al., ACS Catal. 9 (2019) 8555–8560. doi: 10.1021/acscatal.9b02747
Y. Zhang, H.C. Shen, Y.Y. Li, et al., Chem. Commun. 55 (2019) 3769–3772. doi: 10.1039/c9cc01379b
D. Zhu, Z. Jiao, Y.R. Chi, et al., Angew. Chem. Int. Ed. 59 (2020) 5341–5345. doi: 10.1002/anie.201915864
Y.W. Chen, Y. Liu, H.Y. Lu, et al., Nat. Commun. 12 (2021) 5626. doi: 10.1038/s41467-021-25978-6
Q.Y. Liao, C. Ma, Y.C. Wang, et al., Chin. Chem. Lett. 34 (2023) 108371. doi: 10.1016/j.cclet.2023.108371
H.Z. Miao, Y. Liu, Y.W. Chen, et al., Synlett 34 (2023) 451–456. doi: 10.1055/a-1916-2937
X. Wang, H.Z. Miao, G.Q. Lin, Z.T. He, Angew. Chem. Int. Ed. 62 (2023) e202301556. doi: 10.1002/anie.202301556
X.X. Chen, H. Luo, Y.W. Chen, et al., Angew. Chem. Int. Ed. 62 (2023) e202307628. doi: 10.1002/anie.202307628
Y.L. Su, L.L. Li, X.L. Zhou, et al., Org. Lett. 20 (2018) 2403–2406. doi: 10.1021/acs.orglett.8b00740
X. Fang, Q. Li, R. Shi, et al., Org. Lett. 20 (2018) 6084–6088. doi: 10.1021/acs.orglett.8b02481
L. Liao, R. Jana, K.B. Urkalan, et al., J. Am. Chem. Soc. 133 (2011) 5784–5787. doi: 10.1021/ja201358b
V. Saini, M.S. Sigman, J. Am. Chem. Soc. 134 (2012) 11372–11375. doi: 10.1021/ja304344h
M.S. McCammant, T. Shigeta, M.S. Sigman, Org. Lett. 18 (2016) 1792–1795. doi: 10.1021/acs.orglett.6b00517
D. Wu, H. Pang, G. Yin, Chin. Chem. Lett. 34 (2023) 108087. doi: 10.1016/j.cclet.2022.108087
R.X. Liang, Y.X. Jia, Acc. Chem. Res. 55 (2022) 734–745. doi: 10.1021/acs.accounts.1c00781
X.S. Zhang, Y.P. Han, Y. Zhang, et al., Adv. Synth. Catal. 365 (2023) 2436–2466. doi: 10.1002/adsc.202300476
A.V. Narsaiah, P. Narsimha, Med. Chem. Res. 21 (2012) 538–541. doi: 10.1007/s00044-011-9556-x

Scheme 2 Scope of assembled hydroamination. The reaction was conducted in 0.1 mmol scale. Isolated yield.

Scheme 3 Scope of assembled hydroalkylation. Isolated yield. Prepared through assembled hydroalkylation and decarboxylation. See Supporting information for details.

Scheme 4 Assembled hydrofunctionalization cascade. Isolated yield. See Supporting information for detailed reaction conditions.
Table 1. Reaction development for assembled hydrofunctionalization.a
|
||||
| Entry | [Pd] | Solvent | Base | Yield (%) |
| 1 | Pd(OAc)2 | THF | KHCO3 | 5 |
| 2 | Pd(OAc)2 | DCM | KHCO3 | Trace |
| 3 | Pd(OAc)2 | CH3CN | KHCO3 | Trace |
| 4 | Pd(OAc)2 | DMF | KHCO3 | Trace |
| 5 | Pd(OAc)2 | PhEt | KHCO3 | 3 |
| 6 | Pd(OAc)2 | DMSO | KHCO3 | 46 |
| 7 | Pd(PPh3)4 | DMSO | KHCO3 | 29 |
| 8 | Pd[P(tBu)3]2 | DMSO | KHCO3 | 32 |
| 9 | Pd(PPh3)2Cl2 | DMSO | KHCO3 | 40 |
| 10 | Pd2(dba)3 | DMSO | KHCO3 | 53 |
| 11b | Pd2(dba)3 | DMSO | KHCO3 | <48 |
| 12 | Pd2(dba)3 | DMSO | K2CO3 | 48 |
| 13 | Pd2(dba)3 | DMSO | Na2CO3 | 40 |
| 14 | Pd2(dba)3 | DMSO | K3PO4 | 44 |
| 15 | Pd2(dba)3 | DMSO | Et3N | 10 |
| 16c | Pd2(dba)3 | DMSO | Mixture | 63 |
| 17c,d | Pd2(dba)3 | DMSO | Mixture | <54 |
| 18c,e | Pd2(dba)3 | DMSO | Mixture | 71 |
| a The reaction was carried out in 0.10 mmol scale. The yield was determined by 1H NMR. b A series of ligands were used and see Supporting information for details. c Both K2CO3 (1.5 equiv.) and KHCO3 (1.0 equiv.) were used. d At 60 or 100 ℃. e The reaction time was 24 h and isolated yield was shown. |
||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

