College of Safety and Environmental Engineering, Shandong University of Science and Technology, Qingdao 266590, China
b.
Shandong Key Laboratory of Water Pollution Control and Resource Reuse, School of Environmental Science and Engineering, Shandong University, Qingdao 266237, China
Received Date:
14 April 2025 Accepted Date:
25 September 2025 Revised Date:
01 August 2025 Available Online:
15 May 2026
Abstract:
In light of the prevalent issues associated with metal ion dissolution, secondary pollution, and poor stability in traditional metal-based Fenton catalysts, this study innovatively developed a metal-free carbon-based catalyst co-doped with Si-O bonds and graphitic nitrogen using natural diatomite as the precursor. By leveraging the synergistic effects of Si-O bonds and graphitic nitrogen, the electronic structure of the carbon matrix was effectively modulated, establishing an efficient electron transport channel for peroxymonosulfate (PMS) activation. Results showed that the Fenton-like performance of the resulting catalysts was far superior to those of traditional metal catalysts and can be comparable to various single-atom catalysts. Both the radical and 1O2 pathways exhibited a negligible role in the metal-free Si-O/N@DM/PMS systems. In contrast, electron transfer process (ETP) was the predominate oxidation pathway for acetaminophen (PCM) degradation in the Si-O/N@DM/PMS systems. To facilitate engineering applications, we further designed a proton membrane reactor integrated with a four-channel PMS system, which could introduce an enlarged ETP pathway for pollutant degradation; this addresses the key issues of both sulfate pollution and metal leaching in water caused by traditional metal-based Fenton systems.
Antibiotic pollutants are accumulating in global water bodies at an alarming rate of 15%−30% annually, posing a significant ecological threat [1–4]. According to reports from World Health Organization, antibiotics can be consistently detected in major surface waters in China throughout the year [3–6], with concentrations of sulfonamides, tetracyclines, and quinolones frequently reaching micrograms per liter (µg/L). The primary causes of this pollution trend include: first, untreated wastewater from medical institutions, which contains substantial amounts of antibiotic metabolites; second, the excessive use of prophylactic drugs in intensive farming, leading to tens of thousands of tons of veterinary antibiotics being released into the environment annually [7–9]. These pollutants migrate between different media via the water cycle and exhibit biomagnification through the food chain [10]. Consequently, addressing antibiotic pollution has become an urgent priority [11,12].
Advanced oxidation technology based on peroxides activation has become the focus of research in the field of antibiotic degradation due to its strong oxidation capacity (oxidation potential 2.5–3.1 V) and wide pH adaptation range [13–17]. The core of this technology is to stimulate the decomposition of peroxymonosulfate (PMS) through energy input (light, heat, electricity) or catalyst action to produce highly reactive species (such as sulfate radicals (SO4•−), hydroxyl radicals (•OH), singlet oxygen (1O2) or other nonradical pathways [18–22]. Recently, metal-based catalysts, especially the iron-based catalysts and cobalt-based catalysts, have been intensively used for peroxide activation [23–27]. Iron-based materials, despite their low cost, suffer pH-sensitive defects and the efficiency of the Fe2+/Fe3+ cycle plummets in near neutral conditions [24,25]. Although the cobalt-based catalyst maintained a very high antibiotic degradation rate in a wide pH range, many samples confirmed that the cobalt ion dissolution concentration could reach to mg/L scale, far exceeding the standard of drinking water. By anchoring metal atoms on nitrogen-doped carbon substrate, single-atom catalyst can theoretically achieve 100% atom utilization rate with superior Fenton-like activity than conventional metal-based catalysts. For example, the degradation rate constant of Fe-N4 catalyst for antibiotic pollutants can reach > 0.3 min−1 [3], but metal dissolution still occurs under long-term operation [28,29].
Carbon-based catalysts have become the focus of research because of their environmental friendliness [30,31], but their catalytic performances are generally weaker than that of metal-based materials, mainly due to the insufficient density of surface-active sites and low electron transport efficiency [32,33]. It was reported that doping relatively weak electronegative heteroatoms, such as N, B, O, S, Si can effectively regulate the electronic structure of carbonaceous materials and generate new active sites, thus significantly improving the catalytic performance of carbonaceous materials [34–36]. In the earlier phase of our study, Si-O bonds were introduced into the carbon framework, leading to a significant enhancement in PMS activation efficiency [34,35]. Nevertheless, we have not yet explored the potential synergistic effects of coexisting graphite-like nitrogen on this activation process. In fact, the sp2 hybrid structure in graphitized carbon can activate PMS by conjugated π-electron systems [36,37]. Basically, this study will explore the synergic mechanism of silico-oxygen bond and graphitized N in carbon-based carriers: the silico-oxygen bond as an electron donor promotes interfacial charge transfer, while the graphitized region works together to optimize the coupling efficiency through delocalized π-electron stable reaction intermediates, providing a new idea for the construction of high efficiency metal-free catalysts.
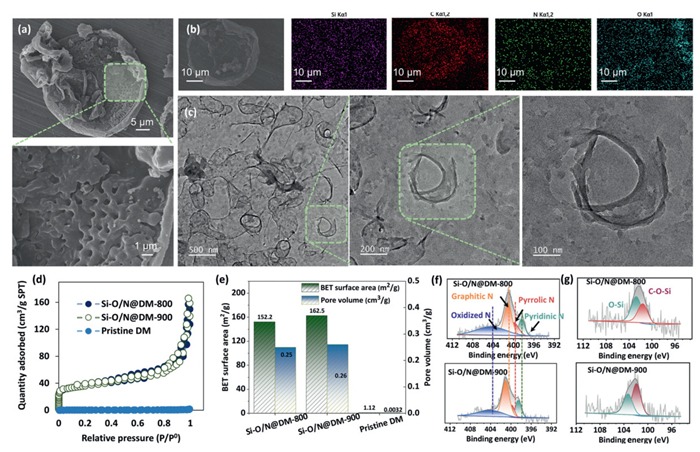
Diatomite (DM) was used as the silicon source and carrier, and two catalysts (Si-O/N@DM-800 and Si-O/N@DM-900) were prepared by adjusting the temperature for modulating the graphitic N fraction in the resulting catalysts. Characterization of the resulting catalysts was shown in Fig. 1. Scanning electron microscopy (SEM) showed that the structure of Si-O/N@DM-900 was based on the morphology of DM (Figs. 1a and b and Fig. S1 in Supporting information). The transmission electron microscope (TEM) image of the Si-O/N@DM-900 exhibited that the resulting catalyst has a regular pore structure with pore size between 0.25 and 1 µm (Fig. 1c). The BET surface areas and pore volumes of both Si-O/N@DM-800, and Si-O/N@DM-900 were detected, and these catalysts exhibited the similar pore parameters with BET surface area of 152.2–162.5 m2/g, and pore volume of 0.25–0.26 cm3/g (Figs. 1d and e). X-ray photoelectron spectroscopy (XPS)results revealed that the two catalysts were predominantly composed of the carbon element (exceeding 60 at %), along with small amounts of other elements, such as Si, O, and C (Figs. S2 and S3 in Supporting information). The XPS N 1s exhibited that the fraction of graphitic N in the Si-O/N@DM-900 was much higher than that in Si-O/N@DM-800 (Fig. 1f). Since the higher fraction of graphitic N in the resulting materials always means the higher amounts of large Π bonds, which would regulate the surface electronic structure for reactants adsorption and charge transfer [33,34]. In addition, the fraction of C–O-Si bonds in the Si-O/N@DM-900 was also higher than that in Si-O/N@DM-800 (Fig. 1g). The C–O-Si bonds represented the interaction of Si-O with the carbon framework, which means more O-Si bonds have been anchored onto the carbon substance in the Si-O/N@DM-900. As a result, both the fractions of C–O-Si bonds and graphitic N in the Si-O/N@DM-900 could be enhanced via the temperature regulation.
Figure 1
Figure 1.
(a) SEM images of Si-O/N@DM-900. (b) Elemental mappings of Si-O/N@DM-900. (c) TEM images of Si-O/N@DM-900. (d) Quantity adsorption of N2 by different catalysts. (e) BET surface areas and pore volumes of both Si-O/N@DM-800, and Si-O/N@DM-900. (f) XPS N 1s of Si-O/N@DM-800 and Si-O/N@DM-900. (g) Si 2p of Si-O/N@DM-800 and Si-O/N@DM-900.
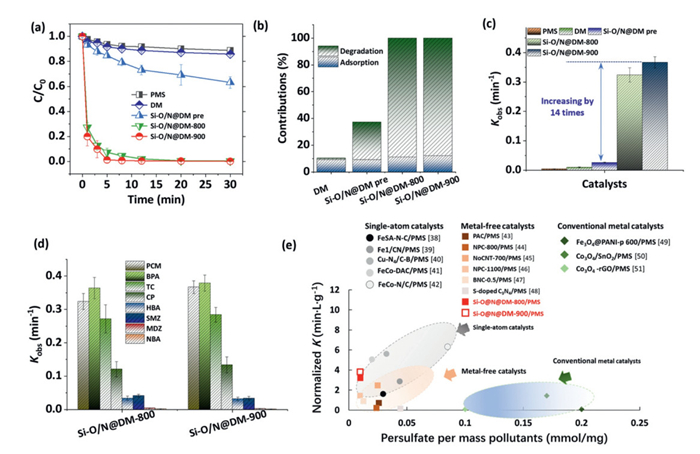
Degradation of acetaminophen (PCM) by different catalyst samples (e.g., pristine DM, Si-O/N@DM pre, Si-O/N@DM-800, and Si-O/N@DM-900) as well as PMS alone was shown in Fig. 2a. Only 11.5% of PCM was degraded by the PMS (0.1 mmol/L) alone system. In contrast, both Si-O/N@DM-800, and Si-O/N@DM-900 could achieve 100% of PCM removal with 8 min with PMS usage of 0.1 mmol/L. Si-O/N@DM-900 with higher graphitic N contribution exhibited the relatively higher PMC uptake, which indicated that the synergy of Si-O binding and graphitic N would facilitate the degradation process. However, Si-O/N@DM pre (prepared at 800 ℃) and DM only achieved 36.9% and 12.9% of PCM removal, respectively. The adsorption of PCM by these catalysts was also evaluated and were shown in Fig. S4 (Supporting information). The adsorption capacities of these catalysts were approximately 9%. As a result, the adsorption process contributed to < 12% of the total removal capacity for both Si-O/N@DM catalysts (Fig. 2b). The degradation rates (Kobs) of these catalysts were further calculated (Fig. 2c), which indicated that the Kobs result (0.367 min-1) of Si-O/N@DM-900 was almost 14 times higher than that of Si-O/N@DM pre. These results confirmed the superior catalytic performances of Si-O/N@DM for pollutant degradation. Degradation of PCM in terms of pH conditions showed that the Si-O/N@DM-900 was stable in range pH conditions for the degradation processes (Fig. S5 in Supporting information), which exhibited its strong anti-interference capacity towards the environmental media [13].
Figure 2
Figure 2.
(a) Degradation of PCM by different catalyst samples as well as PMS alone. (b) Contribution of degradation and adsorption to the PCM removal in different systems. (c) Degradation rates (Kobs) of different catalysts. (d) Degradation curves of different pollutants by the Si-O/N@DM-800, and Si-O/N@DM-900. (e) Comparison of degradation performances with single-atom catalysts, metal-free catalysts, and conventional metal oxides [38–51]. Reaction condition: [catalyst] = 0.1 g/L, [PMS] = 0.1mmol/L, [pollutants] = 10 mg/L.
Furthermore, degradation of different pollutants by the Si-O/N@DM-800, and Si-O/N@DM-900 was conducted in a low catalyst usage (0.1 g/L) and PMS usage (0.1 mmol/L) condition (Fig. 2d, and Fig. S6 in Supporting information). Results shown in Fig. 2d indicated that both catalysts exhibited similar degradation performances towards different pollutants. Degradation of electron-donating pollutants (e.g., PCM, bisphenol A (BPA), tetracycline (TC)) exhibited a higher Kobs range (> 0.25 min-1). In addition, comparison of degradation performances with single-atom catalysts, metal-free catalysts, and conventional metal oxides was given in Fig. 2e. Results indicated that the Si-O/N@DM-800 and Si-O/N@DM-900 exhibited a superior degradation activity similar to those of single-atom catalysts, and far exceeded those of metal-free catalysts and metal oxides [11,13]. These results indicated that the synergy between Si-O bonding and graphitic N enables exceptional Fenton-like activity rivaling single-atom catalysis. In addition, Table S1 presents comparative data on preparation costs, active site density, single-site efficiency, and microscopic reaction mechanisms. These data demonstrate that Si-O/N@DM-800 and Si-O/N@DM-900 exhibit advantages across all evaluated parameters compared to recently reported catalysts for PMS activation.
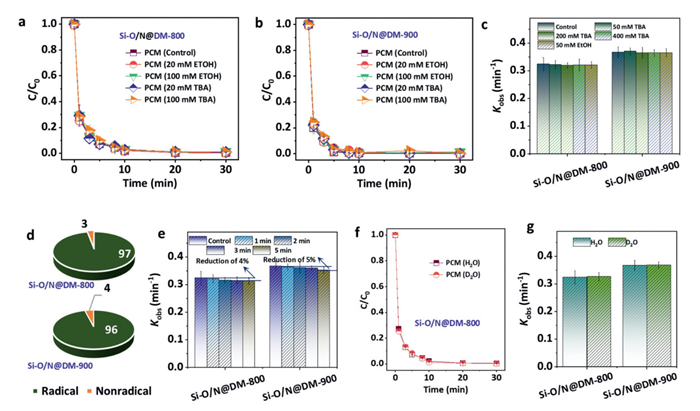
Degradation mechanism of the PCM by the Si-O/N@DM-800/PMS and Si-O/N@DM-900/PMS systems was assessed via the quenching tests. Results showed that the degradation rates of PCM by different concentrations of EtOH and TBA (20–100 mmol/L) was almost in a constant range in both Si-O/N@DM-800/PMS and Si-O/N@DM-900/PMS systems (Figs. 3a-c). Contribution of radical/nonradical pathways towards the PCM degradation in the Si-O/N@DM-800/PMS and Si-O/N@DM-900/PMS systems were identified (Fig. 3d), and the results showed that radical oxidation only contributed to 3%−4% of PCM degradation [3]. These results indicated that the degradation of PCM in the Si-O/N@DM/PMS systems were mainly based on the nonradical pathways. This phenomenon was further confirmed by the premixing tests (1–5 min), as shown in Fig. 3e. The premixture of PMS and Si-O/N@DM-800 (or Si-O/N@DM-900) showed only slight effect on the PCM removal, which indicated that the pre-consumption of radicals via activating PMS by the catalyst would not affect the degradation performances. As a result, radical oxidation was not essential to the PCM degradation in the Si-O/N@DM-800/PMS and Si-O/N@DM-900/PMS systems.
Figure 3
Figure 3.
(a-c) Degradation rates of PCM by different concentrations of EtOH and TBA (20–100 mmol/L) in both Si-O/N@DM-800/PMS and Si-O/N@DM-900/PMS systems (mM =mmol/L). (d) Contribution of radical/nonradical pathways towards the PCM degradation. (e) Kobs results in premixing tests (1–5 min). (f-g) PCM degradation using the D2O as the probe. Reaction condition: [catalyst] = 0.1 g/L, [PMS] = 0.1 mmol/L, [pollutants] = 10 mg/L.
Furthermore, the role of 1O2 on the PCM degradation was also determined via using the D2O as the probe, which showed that the PCM degradation in the D2O solution almost kept constant with that in H2O solution (Figs. 3f and g, and Fig. S7 in Supporting information) [3,34]. This result indicated that the 1O2 pathway could be excluded in the Si-O/N@DM-800/PMS and Si-O/N@DM-900/PMS systems. Since both the radical and 1O2 pathways exhibited a negligible role in the metal-free Si-O/N@DM/PMS systems, other pathways (i.e., electron transfer process (ETP)) should be considered in resulting catalytic systems.
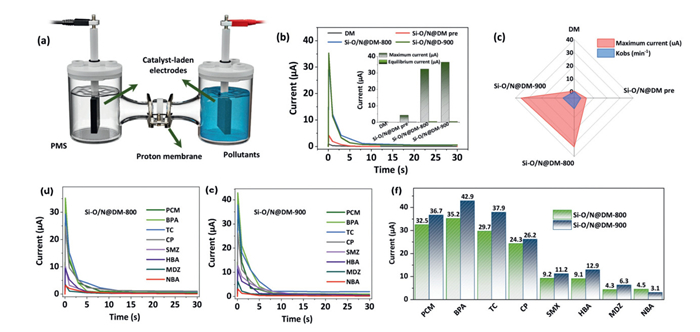
Electrochemical tests were used for identifying the ETP pathway in the Si-O/N@DM-800/PMS and Si-O/N@DM-900/PMS systems. Since the degradation of pollutants could be conducted by the electron transfer rather than the direct contact between the pollutants and PMS or radicals triggered by PMS, proton membrane system with two separated units (PMS system and pollutant system, 50 mL for each unit) connecting via the electric wire and proton membrane was designed for the assessing the ETP oxidation (Fig. 4a). The electrode was coated with different catalysts, and results showed that both Si-O/N@DM-800/PMS and Si-O/N@DM-900 exhibited a stronger current (32.5–36.7 µA), as shown in Fig. 4b. In contrast, Si-O/N@DM-pre and DM exhibited a very low current, and the current change of these catalyst-coated electrodes also corresponded well to their degradation performances towards PCM (Fig. 4c). These results confirmed the strong ETP pathway induced by the Si-O/N@DM-800 and Si-O/N@DM-900. The current changes with the addition of different catalysts in both Si-O/N@DM-800 and Si-O/N@DM-900 laden electrodes were shown in Figs. 4d and e. Results exhibited that the electron-donating pollutants could trigger stronger ETP oxidation so as to achieve a higher degradation performance (Fig. 4f). In addition, Si-O/N@DM-900 exhibited a stronger ETP oxidation towards all kinds of pollutants, which indicated Si-O binding integrated with graphitic N could facilitate the electron transfer for PCM degradation.
Figure 4
Figure 4.
(a) ETP-based system using proton membrane. (b) Current change by using different catalyst-coated electrodes. (c) Radar map of the catalytic performances and currents of different catalysts. (d) Curves of current changes for different pollutants by using the Si-O/N@DM-800 coated electrode. (e) Curves of current changes for different pollutants by using the Si-O/N@DM-900 coated electrode. (f) Comparisons of current changes for different pollutants between the Si-O/N@DM-800 and Si-O/N@DM-900 coated electrode.
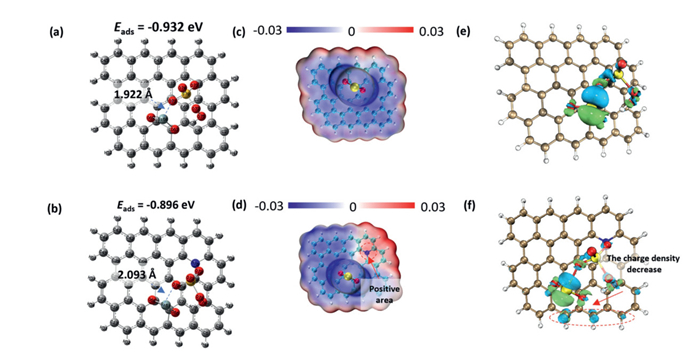
Density functional theory (DFT) calculations were employed to investigate the interactions between PMS and Si-O/N@DM, with the results presented in Fig. 5. The adsorption configurations of PMS on Si-O@DM and Si-O/N@DM are shown in Figs. 5a and b. The adsorption energies (Eads) of PMS on the Si-O@DM and Si-O/N@DM catalysts were found to be similar, with values of −0.932 eV and −0.896 eV, respectively. Additionally, the Si-O bond lengths between PMS and the two catalysts were also comparable, measuring 1.992 Å and 2.093 Å, respectively. These results indicate strong interactions between silicon atoms and the oxygen atom in PMS [52]. The electrostatic potential (ESP) distribution for Si-O@DM and Si-O/N@DM is depicted in Figs. 5c and d, showing negative potentials around oxygen, silicon, and carbon atoms, while nitrogen exhibited a positive ESP region. The positive ESP value of nitrogen suggests that its incorporation enhances the activation of the carbon surface, potentially synergizing with silicon atoms to promote the activation of PMS [52,53]. The differential charge density analysis (Figs. 5e and f) reveals partial electron transfer from PMS to both Si-O@DM and Si-O/N@DM, confirming the activation of PMS by these catalysts. The reduction in charge density on the carbon lattice further indicates that nitrogen facilitates electron transfer. The interaction region indicator (IRI), a wave function analysis method, provides insights into bonding conditions and weak interactions within molecular structures [54]. In this study, IRI surfaces were generated using the Multiwfn wave function analysis software [55]. Chemical bonds and weak interactions are visualized using the standard coloring method, as shown in Fig. S8 (Supporting information). Further analysis via the IRI method revealed that chemical bonds form between silicon atoms and oxygen atoms in PMS, while van der Waals interactions occur between PMS and carbon atoms. These combined interactions enhance the adsorption and activation of PMS on the Si-O/N@DM surface.
Figure 5
Figure 5.
Configuration of (a) Si-O@DM and (b) Si-O/N@DM. ESP of (c) Si-O@DM and (d) Si-O/N@DM. Differential charge of PMS adsorbed on (e) Si-O@DM and (f) Si-O/N@DM.
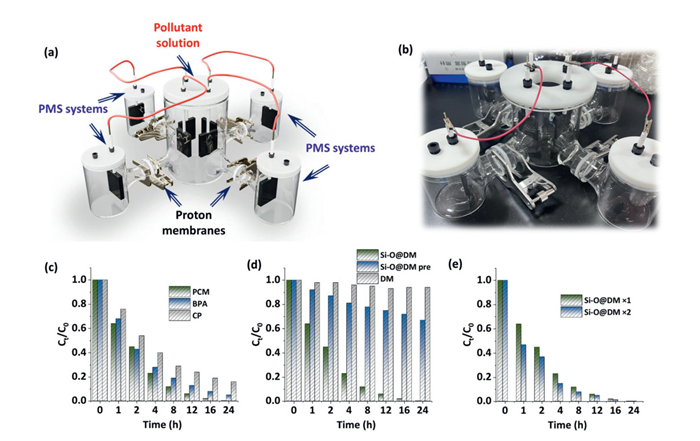
To identify the potential application of ETP pathway for enlarged system, the degradation of various pollutants was further conducted in an enlarged ETP-based modules with one treating system (1 L) connecting with four PMS systems (200 mL each) via the proton membranes (Fig. 6a). The actual photo of resulting module was given in Fig. 6b. The electrode (40 mm × 40 mm × 3 mm) was coated with the Si-O/N@DM-900 catalyst. This treating module could (i) solve the problem of unstable electron transfer process due to insufficient stability of agar used in traditional salt bridge device, and (ii) avoid the release of SO42- into the treating system. The degradation of various pollutants via the resulting module indicated that the PCM could be completely removed via 24 h of reaction, while only 5% and 12% of BPA and p-chlorophenol (CP) were still left after 24 h (Fig. 6c). Comparison of different catalysts (Si-O/N@DM-900, Si-O/N@DM-800, Si-O/N@DM pre, and DM) for the degradation of PCM was shown in Fig. 6d and Fig. S10 (Supporting information). The pristine DM exhibited a very low PCM removal (< 5%) via 24 h, and the removal performance could be increased to approximately 35% by using the Si-O/N@DM pre. In contrast, the removal performance of Si-O/N@DM-800 was only slight lower than that of Si-O/N@DM-900 (Fig. S9 in Supporting information). These results indicated that the inert SiO2 must be etched away; otherwise, it will substantially affect the removal efficiency.
Figure 6
Figure 6.
(a, b) Enlarged ETP-based modules with one treating system (1 L) connecting with four PMS systems (200 mL each) via the proton membranes. (c) Degradation of various pollutants via the resulting module. (d) Comparison of different catalysts (Si-O/N@DM, Si-O/N@DM pre, and DM) for the degradation of PCM. (e) Degradation of PCM via increasing the amount of Si-O/N@DM catalyst coated on the surface of electrode.
The amount of Si-O/N@DM-900 catalyst coated on the surface of electrode was further increased by 2 times, and the results were shown in Fig. 6e. The removal efficiency could be improved to a certain extent; however, it did not increase exponentially with the addition of catalyst load. This phenomenon may be attributed to the overlay of catalyst particles, which partially obscured the active sites [13]. Consequently, enhancing the electrode area or increasing the number of electrodes would likely be more effective in improving catalytic removal capacity rather than the increasing catalyst loading. It is worth noting that, although this study has demonstrated good application potential at the laboratory scale, scaling-up proton exchange membrane reactors must carefully consider inherent challenges such as durability and longevity issues, material costs and resource limitations, as well as system integration complexities.
In summary, the Si-O/N@DM-800 and Si-O/N@DM-900 catalysts demonstrated superior degradation activities comparable to those of single-atom catalysts and significantly surpassed those of metal-free catalysts and metal oxides. These findings suggest that the synergistic effect between Si-O bonding and graphitic nitrogen enhances Fenton-like activity to levels rivaling single-atom catalysis. Moreover, this synergy triggers robust ETP oxidation of electron-donating pollutants. Additionally, we designed a proton membrane reactor integrated with a four-channel PMS system, which not only expands the ETP pathway for pollutant degradation but also effectively addresses sulfate pollution issues in water caused by traditional Fenton systems.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Lifei Hou: Writing – original draft, Software, Formal analysis, Data curation, Conceptualization. Siyuan You: Writing – original draft, Visualization, Data curation, Conceptualization. Rui Li: Data curation. Haoyun Lu: Software, Data curation. Yanan Shang: Writing – review & editing, Supervision, Funding acquisition, Conceptualization. Xing Xu: Writing – review & editing, Supervision, Conceptualization.
Acknowledgments
The research work was supported by National Natural Science Foundation of China (Nos. 52170086, 52300056) and Natural Science Foundation of Shandong Province (Nos. ZR2021ME013, ZR2023QE274). The authors also want to thank Conghua Qi from Shiyanjia Lab (www.shiyanjia.com).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111903.
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580–592. doi: 10.1002/jcc.22885
Figure 1
(a) SEM images of Si-O/N@DM-900. (b) Elemental mappings of Si-O/N@DM-900. (c) TEM images of Si-O/N@DM-900. (d) Quantity adsorption of N2 by different catalysts. (e) BET surface areas and pore volumes of both Si-O/N@DM-800, and Si-O/N@DM-900. (f) XPS N 1s of Si-O/N@DM-800 and Si-O/N@DM-900. (g) Si 2p of Si-O/N@DM-800 and Si-O/N@DM-900.
Figure 2
(a) Degradation of PCM by different catalyst samples as well as PMS alone. (b) Contribution of degradation and adsorption to the PCM removal in different systems. (c) Degradation rates (Kobs) of different catalysts. (d) Degradation curves of different pollutants by the Si-O/N@DM-800, and Si-O/N@DM-900. (e) Comparison of degradation performances with single-atom catalysts, metal-free catalysts, and conventional metal oxides [38–51]. Reaction condition: [catalyst] = 0.1 g/L, [PMS] = 0.1mmol/L, [pollutants] = 10 mg/L.
Figure 3
(a-c) Degradation rates of PCM by different concentrations of EtOH and TBA (20–100 mmol/L) in both Si-O/N@DM-800/PMS and Si-O/N@DM-900/PMS systems (mM =mmol/L). (d) Contribution of radical/nonradical pathways towards the PCM degradation. (e) Kobs results in premixing tests (1–5 min). (f-g) PCM degradation using the D2O as the probe. Reaction condition: [catalyst] = 0.1 g/L, [PMS] = 0.1 mmol/L, [pollutants] = 10 mg/L.
Figure 4
(a) ETP-based system using proton membrane. (b) Current change by using different catalyst-coated electrodes. (c) Radar map of the catalytic performances and currents of different catalysts. (d) Curves of current changes for different pollutants by using the Si-O/N@DM-800 coated electrode. (e) Curves of current changes for different pollutants by using the Si-O/N@DM-900 coated electrode. (f) Comparisons of current changes for different pollutants between the Si-O/N@DM-800 and Si-O/N@DM-900 coated electrode.
Figure 5
Configuration of (a) Si-O@DM and (b) Si-O/N@DM. ESP of (c) Si-O@DM and (d) Si-O/N@DM. Differential charge of PMS adsorbed on (e) Si-O@DM and (f) Si-O/N@DM.
Figure 6
(a, b) Enlarged ETP-based modules with one treating system (1 L) connecting with four PMS systems (200 mL each) via the proton membranes. (c) Degradation of various pollutants via the resulting module. (d) Comparison of different catalysts (Si-O/N@DM, Si-O/N@DM pre, and DM) for the degradation of PCM. (e) Degradation of PCM via increasing the amount of Si-O/N@DM catalyst coated on the surface of electrode.

 DownLoad:
DownLoad:

 下载:
下载:







 下载:
下载:
