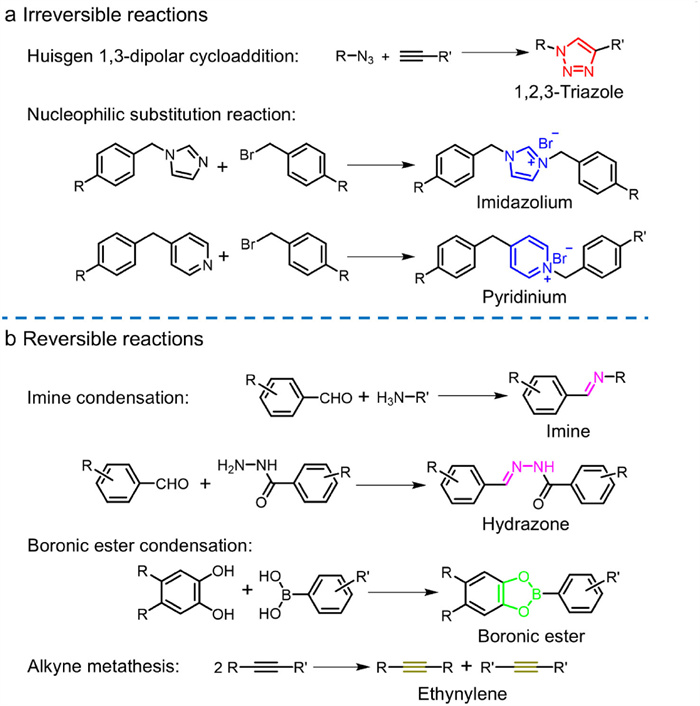
Figure 1.
Summary of reactions used to synthesize interlocked covalent organic cages: (a) irreversible reactions and (b) reversible reactions.
Interlocked covalent organic cages: Design, synthesis, and self-assembly
Bin Yao , Yao Bu , Hongfei Sun , Guowang Li , Xianying Wu , Wei Wang

Interlocked and entwined behaviors are widely observed in biomacromolecules such as oligonucleotides and proteins [1]. To gain fundamental insights into the structure-function relationships of these bioactive macromolecules, diverse mechanically interlocked molecules (MIMs) are prepared, including rotaxanes [2-5], molecular knots [6,7], and catenanes [8-10]. Catenanes, generally composed of two or more interlocked macrocycles, are an important class in MIM family to construct artificial switches, rotary motors, and sensors [11,12]. In catenane structures, the interlocked macrocycles are not directly linked by chemical bonds, and one or more covalent bonds require disconnection to make them individual molecular entities. Since Frisch and Wasserman initially reported the first [2]catenane via an acyloin condensation reaction and investigated its chemical topology in the early 1960s [13,14], significant progresses have been witnessed in this area. Many methods to synthesize catenanes have been developed, and higher order [n]catenanes or entwined catenanes have also been prepared [15-17].
For the first forty years of catenane history, researchers focused primarily on different interlocked rings. Until 1999, three-dimensional (3D) catenane, that is interlocked cage, become a member of catenane family. Fujita et al. [18] designed and synthesized two triply interlocked coordination cages through coordination of Pd2+ or Pt2+ cations with two distinguished pyridine derivatives of triazine. The driving force for interlocked dimers over their monomeric cage counterparts was found to be π–π stacking interactions between the triazine rings. Metal−ligand coordination bonds are endowed with the advantages of high directionality, bonding energy, and dynamic tunability [19-21], thus metal coordination interlocked cages proliferated rapidly following this pioneer work; examples bearing different transition metal ions and various topologies have since been synthesized [22-26].
In sharp contrast, purely organic interlocked cages are paid little attention, primarily because of the great challenge to synthesize. There are three reasons for this difficulty. Firstly, conversion and selectivity are lower for organic reactions than those for coordination ones, since there are more competing side reactions. Secondly, the dimensional stability of pure organic cages is relatively low due to their lack of highly oriented, rigid metal coordination bonds. Thirdly, the interactions in purely organic systems are normally weaker than those in metal-organic architectures, leading to weak driving forces to induce interlocked structure. However, interlocked covalent organic cages have their unique advantages. There is no risk of heavy metal toxicity in biological applications of purely organic cages. Choosing suitable monomers gives a tunable, and much wider, range of reversibility in organic reactions. Critically, interlocked organic cages move more freely during self-assembly, resulting in more sophisticated supramolecular structures. In view of these special superiorities, tremendous efforts finally lead to the first preparation of interlocked covalent organic cages in 2009 [27].
Since then, various reversible and irreversible reactions (Fig. 1) have been employed to prepare interlocked covalent organic cages bearing different topologies and symmetries (timeline shown in Fig. S1 in Supporting information). Fig. S2 (Supporting information) represents the topology design for these interlocked cages. Multidentate monomers, generally containing C3- and C2-symmetry and bearing various functional groups, were utilized to construct interlocked cages with different topological structures, such as interlocked trigonal-prism, trigonal-bipyramid, tetrahedron, cube, and polyhedron. Surprisingly, when monomeric cages were assembled into interlocked structures, the resulting interlocked organic cages were mostly observed to adopt triply interlocked or interpenetrated patterns in spite of the different topologies, functional groups, and sizes of these monomers. Theoretically, the triple interlocked structures were a result of delicate balance of numerous interactions, including π-π interactions, hydrogen bonding, dipole-dipole interactions.
Remarkably, catenanes [28,29], organic cages [30-35], and interlocked coordination cages [36-39] have been well reviewed in the past. Although interlocked covalent organic cage is occasionally mentioned [36,40], only a limited number of cases are covered, thus no reviews systematically summarize their design, synthesis, and self-assemblies. Given their unique structural features and significant contributions to MIM fields, this article systematically summarizes the recent progresses of interlocked covalent organic cages. Notably, the assignment of interlocked structures was verified by different analytical techniques, including 1D nuclear magnetic resonance (1H NMR and 13C NMR), gel permeation chromatography (GPC), electrospray ionization mass spectrometry (ESI-MS) or matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS), single-crystal X-ray diffraction (SC-XRD), and various 2D NMR experiments, such as 1H-1H correlation spectroscopy (1H-1H COSY), diffusion ordered spectroscopy (DOSY), nuclear overhauser effect spectroscopy (NOESY), heteronuclear single quantum coherence spectroscopy (HSQC), and heteronuclear multiple bond correlation spectroscopy (HMBC). SC-XRD provides the most definitive evidence of interlocked structure, but not all cages form crystals yielding single-crystal diffraction results with sufficient resolution. Unless otherwise specified, the characterization details of interlocked structures will not be specially discussed since each work generally uses different combinations of the techniques listed above. This review primarily focuses on proper selection of reactions and monomers, careful topological design, critical analysis of driving force as well as detailed mechanisms in constructing interlocked structure, and specific self-assembly behaviors in single crystals.
To resolve the above-mentioned difficulties in preparing interlocked covalent organic cages, Beer et al. [27] innovatively designed the first one (4-SO42− in Fig. 2) through the combination of click chemistry and templated synthesis. The click reaction [41] is clean, fast, efficient, and highly selective; template-assisted methodology [28] is widely utilized in preparing mechanically interlocked rings because it provides sufficient interactions to organize the precursor, which must be multi-armed, into a suitable conformation. As illustrated in Fig. 2a, the interlocked cage was prepared by organizing two three-armed components to proper assembly followed by capping two ends. For the preorganization stage, tris-urea functionalized ligand (1 in Fig. 2b) was combined with 0.5 equiv. of sulfate anion (SO42–); SO42– was selected because of its tetrahedral coordination mode and strong binding affinity via multiple hydrogen bonds toward urea functional groups, providing excellent template effect. Afterward, the three azide motifs at two ends of the precursor were capped with 1,3,5-tris(prop-2-ynyloxy)benzene (2) via cupper-catalyzed click reaction of Huisgen 1,3-dipolar cycloaddition on account of its unique features of high conversion and high selectivity [42,43]. Indeed, the preorganization and click reaction were conducted in a one-pot pattern. The triply interlocked capsule (4-SO42−) was isolated with a moderate yield of 18% by preparative TLC (for a summary of the synthesis and molecular configuration, see Table S1 in Supporting information), and its interlocked structure was undoubtedly confirmed by 1H NMR, ESI-MS, and DOSY spectra. To validate the template effect of SO42−, a synthesis substituting it for Cl− was attempted under the same conditions, however, only non-interlocked product (3-Cl−) was detected.
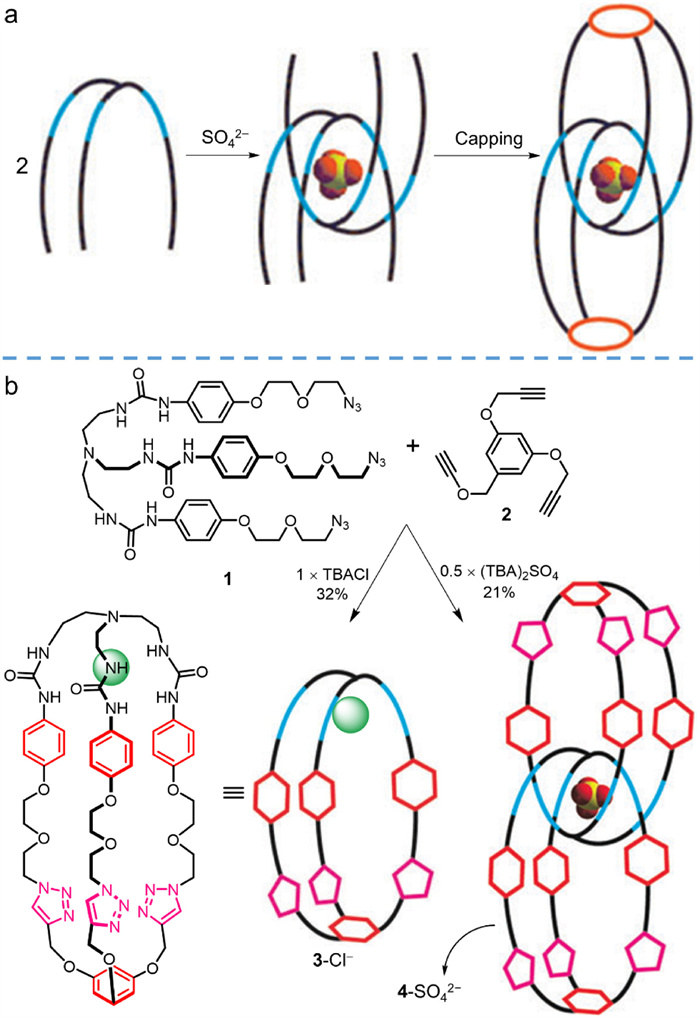
This work supplies an outstanding benchmark for the synthesis of mechanically interlocked covalent organic cages. Nevertheless, the molecular skeleton of 4-SO42− is flexible, rendering it difficult to keep its conformation and size stable. The challenge to explore new efficient reactions and effective building blocks to construct shape-persistent interlocked covalent organic cages remains.
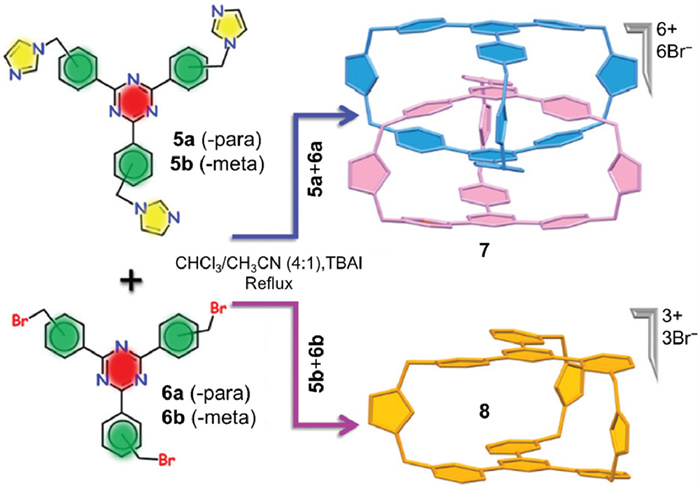
The quest to fabricate interlocked covalent organic cages from irreversible organic reaction encountered diverse challenges, and the next breakthrough would not occur for over a decade. In 2021, Mukherjee et al. [44] found another efficient irreversible reaction that yielded an interlocked imidazolium cage (7 in Fig. 3). 7 was prepared from nucleophilic substitution [45,46] of dilute 5a and 6a (0.235 mmol/L in CHCl3/CH3CN) under refluxing conditions, with tetra-n-butylammonium iodide (TBAI) as a catalyst. 7 was easily isolated with a considerable yield of 26% because of its insolubility in reaction mixture. It was supposed that π–π interactions were the pivotal force driving interlocking, so a mechanism involving a preorganized pathway was proposed. In this mechanism, 5a and 6a pre-organize into a structurally entangled intermediate through π–π stacking, then six imidazolium rings simultaneously cap to form the interlocked structure. To confirm the mechanism, a guest molecule, either 2,4,6-triphenyl-1,3,5-triazine or pyrene, was added to the reaction solution; either prevented the formation of 7. Furthermore, two meta reaction substrates (5b and 6b in Fig. 3) were also tested for interlocked product, but only non-interlocked monomeric cage 8 was detected. Predictably, introducing guest molecules or meta substrates disrupted the π–π stacking between reactants, preventing entangled intermediates from forming.
Highly charged interlocked structures like 7 are soluble in aqueous media, so they supply an excellent platform to investigate the structure-function relationships of interlocked species in biological systems. However, Mukherjee et al. were unable to acquire single crystals of the interlocked cage or entangled intermediate. Starting from similar structures (9 and 6a in Fig. 4a), Stoddart et al. fabricated interlocked pyridinium cages, proposed a much clearer suit[3]ane-based interlocked mechanism [47,48], and elucidated the key intermediates. Unlike the reaction combining 5a and 6a, nucleophilic substitution of 9 and 6a yielded three different cages: one monomeric cage, 10·3Br (41%), and two interlocked cages, the isomers 11a·6Br (23%) and 11b·6Br (13%). Three species were effectively isolated by chromatographic purification, and the triply interlocked cages were well characterized in both solution and solid states, with single crystals prepared by slow vapor diffusion. Taking 11a6+ as an example (Fig. 4b), SC-XRD indicated the four triphenyltriazine platforms adopt an efficient quadruple stacking mode with plane-to-plane distances ranging from 3.3 Å to 3.5 Å, characteristic of efficient π–π stacking interactions. The dihedral angles of two partner cages were close to 60o, alleviating the intensive Coulombic repulsion between charged pyridinium functional groups.
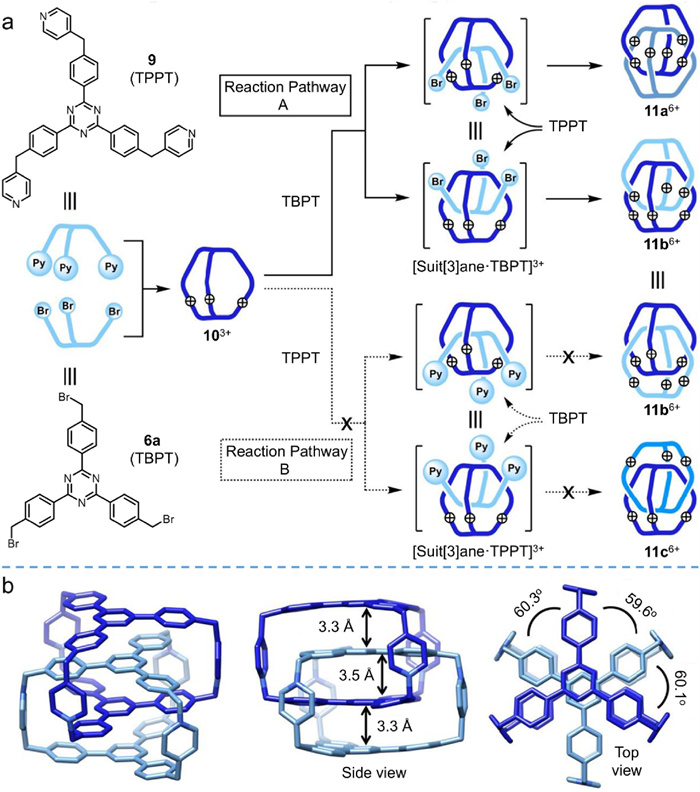
As a matter of fact, the nucleophilic substitution reaction of 9 and 6a would theoretically produce three configurations: 11a·6Br, 11b·6Br, and 11c·6Br (Fig. 4a). 11c·6Br is thermodynamically favored because the charged pyridiniums are most distant from one another. However, only 11a·6Br and 11b·6Br were yielded, indicating that the reaction was kinetically controlled. In contrast to the formation of 7, an encapsulated interlocking pathway containing a suit[3]ane-based intermediate was proposed to rationalize the interlocked formation of 11a6+ and 11b6+. In the first stage of this mechanism, a monomeric cage 103+ is formed from 9 and 6a. Whereafter, a monomer 6a is trapped inside the 103+ cavity to yield [Suit[3]ane.TBPT]3+ (reaction pathway A in Fig. 4a); steric hindrance prevents the competing pathway B (Fig. 4a) in which 9 is trapped. Finally, another monomer 9 reacts with the trapped 6a at two opposite directions of the monomeric cage, generating 11a3+ and 11b3+. Key intermediates were captured and monitored to validate this mechanism. As illustrated in Fig. S3a (Supporting information), the monomeric cage 10·3Br was respectively mixed with 9 and 6a to capture different intermediates, but only [Suit[3]ane.TBPT.3Br] was detected by 1H NMR. Furthermore, after counterion exchange with trifluoroacetic acid (TFA), the resulting Suit[3]ane.TBPT.3CF3CO2 could be isolated by reversed-phase column chromatography. On the contrary, the intermediate characteristic of pathway B, [Suit[3]ane.TPPT.3Br], was not detected. In addition, Suit[3]ane.TPyPT.6CF3CO2 was synthesized by nucleophilic substitution of [Suit[3]ane.TBPT.3Br] with pyridine followed by counterion exchange (Fig. S3b in Supporting information). These results imply that only 6a (TBPT), with less steric hindrance, could be captured by the inner cavity of the monomeric cage 10·3Br.
The biggest advantage of irreversible bond formation is the good chemical stability of interlocked cages it forms. Nevertheless, this approach also has certain disadvantages. Irreversible reactions cannot self-correct if a wrong connection was made, limiting the preparation of large cages. In the above discussed examples, only six new chemical bonds were formed during cage synthesis; there are no reports with a larger number of bonds formed. Moreover, low reversibility leads to more side reactions, lowering yields and requiring complex purification processes.
Interlocked covalent organic cages combine of catenanes and covalent organic cages. The history of both is closely linked to dynamic covalent chemistry [49-51]. The highly reversible character of dynamic covalent chemistry ensures that the bonds newly formed in interlocked structures or cages are capable of error-checking and self-healing ability, allowing functional groups to be adjusted to their proper positions and orientations [52,53]. Together with suitable weak interactions tuned to provide favorable thermodynamic or kinetic conditions, interlocked covalent organic cages can be prepared through a variety of highly reversible chemical reactions.
Several key factors, including conversion, selectivity, reversibility and robustness, are worthy of careful consideration when exploring efficient reactions to construct interlocked covalent molecular cages. Topological diversity and conveniently synthesizable monomers are also essential. Schiff-base chemistry [54-58], generally originating from an aldehyde and an amine, has been widely applied to synthesize various organic monomeric cages bearing different topologies and sizes owing to its outstanding conversion, selectivity, reversibility, and robustness [59-62]. Moreover, derivatization of aldehyde or amine is also easy. Assisted with favorable non-covalent interactions, Schiff-base chemistry shows great promise for synthesizing interlocked molecular cages.
Cooper et al. [63] reported the first series, 14a–c, of interlocked imine cages synthesized by reversible imine condensation with no additional templates (Fig. 5). Starting from the C3-symmetric aldehyde derivative 1,3,5-triformylbenzene (12) and aliphatic amines, imine condensation was conducted in CH3CN at room temperature with TFA as a catalyst. After standing for 7 days, solid products were directly filtered and redissolved in CH2Cl2 to separate insoluble precipitates; evaporation of CH2Cl2 yielded pure products at high yields (40%−60%). Imine condensation tolerated monomers 13a−c, each bearing different substituents; even reactions with mixtures of monomers were successful. The TFA catalyst was believed to play a vital role in producing the interlocked species, since only monomeric cages 15a−c were yielded in its absence. The interlocked cages were further demonstrated to be more thermodynamically favored as the monomeric 15c transformed spontaneously into its interlocked counterpart 14c after 50 days in p-xylene without any catalyst. The acid catalyst principally accelerates the dynamic equilibrium rate of reversible imine formation. This finding demonstrates that the interlocked product can be more thermodynamically stable, in some cases, than its monomeric counterpart.
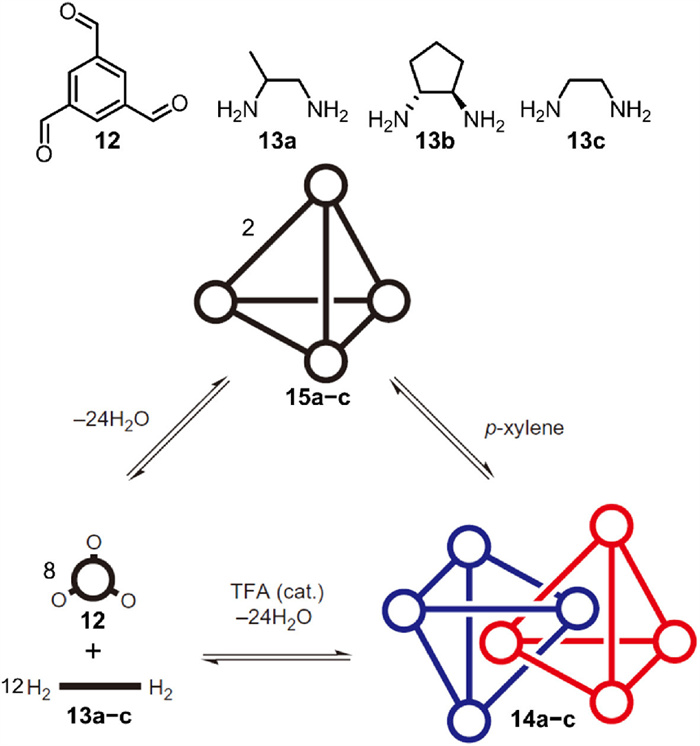
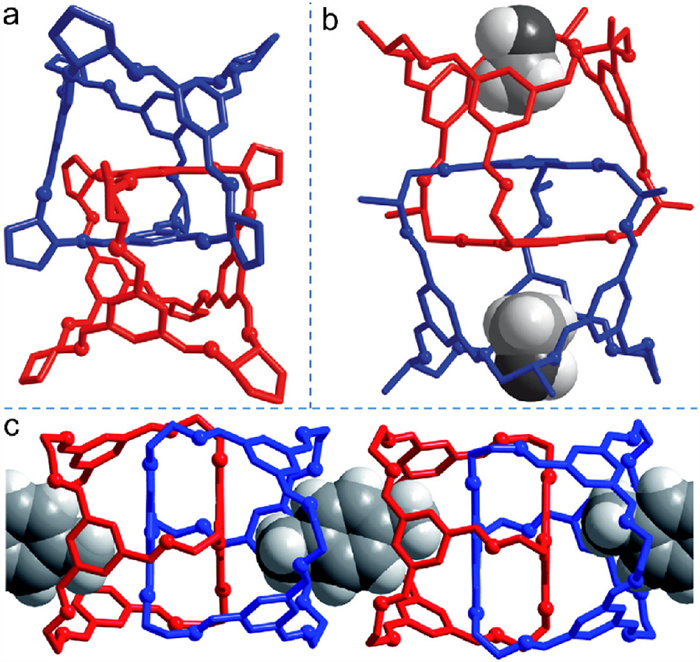
Significantly, single crystals of 14a−c suitable for X-ray diffraction were grown. As outlined in Fig. 6a, the interlocked cage is triply interlocked by two tetrahedral cages, in which three of the four windows in every cage are penetrated by the bisimine linkers. Moreover, each cage accommodates an aromatic ring from its interlocked partner in its inner cavity. The interlocked structure is stabilized by π–π stacking interactions and short aryl-H···C=N contacts between the encaged aromatic ring, and the penetrating imine bonds provide additional stabilization. Each interlocked species has two open, non-penetrated windows at the opposing ends of interlocked dimer, forming two exterior cavities to accommodate other guest molecules. For example, 14a accommodates two methanol molecules (Fig. 6b), and two cavities from adjacent 14c interlocked dimers could share a p-xylene molecule, leading to supramolecular host–guest assembly (Fig. 6c).
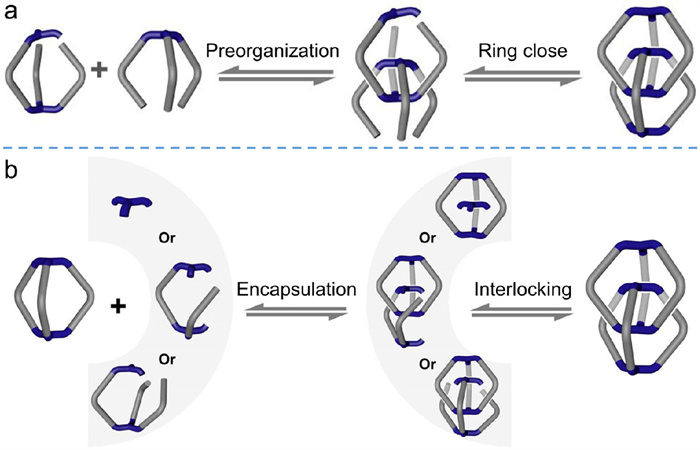
Although Cooper et al. synthesized interlocked imine cages successfully and established π–π stacking interactions as the main driving forces for their formation, detailed catenation mechanism of these interlocked cages is unaddressed. Fig. 7 represents two typical mechanisms for the formation of interlocked cages. One is a preorganized interlocking pathway (Fig. 7a), in which the interlocked cage is formed from a preorganized entangled structure (as in the case of 4-SO42−). The other is an encapsulated interlocking pathway (Fig. 7b). In this pathway, an initial monomeric cage forms; a reactive substrate or intermediate is encapsulated into its cavity; and a final interlocking step generates the catenated structure. Unfortunately, the intermediates involved in Schiff-base chemistry are not synthetically accessible because of high dynamic and reversible features of imine condensation. Zhang et al. [64] creatively designed a series of monomers and investigated the intermediate processes of reversible imine condensation with a cage-breaking experiment assisted by theoretical calculations. This work elucidated the structural influence of building blocks, enthalpic and entropic contributions to intermediate formation, solvent effects, and mechanisms of interlocking.
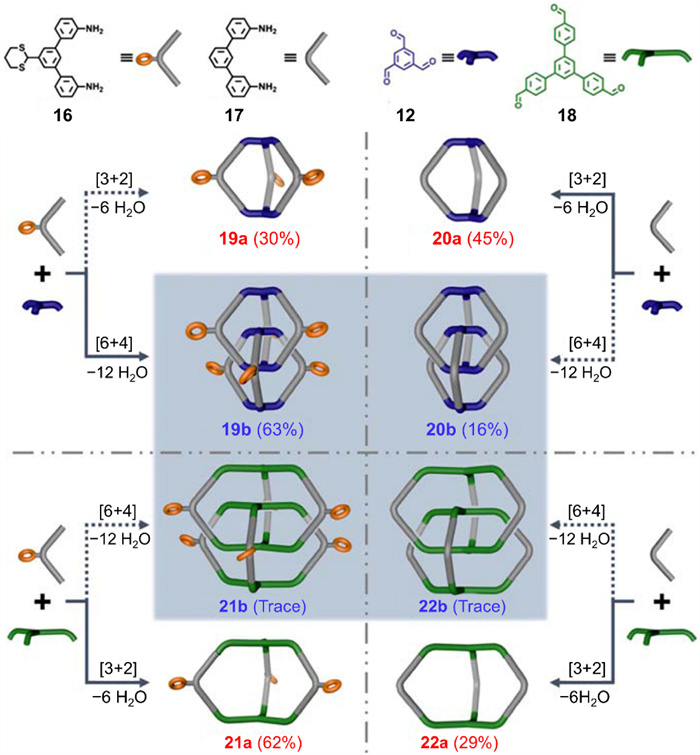
In Zhang’s study, two C2-symmetric aromatic amines (16 and 17 in Fig. 8) were selected as linkers, and two C3-symmetric aromatic trialdehydes (12 and 18 in Fig. 8) were selected as panels. 16 has a sterically hindered dithiane group and 18 bears a flexible skeleton. Cycloimination reactions were carried out using TFA and CHCl3 as a catalyst and a solvent, respectively, and reaction results were indicated by 1H NMR and MALDI-TOF MS. Besides, monomeric or interlocked cages were isolated via flash column chromatography when the yields were considerable. As depicted in Fig. 8, the product distribution of monomeric and interlocked species varies significantly between different amine–trialdehyde combinations. Combining the rigid panel 12 with 16 produced a high yield (63%) of the triply interlocked cage 19b, confirmed by SC-XRD, while combining 12 and 17 produced the interlocked species 20b in much lower yield (16%). Combining the flexible panel 18 with either linker yielded trace amounts of interlocked products, observable only by MALDI-TOF MS. These results imply that a sterically hindered group in the linker and a rigid panel favor the formation of interlocked cages.
To reasonably explain the experimental observations, cage-breaking experiments were performed by adding excess panels to the reaction mixture and tracking intermediates via 1H NMR and MALDI-TOF MS. Moreover, the stability of intermediates was compared by theoretical calculations. In the reaction combining 12 and 16, 12⊂19a was confirmed as the dominant intermediate stabilized by π–π stacking between aromatic panels and triple hydrogen bonds between the carbonyl groups of 12 and the alkyl bonds of the m-terphenyl linker. 12⊂19a was also predicted to be enthalpically favored, leading to a high yield of the interlocked product. The intermediate 12⊂20a, formed in the combination of 12 and 17, was predicted to be less stable than 12⊂19a; it also faced competition from entropically favored uncaged intermediates, yielding more polyimine precipitates (56%) and a low yield (16%) of the interlocked product. These observations reveal that imine reactions thus undergo an encapsulated interlocking pathway when generating interlocked products: a monomeric cage formed first, subsequently captured a panel (12) to afford an enthalpically favored encapsulated intermediate supplying a template-facilitated interlocking mechanism, and finally the interlocked dimer formed gradually. Steric hindrance in the bulky linker improves the solubility of intermediates and prevents them from forming aggregated precipitates. Monomeric cages were the preponderous final products synthesized from the flexible panel 18, and the intermediates contained more disordered segment. Simulations revealed that the intermediates 18⊂21a and 18⊂22a were thermodynamically unfavorable due to the lower enthalpy of solvent-containing monomeric cages. These results are unsurprising since flexible building blocks generally disfavor shape-persistent organic cages.
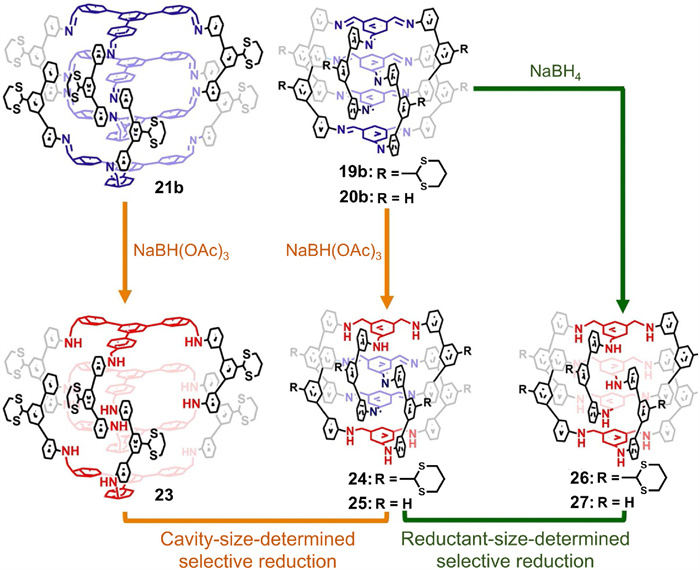
Having ascertained the volume of interlocked cages’ inner cavity by theoretical simulations, catenane with dissymmetric cages (denoted as CDCs) was further prepared via a strategy of selective space-discriminative reduction [65]. As represented in Fig. 9, both the inner cavity volume and the size of reductant play vital roles in determining the final reduction products. For example, CDC 24 was only fabricated from 19b when the size of its reductant (NaBH(OAc)3) did not match the spatial volume of its inner cavity. The bulky NaBH(OAc)3 could not reach 19b’s inner cavity, so it selectively reduced the imine motifs on the outer surface of the interlocked cage. In cases where the reductant was smaller than the inner cavity (e.g., 20b with NaBH4 as a reductant or 21b with NaBH(OAc)3 as reductant), the reductant freely passed into the inner cavity and had access to both exterior and interior imine bonds, leading to non-selective reduction and catenane with symmetric cages (denoted as CSCs).
Compared to the rigid imine bonds in CSCs, the saturated amine bonds endow CDCs with more molecular flexibility at the two end positions of the interlocked cage. Integrating with the effects of lateral substituents, CDCs and CSCs reveal significant differences in their assemblies [66]. 19b was selected as a typical CSC and 24 as a typical CDC. 28, a CDC bearing more steric hindrance, was also synthesized by incorporating additional methyl groups on the periphery (Fig. 10). The self-assembly properties of these interlocked cages in solid states were systematically investigated by SC-XRD. 19b, which features four rigid imine panels, adopts an irregular propeller-like primary structure (Fig. 10a). The m-terphenyl blades twist in different twisting directions (top view), but the relatively rigid imine bonds align the four panels parallel to one another (side view). Two primary structures subsequently aggregate into a supramolecular dimer (secondary structure) in opposite orientations. It is noteworthy that intermolecular π–π stacking between adjacent amine panels is precluded, since the interlocked cages are offset by 9.4 Å. The supramolecular dimers pack together to generate a staggered helical column (tertiary structure), which further organizes into a triclinic crystalline supramolecular (quaternary) structure. The primary and secondary structures of 24 are similar to those of 19b, with an analogous irregular propeller-like configuration and a dimer assembly featuring opposite orientations. However, the increased flexibility of 24 leads to more disorder in the two exterior amine panels, so the supramolecular dimers are bricklike structure (secondary structure, Fig. 10b). These bricklike dimers self-assemble into a monoclinic crystalline phase (tertiary structure) with the lowest symmetry order; it is a continuous, 3D wave- or zigzag-like plank. 28 has a highly symmetric primary structure (Fig. 10c): the m-terphenyl blades of each partner cage rotate in the same directions, and four panels align in despite being composed of different functional groups. This symmetric propeller-like structure self-assembles into supramolecular columns (secondary structure). The interlocked cages in the columns superimpose on one another regularly, facilitating π–π stacking interactions between adjacent amine panels. Finally, the supramolecular columns self-organize into a columnar hexagonal lattice (trigonal crystalline phase, tertiary structure) with the highest symmetry order.
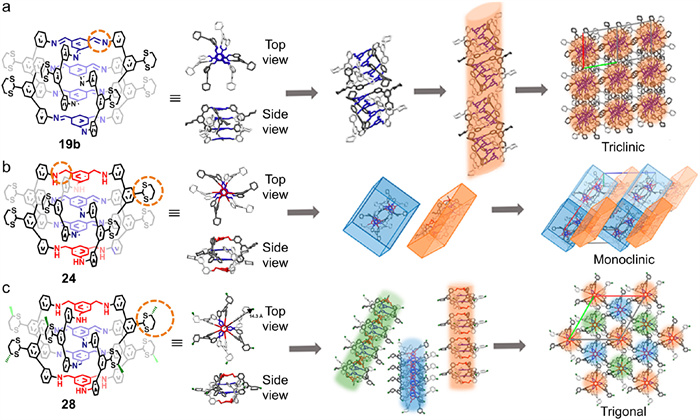
In short, the interlocked 19b cages self-assemble into triclinic crystalline bearing four-level well-defined hierarchical structures, while 24 and 28 self-organize with three-level hierarchical structures, culminating in monoclinic and trigonal crystalline phases, respectively. Significantly, a subtle structural change in interlocked cage changes their primary structure topology dramatically, which further causes the different hierarchical self-assembly behaviors in higher-level assemblies. Interlocked cages’ panels and linkers thus provide a powerful toolbox for designing and constructing diverse complex molecules to explore novel supramolecular structures.
The methyl groups of the dithiane rings in compound 28 are R configurations. Actually, 28 also features other chiral centers, which are the C atoms directly connected to sulfur atoms. The precursors of 29 are also not racemic ((R,S)-29 and (R,R)-29 in Fig. 11a), but the configuration of (R,S)-29 is enantiomeric excess. Nevertheless, only one configuration of interlocked cage was detected in the single-crystal of 28, meaning that a spontaneous chiral resolution occurred during the reaction and crystallization processes. As a consequence, Zhang et al. [67] exquisitely designed a series of experiments to elucidate the spontaneous resolution process. Astonishingly, a pair of homochiral entities were resolved out of 720 possible stereoisomers.
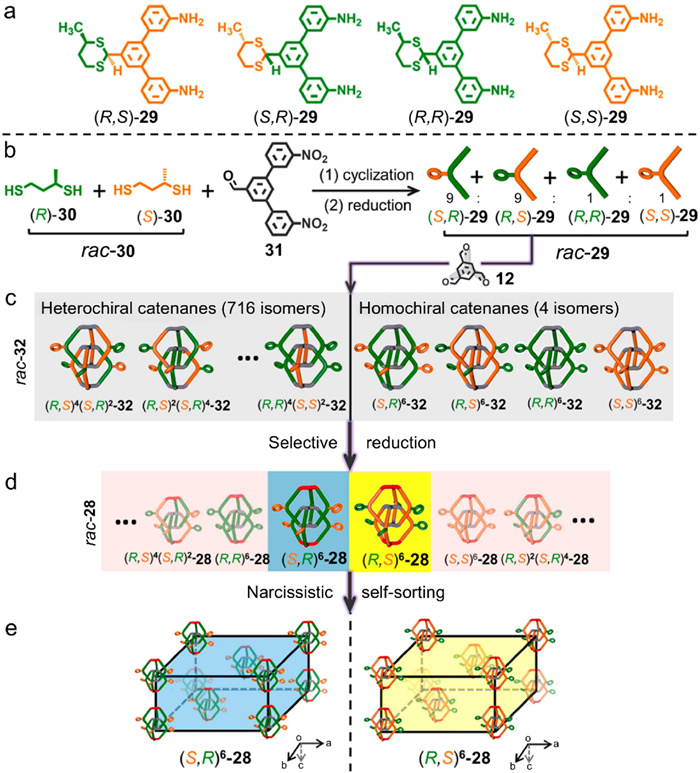
This spontaneous chiral resolution is achieved via two key processes: molecular diastereomeric enrichment followed by narcissistic self-sorting at the supramolecular level (Figs. 11b−e).
Diastereomeric enrichment took place during the preparation of precursor 29 from racemic 30 and 31. The four enantiomers were not equally generated but formed in a proportion of 9:9:1:1 for (R,S)-29, (S,R)-29, (R,R)-29, and (S,S)-29, respectively. This uneven distribution is ascribed to the increased stability of (S,R)-29 and (R,S)-29, in which the chiral methyl groups adopt equatorial positions. The diastereomeric enrichment was demonstrated by a combination of chiral high performance liquid chromatography (HPLC) and circular dichroism (CD). Diastereomeric enrichment at the molecular level is transferred to the interlocked cages when the enriched mixture is utilized as reaction raw materials. After undergoing imine condensation with 12 followed by selective reduction using NaBH(OAc)3 reductant as described previously, racemic CSC 32 and CDC 28 were successively prepared. Narcissistic self-sorting occurred in the crystallization of racemic CDC 28, with only a pair of (R,S)6-28 or (S,R)6-28 crystals were produced individually. Starting from racemic CSC 32, however, only indiscriminate crystallization of enantiomers was observed; no homochiral crystals were formed.
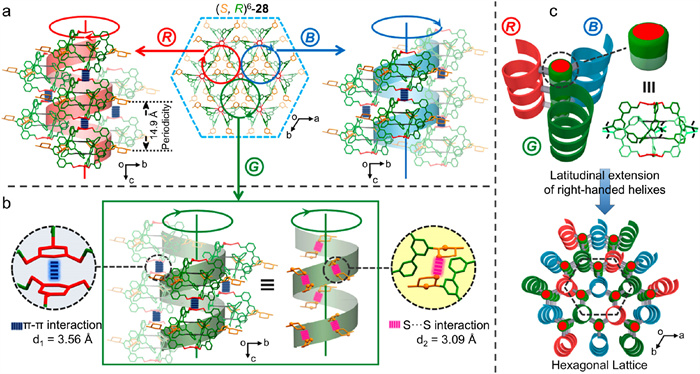
The simultaneous crystallization of different diastereomers might be attributed to the lack of long-range directional interactions between the interlocked cages since the longitudinal π–π stacking interactions are disrupted (as in the case of 19b) when all the panels are connected to other building blocks by rigid imine motifs. On the contrary, the flexible exterior amine panels of 28 are deformable upon crystallization, leading to a highly symmetric conformation that stacks in an eclipsed style through π–π stacking interactions during crystallization. Fig. 12 shows the narcissistic self-sorting of homochiral enantiomers of (S,R)6-28 upon crystallization. (S,R)6-28 stacks in an eclipsed fashion to form right-handed helical supramolecular columns. The helical assembly features both π–π stacking along the c-axis and helically arranged S···S interactions between adjacent dithiane moieties. Only the homochiral (S,R)6-28 could guarantee this exclusive helical arrangement through two synergetic weak interactions. The helical supramolecular columns are further interwoven into a columnar hexagonal lattice via latitudinal extension, achieving spontaneous resolution of homochiral enantiomer from multicomponent enantiomers. Doubtlessly, the coexistence of 28’s interior rigid imine bonds and exterior flexible amine bonds plays an indispensable role in the narcissistic self-sorting during crystallization. The interior rigid imines give stability to the molecular skeleton of 28, while the exterior amines endow the interlocked cage with enough flexibility to adjust the molecular conformation during molecular packing, facilitating effective π–π stacking and S···S interactions. The spontaneous self-sorting resolution process provides not only a reliable route to guide a sophisticated system toward the generation of a specific product or products from a family of equally probable multiple possibilities but also a novel strategy to understand the various self-assembly pathways in natural systems [54,68].
In the above discussed interlocked cages synthesized by imine condensations, only the rigid 1,3,5-triformylbenzene (12) panel was successfully assembled into interlocked cages. The flexible panel 18 yielded only traces of interlocked species requiring detection by MALDI-TOF MS, likely due to its more flexible molecular backbone. Indeed, competition from solvents is not the only impacting factor; the mismatch between the size of molecular conformations and steric hindrance are also worthy of consideration. To resolve this question, Zhang et al. [69] carefully designed a series of monomers with different molecular lengths and steric hindrance to regulate their noncovalent interactions. They not only synthesized various interlocked dimers with high conversion rates using flexible 18 as key panel, but also successfully fabricated an interwoven trimeric cage-catenane (43b in Fig. 13) with topological chirality [70].
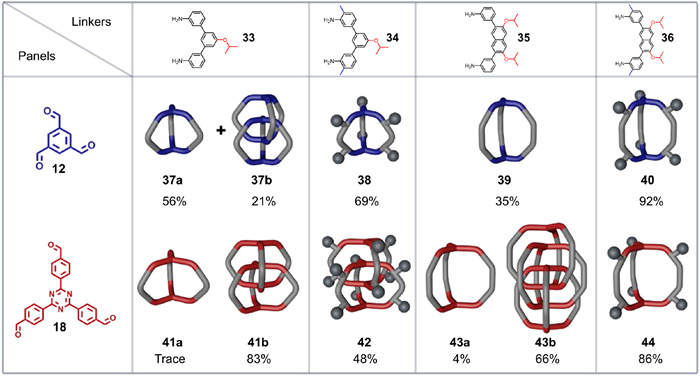
Before experimental implementation, the probability of forming interwoven species was predicted using a refined statistical model considering the template effect supplied by π-π stacking. Combining 18 and 35 was predicted to form predominantly an interwoven trimeric cage-catenane rather than its chain-like interlocked counterpart because of their molecular length, steric hindrance, and solubility. On the basis of this prediction, cycloimination reaction of 18 and 35 was performed in similar conditions as previously mentioned, with TFA catalyst and CHCl3 solvent. As predicted, the interwoven trimeric catenane was purified in high yield (62%) using recycling preparative GPC by virtue of the big discrepancy in hydrodynamic volume of the different species. Single-crystal X-ray diffraction demonstrated an interwoven trimeric catenane structure in which every monomeric cage is triply interlocked and each window of a cage is penetrated by two twisted linkers; six triphenylbenzene panels are aligned in parallel, with a rotation angle close to 40o and a distance of approximately 3.6 Å. The short distances ensure efficient π-π stacking interactions between adjacent triphenylbenzene panels, providing an advantageous template effect to prevent the formation of chain-like catenanes and leading to the predominance of interwoven trimeric cage-catenane.
Precursors’ molecular structure also impacts the distribution of monomeric and interlocked cages (Fig. 13). Starting from the rigid panel 12, only 33 produced an interlocked cage product; other combinations yielded only monomeric cages, probably due to the large steric hindrance of 34−36. From the larger panel 18, product distributions varied significantly between different combinations. In general, increased steric hindrance (in, e.g., 34 and 36) decreases the conversion rate of interlocked cage and favors the formation of monomeric cages. Interlocked species require the combination of various factors, including π–π stacking between panels, linkers’ length, and steric effects. The enhanced π–π stacking interactions of panels is the main driving force for facilitating the interlocking of monomeric cage. The length together with the steric hindrance of linker determine how many panels can be accommodated in the inner cavity of monomeric cage and how many linkers could penetrate a window forming between two linkers, resulting in interlocked dimer and interwoven trimeric catenane. Remarkably, the interwoven trimeric catenane 43b has two configurations that cannot be converted to each other without bond fission, since every cage interlocks with the other two (Fig. S4 in Supporting information). Herein, in contrast to the chain-like counterpart presenting topological achirality by virtue of the relatively free rotation conditions, the interwoven trimeric catenane 43b would exhibit topological chirality [70]. Successfully, two enantiomers were isolated via preparative chiral HPLC, and the topological chirality were undoubtedly proved by CD spectroscopy. This probability analysis-assisted reasonable design strategy opens a new perspective for fabricating topologically sophisticated structures.
Zhao et al. [71] provided an alternative strategy to incorporate flexible 18 into interlocked cages, using a substituent-free aromatic linker. Without steric hindrance effect from substituents, more other interactions need to be incorporated, thus they took multiple π–π interactions between aromatic chromophores into consideration. As shown in Fig. 14a, C3-symmetric 18 and C2-symmetric 45 were combined to produce a trigonal-prismatic interlocked cage 46 under the catalysis of AcOH. Pure interlocked product could be easily isolated in a relatively high yield (45%) via simple dissolution and precipitation operations due to the solubility of different species. The size and structure of C3-symmetric monomers had a considerable effect on the final configurations of the cages. In contrast to the previous examples, only monomer 18 bearing flexible triphenylbenzene panel could produce interlocked product, while other monomers with benzene or triphenylamine cores yielded only monomeric cages. The different reaction results are attributed to the discrepancy in π–π interaction patterns between different building blocks.
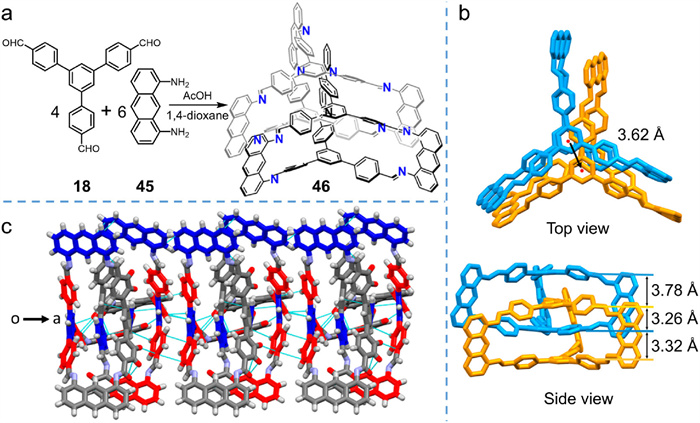
In contrast to 43b, which only has π–π stacking interactions between the panels, the crystal structure of 46 (Figs. 14b and c) features multiple π–π interaction modes that synergistically contribute to interlocked cage formation. The two interlocked partner cages adopt a slippage staggered geometry with a slipped distance of 3.62 Å (Fig. 14b). The triphenylbenzene panels supply multiple π–π interactions, including not only π–π stacking interactions (between the blue benzene of triphenylbenzene in Fig. 14c), but also T-shaped π–π interactions (between the red moieties in Fig. 14c). The anthracene linkers also play indispensable roles, exhibiting T-shaped π–π interactions between triphenylbenzene and anthracene or π–π stacking interactions between anthracene units. The various types of π–π interactions induce close stacking of triphenylbenzene and anthracene units, restrict free rotation of the partner cages, and cause slippage of the triphenylbenzene subunits. In other panels, owing to the small monomeric cage inner cavity (with benzene panel) or to deficient π–π interactions (with triphenylamine panel), only monomeric cages were afforded. Finally, the interlocked cages 46 self-assemble in a “head-to-tail” style along the b axis, generating a two-dimensional layer mode in the bc plane, and the layers are repeated along the a axis to form the single crystal of 46.
Examining the imine-based interlocked cages discussed above, they have one common characteristic: π–π interactions are the dominant forces driving interlocking. In addition, the topology of monomeric cages is generally trigonal-bipyramid or trigonal-prism (summary of molecular configuration, see Table S1). Understanding the driving forces behind the formation of interlocked cage catenanes allows improved selectivity between monomeric and interlocked cages, and expanding topological diversity helps to probe the deep structure-performance relationship in MIMs.
Mastalerz et al. [72] offered various possibilities to construct cubic interlocked cages using other weak interactions rather than π–π interactions. As depicted in Fig. 15a, imine condensation of C3-symmetric tribenzotriquinacene (TBTQ)-salicylaldehyde (+)-(M)-47 and C2-symmetric amine 48 was implemented in chlorobenzene with TFA as a catalyst, yielding a cubic monomeric cage ((M)-49) and its interlocked dimer ((M)-50) simultaneously. The cage-catenane product was isolated by recycling GPC, and the interlocked structure was demonstrated as quadruply interlocked catenane by SC-XRD (Fig. 15b). Moreover, the driving force for the formation of interlocked cage could be elucidated from its single crystal structure. In view of the unique molecular orientation and the great distance between aromatic rings, π–π interactions are completely excluded. The main driving force was subsequently ascribed to the weak hydrogen bonding interactions. The hydrogen bonds (interactions A) between the oxygen atoms of hydroxy groups and the aromatic protons of the TBTQ-units are well shown in Fig. 15c, the spacings (2.5−2.7 Å) and angles (158o−177o) of which nicely fall in the proper regions of weak hydrogen bonds. Though weak in intensity, the larger quantity (16 in each interlocked cage) makes their overall effect substantial. Further integrating with other weak interactions, such as dipole-dipole interactions between the imine-hydrogen atoms (interactions B in Fig. 15c) and attractive interactions between the neighboring oxygen atoms (interactions C in Fig. 15c), these weak interactions synergistically stabilize the interlocked structure.
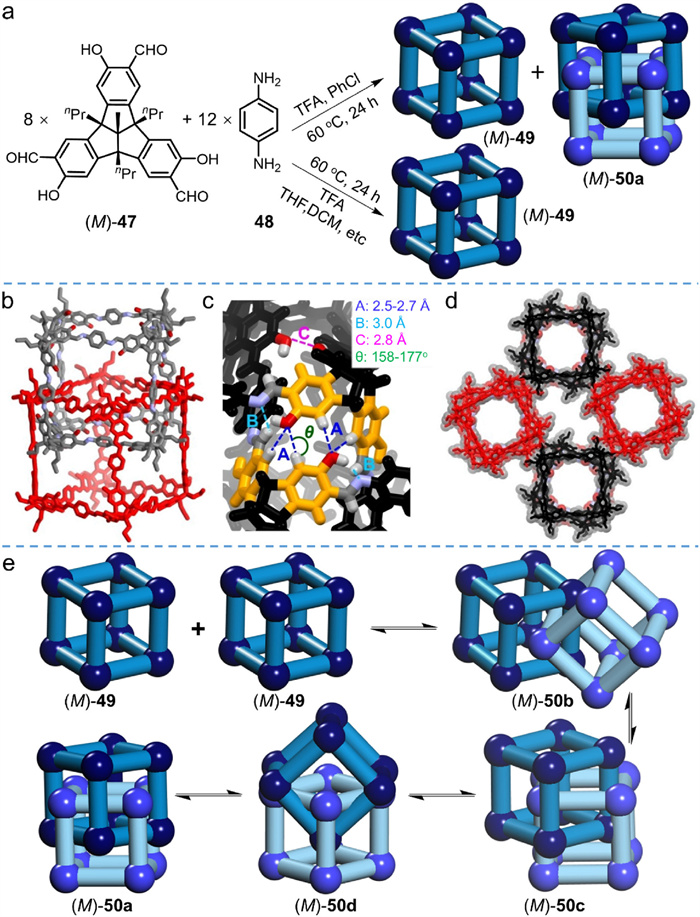
The hydrogen bond-mediated interlocking mechanism is nicely consistent with an observed inverse correlation between solvent polarity and the ratio of interlocked to monomeric cages formed. Less polar solvents (e.g., chlorobenzene, benzene, and toluene) were profitable for the formation of interlocked cages, while more polar solvents (e.g., nitrobenzene, dichloromethane, and 1,2-dichlorobenzene) promoted the formation of monomeric cages. In solvents such as 1,4-dioxane and tetrahydrofuran with strong hydrogen bond receptors, only single monomeric cages were observed, demonstrating the indispensable role of hydrogen bonds in the formation of interlocked structure. The quadruply interlocked dimers further self-assemble into molecular stacks (Fig. 15d) driven and stabilized by intermolecular hydrogen bonds and weak van der Waals interactions between the alkyl chains and heteroatoms. Theoretically, this packing mode is expected to produce a highly porous assembly with two distinctive pores in the solid state. Nevertheless, the porosity was not observed, probably due to the loss of crystallinity and porosity during desolvation process owing to weak interactions.
The interlocked mechanism was further investigated by scrambling experiments. Two monomeric cages, deuterium-labeled at all corners or all linkers respectively, were synthesized; each deuterium-labeled monomeric cage was mixed with normal monomeric cages under synthetic conditions. Interlocked cages were observed via MALDI-TOF MS in both scrambling experiments, but the distribution of molecular weights differed strongly. The combination of normal and corner-labeled cages gave three main MS signals, corresponding to species undeuterated, semi-deuterated, and fully deuterated at the corners. By contrast, combining normal and linker-labeled cages afforded a broad distribution of MS signals, indicating that the deuterium-labeled and unlabeled linkers underwent exchange. This implies that the interlocked cages form from two preformed monomeric cages. Fig. 15e illustrates the proposed mechanism for the formation of interlocked cage (M)-50a from monomeric cage (M)-49 step by step. TBTQ-corner subunits never fully dissociate from a monomeric cage which would require breaking three imine bonds simultaneously. Instead, one 1,4-phenylenediamine linker is likely to release from monomeric cage before each interlocking, requiring the disconnection of only two imine bonds.
Mastalerz et al. [73] also investigated other weak interactions besides hydrogen bonds that could direct the formation of interlocked cages. As shown schematically in Fig. 16, for the ease of synthesis, imine condensations were carried out by reacting C3-symmetric (−)-(P)-51 with various C2-symmetric 1,4-phenylenediamine derivatives (52a−k) bearing different substituents upon the catalysis of TFA. Taking the combination of (−)-(P)-51 and 52a as an example, the imine condensation gave rise to three kinds of cage products: monomeric cage 53a, an interlocked dimer 53b, and an interlocked trimer 53c. Solvent was found to play a subtle role in regulating the formation of different interlocked species as the relative proportions of three products could be well modulated by adjusting the solvent polarity and reaction temperature. Under optimized conditions, 53a, 53b and 53c were isolated in yields of 76%, 46%, and 80%, respectively. The catenated dimer (trimer) were found to be triply interlocked (interwoven) structure via 1D and 2D NMR. Substituent effects were systematically investigated. Condensation of (−)-(P)-51 with substrates bearing bulky substituents (e.g., 52e and 52f) or small weakly polar groups (e.g., 52h and 52i) provided only monomeric cages (Fig. 16a). The failure to yield interlocked products was attributed to the steric effect of bulky substituents and to insufficient weak interactions from small, but weakly polar, groups. Surprisingly, the imine reaction of (−)-(P)-51 and 52g, which possessing highly polar hydroxyl groups, yielded only monomeric cage, probably owing to the mismatched orientation of functional groups or to spacing issue precluding hydrogen bond formation. The other combinations produced interlocked products while 52a and 52b gave both interlocked dimer and trimer.
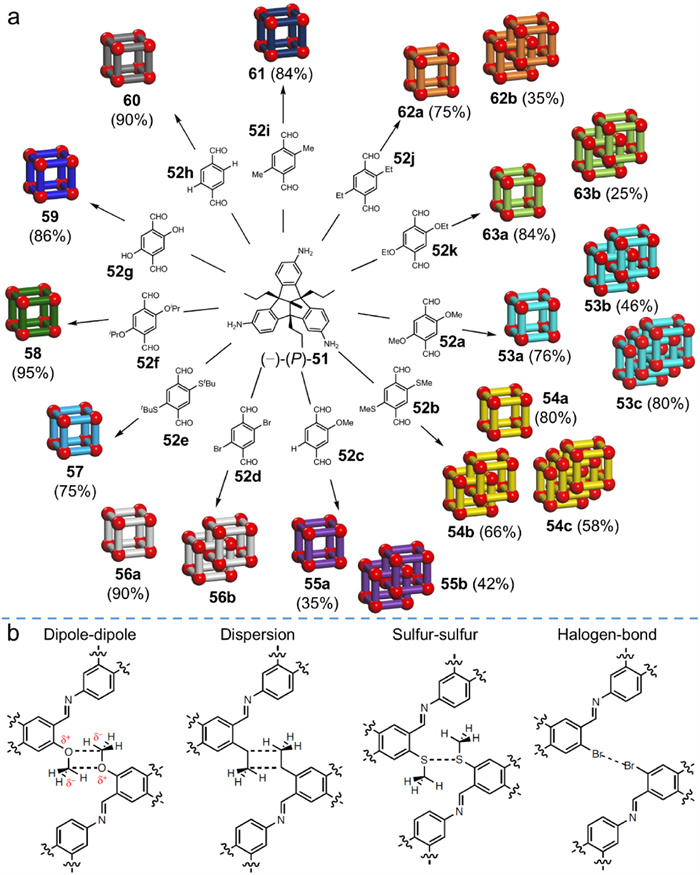
Considering subtle changes in substituents determines the success of synthesizing interlocked products, weak interactions, rather than π–π interactions, were believed to play leading roles. Fig. 16b summarizes the diverse weak interactions that induce the formation of interlocked cages, including dipole-dipole interactions, dispersion interactions, sulfur-sulfur interactions, and halogen bond interactions. A mechanistic investigation on the interlocking process was also performed by mixing unlabeled and 15N-labeled 53a under reaction conditions. The mechanism of interlocking was found to be different from that of (M)-50a. Since MALDI-TOF MS exhibited that the molecular weight distribution showed statistical results, implying monomer 51 was speculated to disconnect from a monomeric cage during the reaction, likely related to the absence of strong hydrogen bonds. The series of works by Mastalerz and coworkers demonstrates that weaker interactions, rather than π–π interactions, are viable as chief driving forces for interlocking processes, opening a broad avenue for fabricating sophisticated MIMs.
By virtue of the highly reversible characteristic and diverse available weak interactions serving as driving forces, imine condensation shows unparalleled advantages in constructing various interlocked organic cages. Nonetheless, a problem should not be overlooked: the imine bonds would undergo hydrolysis upon aqueous conditions. Constructing robust chemical bonds compatible with water is of great importance to mimic biological processes. As discussed above, the hydrophilicity issue could be resolved through introducing charged functional groups. Replacing imine bonds with hydrazone (−C=N−N−) functional groups could overcome the sensitive hydrolysis problem of imine bonds [74]. On one hand, hydrazone bonds are highly reversible under acidic conditions, retaining the ability to realize error-checking and self-correcting. On the other hand, hydrazone C=N bonds are less electrophilic because of the delocalized lone electron pair from the neighbor nitrogen atom and are inert to water [75,76].
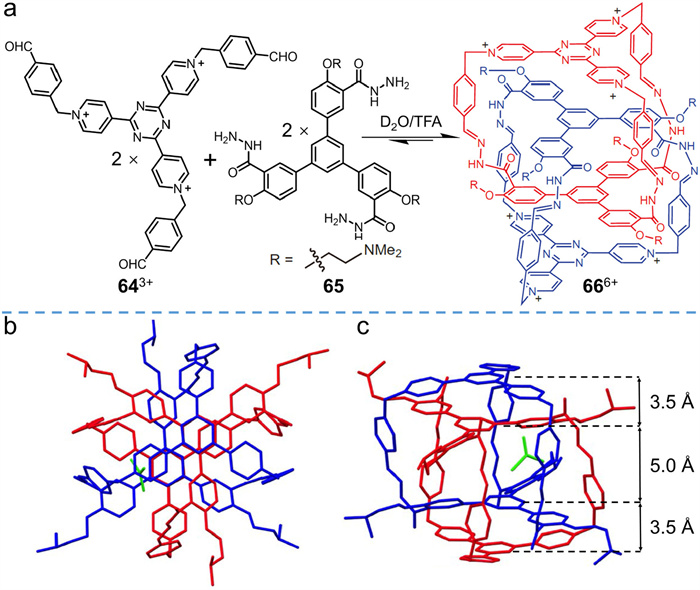
Li et al. [77] prepared an interlocked hydrazone cage via kinetic self-assembly of building blocks in water. In their study, two water-soluble monomers were prepared, including an aldehyde derivative of pyridine salt (643+ in Fig. 17a) and a hydrazine derivative (65 in Fig. 17a). Imine condensation reaction was conducted by mixing two precursors in D2O, facilitating the real-time monitoring of reaction intermediates. Polymers and oligomers were observed at the beginning of the reaction, but the intermediates self-assembled into interlocked species 66 after heating at 70 ℃ for 8 h, as confirmed by 1H NMR, ESI-MS, and HPLC. Single crystals of 66·6Cl were successfully prepared, and SC-XRD analysis (Figs. 17b and c) demonstrated the interlocked structural feature. 66 is triply interlocked and adopts a propeller-like conformation to alleviate steric hindrance. Due to electrostatic repulsion, the tricationic tripyridinium–triazine subunits were found on the outside of the interlocked cage while the two triphenylbenzene panels were encapsulated in the inner cavity of the partner cage. The side view of the crystal structure (Fig. 17c) shows that π–π donor–acceptor interactions (3.5 Å) between benzene and triazine are more favorable than π–π stacking interactions (5 Å) between benzene groups. When the triazine core was replaced with benzene, no interlocked product was observed, further confirming that π–π donor–acceptor interactions are one of the main forces driving interlocking. Hydrophobic effects are also important, since the interlocked dimer disassembled into monomeric cages in strongly polar organic solvents (e.g., DMSO or MeCN). Synergy between the two driving forces ensures that the intermediates self-assemble into interlocked cages under aqueous conditions.
Boronic esters, famous for their high reversibility during the condensation of boronic acid and catechol derivatives, are widely applied for constructing various covalent organic frameworks [78,79]. Through exquisite molecular design, boronic ester condensation also provides a reliable route for covalent molecular cages. Organic molecular cages of different topologies (e.g., tetrahedron, cube, trigonal-bipyramid, octahedron, and cuboctahedron) and different sizes have been reported successively [80-85]. While synthesizing a permanent mesoporous covalent organic cage (69 in Fig. 18) endowed with exceptionally high Brunauer–Emmett–Teller (BET) surface area (3758 m2/g) [86], Mastalerz et al. [87] accidentally made an interlocked organic cage 70b.
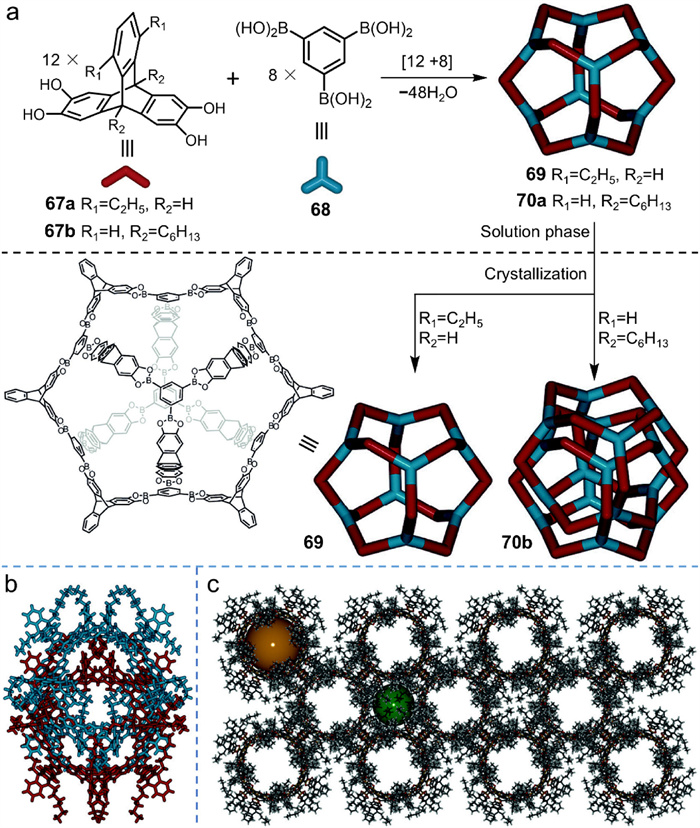
69 was initially fabricated by condensation of triptycene tetraol (67a) and benzene tris(boronic acid) (68). 67a is substituted at 13- and 16-positions that needed tedious synthesis procedure. When the alkyl chains were located instead at the bridgehead positions, synthesizing the modified 67b required only three steps and was much more efficient. Monomeric cage 70a was yielded almost quantitatively in highly dilute chloroform solution. When prepared single crystal of 70a via vapor diffusion method, Mastalerz et al. unexpectedly gained an interlocked cage 70b in a high yield of 62%. Single-crystal X-ray analysis confirmed its quadruply interlocked structure (Fig. 18b). As a contrast, 69 still revealed monomeric cage configuration in the crystal formation although it has the same molecular skeleton as 70b except for containing different alkyl substituents. This implies that the dispersion interactions may be the driving force for the formation of interlocked cage. The short contacts between the alkyl chains and the aromatic rings in 70b support this hypothesis. The alkyl substituents interact with each other or with aromatic rings in the interlocked structure, providing sufficient negative enthalpy to compensate the entropy loss during crystal formation. In addition, 70b is the first reported example of porous molecular catenane. As portrayed in Fig. 18c, the interlocked cages assemble with two different pore types: one intrinsic pore (2.0 nm, orange sphere in Fig. 18c) formed inside the interlocked cage and one extrinsic pore (1.4 nm, brown sphere in Fig. 18c) found between the self-assembled interlocked cages. Further pore distribution analysis from BET experiment agreed well with the single-crystal structure. Significantly, the BET surface area of 70b was measured up to 1742 m2/g (Langmuir model), representing one of the highest values observed for porous organic cages.
In this case, interlocking proceeds by a kinetically interrupted thermodynamic mechanism, while the driving force for interlocking arises from crystalline close packing. Remarkably, 96-fold condensation of 40 molecules are required for the formation of interlocked cage 70b in a one-pot reaction, highlighting the extremely high conversion and high selectivity of boronic ester condensation. Nevertheless, the interlocked cage was only observed in the crystalline form and it rapidly transformed into monomeric cage upon redissolution, likely due to the lability of B−O bonds. Given its vulnerability, research about boronic ester-based interlocked organic cages has not been further developed.
Considering the labile stability of imine or B−O bonds, it is urgent to improve the chemical stability of linkers while maintaining their reversibility. Alkyne metathesis, because of its high rigidity and linearity of ethynylene bonds, has recently emerged as a feasible dynamic covalent reaction to construct conjugated shape-persistent organic cages [88-90]. However, due to high self-exchange and non-directional characteristics, alkyne metathesis is so complicated that it mainly provides non-interlocked cages [91-93]. It was not until 2015 that Zhang et al. [94] designed two peculiar monomers to gain the desired interlocked aryleneethynylene cages (73b and 74b in Fig. 19a).
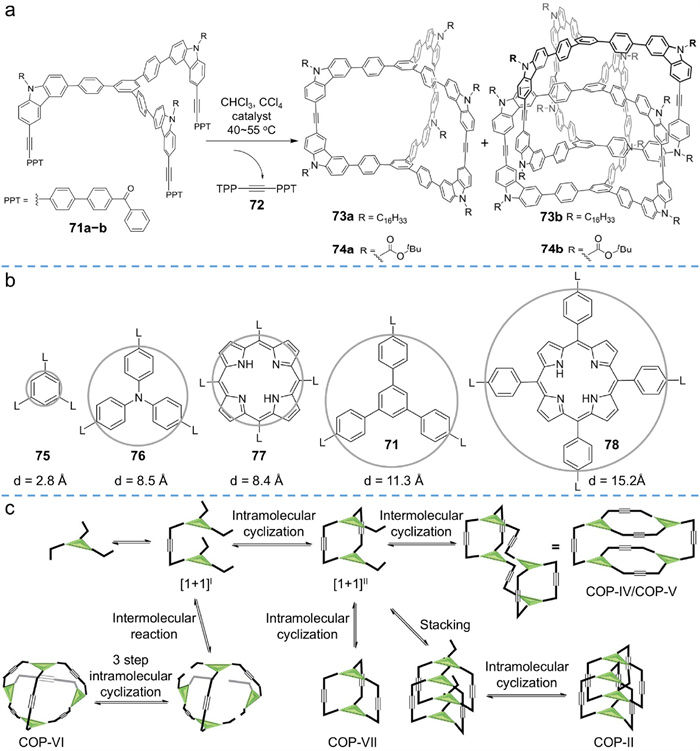
In their study, two C3-symmetric monomers (71a and 71b) were designed, both of which are composed of one panel (a triphenyl benzene as face) and three identical carbazole arms (as edges). 3,6-Disubstitued carbazole motifs lock the face-to-edge angle at around 90o, ensuring proper orientation of the functional groups. The alkyne metathesis was catalyzed by a high active multidentate molybdenum carbyne catalyst, and the equilibrium was driven to favor product formation by precipitating insoluble byproduct 72.
Different from the examples, the interlocked products 73b (90% combined yield with 73a) and 74b (59%) were predominantly formed over their non-interlocked counterparts 73a and 74a (6%). Real-time analysis of the reaction process showed that 74b was predominantly yielded within 30 min and no obvious conversion of monomeric cages into interlocked species was found, indicating that the interlocked species were thermodynamically favored. By virtue of the polar tBoc groups, 74a and 74b were successfully isolated via flash column chromatography. Solvent effects were also tested. CCl4/CHCl3 promoted the formation of interlocked cages, while toluene promoted monomeric cages, probably due to the solvent template effect.
To gain profound insight into the probable mechanism of interlocking, more monomers (75−78 in Fig. 19b) bearing different topologies or different sizes were tested [95]. Under the same conditions, the alkyne metathesis of 75−78 containing benzene, triphenylamine, porphyrin, and tetraphenyl porphyrin panels led to a tetrameric cage with D2h symmetry (COP-Ⅳ in Fig. 19c), a dimer cage with D3h symmetry (COP-Ⅶ in Fig. 19c), a dimer cage with D4h symmetry, and unknown precipitates, respectively. Bearing similar shape but different sizes, 71, 75, and 76 gave extremely different molecular cage assemblies, implying the significant effect of building blocks on the topological structure of molecular cages. Possible mechanisms for the formation of different molecular topologies are enumerated in Fig. 19c. Taking monomer with C3-symmetry as example, the initial stage of the reaction was a bimolecular process, yielding a [1+1]Ⅰ dimer. Subsequently, the formation of the intermediate [1+1]Ⅱ is kinetically favored over the production of COP-Ⅵ, which requires one intermolecular reaction and a further three steps of intramolecular cyclization. For 75 bearing small size benzene as panel, formation of the [1+1]Ⅱ intermediate is preferred due to steric effects, resulting in tetramer monomeric cages (COP-Ⅳ or COP-Ⅴ). In regard of 76 bearing more molecular flexibility, intramolecular cyclization is more kinetically preferred, leading to monomeric cage (COP-Ⅶ). In terms of 71, the interlocked species COP-Ⅱ is more thermodynamically favorable than the kinetically controlled product. The entropy loss in the interlocked process might be compensated by the enthalpy gain from the weak intermolecular interactions arising from the π–π stacking interactions between aromatic chromophores.
The high rigidity of ethynylene bond endows the molecular cage with high shape-persistency. The robust ethynylene linkages also provide the interlocked cage with excellent chemical stability since their disconnection and formation usually need the usage of a catalyst. As deduced from Fig. 19c, the non-directional alkyne metathesis is pretty complicated, during which both thermodynamic and kinetic factors could play vital roles in determining the final assemblies. Assessing the structure-activity relationships between monomers and products accurately is challenging because of the diversity and complexity of alkyne metathesis reaction. 73b and 74b were the only examples of interlocked aryleneethynylene cages. Thus, more detailed mechanism analysis and careful molecular design are worth exploring in depth.
The high reversibility of dynamic covalent chemistry endows the resulting chemical bonds with outstanding error-checking and self-correcting capability. Nonetheless, it means that the corresponding bonds are associated with less chemical stability (e.g., imine and boronic ester), surviving only over a narrow pH or concentration window. In addition, most of the above examples led to products insoluble in water, making it difficult to mimic real biological processes. Developing more adaptable reactions thus remains an important task.
High structural and topological complexity of interlocked covalent organic cages is favorable to mimic the mechanically interlocked behaviors of biomolecules in living organisms. Given that interlocking faces many competing side reactions, fabrication of interlocked covalent organic cages is a very attractive but challenging job. Several factors, including proper molecular configuration and length, suitable functional groups, and helpful solubilizing units, are critical for selecting monomers used in the design of interlocked organic cages. Proper molecular configuration and length unlocks desired topologies as well as provides large enough inner cavities and windows, so that partner cage subunits or penetrating linkers can be accommodated. Suitable functional groups generate adequate weak interactions, directing the formation of interlocked structures. Helpful solubilizing units provide steric hindrance to prevent the formation of precipitating polymers or oligomers. Both irreversible and reversible reactions have been demonstrated to be useful for the synthesis of interlocked cages, while the former yields hydrophilic interlocked cages with stable chemical bonds, the latter generally has a higher conversion rate. Schiff-base chemistry is the most successful reversible reaction for constructing interlocked organic cages, and it has been used to fabricate various chemically and topologically interlocked cages. Highlights of the Schiff-base chemistry include the interwoven trimer 53c isolated at 80% yield and 66·6Cl prepared quantitatively in solution.
Most interlocked structures are more thermodynamically stable than their monomeric cage counterparts. Several weak interactions, including π–π interactions, hydrogen bonds, dipole−dipole interactions, dispersion interactions, and solvent template effect, play key roles in inducing the formation of interlocked species instead of monomeric cages. Stacking, T-shaped, and donor–acceptor π–π interactions are the primary force driving interlocking. Several mechanisms of interlocking were investigated, including encapsulated interlocking, preorganized interlocking, interlocking step-by-step from individual monomeric cages, and self-assembly from linear polymer or oligomer intermediates. Encapsulated interlocking pathway is commonly found, which involves in forming a monomeric cage first, then encapsulating a building block in its inner cavity via weak interactions. Interlocked covalent organic cages can self-assemble hierarchically into various supramolecular structures: monoclinic, triclinic, and trigonal crystalline phases as well as porous organic cages. Subtle structural differences exert dramatic influence on the properties of these assemblies, highlighting the high motional freedom of the organic backbones. Understanding the factors important in the design of interlocked organic cages provides insights valuable for designing more sophisticated molecules and understanding of biological interlocking.
Remarkable achievements have made on interlocked covalent organic cages over the past decade. Nevertheless, compared to well-investigated organic monomeric cages, interlocked rings, and interlocked coordination cages, research on interlocked covalent organic cages is still in its infancy. Much effort is still needed to understand this significant and challenging structure completely. Firstly, more reactions and novel topologies should be developed. Many have already been achieved in monomeric and interlocked coordination cages, so the time is ripe to expand these foundational building blocks for interlocked covalent organic cages. Secondly, deeper analysis of the many driving forces and mechanisms underlying the formation of interlocked organic cages is required. A computational model to evaluate the strength of these driving forces in guiding the formation of interlocked structure would be timely. Faster and more accurate methods are needed to capture reaction intermediates and to elucidate interlocking mechanisms. Thirdly, enantiomers and more challenging structures should be synthesized with more regularity. Interlocked organic cages usually feature enantiomers with different configurations or topological chirality. Controlling the generation of more challenging enantiomers through subtly regulating weak interactions is a difficult task [96,97]. Fabricating polymerized interlocked organic cages is also an attractive challenge, as polymerized species have been constructed in poly[n]catenanes [98-100] and interlocked coordination cages [101]. Finally, more accurate characterization technologies and wide applications are worthy of further exploration. In particular, more real-time means to precisely probe the structural changes in the intermediate processes are urgently needed to be developed. In addition, considering the unique structural features, attractive dynamic behaviors, and versatile self-assembly capacity of interlocked organic cages, they would show wide potential applications in separation, recognition, and catalysis [40,102,103], and more efforts towards these promising directions are highly demanded.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Bin Yao: Writing – review & editing, Writing – original draft, Conceptualization. Yao Bu: Writing – review & editing, Writing – original draft. Hongfei Sun: Writing – review & editing, Writing – original draft. Guowang Li: Writing – review & editing, Writing – original draft. Xianying Wu: Writing – review & editing, Writing – original draft. Wei Wang: Writing – review & editing, Conceptualization.
This research was supported by the Science and Technology Research Program of Chongqing Municipal Education Commission (No. KJQN202400807) and Natural Science Foundation of Shanghai (No. 23ZR1419600).
Supplementary material associated with this article can be found, in the online version, at doi:
R.S. Forgan, J.P. Sauvage, J.F. Stoddart, Chem. Rev. 111 (2011) 5434–5464. doi: 10.1021/cr200034u
S. Erbas-Cakmak, D.A. Leigh, C.T. McTernan, et al., Chem. Rev. 115 (2015) 10081–10206. doi: 10.1021/acs.chemrev.5b00146
E.M.G. Jamieson, F. Modicom, S.M. Goldup, Chem. Soc. Rev. 47 (2018) 5266–5311. doi: 10.1039/c8cs00097b
L. Chen, X. Sheng, G. Li, et al., Chem. Soc. Rev. 51 (2022) 7046–7065. doi: 10.1039/d2cs00202g
X.-Q. Xu, X.-Q. Wang, W. Wang, Chin. Chem. Lett. 34 (2023) 107665.
Y. Liu, M. O’Keeffe, M.M.J. Treacy, et al., Chem. Soc. Rev. 47 (2018) 4642–4664. doi: 10.1039/c7cs00695k
Z. Ashbridge, S.D.P. Fielden, D.A. Leigh, et al., Chem. Soc. Rev. 51 (2022) 7779–7809. doi: 10.1039/d2cs00323f
Z. Niu, H.W. Gibson, Chem. Rev. 109 (2009) 6024–6046. doi: 10.1021/cr900002h
A. Saura-Sanmartin, C.A. Schalley, Chem 9 (2023) 823–846.
D. Dattler, G. Fuks, J. Heiser, et al., Chem. Rev. 120 (2020) 310–433. doi: 10.1021/acs.chemrev.9b00288
G. Gil-Ramírez, D.A. Leigh, A.J. Stephens, Angew. Chem. Int. Ed. 54 (2015) 6110–6150. doi: 10.1002/anie.201411619
M.J. Langton, P.D. Beer, Acc. Chem. Res. 47 (2014) 1935–1949. doi: 10.1021/ar500012a
E. Wasserman, J. Am. Chem. Soc. 82 (1960) 4433–4434. doi: 10.1021/ja01501a082
H.L. Frisch, E. Wasserman, J. Am. Chem. Soc. 83 (1961) 3789–3795. doi: 10.1021/ja01479a015
G. Liu, P.M. Rauscher, B.W. Rawe, et al., Chem. Soc. Rev. 51 (2022) 4928–4948. doi: 10.1039/d2cs00256f
B.T. Shahraki, S. Maghsoudi, Y. Fatahi, et al., Coordin. Chem. Rev. 423 (2020) 213484.
H.Y. Au-Yeung, Y. Deng, Chem. Sci. 13 (2022) 3315–3334. doi: 10.1039/d1sc05391d
M. Fujita, N. Fujita, K. Ogura, et al., Nature 400 (1999) 52–55.
W.X. Gao, H.J. Feng, B.B. Guo, et al., Chem. Rev. 120 (2020) 6288–6325. doi: 10.1021/acs.chemrev.0c00321
E.G. Percástegui, V. Jancik, Coordin. Chem. Rev. 407 (2020) 213165.
Q.Y. Hong, B. Huang, M.X. Wu, et al., Nat. Commun. 16 (2025) 2484.
D. Luo, B. Pan, J. Zhang, et al., Chin. Chem. Lett. 32 (2021) 1397–1399.
T.R. Schulte, J.J. Holstein, L. Schneider, et al., Angew. Chem. Int. Ed. 59 (2020) 22489–22493. doi: 10.1002/anie.202010995
Y. Wang, Y. Zhang, Z. Zhou, et al., Nat. Commun. 11 (2020) 2727.
Y. Li, H. Jiang, W. Zhang, et al., J. Am. Chem. Soc. 146 (2024) 3147–3159. doi: 10.1021/jacs.3c10734
J. Martí-Rujas, Commun. Chem. 8 (2025) 92.
Y. Li, K.M. Mullen, T.D. Claridge, et al., Chem. Commun. 45 (2009) 7134–7136. doi: 10.1039/b915548a
Q. Chen, K. Zhu, Chem. Soc. Rev. 53 (2024) 5677–5703.
N.H. Evans, P.D. Beer, Chem. Soc. Rev. 43 (2014) 4658–4683. doi: 10.1039/c4cs00029c
G. Montà-González, F. Sancenón, R. Martínez-Mañez, et al., Chem. Rev. 122 (2022) 13636–13708. doi: 10.1021/acs.chemrev.2c00198
X. Yang, Z. Ullah, J.F. Stoddart, et al., Chem. Rev. 123 (2023) 4602–4634. doi: 10.1021/acs.chemrev.2c00667
R.A. Borse, Y.X. Tan, D. Yuan, et al., Energy Environ. Sci. 17 (2024) 1307–1329. doi: 10.1039/d3ee03360k
T. Hasell, A.I. Cooper, Nat. Rev. Mater. 1 (2016) 16053.
J. Li, Y. Yang, J. Yang, et al., Coordin. Chem. Rev. 523 (2025) 216260.
I.A. Rather, F. Wang, R. Siddiqui, et al., Coordin. Chem. Rev. 535 (2025) 216648.
M. Frank, M.D. Johnstone, G.H. Clever, Chem. Eur. J. 22 (2016) 14104–14125. doi: 10.1002/chem.201601752
S. La Cognata, A. Miljkovic, R. Mobili, et al., ChemPlusChem 85 (2020) 1145–1155. doi: 10.1002/cplu.202000274
S.L. Huang, T.S.A. Hor, G.X. Jin, Coordin. Chem. Rev. 333 (2017) 1–26. doi: 10.1145/2897512
R. Zhu, J. Ding, L. Jin, et al., Coordin. Chem. Rev. 389 (2019) 119–140.
E. Du, X. Tang, W. Zhang, et al., Nat. Rev. Chem. 9 (2025) 506–522. doi: 10.1038/s41570-025-00721-7
K.D. Hänni, D.A. Leigh, Chem. Soc. Rev. 39 (2010) 1240–1251.
R. Peng, Y. Xu, Q. Cao, Chin. Chem. Lett. 29 (2018) 1465–1474.
Y. Liu, W. Zhao, C.H. Chen, et al., Science 365 (2019) 159–161. doi: 10.1126/science.aaw5145
D. Chakraborty, R. Modak, P. Howlader, et al., Chem. Commun. 57 (2021) 3995–3998. doi: 10.1039/d1cc00627d
T. Jiao, K. Cai, Z. Liu, et al., Chem. Sci. 10 (2019) 5114–5123. doi: 10.1039/c9sc00591a
Y. Shi, K. Cai, H. Xiao, et al., J. Am. Chem. Soc. 140 (2018) 13835–13842. doi: 10.1021/jacs.8b08555
Y. Wu, Q.H. Guo, Y. Qiu, et al., Proc. Natl. Acad. Sci. U. S. A. 119 (2022) e2118573119.
X.Y. Chen, D. Shen, K. Cai, et al., J. Am. Chem. Soc. 142 (2020) 20152–20160. doi: 10.1021/jacs.0c09896
Y. Jin, Q. Wang, P. Taynton, et al., Acc. Chem. Res. 47 (2014) 1575–1586. doi: 10.1021/ar500037v
Y. Jin, C. Yu, R.J. Denman, et al., Chem. Soc. Rev. 42 (2013) 6634–6654. doi: 10.1039/c3cs60044k
J. Yu, M. Gaedke, F. Schaufelberger, Eur. J. Org. Chem. 26 (2022) e202201130.
W. Drożdż, A. Ciesielski, A.R. Stefankiewicz, Angew. Chem. Int. Ed. 62 (2023) e202307552.
F.B.L. Cougnon, A.R. Stefankiewicz, S. Ulrich, Chem. Sci. 15 (2024) 879–895. doi: 10.1039/d3sc05343a
K. Acharyya, P.S. Mukherjee, Angew. Chem. Int. Ed. 58 (2019) 8640–8653. doi: 10.1002/anie.201900163
M.E. Belowich, J.F. Stoddart, Chem. Soc. Rev. 41 (2012) 2003–2024. doi: 10.1039/c2cs15305j
X. Cao, Y. Jin, H. Wang, et al., Chin. Chem. Lett. 35 (2024) 109201.
Y.L. Lai, H.J. Zhang, J. Su, et al., Chin. Chem. Lett. 34 (2023) 107686.
Y. Pang, M. Wang, N.-H. Yang, et al., Chin. Chem. Lett. 35 (2024) 109575.
K. Su, W. Wang, S. Du, et al., J. Am. Chem. Soc. 142 (2020) 18060–18072. doi: 10.1021/jacs.0c07367
Q. Chen, Z. Li, Y. Lei, et al., Nat. Commun. 14 (2023) 4627.
A.U. Malik, F. Gan, C. Shen, et al., J. Am. Chem. Soc. 140 (2018) 2769–2772. doi: 10.1021/jacs.7b13512
L. Feng, Y.X. Tan, E.S.M. El-Sayed, et al., CCS Chem. 6 (2024) 2264–2274. doi: 10.31635/ccschem.024.202303625
T. Hasell, X. Wu, J.T. Jones, et al., Nat. Chem. 2 (2010) 750–755. doi: 10.1038/nchem.739
S. Xu, P. Li, Z.-Y. Li, et al., CCS Chem. 3 (2021) 1838–1850. doi: 10.31635/ccschem.020.202000360
P. Li, S. Xu, C. Yu, et al., Angew. Chem. Int. Ed. 59 (2020) 7113–7121. doi: 10.1002/anie.202000442
Z. Sun, P. Li, S. Xu, et al., J. Am. Chem. Soc. 142 (2020) 10833–10840. doi: 10.1021/jacs.0c03330
P. Li, Z. Sun, J. Chen, et al., J. Am. Chem. Soc. 144 (2022) 1342–1350. doi: 10.1021/jacs.1c11452
C. Chen, S. Zhang, Acc. Chem. Res. 58 (2025) 583–598. doi: 10.1021/acs.accounts.4c00754
L. Chen, Z. Chen, W. Wang, et al., J. Am. Chem. Soc. 146 (2024) 30303–30313. doi: 10.1021/jacs.4c10104
W. Wang, S. Zhou, X. Yu, et al., CCS Chem. 6 (2024) 2084–2109. doi: 10.31635/ccschem.024.202404398
D. Wang, L. Zhang, Y. Zhao, J. Org. Chem. 87 (2022) 2767–2772. doi: 10.1021/acs.joc.1c02688
P. Wagner, F. Rominger, J.H. Gross, et al., Angew. Chem. Int. Ed. 62 (2023) e202217251.
B.P. Benke, T. Kirschbaum, J. Graf, et al., Nat. Chem. 15 (2023) 413–423. doi: 10.1038/s41557-022-01094-w
T. Jiao, G. Wu, Y. Zhang, et al., Angew. Chem. Int. Ed. 59 (2020) 18350–18367. doi: 10.1002/anie.201910739
M. Yang, F. Qiu, E.-S.M. El-Sayed, et al., Chem. Sci. 12 (2021) 13307–13315. doi: 10.1039/d1sc04531h
O. Vestrheim, M.E. Schenkelberg, Q. Dai, et al., Org. Chem. Front. 10 (2023) 3965–3974. doi: 10.1039/d3qo00480e
H. Li, H. Zhang, A.D. Lammer, et al., Nat. Chem. 7 (2015) 1003–1008. doi: 10.1038/nchem.2392
K. Geng, T. He, R. Liu, et al., Chem. Rev. 120 (2020) 8814–8933. doi: 10.1021/acs.chemrev.9b00550
A.P. Côté, A.I. Benin, N.W. Ockwig, et al., Science 310 (2005) 1166–1170. doi: 10.1126/science.1120411
S.M. Elbert, N.I. Regenauer, D. Schindler, et al., Chem. Eur. J. 24 (2018) 11438–11443. doi: 10.1002/chem.201802123
M. Rondelli, S. Delgado-Hernández, A.H. Daranas, et al., Chem. Sci. 14 (2023) 12953–12960. doi: 10.1039/d3sc02920d
S. Klotzbach, F. Beuerle, Angew. Chem. Int. Ed. 54 (2015) 10356–10360. doi: 10.1002/anie.201502983
S. Ivanova, E. Koster, J.J. Holstein, et al., Angew. Chem. Int. Ed. 60 (2021) 17455–17463. doi: 10.1002/anie.202102982
K. Ono, K. Johmoto, N. Yasuda, et al., J. Am. Chem. Soc. 137 (2015) 7015–7018. doi: 10.1021/jacs.5b02716
N. Schäfer, L. Glanz, A. Lützen, et al., Org. Chem. Front. 12 (2025) 1763–1771. doi: 10.1039/d4qo02012j
G. Zhang, O. Presly, F. White, et al., Angew. Chem. Int. Ed. 53 (2014) 1516–1520. doi: 10.1002/anie.201308924
G. Zhang, O. Presly, F. White, et al., Angew. Chem. Int. Ed. 53 (2014) 5126–5130. doi: 10.1002/anie.201400285
S. Huang, Z. Lei, Y. Jin, et al., Chem. Sci. 12 (2021) 9591–9606. doi: 10.1039/d1sc01881g
A. Fürstner, J. Am. Chem. Soc. 143 (2021) 15538–15555. doi: 10.1021/jacs.1c08040
M. Cui, G. Jia, J. Am. Chem. Soc. 144 (2022) 12546–12566. doi: 10.1021/jacs.2c01192
C. Zhang, Q. Wang, H. Long, et al., J. Am. Chem. Soc. 133 (2011) 20995–21001. doi: 10.1021/ja210418t
Q. Wang, C. Zhang, B.C. Noll, et al., Angew. Chem. Int. Ed. 53 (2014) 10663–10667. doi: 10.1002/anie.201404880
B. Bishop, S. Huang, H. Chen, et al., Chin. Chem. Lett. 35 (2024) 109966.
Q. Wang, C. Yu, H. Long, et al., Angew. Chem. Int. Ed. 54 (2015) 7550–7554. doi: 10.1002/anie.201501679
Q. Wang, C. Yu, C. Zhang, et al., Chem. Sci. 7 (2016) 3370–3376.
J.R.J. Maynard, S.M. Goldup, Chem 6 (2020) 1914–1932.
P. Montes-Tolentino, A.S. Mikherdov, C. Drechsler, et al., Angew. Chem. Int. Ed. 64 (2025) e202423810.
S. Datta, Y. Kato, S. Higashiharaguchi, et al., Nature 583 (2020) 400–405. doi: 10.1038/s41586-020-2445-z
Q. Wu, P.M. Rauscher, X. Lang, et al., Science 358 (2017) 1434–1439. doi: 10.1126/science.aap7675
W. Wang, Z. Chen, S. Zhang, J. Am. Chem. Soc. 147 (2025) 20082–20091. doi: 10.1021/jacs.5c05684
L. Cheng, C. Liang, W. Liu, et al., J. Am. Chem. Soc. 142 (2020) 16218–16222. doi: 10.1021/jacs.0c08117
T. Li, Y. Pan, L. Ding, et al., Chem. Synth. 4 (2024) 35.
R. Wang, Y. Liang, J. Rebek, et al., Chin. Chem. Lett. 35 (2024) 109228.

Figure 1 Summary of reactions used to synthesize interlocked covalent organic cages: (a) irreversible reactions and (b) reversible reactions.

Figure 2 (a) Schematic diagram of the proposed mechanism of the anion templated synthesis process for interlocked structure. (b) Synthesis of monomeric cages 3-Cl− and interlocked cage 4-SO42− assisted with different anions by copper(Ⅰ) catalyzed Huisgen 1,3-dipolar cycloaddition reaction. Adapted with permission [27]. Copyright 2009, the Royal Society of Chemistry.

Figure 3 (a) Selective synthesis of interlocked cage 7 and monomeric cage 8 by choosing proper configuration of monomers. Adapted with permission [44]. Copyright 2021, the Royal Society of Chemistry.

Figure 4 Schematic illustration of the suit[3]ane-based approach for the synthesis of interlocked cages 11a6+ and 11b6+. (b) X-ray single-crystal structure of 11a6+. Adapted with permission [47]. Copyright 2022, National Academy of Sciences of the United States of America.

Figure 5 Synthesis of interlocked imine cages 14a−c by imine condensations. Adapted with permission [63]. Copyright 2010, Springer Nature.

Figure 6 Crystal structures from single crystal data: (a) 14b, (b) 14a accommodating methanol guests, and (c) supramolecular assembly from 14c and p-xylene via host–guest interactions. Adapted with permission [63]. Copyright 2010, Springer Nature.

Figure 7 Two general pathways to generate interlocked cages: (a) preorganized interlocking pathway and (b) encapsulated interlocking pathway. Adapted with permission [65]. Copyright 2020, Wiley-VCH.

Figure 8 Summary of the product distributions of monomeric and interlocked cages produced by cycloimination of different diamine linkers and different trialdehyde panels. Adapted with permission [64]. Copyright 2021, Chinese Chemical Society.

Figure 9 Schematic illustration of the synthesis of catenanes with dissymmetric cages via selective space-discriminative reduction strategy. Adapted with permission [65]. Copyright 2020, Wiley-VCH.

Figure 10 Controlled self-assembly of (a) 19b into triclinic crystalline phase, (b) 24 into continuous wavelike plank, and (c) 28 into trigonal crystalline phase with well-defined hierarchical structures. The amine panels are labeled in red, and imine panels are labeled in blue. Adapted with permission [66]. Copyright 2020, American Chemical Society.

Figure 11 (a) Chemical structures of the four enantiomers of 29. Schematic diagram of the spontaneous resolution of racemic interlocked cages: (b) synthesis of isomeric precursors (rac-29) from rac-30, (c) construction of interlocked cages (rac-32) by imine condensation, (d) selective reduction of rac-32 to rac-28, and (e) two homochiral enantiomers gained from narcissistic self-sorting upon crystallization. Adapted with permission [67]. Copyright 2022, American Chemical Society.

Figure 12 Schematic representation of the narcissistic self-sorting of homochiral (S,R)6-28 upon crystallization: (a) right-handed helical assembly gained from interlocked cage (S,R)6-28, (b) π–π stacking interactions and S...S interactions in the helical assembly, and (c) hexagonal crystalline lattice structure assembled from right-handed supramolecular columns. Adapted with permission [67]. Copyright 2022, American Chemical Society.

Figure 13 Summary of the product distributions of cage species from imine condensations of different panels and linkers. Adapted with permission [69]. Copyright 2024, American Chemical Society.

Figure 14 (a) Synthesis of interlocked cage 46 via template-free and one-pot imine condensation reaction. Structure analysis of 46: (b) top view and side view of an interlocked cage and (c) crystal-packing diagram of 46 along the oa axis. Adapted with permission [71]. Copyright 2022 American Chemical Society.

Figure 15 (a) Schematic illustration of the synthesis of monomeric cubic cage (M)-49 and interlocked cage (M)-50a with TFA as catalyst in different types of solvents. (b) X-ray single crystal structure of (M)-50a. (c) Different interactions and angles within the two partner cages in an interlocked catenane. (d) Packing pattern of (M)-50a in the solid state. (e) Proposed mechanism for the formation of quadruply interlocked catenane (M)-50a from monomeric cage (M)-49 step by step. Adapted with permission [72]. Copyright 2020, Wiley-VCH.

Figure 16 (a) Summary of the product distributions of the imine condensations of (−)-(P)-51 and various dialdehydes (52a−k). (b) Summary of diverse weak interactions leading to the cage-catenane formations. Adapted with permission [73]. Copyright 2023, Springer Nature.

Figure 17 (a) Synthesis of interlocked cage 666+ bearing hydrazone linkers in acidic aqueous solution. (b) Top and (c) side view of 66·6Cl deduced from SC-XRD analysis. Adapted with permission [77]. Copyright 2015, Springer Nature.

Figure 18 (a) Synthesis of boronic ester cages 69 and 70a via [12+8] condensation and the formation of catenated cage 70b upon crystallization (For clarity, alkyl chains are selectively omitted in the cage structure). Crystal structure of (b) interlocked cage and (c) their assemblies in the solid state. The orange sphere and the green sphere represent two cavities in the assembly. Adapted with permission [87]. Copyright 2014, Wiley-VCH.

Figure 19 (a) Synthesis of interlocked aryleneethynylene cages 73b and 74b via alkyne metathesis. (b) Different monomers bearing various panels tested for covalent organic polyhedrons. (c) Possible reaction pathways of three-arm alkynyl derivatives for different covalent organic polyhedrons. (a) is adapted with permission [94]. Copyright 2015, Wiley-VCH. (b) and (c) are adapted with permission [95]. Copyright 2016, the Royal Society of Chemistry.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: