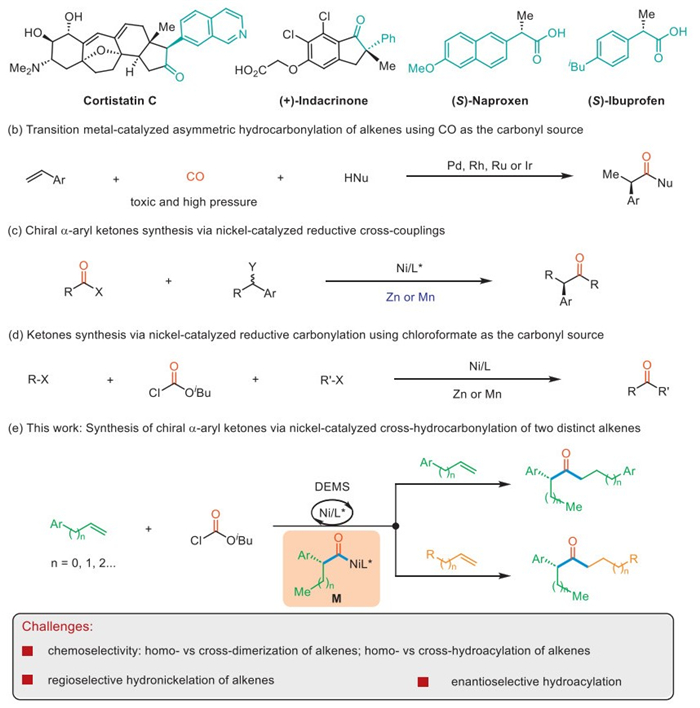
Figure 1.
Strategies for the synthesis of chiral α-aryl ketones.
Enantioselective synthesis of α-aryl ketones via cross-hydroacylation of two distinct alkenes
Yongli Liu , Dan Xu , Yuanyuan Ping , Nengzhong Wang , Wangqing Kong

Chiral α-aryl carbonyl moieties are privileged structural motifs that are widely present in various biologically active natural products and pharmaceuticals. For example, the biologically active natural products cortistatin C and (+)-indacrinone both contain an α-aryl ketone unit; (S)-Naproxen and (S)-ibuprofen are a class of non-steroidal anti-inflammatory drugs containing an α-aryl carboxylic acid unit (Fig. 1a). The best-known strategy to access this structure motif is transition metal-catalyzed or base-mediated α-arylation of enolates [1–4]. However, such strategy often suffers from daunting challenges in accessing α-aryl ketones containing α-tertiary stereocenters because they are base-sensitive and easily racemized via enolization [5]. Alkenes are among the most readily available and abundant feedstocks, and many powerful synthetic strategies in organic synthesis utilize alkenes as ideal starting points. Transition metal-catalyzed hydro-carbonylation of alkenes is a fundamental transformation in the asymmetric synthesis of α-aryl carbonyl compounds [6,7] and has been widely used in academia and industry (Fig. 1b). However, most of these processes rely heavily on rare and precious metals (such as Pd, Rh, Ru or Ir) as catalysts and use high-pressure, toxic and flammable CO gas as the carbonyl source [8–12]. Therefore, the exploration of a novel and mild alkene carbonylation processes to access chiral α-aryl carbonyl compounds using earth-abundant transition metals and simple, easy-to-operate and less toxic CO surrogates is highly sought after.
In the past few decades, the nickel-catalyzed reductive cross-coupling reaction pioneered by the Weix and Gong groups has received widespread attention [13–24], and the reductive cross-coupling of alkyl halides with carboxylic acid derivatives has become an effective strategy for ketone synthesis [25–36]. Subsequently, Reisman [37], Melchiorre [38], Zhu [39,40], and others [41,42] achieved the synthesis of chiral α-aryl ketones through Ni-catalyzed enantioselective cross-couplings (Fig. 1c). However, these methods rely on the use of secondary benzyl chlorides or acyl chlorides or anhydrides as acyl coupling agents, which are often unstable or require additional preparation steps. In addition, these strategies require the use of stoichiometric amounts of metal reducing agents, such as manganese or zinc. On the other hand, chloroformates are attractive as a safe CO source in organic synthesis due to their low cost, good availability, stability, and ease handling. In 2019, Hu and our group independently developed nickel-catalyzed reductive carbonylation reaction, using chloroformate as a CO source to synthesize dialkyl ketones (Fig. 1d) [43–46]. Inspired by the recent work of Hu [47], Fu [48], Zhu [49], etc. [50–62] on NiH-catalyzed hydrofunctionalization of alkenes [63–65] and building on our ongoing interest in Ni-catalyzed enantioselective difunctionalization of alkenes [66–72], we hypothesized that the in situ generated Ni-H species underwent oxidative addition with chloroformate followed by hydrometallation/chain-walking with aryl-substituted alkenes. Subsequent, CO extrusion and reinsertion would form the acyl-nickel intermediate M. The desired α-aryl ketones would then be obtained through migratory insertion of the acyl-Ni species M into another terminal alkene followed by reductive elimination (Fig. 1e). While we were preparing the manuscript, Shu et al. reported a similar cross-hydroacylation of olefins using chloroformates as a CO source [63–65].
To achieve this attractive transformation, several challenges need to be overcome. First, Ni-catalyzed alkene cross-dimerization have been reported to afford linear or branched alkyl-alkyl coupling products [73,74], that are highly competitive with the desired alkene cross-hydroacylation reaction. Second, controlling the regioselectivity of the alkene hydronickelation process is a challenging task. Moreover, two distinct alkenes and chloroformate must be assembled in a highly chemo-, regio- and enantioselective manner. Herein we report the successful implementation of this concept to the synthesis of enantioenriched α-aryl ketones via cross-hydroacylation of two distinct alkenes using isobutyl chloroformate as a safe CO source and electron acceptor.
We initiated our investigations by studying the enantioselective cross-hydroacylation of aryl alkene 1 and unactivated alkene 2 using ClCO2iBu as a CO source and electron acceptor (Table 1). After extensive evaluation of the reaction parameters, it was found that α-aryl ketone 3 was obtained in 63% yield and 95% ee at 0 ℃ using NiI2 as the nickel source, C2-symmetric bis-oxazoline L2 as the chiral ligand, diethoxymethylsilane (DEMS) as the hydride source, and KF as the additive in a mixture solvent of NMP and Et2O (4:1) (entry 1). The geminal disubstitution on the tether of the bis(oxazoline) ligands is critical for regulating the efficiency and enantioselectivity of the reaction (entries 2–5). The steric hindrance of the substituents at the C4 position of the bis(oxazoline) ligand leads to a significant decrease in the enantioselectivity (entry 6). Inferior results were observed when using the bulky ClCO2iPr or linear ClCO2nBu, while product 3 could not be obtained when using ClCO2Ph as the CO source (entries 7–9). We further found that silanes also have a significant effect on the reaction outcome. Dimethoxymethylsilane (DMMS) and polymethylhydrosiloxane (PMHS) gave slightly lower yields and ee values, while (MeO)3SiH and (Me2SiH)2O failed to provide the desired product (entries 10–13). When NiBr2(dme) was used instead of NiI2, the yield of product 3 was not affected, but the enantioselectivity decreased significantly to 63% ee (entry 14). In addition, when Ni(acac)2 or Ni(cod)2 was used as a catalyst, the target product 3 was not obtained (entry 15), and the ee value decreased significantly when the reaction was carried out at room temperature (entry 16).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||
| Entry | Variants | Yield (%)b of 3 | ee (%)c of 3 |
| 1 | None | 63 | 95 |
| 2 | L1 instead of L3 | Trace | – |
| 3 | L2 instead of L3 | 51 | 89 |
| 4 | L4 instead of L3 | 45 | 96 |
| 5 | L5 instead of L3 | Trace | – |
| 6 | L6 instead of L3 | 33 | 17 |
| 7 | ClCO2iPr instead of ClCO2iBu | 22 | 95 |
| 8 | ClCO2nBu instead of ClCO2iBu | 43 | 95 |
| 9 | ClCO2Ph instead of ClCO2iBu | Trace | – |
| 10 | DMMS instead of DEMS | 61 | 83 |
| 11 | PMHS instead of DEMS | 26 | 88 |
| 12 | (MeO)3SiH instead of DEMS | Trace | – |
| 13 | (Me2SiH)2O instead of DEMS | Trace | – |
| 14 | NiBr2(dme) instead of NiI2 | 60 | 63 |
| 15 | Ni(COD)2 or Ni(acac)2 instead of NiI2 | Trace | – |
| 16 | Room temperature instead of 0 ℃ | 38 | 65 |
| a Reaction conditions: 1 (0.1 mmol), ClCO2iBu (0.4 mmol), 2 (0.5 mmol), NiI2 (10 mol%), L3 (20 mol%), DEMS (0.4 mmol), KF (0.2 mmol) in 1.0 mL solvent (NMP:Et2O = 4:1) at 0 ℃. b Isolated yield. c Determined by HPLC using a chiral stationary phase. |
|||
With the optimal conditions in hand, we sought to examine the generality of the enantioselective cross-hydroacylation of aryl alkenes and unactivated alkenes with ClCO2iBu. As shown in Fig. 2, aryl alkenes bearing various functional groups at the para-position of the aromatic ring, such as ether (6), fluorine (7), trifluoromethyl (10), ester (11 and 13), nitrile (12), and sulfone (14), were readily accommodated. Notably, we found that aryl chloride and aryl bromide (8 and 9), which are susceptible to Ni-catalyzed cross-couplings, remained intact, opening up additional avenues for further derivatization of the resulting products. The meta- and ortho-substituted aryl alkenes proceeded smoothly to provide the corresponding products 15–17 in good yields and excellent enantioselectivities. In addition, heteroaryl-substituted styrenes were also investigated. Naphthalene (18), dibenzofuran (19), dibenzothiophene (20), indole (21), carbazole (22), morpholine (23) could be successfully incorporated into the corresponding products in satisfactory yields with very high enantiomeric induction. Aryl alkene derived from estrone (24) has no adverse effect on efficiency and diastereoselective (>20/1, dr). The reaction is not limited to terminal olefins, but internal olefins are also suitable substrates. Although the yields are slightly reduced, excellent enantioselectivities can still be achieved (25–26).
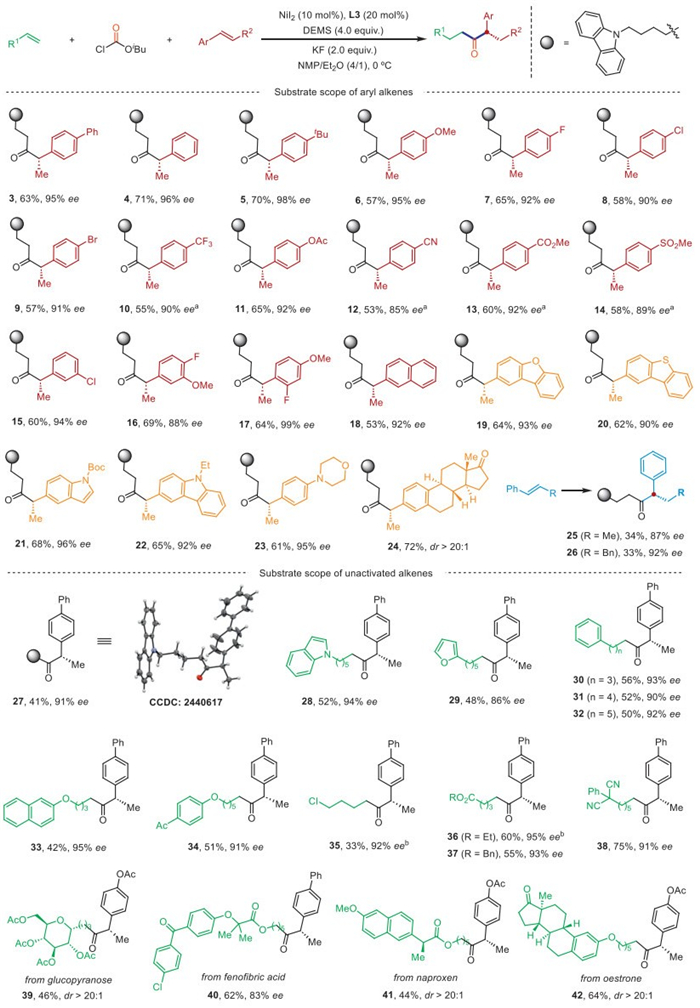
Next, the substrate scope with respect to unactivated alkene was investigated. Alkenes containing various (aromatic)heterocycles, such as carbazole (27), indole (28), furan (29), and phenyl (30–32), reacted smoothly to give the corresponding products in synthetically useful yields and excellent enantioselectivities. X-ray crystallographic analysis unequivocally confirmed the S configuration of 27 (CCDC 2440617), and the corresponding absolute configurations of the other products were determined by analogy. Alkene bearing various synthetically useful functional groups, such as ether (33–34), ester (36–37), and nitrile (38), were well tolerated. Remarkably, the alkyl chloride (35) survived intact and served as a versatile synthetic handle for further functionalization. Moreover, alkenes derived from complex biologically important molecules, including glucopyranose (39), fenofibric acid (40), naproxen (41), and estrone (42) also reacted smoothly to afford the target products with excellent diastereoselectivities.
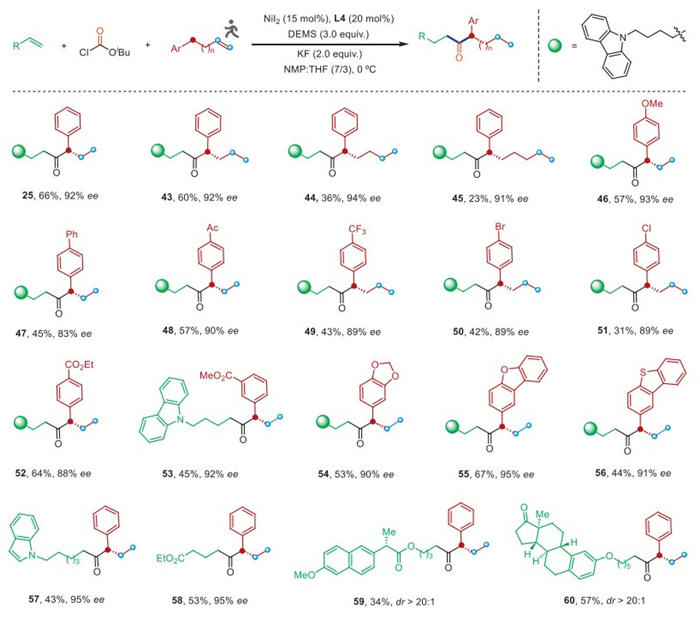
Next, the enantioselective remote cross-hydroacylation of an aryl-substituted alkene and another unactivated alkene with ClCO2iBu was explored (Fig. 3) [75–88]. We first tested aryl-substituted alkenes with different chain lengths between the starting C═C bond and the remote aromatic group and found that the longer the migration distance, the lower the yield of products (25, 43–45), but fortunately, the enantioselectivity of these products was not affected (>91% ee). Aryl-substituted alkenes bearing a variety of substituents at the para- or meta-position on the aromatic ring, including ether (46), phenyl (47), ketone (48), trifluoromethyl (49), and esters (52 and 53), all reacted smoothly to afford the desired products in synthetically useful yields and high enantioselectivities. Substituents that can be used for further cross-coupling, such as aryl bromide (50) and aryl chloride (51), remained intact. Heteroaryl-substituted alkenes, such as dibenzothiophene and dibenzofuran, can also serve as competent substrates (54–56). Unactivated terminal alkenes containing a variety of functional groups were readily accommodated, including indole (57), ester (58), and complex biologically important molecules such as naproxen (59) and estrone (60), affording the corresponding products with excellent enantioselectivity.
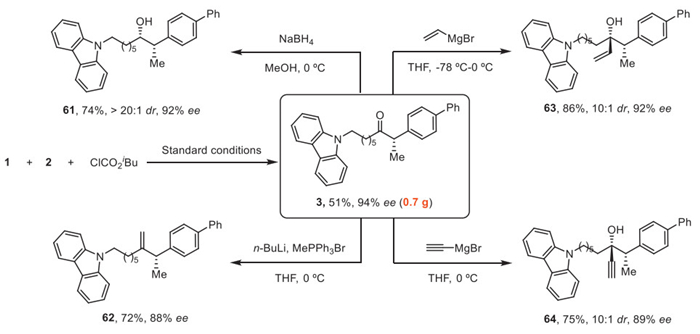
We carried out the reaction of alkenes 1 with 2 and ClCO2iBu on a 3 mmol scale to afford chiral α-aryl ketone 3 in 51% yield (0.7 g) and 94% ee, revealing the practical applicability of this Ni-catalyzed cross-hydroacylation of alkenes. Diastereoselective reduction of ketone 3 using NaBH4 gave alcohol 61 containing two adjacent stereocenters in 74% yield. The ketone moiety of 3 was olefinated with PPh3MeBr via Wittig reaction to afford the new alkene 62 in 72% yield. In addition, nucleophilic addition of a vinyl Grignard reagent onto 3 afforded the tertiary alcohol 63 in 86% yield with a diastereoselectivity of 10/1. Alkynylation of 3 gave the corresponding propargyl alcohol 64 in 75% yield with a diastereoselectivity of 10/1 (Fig. 4).
To gain insight into the cross-hydroacylation reaction of alkenes, a series of mechanistic experiments were performed. The reaction of unactivated alkene 2 with ClCO2iBu failed to produce the expected product. Most of the alkene 2 was recovered (82%), with only a small amount being reduced to the alkane product 65 (Fig. 5a). In addition, control experiments were performed to investigate the reaction sequence of aryl alkenes and unactivated alkenes with Ni-H species. 5 equiv. of unactivated alkene 2 were required to generate chiral α-aryl ketone 3 with satisfactory conversion and yield. Notably, the reaction of unactivated alkene 2 and aryl alkene 1 in a 1:1 ratio produced only trace amounts of product 3, whereas ketone 66 was obtained in 21% yield with 90% ee (Fig. 5b). By slightly modification of the reaction conditions, the reaction of aryl alkene 1 with ClCO2iBu gave product 66 in 50% yield and 90% ee (Fig. 5c). We further expanded the substrate scope and found that both electron-rich and electron-deficient substituted aryl alkenes could react smoothly to give the corresponding products 66–69 in good yields and high enantioselectivities. These results clearly indicate that aryl alkenes preferentially react with Ni-H species rather than unactivated alkenes.
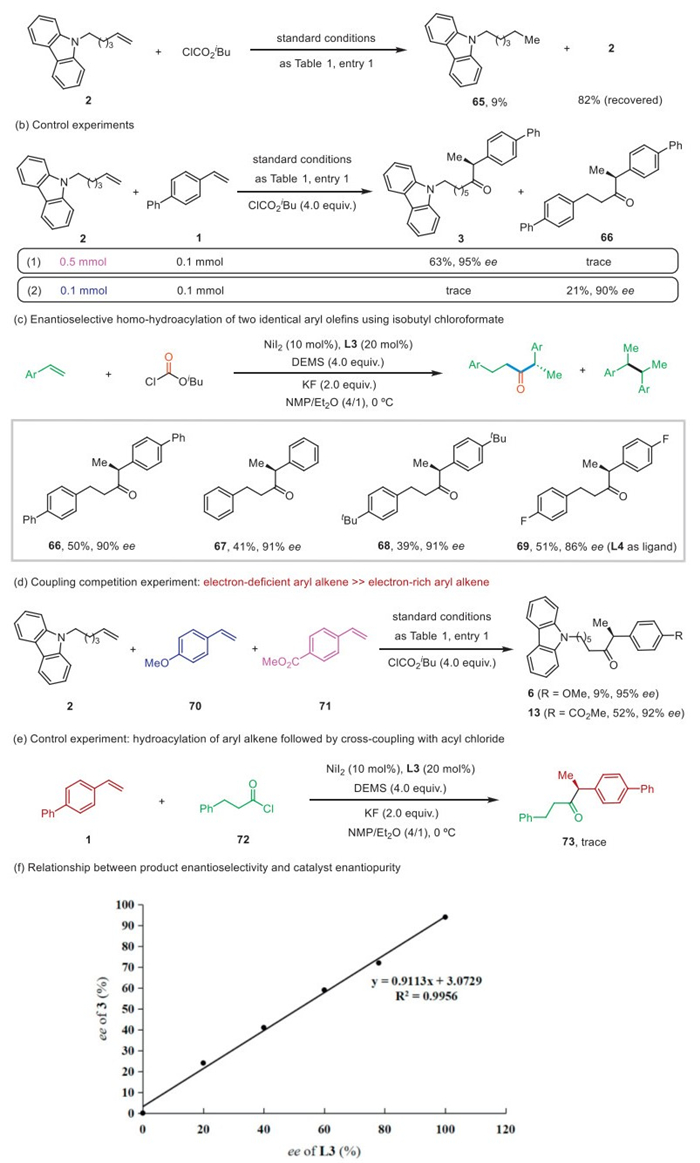
The competitive reaction of unactivated alkene 2 with electron-deficient aryl alkene 70 and electron-rich aryl alkene 71 was further investigated, with product 13 being the dominant product. This result may indicate that electron-deficient aryl alkenes are more reactive than electron-rich aryl alkenes when reacting with Ni-H species (Fig. 5d).
The reaction of aryl alkene 1 with acyl chloride 72 gave only a trace amount of product 73 (Fig. 5e). These results may suggest that the acyl-nickel intermediate generated from the hydronickelation of unactivated alkene followed by CO insertion may be not the key intermediate for this transformation. This is completely different from the mechanism of reductive carbonylation of N-vinyl indoles with unactivated alkenes to produce α-N-heteroaromatic ketones [63–65]. Furthermore, we observed a linear relationship between the enantiomeric excess of product 3 and the enantiomeric purity of ligand L3, suggesting that a 1:1 ratio of nickel to ligand was involved in the enantiomeric determination step (Fig. 5f).
To shed light on the hydrometallation process of alkenes, we synthesized deuterated alkenes 1-D2 and 2-D2 and performed a series of deuterium labeling experiments under standard conditions (Fig. 6). Interestingly, deuterium scrambling was observed at the terminal position of the alkenes (Figs. 6a-c, both aryl alkene and unactivated alkene). In addition, the reaction of alkene 1 with ClCO2iBu using Ph2SiD2 instead of DEMS as the hydride source afforded product 66 in 21% yield with 91% ee. Notably, in addition to the chiral carbon stereocenter at the α-position to the carbonyl group, deuterium is delivered to three sites of the product 66 (Fig. 6d). Taking together, these results suggest that the migratory insertion of Ni-H species into aryl alkene is reversable and highly regiospecific.

Based on the above experimental results and previous studies [47–65], a plausible reaction mechanism is proposed (Fig. 7). First, oxidative addition of ClCO2iBu to catalytically active Ni(0) species A affords Ni(Ⅱ) intermediate B. In the presence of DEMS and KF, Ni-H species C would be generated from B. Reversible and regioselective hydrometallation/chain walking of Ni-H species C into an aryl alkene affords alkyl-Ni(Ⅱ) species D, which would participate in a rapid equilibrium involving CO extrusion and reinsertion to ultimately form the acyl-Ni intermediate E. An intramolecular migratory insertion of the acyl-Ni species E into another alkene affords the alkyl-nickel(Ⅱ) intermediate F, which further undergoes transmetalation reaction with DEMS with assistance of a base to generate alkyl-Ni-H species G. The alkyl-Ni-H intermediates G and G' can be interconverted via β-H elimination and reinsertion process (chain walking), which is supported by the deuterium labeling experiments in Fig 6d. Finally, the alkyl-Ni-H species undergoes reductive elimination to form α-chiral ketones and catalytically active Ni(0) species H, thus completing the catalytic cycle. This mechanism is different from that reported by Shu et al., [65] and we believe that Ni-H will preferentially react with more active aryl alkenes rather than alkyl alkenes.

In summary, we report here a nickel-catalyzed enantioselective cross-hydroacylation of aryl alkenes with unactivated alkenes using ClCO2iBu as a safe CO source and electron acceptor. Two distinct alkenes and chloroformates are assembled efficiently in a chemo-, regio- and enantioselective manner, providing a scalable, environmentally friendly alternative for the synthesis of enantiomerically enriched α-aryl ketones. This reaction uses abundant and readily available starting materials and proceeds under mild reaction conditions without the use of toxic CO gas or metal carbonyl reagents.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yongli Liu: Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Dan Xu: Methodology, Investigation, Formal analysis, Data curation. Yuanyuan Ping: Project administration, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation. Nengzhong Wang: Methodology, Investigation, Funding acquisition, Formal analysis, Data curation. Wangqing Kong: Writing – review & editing, Writing – original draft, Supervision, Resources, Project administration, Methodology, Investigation, Funding acquisition, Conceptualization.
This project was supported by the National Natural Science Foundation of China (No. 22171215 to W.K., No. 22301225 to Y.P., and No. 22471203 to W.K.). Open Research Fund of State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University. We thank the Core Facility of Wuhan University for X-ray single crystal diffraction analysis.
Supplementary material associated with this article can be found, in the online version, at doi:
Y.J. Hao, X.S. Hu, Y. Zhou, et al., ACS Catal. 10 (2020) 955–993. doi: 10.1021/acscatal.9b04480
C.C.C. Johansson, T.J. Colacot, Angew. Chem. Int. Ed. 49 (2010) 676–707. doi: 10.1002/anie.200903424
P.M. Lundin, J. Esquivias, G.C. Fu, Angew. Chem. Int. Ed. 48 (2009) 154–156. doi: 10.1002/anie.200804888
S. Lou, G.C. Fu, J. Am. Chem. Soc. 132 (2010) 1264–1266. doi: 10.1021/ja909689t
M. Orlandi, M. Escudero-Casao, G. Licini, Synthesis 53 (2021) 4559–4566. doi: 10.1055/a-1560-5245
J. Peng, X. Liu, L. Li, et al., Sci. China Chem. 65 (2022) 441–461. doi: 10.1007/s11426-021-1165-6
J. Li, Y. Shi, Chem. Soc. Rev. 51 (2022) 6757–6773. doi: 10.1039/d2cs00150k
C.F.J. Barnard, Organometallics 27 (2008) 5402–5422. doi: 10.1021/om800549q
Q. Liu, H. Zhang, A. Lei, Angew. Chem. Int. Ed. 50 (2011) 10788–10799. doi: 10.1002/anie.201100763
A. Parenty, X. Moreau, G. Niel, et al., Chem. Rev. 113 (2013) 1–35.
S. Sumino, A. Fusano, T. Fukuyama, et al., Acc. Chem. Res. 47 (2014) 1563–1574. doi: 10.1021/ar500035q
J. Peng, F. Wu, X.F. Wu, Chem. Rev. 119 (2019) 2090–2127. doi: 10.1021/acs.chemrev.8b00068
C.E.I. Knappke, S. Grupe, D. Gartner, et al., Chem. Eur. J. 20 (2014) 6828–6842. doi: 10.1002/chem.201402302
S.Z. Tasker, E.A. Standley, T.F. Jamison, Nature 509 (2014) 299–309. doi: 10.1038/nature13274
D.J. Weix, Acc. Chem. Res. 48 (2015) 1767–1775. doi: 10.1021/acs.accounts.5b00057
E.L. Lucas, E.R. Jarvo, Nat. Rev. Chem. 1 (2017) 0065. doi: 10.1038/s41570-017-0065
J. Liu, Y. Ye, J.L. Sessler, et al., Acc. Chem. Res. 53 (2020) 1833–1845. doi: 10.1021/acs.accounts.0c00291
J. Diccianni, Q. Lin, T. Diao, Acc. Chem. Res. 53 (2020) 906–919. doi: 10.1021/acs.accounts.0c00032
Q. Pan, Y. Ping, W. Kong, Acc. Chem. Res. 56 (2023) 515–535. doi: 10.1021/acs.accounts.2c00771
Y. Ping, H. Song, W. Kong, Chin. J. Org. Chem. 42 (2022) 3302–3321. doi: 10.6023/cjoc202205046
Y. Ping, W. Kong, Synthesis 52 (2020) 979–992. doi: 10.1055/s-0039-1690807
W. Xue, X. Jia, X. Wang, et al., Chem. Soc. Rev. 50 (2021) 4162–4184. doi: 10.1039/d0cs01107j
D.A. Everson, R. Shrestha, D.J. Weix, J. Am. Chem. Soc. 132 (2010) 920–921. doi: 10.1021/ja9093956
X. Yu, T. Yang, S. Wang, et al., Org. Lett. 13 (2011) 2138–2141. doi: 10.1021/ol200617f
A.C. Wotal, D.J. Weix, Org. Lett. 14 (2012) 1476–1479. doi: 10.1021/ol300217x
F. Wu, W. Lu, Q. Qian, et al., Org. Lett. 14 (2012) 3044–3047. doi: 10.1021/ol3011198
H. Yin, C. Zhao, H. You, et al., Chem. Commun. 48 (2012) 7034–7036. doi: 10.1039/c2cc33232a
A.H. Cherney, N.T.S. Kadunce, E. Reisman, J. Am. Chem. Soc. 135 (2013) 7442–7445. doi: 10.1021/ja402922w
T. Moragas, A. Correa, R. Martin, Chem. Eur. J. 20 (2014) 8242–8258. doi: 10.1002/chem.201402509
C. Zhao, X. Jia, X. Wang, et al., J. Am. Chem. Soc. 136 (2014) 17645–17651. doi: 10.1021/ja510653n
X. Jia, X. Zhang, Q. Qian, et al., Chem. Commun. 51 (2015) 10302–10305. doi: 10.1039/C5CC03113C
J. Gu, X. Wang, W. Xue, et al., Org. Chem. Front. 2 (2015) 1411–1421. doi: 10.1039/C5QO00224A
M. Zheng, W. Xue, T. Xue, et al., Org. Lett. 18 (2016) 6152–6155. doi: 10.1021/acs.orglett.6b03158
X. Wang, Y. Dai, H. Gong, Top. Curr. Chem. 374 (2016) 43. doi: 10.1007/s41061-016-0042-2
J. Wang, B.P. Cary, P.D. Beyer, et al., Angew. Chem. Int. Ed. 58 (2019) 12081–12085. doi: 10.1002/anie.201906000
Y. Sun, L. Su, W. Tong, et al., Synlett 32 (2021) 1762–1766. doi: 10.1055/a-1550-7935
A.H. Cherney, N.T. Kadunce, S.E. Reisman, J. Am. Chem. Soc. 135 (2013) 7442–7445. doi: 10.1021/ja402922w
E. Gandolfo, X. Tang, S.R. Roy, et al., Angew. Chem. Int. Ed. 58 (2019) 16854–16858. doi: 10.1002/anie.201910168
J. Chen, S. Zhu, J. Am. Chem. Soc. 143 (2021) 14089–14096. doi: 10.1021/jacs.1c07851
X. Jiang, F. Sheng, Y. Zhang, et al., J. Am. Chem. Soc. 144 (2022) 21448–21456. doi: 10.1021/jacs.2c10785
L. Huan, X. Shu, W. Zu, et al., Nat. Commun. 12 (2021) 3536. doi: 10.1038/s41467-021-23887-2
R. Oost, A. Misale, N. Maulide, Angew. Chem. Int. Ed. 55 (2016) 4587–4590. doi: 10.1002/anie.201600597
A. Rérat, C. Michon, F. Agbossou-Niedercorn, et al., Eur. J. Org. Chem. 2016 (2016) 4554–4560. doi: 10.1002/ejoc.201600738
R. Shi, X. Hu, Angew. Chem. Int. Ed. 58 (2019) 7454–7458. doi: 10.1002/anie.201903330
S. Xu, K. Wang, W. Kong, Org. Lett. 21 (2019) 7498–7503. doi: 10.1021/acs.orglett.9b02788
H. Chen, H. Yue, C. Zhu, et al., Angew. Chem. Int. Ed. 61 (2022) e202204144. doi: 10.1002/anie.202204144
J. Breitenfeld, R. Scopelliti, X. Hu, Organometallics 31 (2012) 2128–2136. doi: 10.1021/om201279j
X. Lu, B. Xiao, Z. Zhang, et al., Nat. Commun. 7 (2016) 11129. doi: 10.1038/ncomms11129
Y.L. He, Y.L. Cai, S.L. Zhu, J. Am. Chem. Soc. 139 (2017) 1061–1064. doi: 10.1021/jacs.6b11962
F. Zhou, J. Zhu, Y. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 4058–4062. doi: 10.1002/anie.201712731
Z.B. Wang, H.L. Yin, G.C. Fu, Nature 563 (2018) 379–383. doi: 10.1038/s41586-018-0669-y
F. Zhou, Y. Zhang, X. Xu, et al., Angew. Chem. Int. Ed. 58 (2019) 1754–1758. doi: 10.1002/anie.201813222
S. He, J. Wang, Y. Li, et al., J. Am. Chem. Soc. 142 (2020) 214–221. doi: 10.1021/jacs.9b09415
Z. Yang, G.C. Fu, J. Am. Chem. Soc. 142 (2020) 5870–5875. doi: 10.1021/jacs.0c01324
D. Qian, S. Bera, X. Hu, J. Am. Chem. Soc. 143 (2021) 1959–1967. doi: 10.1021/jacs.0c11630
J. Wang, Y. Li, W. Nie, et al., Nat. Commun. 12 (2021) 1313. doi: 10.1038/s41467-021-21600-x
S. Wang, J. Zhang, T. Zhang, et al., Nat. Commun. 12 (2021) 2771. doi: 10.1038/s41467-021-22983-7
S. Cuesta-Galisteo, J. Schorgenhumer, X. Wei, et al., Angew. Chem. Int. Ed. 60 (2021) 1605–1609. doi: 10.1002/anie.202011342
S. Sun, Y. Cai, D. Zhang, et al., J. Am. Chem. Soc. 144 (2022) 1130–1137. doi: 10.1021/jacs.1c12350
X.X. Wang, X. Lu, Y. Li, et al., Sci. China Chem. 63 (2020) 1586–1600. doi: 10.1007/s11426-020-9838-x
Z. Zhang, S. Bera, C. Fan, et al., J. Am. Chem. Soc. 144 (2022) 7015–7029. doi: 10.1021/jacs.1c13482
Y. Wang, Y. He, S. Zhu, Acc. Chem. Res. 55 (2022) 3519–3536. doi: 10.1021/acs.accounts.2c00628
X. Chen, Q. Yu, W. Shu, Angew. Chem. Int. Ed. 64 (2025) e202423426. doi: 10.1002/anie.202423426
Y. Du, X. Chen, Y. Li, et al., Nat. Commun. 16 (2025) 4163. doi: 10.1038/s41467-025-57967-4
Y. Du, J. Lin, Y. Li, et al., J. Am. Chem. Soc. 147 (2025) 18944–18952. doi: 10.1021/jacs.5c03451
K. Wang, Z. Ding, Z. Zhou, et al., J. Am. Chem. Soc. 140 (2018) 12364–12368. doi: 10.1021/jacs.8b08190
Y. Ping, K. Wang, Q. Pan, et al., ACS Catal. 9 (2019) 7335–7342. doi: 10.1021/acscatal.9b02081
T. Ma, Y. Chen, Y. Li, et al., ACS Catal. 9 (2019) 9127–9133. doi: 10.1021/acscatal.9b03172
X. Chen, J. Yue, K. Wang, et al., Angew. Chem. Int. Ed. 60 (2021) 14068–14075. doi: 10.1002/anie.202102769
Q. Pan, Y. Ping, Y. Wang, et al., J. Am. Chem. Soc. 143 (2021) 10282–10291. doi: 10.1021/jacs.1c03827
Q. Pan, K. Wang, W. Xu, et al., J. Am. Chem. Soc. 146 (2024) 15453–15463. doi: 10.1021/jacs.4c03745
S. Hu, X. Wang, T. Wu, Angew. Chem. Int. Ed. 64 (2025) e202413892. doi: 10.1002/anie.202413892
L. Cheng, J. Liu, Y. Chen, et al., Nat. Synth. 2 (2023) 364–372. doi: 10.1038/s44160-023-00239-0
P. Yang, H. Zhao, X. Chen, et al., Nat. Synth. 3 (2024) 1360–1368. doi: 10.1038/s44160-024-00609-2
W. Lee, C. Wang, Y. Lin, et al., Org. Lett. 15 (2013) 5358–5361. doi: 10.1021/ol402644y
J. Bair, Y. Schramm, A. Sergeev, et al., J. Am. Chem. Soc. 136 (2014) 13098–13101. doi: 10.1021/ja505579f
L. Xiao, L. Cheng, W. Feng, et al., Angew. Chem. Int. Ed. 57 (2018) 461–464. doi: 10.1002/anie.201710735
H. Lv, L. Xiao, D. Zhao, et al., Chem. Sci. 9 (2018) 6839–6843. doi: 10.1039/c8sc02101e
L. Cheng, M. Li, L. Xiao, et al., J. Am. Chem. Soc. 140 (2018) 11627–11630. doi: 10.1021/jacs.8b09346
Y. Chen, B. Shuai, X. Xu, et al., J. Am. Chem. Soc. 141 (2019) 3395–3399. doi: 10.1021/jacs.8b13524
W. Zhang, X. Yang, J. Ma, et al., J. Am. Chem. Soc. 141 (2019) 5628–5634. doi: 10.1021/jacs.9b00931
X. Lv, C. Fan, L. Xiao, et al., CCS Chem. 1 (2019) 328–334. doi: 10.31635/ccschem.019.20190026
Y. Li, Y. Luo, L. Peng, et al., Nat. Commun. 11 (2020) 417. doi: 10.1007/s10815-019-01652-1
Y. He, C. Liu, L. Yu, et al., Angew. Chem. Int. Ed. 59 (2020) 9186–9191. doi: 10.1002/anie.202001742
W. Shao, C. Besnard, L. Guénée, et al., J. Am. Chem. Soc. 142 (2020) 16486–16492. doi: 10.1021/jacs.0c08319
J. Marcum, T. Taylor, S.J. Meek, Angew. Chem. Int. Ed. 59 (2020) 14070–14075. doi: 10.1002/anie.202004982
R. Yu, S. Rajasekar, X. Fang, Angew. Chem. Int. Ed. 59 (2020) 21436–21441. doi: 10.1002/anie.202008854
Z. Li, Y. Fu, R. Deng, et al., Angew. Chem. Int. Ed. 59 (2020) 23306–23312. doi: 10.1002/anie.202010840

Figure 2 Enantioselective cross-hydrocarbonylation of aryl alkenes and unactivated alkenes with ClCO2iBu. Yields are of isolated products. Enantiomeric excesses of major enantiomer were determined by HPLC on a chiral stationary phase. a The reaction was performed at −10 ℃. b L4 was used.

Figure 3 Enantioselective remote hydrocarbonylation of aryl-substituted alkenes and unactivated alkenes with ClCO2iBu. Reaction conditions: unactivated alkene (0.5 mmol), ClCO2iBu (0.4 mmol), aryl-substituted alkene (0.1 mmol), NiI2 (15 mol%), L4 (20 mol%), DEMS (0.3 mmol), KF (0.2 mmol) in 0.5 mL NMP/THF (7/3) at 0 ℃. Yields are of isolated products. Enantiomeric excesses of major enantiomer were determined by HPLC on a chiral stationary phase.
Table 1. Optimization of reaction conditions.a
|
|||
| Entry | Variants | Yield (%)b of 3 | ee (%)c of 3 |
| 1 | None | 63 | 95 |
| 2 | L1 instead of L3 | Trace | – |
| 3 | L2 instead of L3 | 51 | 89 |
| 4 | L4 instead of L3 | 45 | 96 |
| 5 | L5 instead of L3 | Trace | – |
| 6 | L6 instead of L3 | 33 | 17 |
| 7 | ClCO2iPr instead of ClCO2iBu | 22 | 95 |
| 8 | ClCO2nBu instead of ClCO2iBu | 43 | 95 |
| 9 | ClCO2Ph instead of ClCO2iBu | Trace | – |
| 10 | DMMS instead of DEMS | 61 | 83 |
| 11 | PMHS instead of DEMS | 26 | 88 |
| 12 | (MeO)3SiH instead of DEMS | Trace | – |
| 13 | (Me2SiH)2O instead of DEMS | Trace | – |
| 14 | NiBr2(dme) instead of NiI2 | 60 | 63 |
| 15 | Ni(COD)2 or Ni(acac)2 instead of NiI2 | Trace | – |
| 16 | Room temperature instead of 0 ℃ | 38 | 65 |
| a Reaction conditions: 1 (0.1 mmol), ClCO2iBu (0.4 mmol), 2 (0.5 mmol), NiI2 (10 mol%), L3 (20 mol%), DEMS (0.4 mmol), KF (0.2 mmol) in 1.0 mL solvent (NMP:Et2O = 4:1) at 0 ℃. b Isolated yield. c Determined by HPLC using a chiral stationary phase. |
|||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




