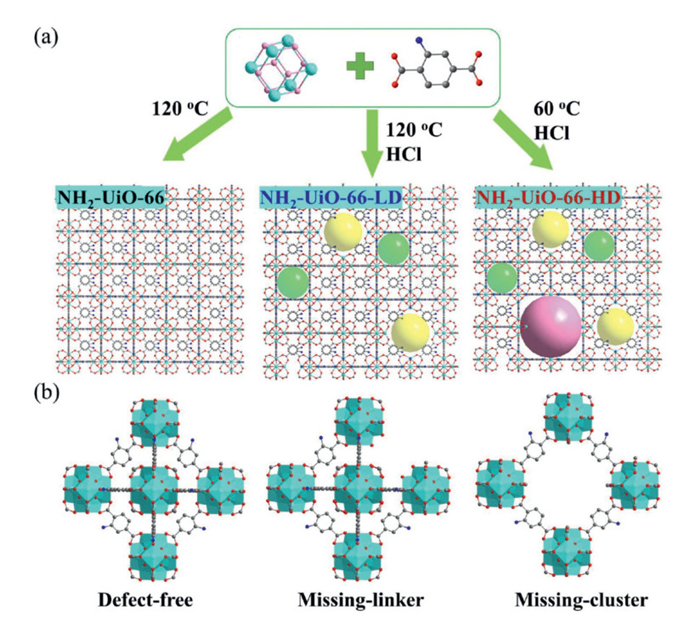
Figure 1.
(a) Schematic illustration of the fabrication process for pristine NH2-UiO-66 and defective NH2-UiO-66, (b) causes of defects.
Enhanced PFOA removal via defect engineering in NH2-UiO-66
Shiyu Wei , Xiang Li , Chao Huang , Dongmei Chen , Shunlin Zhang , Bixue Zhu

Per- and polyfluoroalkyl substances (PFASs), renowned for their thermal stability, hydrophobicity, and oleophobicity, have been widely used since the 1940s in industrial and consumer products [1]. However, their extreme persistence, bioaccumulation potential, and toxicity pose severe environmental and health risks [2]. Among PFASs, perfluorooctanoic acid (PFOA) is a pervasive contaminant detected in water systems, a major exposure pathway for humans, necessitating urgent development of efficient removal technologies [3]. While methods like adsorption, electrochemical oxidation, photocatalysis, and bioremediation have been explored, with adsorption emerging as the most viable due to its cost-effectiveness, simplicity, and scalability [4–11]. Still, conventional adsorbents (e.g., activated carbon, resins, carbon nanotubes) suffer from limited efficiency and poor regenerability, underscoring the need for advanced materials with high capacity and reusability [12,13].
Metal-organic frameworks (MOFs), crystalline porous materials built from metal clusters and organic linkers, offer exceptional surface areas, structural tunability, and functional diversity, positioning them as superior adsorbents for PFASs [14–17]. Their adsorption mechanisms primarily encompass noncovalent interactions (electrostatic interactions, hydrogen bonds, hydrophobic interactions, and van der Waals forces) with PFASs [4]. The anionic nature of PFASs (from terminal -COOH/-SO3H groups), and their fluorine-rich hydrophobic chains, drives complex interactions with MOFs, which can be amplified through strategic functionalization [18]. Organic linkers in MOFs often bear -F, -COOH, -OH, or -NH2 groups, serving as hydrogen-bond donors/acceptors to enhance PFAS affinity [19–22]. For instance, Zhou et al. demonstrated that zirconium-based MOFs (PCN-1001/1002) with free hydroxyl sites achieved 632 mg/g PFOA adsorption via hydrogen bonding and Lewis acid-base interactions [23].
Unsaturated metal sites in MOFs further improve PFAS capture. Ma et al. compared PFOS adsorption across MIL-53(Al), MIL-53(Fe), and MIL-101(Cr), revealing MIL-53(Al)'s superiority due to its high density of unsaturated Al3+ sites [24]. Similarly, Zhou et al. developed PCN-999, a Zr-based MOF with dual Zr6 and (Zr6)2 clusters, achieving unprecedented PFOA uptake (1089 mg/g) via unsaturated coordination sites that strengthened physical adsorption [25]. However, intentionally engineering MOFs with unsaturated metal centers remains challenging.
Defect engineering, a strategy to introduce controlled imperfections, has recently emerged to optimize MOF properties, enabling tailored active sites for enhanced performance in environmental and energy applications [26–28]. This study innovatively integrates defect engineering with amino (-NH2) functionalization to synergistically combine coordination sites, N-H···F hydrogen bonding, and electrostatic interactions within a MOF. It synthesizes NH2-UiO-66 derivatives (NH2-UiO-66, -LD, and -HD) with tunable defect densities for efficient PFOA removal. The highly defective NH2-UiO-66-HD achieved 95% PFOA adsorption within 30 min, with a maximum capacity of 739.31 mg/g (1.79 mmol/g), outperforming most reported adsorbents. Crucially, defect-induced unsaturated Zr sites coordinated with PFOA's -COO⁻ groups, synergizing with N–H···F hydrogen bonds (from amino groups) and electrostatic interactions (via protonated -NH3+) to drive rapid, robust adsorption. The material retained > 90% efficiency over seven regeneration cycles, demonstrating exceptional stability. This work establishes defect engineering as a transformative approach to unlock MOFs' potential by exposing unsaturated metal centers, offering a sustainable solution for PFAS-contaminated water remediation.
During NH2-UiO-66 synthesis, modulators and reaction temperatures critically influence crystallization rates and pathways (Fig. 1a), often inducing defects via incomplete framework assembly (Fig. 1b) [26]. Powder X-Ray diffraction (PXRD) analysis (Fig. 2a) confirmed that NH2-UiO-66, NH2-UiO-66-LD (low defect), and NH2-UiO-66-HD (high defect) retained the parent structure. However, NH2-UiO-66-HD exhibited weakened (111)/(200) and intensified (220)/(600)/(711) peaks, reflecting altered crystal size/orientation [29]. The field emission scanning electron microscope (FESEM) image in Fig. 2c further confirms this observation. NH2-UiO-66 exhibits a relatively perfect octahedral shape with an average size of about 100 nm. NH2-UiO-66-LD also has an average size of approximately 100 nm, but its octahedral particle morphology is much worse. NH2-UiO-66-HD presents irregular granular crystals with an average size of around 50 nm. These findings indicate that synthetic conditions significantly impact the crystallinity and morphology of NH2-UiO-66 materials, and in turn affect their physical properties, such as pore structure and thermal stability [30–33].
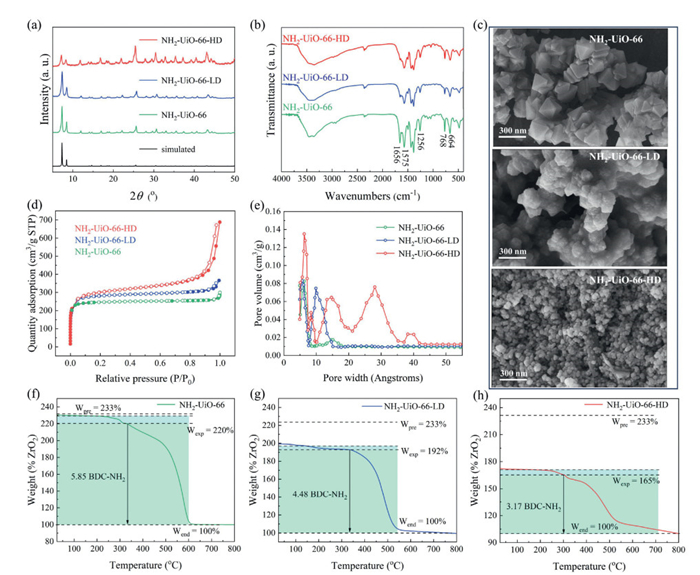
FT-IR spectra (Fig. 2b) identified functional groups, N–H (664/768 cm-1), C–H (1256 cm-1), and C=O (1656 cm-1) bonds. N2 adsorption isotherms (Fig. 2d) revealed pore variations, pristine NH2-UiO-66 showed microporous Type Ⅰ behavior, while NH2-UiO-66-LD and -HD exhibited enhanced adsorption from framework defects. NH2-UiO-66-HD's Type Ⅱ isotherm indicated monolayer adsorption, with mesopore volumes rising from 23.48% (pristine) to 65.31% (high-defect) (Table S3 in Supporting information). Pore size distributions (Fig. 2e) confirmed mesopore expansion (20–42 nm) in NH2-UiO-66-HD versus narrower pores (≤17 nm) in less defective variants.
TGA profiles (Figs. 2f-h) delineated thermal stability. All samples showed three-stage decomposition (physical desorption < 150 ℃, Zr6 node dehydroxylation at 150–300 ℃, framework collapse > 300 ℃). NH2-UiO-66 and -LD exhibited framework degradation above 340 ℃, whereas NH2-UiO-66-HD degraded earlier (300 ℃), correlating defect density with reduced thermal resilience. Residual ZrO2 post-800 ℃ accounted for 120 wt% (pristine), 92 wt% (LD) and 65 wt% (HD) losses, corresponding to linker-to-cluster ratios of 5.85, 4.48 and 3.17. Ideal NH2-UiO-66 features six BDC-NH2 linkers per [Zr6O4(OH)4] node; reduced coordination numbers in defective variants confirmed missing linkers and cluster losses, amplifying structural defects. Furthermore, the PXRD diffraction data of defective NH2-UiO-66 were Rietveld refined according to linker-to-cluster ratios of 4.48 and 3.17. The PXRD Rietveld refinement fitted well (Figs. S1 and S2, Tables S1 and S2 in Supporting information) [34,35]. At the same time, the surface of defective NH2-UiO-66 was positively charged in acidic pH ranges, and NH2-UiO-66 exhibited stronger positive charge (Fig. S3 in Supporting information). The pHzp values of NH2-UiO-66, NH2-UiO-66-LD, and NH2-UiO-66-HD were 7.4, 6.6, and 6.4, respectively. This indicates that amino-functionalized MOFs exert a positive effect on adsorbing anionic groups of PFASs in acidic conditions via strong electrostatic forces.
The defective NH2-UiO-66 MOFs, with abundant amino groups, a hierarchical pore structure, and unsaturated Zr sites, show great potential for efficient PFOA adsorption. Static adsorption experiments were conducted by placing MOFs in a PFOA solution (10 mL, 1000 mg/L, pH 4) and stirring for 4 h. Results indicated that adsorption capacity increased with defect degree: 249.27 mg/g for NH2-UiO-66, 362.41 mg/g for NH2-UiO-66-LD, and 579.27 mg/g for NH2-UiO-66-HD (Fig. 3a and Table S4 in Supporting information). NH2-UiO-66-HD's higher affinity is likely attributed to its rich open coordination sites and hierarchical pore structure.
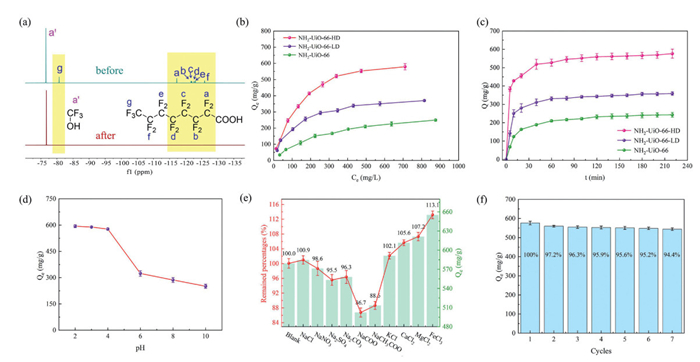
Adsorption isotherms (Figs. S4-S6 in Supporting information) were used to study PFOA adsorption models and capacities. Fig. 3b shows that as the initial concentration of PFOA increases, the adsorbed amount also increases. Langmuir and Freundlich models were used to fit isotherm data (Fig. S7 in Supporting information), with parameters in Table S4. The Langmuir model shows higher correlation coefficients (R2 > 0.9915), suggesting monolayer adsorption. NH2-UiO-66-HD has maximum PFOA adsorption capacity is 739.31 mg/g (1.79 mmol/g), one of the highest values among various adsorbents including MOFs and others like MWNT, FCX4-P, DFB-CDP, activated carbon, and fully silicated beta (β) (Table S6 in Supporting information) [36–41].
PFOA adsorption kinetics (Figs. S8-S10 in Supporting information) show similar behaviors: Rapid adsorption within the first 30 min, then slower until equilibrium. NH2-UiO-66 reaches 95% saturation (236.3 mg/g) in 45 min, NH2-UiO-66-LD (347.3 mg/g) in 40 min and NH2-UiO-66-HD (545.5 mg/g) in 30 min (Fig. 3c). Pseudo-first-order and pseudo-second-order models were applied (Fig. S11 in Supporting information). The pseudo-second-order model fits the data well (R2 > 0.9918), indicating chemically controlled adsorption (Table S5) [17]. Possible mechanisms include hydrogen bonding, electrostatic attraction and coordination interactions. Intraparticle diffusion modeling shows multiple linear relationships, indicating multi-step adsorption process (Table S5) [42]. The adsorption process can be divided into film diffusion, intraparticle diffusion, and surface attachment, with rate constants in the order ki > kii > kiii. High initial rate suggests surface adsorption, and lines not passing through origin imply other processes are involved [43].
Other factors significantly influence NH2-UiO-66 adsorption. NH2-UiO-66-HD, the optimal adsorbent, was selected to study pH effects, ion competition, and reusability. As the pH increased from 4 to 6 and then to 10, the adsorption capacity of perfluorooctanoic acid (PFOA) decreased sharply from 579.27 mg/g to 321 mg/g and then to 251 mg/g (Fig. 3d and Fig. S12 in Supporting information). Since PFOA (pKa = −0.5) remains deprotonated under experimental conditions [44], the pH-dependent adsorption arises from amino group protonation below pH 6, enhancing pore positive charge density and electrostatic interactions. Ion competition experiments revealed metal cations (Na+, K+, Ca2+, Mg2+, Fe3+) enhanced adsorption, with Fe3+ increasing efficiency by 113% (Fig. 3e and Fig. S13 in Supporting information). Metal cations influence PFOA adsorption through ion strength, bridging, ion exchange capacity, competitive adsorption, and surface charge neutralization [15]. In the defective NH2-UiO-66 system, Zr6 cluster loss causes BDC-NH2 to dissociate into carboxylate anions (-COO⁻), negatively charging the material surface. Metal cations can neutralize these charges, reducing electrostatic repulsion and allowing adsorbates (e.g., PFOA) easier access to the adsorbent surface. They can also bridge PFOA anions to carboxylate anion sites, enhancing adsorption. Conversely, anions (Cl-, NO3-, SO42-, CO32-, COO-, CH3COO-) showed inhibitory effects, particularly formate (86.7% of control) and acetate (88.5%), suggesting competitive ligand interactions with unsaturated metal centers during PFOA coordination.
The reusability of an adsorbent is crucial for practical applications, especially in environmental remediation, as it directly affects the economic viability and sustainability of the treatment process. Therefore, the recyclability of NH2-UiO-66-HD was investigated (Fig. S14 in Supporting information). After each adsorption cycle, NH2-UiO-66-HD was recovered via filtration, followed by methanol washing and activation to be reused in subsequent PFOA adsorption cycles. Over seven consecutive cycles, NH2-UiO-66-HD maintained a significant portion of its initial adsorption capacity for PFOA (Fig. 3f). Notably, while there was a considerable decrease in adsorption capacity during the second cycle (97.2%), the following five cycles saw capacities largely preserved at over 94.4%. This phenomenon might be attributed to the strong coordination bonds between PFOA and the open coordination sites within NH2-UiO-66-HD, which are not easily disrupted by simple solvent exchange. The excellent retention of adsorption capacity indicates that NH2-UiO-66-HD possesses a long cyclic life. Moreover, the structural integrity and porosity of NH2-UiO-66-HD remained intact after seven cycles, as evidenced by the consistent PXRD patterns (Fig. S16 in Supporting information) and N2 adsorption isotherms (Fig. S17 in Supporting information) before and after cycling. These results further confirm the robustness and stability of NH2-UiO-66-HD under repeated use conditions, highlighting its potential for practical application in wastewater treatment.
NH2-UiO-66-HD was assessed for PFASs removal by immersing it in aqueous solutions of PFBA, PFBS, PFHxA, PFHxS, PFNA, PFDA and PFOS (each at 1000 ppm). High adsorption capacities were observed: PFBA (265.41 mg/g, 1.24 mol/g), PFBS (255 mg/g, 0.85 mol/g), PFHxA (411.40 mg/g, 1.31 mol/g), PFHxS (447.04 mg/g, 1.12 mol/g), PFOS (625 mg/g, 1.25 mol/g), PFNA (675.83 mg/g, 1.46 mol/g), and PFDA (916.49 mg/g, 1.87 mol/g) (Fig. S15 in Supporting information). Fluorinated carboxylic compounds showed higher adsorption than sulfonic acids, with the dicarboxylic PFSEA showing particularly high capacity, likely due to stronger coordination with unsaturated metal sites. Also, short-chain PFASs had lower adsorption than long-chain ones, possibly due to differences in hydrophobicity.
The investigation into the adsorption mechanism of PFOA can guide the development of future adsorbents. Scanning electron microscope (SEM) images revealed NH2-UiO-66 MOFs' particles remained unchanged post-adsorption, indicating unaltered crystallinity (Fig. 4a). Transmission electron microscope (TEM) and energy dispersive spectrometer (EDS) elemental mapping showed three defective NH2-UiO-66 types effectively adsorbed PFOA uniformly within particles (Fig. 4b). FT-IR spectra confirmed pollutant capture by MOFs, with characteristic PFOA peaks shifting post-adsorption, indicating successful loading and interaction, likely N–H···F hydrogen bonding (Fig. 4c). Notably, the characteristic peaks at 1146 and 1202 cm-1, attributed to C–C–C bending vibrations and asymmetric C-F stretching modes of PFOA molecules, exhibit slight shifts post-adsorption, indicating successful loading of PFOA onto the MOFs framework and interaction with it. These interactions may be N–H···F hydrogen bonding interactions. Notably, the peak corresponding to the symmetric C=O stretching mode at 1656 cm-1 for NH2-UiO-66-LD and NH2-UIO-66-HD shows significant shifts after PFOA adsorption, suggesting coordination interactions between the carboxylate groups of PFOA and unsaturated Zr sites on defective NH2-UiO-66 (Fig. 4c).
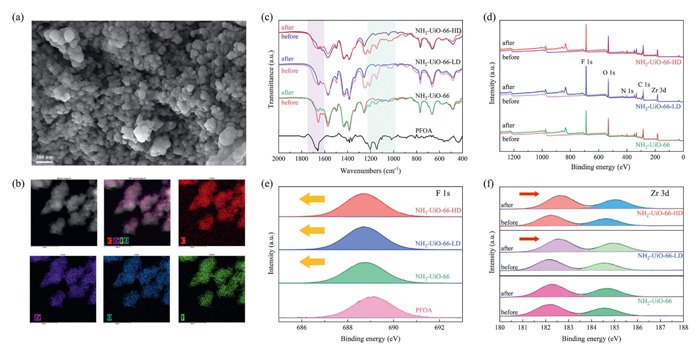
X-ray photoelectron spectroscopy (XPS) analysis further explored the adsorption mechanism (Fig. 4d). A high-intensity F 1s peak post-adsorption indicated successful PFOA encapsulation. The F 1s binding energy of raw PFOA at 689.0 eV decreased after adsorption, implying strong hydrogen bonding between C-F chains and the MOFs framework (Fig. 4e). Conversely, Zr 3d binding energies for NH2-UiO-66-LD and NH2-UiO-66-HD shifted to higher values after adsorption, indicating Zr oxidation (Fig. 4f). Given that PFOA carries a more negative charge compared to coordinated H2O, when PFOA forms coordination bonds with unsaturated Zr sites, the charge density of Zr decreases, leading to an increase in the binding energy of Zr 3d, which suggests that coordination interaction controls the adsorption process. Greater shifts in binding energy were observed for NH2-UiO-66-HD and NH2-UiO-66-LD compared to NH2-UiO-66 due to additional unsaturated Zr sites allowing more coordinated H2O to be replaced by PFOA [45,46].
This study demonstrates that defect engineering in NH2-UiO-66 significantly enhances PFOA adsorption performance. The highly defective NH2-UiO-66-HD achieved a maximum theoretical capacity of 739.31 mg/g (1.79 mmol/g), with rapid kinetics (95% removal within 30 min) and robust regenerability (> 94% retention after 7 cycles). Mechanistic analyses revealed synergistic contributions from N–H···F hydrogen bonding between amino groups and PFOA, electrostatic attraction via protonated -NH3+ under acidic conditions, and coordination interactions between defect-induced unsaturated Zr sites and PFOA's -COO⁻ groups. By strategically exposing unsaturated metal centers through defect modulation, this work establishes a paradigm for optimizing MOF adsorbents, offering a sustainable and scalable solution for PFAS-contaminated water remediation. The findings underscore the critical role of structural design in advancing material functionality for environmental applications.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Shiyu Wei: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation. Xiang Li: Software, Investigation. Chao Huang: Funding acquisition, Conceptualization. Dongmei Chen: Project administration, Investigation. Shunlin Zhang: Writing – review & editing, Supervision, Funding acquisition. Bixue Zhu: Supervision, Project administration.
This work was financially supported by the Guizhou Provincial Key Laboratory Platform Project (No. ZSYS [2025] 008). C. Huang thanks the National Natural Science Foundation of China (No. 22461009). D. Chen thanks the National Natural Science Foundation of China (No. 32560414). S. Zhang thanks the Guizhou Provincial Basic Research General Program (Natural Science) (No. MS[2025]692), the Guizhou Provincial Basic Research Youth Program (Natural Science) (No. QianKeHe Basic-[2024]114), the Guizhou University Cultivation Projects (No. [2022]100), the Science and Technology Innovation Talent Team of Guizhou Province (No. QKHPTRC-CXTD[2023]005), the Science and Technology Innovation Team of Higher Education Department of Guizhou Province (No. QJJ[2023]053).
Supplementary material associated with this article can be found, in the online version, at doi:
M.G. Evich, M.J.B. Davis, J.P. McCord, et al., Science 375 (2022) 9065. doi: 10.1126/science.abg9065
C. Sonne, M.S. Bank, B.M. Jenssen, et al., Science 379 (2023) 887–888. doi: 10.1126/science.adh0934
J. Hu, X. Yang, X. Song, et al., J. Hazard. Mater. 480 (2024) 136283. doi: 10.1016/j.jhazmat.2024.136283
R. Li, N.N. Adarsh, H. Lu, M. Wriedt, Matter 5 (2022) 3161–3193. doi: 10.1016/j.matt.2022.07.028
W. Ji, L. Xiao, Y. Ling, et al., J. Am. Chem. Soc. 140 (2018) 12677–12681. doi: 10.1021/jacs.8b06958
Z. Chen, Y.L. Lu, L. Wang, et al., J. Am. Chem. Soc. 145 (2023) 260–267. doi: 10.1021/jacs.2c09866
A. Román Santiago, S. Yin, J. Elbert, et al., J. Am. Chem. Soc. 145 (2023) 9508–9519. doi: 10.1021/jacs.2c10963
Y. Fang, P. Meng, C. Schaefer, D.R.U. Knappe, Water Res. 230 (2023) 119522. doi: 10.1016/j.watres.2022.119522
S. Sinha, A. Chaturvedi, R.K. Gautam, J.J. Jiang, J. Am. Chem. Soc. 145 (2023) 27390–27396. doi: 10.1021/jacs.3c08352
Y. Wen, Á. Rentería-Gómez, G.S. Day, et al., J. Am. Chem. Soc. 144 (2022) 11840–11850. doi: 10.1021/jacs.2c04341
C. Zhang, K. Yan, C. Fu, et al., Chem. Rev. 122 (2022) 167–208. doi: 10.1021/acs.chemrev.1c00632
N. Kim, J. Elbert, E. Shchukina, X. Su, Nat. Commun. 15 (2024) 8321. doi: 10.1038/s41467-024-52630-w
J. Fang, S. Li, T. Gu, et al., J. Environ. Chem. Eng. 12 (2024) 111833. doi: 10.1016/j.jece.2023.111833
S. Li, J. Ma, J. Cheng, et al., Langmuir 40 (2024) 2815–2829. doi: 10.1021/acs.langmuir.3c02939
P.S. Pauletto, T.J. Bandosz, J. Hazard. Mater. 425 (2022) 127810. doi: 10.1016/j.jhazmat.2021.127810
L.I. FitzGerald, J.F. Olorunyomi, R. Singh, C.M. Doherty, ChemSusChem 15 (2022) 202201136. doi: 10.1002/cssc.202201136
H. Shan, J. Xiao, G. Duan, et al., Univ. Nat. Sci. Ed. 36 (2023) 43–52. doi: 10.1007/s11802-023-5076-9
N. Ilić, K. Tan, F. Mayr, et al., Adv. Mater. 37 (2024) 2413120.
M. Endoh, H. Konno, Chem. Lett. 50 (2021) 1592–1596. doi: 10.1246/cl.210233
Q. Li, S. Zhu, F. Chen, C. Guo, Environ. Res. 211 (2022) 113083. doi: 10.1016/j.envres.2022.113083
S. Zhang, Y. Xie, R.J. Somerville, et al., Small 19 (2023) 2206999. doi: 10.1002/smll.202206999
G. Luo, S. Zhang, S.L. Zhang, et al., J. Mol. Struct. 1299 (2024) 137145. doi: 10.1016/j.molstruc.2023.137145
R.R. Liang, Y. Yang, Z. Han, et al., Adv. Mater. 36 (2024) 2407194. doi: 10.1002/adma.202407194
C. Zhao, Y. Xu, F. Xiao, et al., Chem. Eng. J. 406 (2021) 126852. doi: 10.1016/j.cej.2020.126852
R.R. Liang, S. Xu, Z. Han, et al., J. Am. Chem. Soc. 146 (2024) 9811–9818. doi: 10.1021/jacs.3c14487
Z. Fang, B. Bueken, D.E. De Vos, R.A. Fischer, Angew. Chem. Int. Ed. 54 (2015) 7234–7254. doi: 10.1002/anie.201411540
N.S. Portillo-Vélez, J.L. Obeso, J.A. de los Reyes, et al., Commun. Mater. 5 (2024) 247. doi: 10.1038/s43246-024-00691-1
J. Ren, M. Ledwaba, N.M. Musyoka, et al., Coord. Chem. Rev. 349 (2017) 169–197. doi: 10.1016/j.ccr.2017.08.017
Y. Bai, Y. Dou, L.H. Xie, et al., Chem. Soc. Rev. 45 (2016) 2327–2368. doi: 10.1039/C5CS00837A
S. Yuan, L. Feng, K. Wang, et al., Adv. Mater. 30 (2018) 1704303. doi: 10.1002/adma.201704303
X. Miao, S. Chen, C. Peng, et al., Int. J. Hydrogen Energy 71 (2024) 121–130. doi: 10.1016/j.ijhydene.2024.05.224
R. Han, K. Wang, Q. Jiang, et al., J. Colloid Interface Sci. 671 (2024) 680–691. doi: 10.1016/j.jcis.2024.05.206
X. Zhao, M. Liu, Z. Shang, et al., Energy Fuels 38 (2024) 17939–17947. doi: 10.1021/acs.energyfuels.4c03204
X. Huang, Z. Shang, X. Zhao, et al., J. Energy Storage 102 (2024) 114054. doi: 10.1016/j.est.2024.114054
G. Luo, J. Jiang, S. Wei, et al., Sep. Purif. Technol. 343 (2024) 127133. doi: 10.1016/j.seppur.2024.127133
L. Xiao, Y. Ling, A. Alsbaiee, et al., J. Am. Chem. Soc. 139 (2017) 7689–7692. doi: 10.1021/jacs.7b02381
X. Li, H. Zhao, X. Quan, et al., J. Hazard. Mater. 186 (2011) 407–415. doi: 10.1016/j.jhazmat.2010.11.012
D. Shettć, I. Jahovic, T. Skorjanc, et al., ACS Appl. Mater. Interfaces 12 (2020) 43160–43166. doi: 10.1021/acsami.0c13400
D. Zhang, Q. Luo, B. Gao, et al., Chemosphere 144 (2016) 2336–2342. doi: 10.1016/j.chemosphere.2015.10.124
M. Van den Bergh, A. Krajnc, S. Voorspoels, et al., Angew. Chem. Int. Ed. 59 (2020) 13666-13666. doi: 10.1002/anie.202007703
J. Wang, Z. Li, N. Hu, et al., J. Mater. Chem. A 5 (2017) 22506–22511. doi: 10.1039/C7TA08598B
L. Piai, J.E. Dykstra, M.G. Adishakti, et al., Water Res. 162 (2019) 518–527.
L. Ding, H. Deng, C. Wu, X. Han, Chem. Eng. J. 181 (2012) 360–370.
M.J. Chen, A.C. Yang, N.H. Wang, et al., Microp. Mesop. Mater. 236 (2016) 202–210.
A. Dhakshinamoorthy, M. Alvaro, P. Horcajada, et al., ACS Catal. 2 (2012) 2060–2065. doi: 10.1021/cs300345b
D.W. Kim, H.G. Kim, D.H. Cho, Catal. Commun. 73 (2016) 69–73.

Figure 1 (a) Schematic illustration of the fabrication process for pristine NH2-UiO-66 and defective NH2-UiO-66, (b) causes of defects.

Figure 2 (a) PXRD patterns, (b) FTIR spectra, and (c) SEM images of NH2-UiO-66, NH2-UiO-66-LD and NH2-UiO-66-HD. (d) N2 adsorption-desorption isotherms at 77 K for NH2-UiO-66, NH2-UiO-66-LD, and NH2-UiO-66-HD, where solid circles represent adsorption and hollow circles desorption and (e) pore size distributions. TGA curves of (f) NH2-UiO-66, (g) NH2-UiO-66-LD and (h) NH2-UiO-66-HD.

Figure 3 (a) 19F NMR spectra of a PFOA solution with an initial concentration of 500 ppm before and after treatment with NH2-UiO-66-HD. (b) Adsorption isotherms of NH2-UiO-66, NH2-UiO-66-LD and NH2-UiO-66-HD under conditions: Temperature (T) = 298 K; initial concentration (C0) = 50–1000 mg/L, pH 4, contact time (t) = 4 h. (c) Adsorption capacity as a function of time for NH2-UiO-66, NH2-UiO-66-LD and NH2-UiO-66-HD under conditions: C0 = 1000 mg/L, pH 4, T = 298 K. (d) Influence of pH on the adsorption capacity of NH2-UiO-66-HD under conditions: C0 = 1000 mg/L, T = 298 K, t = 4 h. (e) Impact of various ions on the adsorption capacity of NH2-UiO-66-HD under conditions: C0 = 1000 mg/L, T = 298 K, t = 4 h, pH 4. (f) Adsorption performance of NH2-UiO-66-HD towards PFOA after different cycles under similar conditions.

Figure 4 (a) SEM analysis and (b) EDS elemental mapping of NH2-UiO-66-HD after PFOA adsorption. (c) Effects of PFOA adsorption on the FT-IR spectra of NH2-UiO-66, NH2-UiO-66-LD and NH2-UiO-66-HD. (d) Broad-scan XPS spectra of NH2-UiO-66, NH2-UiO-66-LD and NH2-UiO-66-HD before and after PFOA adsorption. (e) High-resolution F 1s and (f) Zr 3d XPS spectra of NH2-UiO-66, NH2-UiO-66-LD and NH2-UiO-66-HD influenced by PFOA adsorption.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: