Key Laboratory of Pollution Process and Environmental Criteria, Ministry of Education, College of Environmental Science and Engineering, Nankai University, Tianjin 300350, China
b.
Tianjin Key Laboratory of Environmental Technology for Complex Trans-Media Pollution, College of Environmental Science and Engineering, Nankai University, Tianjin 300350, China
c.
Tianjin Advanced Water Treatment Technology International Joint Research Center, College of Environmental Science and Engineering, Nankai University, Tianjin 300350, China
Received Date:
10 July 2025 Accepted Date:
17 September 2025 Revised Date:
11 September 2025 Available Online:
15 May 2026
Abstract:
Chlorine is not only widely used as an important basic chemical, but also shows promising in-situ electrochemical remediation. Unfortunately, its electrochemical production usually relies on expensive noble-metal dimensionally stable anode (DSA). Herein, a high-performance non-noble metal Co3O4/Ti anode was developed by a simple electrodeposition-calcination method, demonstrating a high efficiency in producing active chlorine in a wide pH range (3–11) and at relatively low Cl- concentration close to different real environmental requirements due to its abundant surface area and active sites provided by the interlaced nanosheet structure anode. Compared with commercial DSA, the Co3O4/Ti anode offered significant advantages in terms of Faraday efficiency, electric energy consumption and economic cost, achieving the rate of active chlorine production of 14.97 mg L-1 min-1 in 0.5 mol/L NaCl electrolyte solution (pH 6) with a Faraday efficiency of 96.8% and low energy consumption of 2.49 kWh/kg. Moreover, the robust backbone structure of the anode enabled the Faraday efficiency to be maintained at about 92.2% without deactivation after ten cycles of reaction. In addition, this Co3O4/Ti electrode demonstrated effectiveness in treating organic pollutants and mariculture wastewater and seawater rapid sterilization. This study provides new inspirations for the construction of highly efficient, low-cost, and low energy consumption non-noble metal cobalt-based anode for the in-situ environmental remediation application.
Among the 100 most crucial basic chemicals in the world, chlorine has a wide range of applications in polymer and pharmaceutical manufacturing, textile and pulp bleaching, environmental protection and disinfection [1–3]. Currently, the industrial production of chlorine gas mainly relies on the chlor-alkali industry, which involves electrolyzing a saturated NaCl (5–6 mol/L) solution (pH~2), the chlorine evolution reaction (CER) occurs at the anode [4,5]. However, for every ton of chlorine produced by the traditional chlor-alkali process, nearly 3000 kWh of electricity is consumed, and the energy cost can account for 60%−70% of the total cost, making it one of the major high-energy-consuming industries [6,7]. In addition, the chlor-alkali industry demands substantial investment in the fabrication of electrodes, maintenance of equipment, and transportation and processing of products [8,9]. Therefore, the development of anodes with excellent chlorine evolution performance is of great significance for reducing energy consumption and cost of the chlor-alkali industry.
Currently, noble metal-based dimensionally stable anode (DSA, IrO2 and RuO2) are commonly chosen for the industrial production of chlorine due to their high durability and efficiency [10–12]. However, the noble metals Ru and Ir inevitably lead to higher production costs due to their scarce resources and high prices [13,14]. On the other hand, it is known from the thermodynamic data of anodic reaction that the standard potential of the CER reaction is higher than that of the oxygen evolution reaction (OER), which leads to its poor CER selectivity due to the participation of the side reaction of OER in the competition [15,16].
Therefore, there is an urgent need to develop efficient and low-cost CER anode catalysts [3]. Wang et al. utilized a defect-anchoring strategy to anchor Ir single atoms to amorphous Ti oxides, preparing highly active and durable single-atom Ir1O4 anodes. Its CER performance was greatly enhanced while reducing the amount of noble-metal to lower the cost, and its mass activity (95 mA/mgIr) reached 500 times of that of commercial DSA [9]. Xiao et al. prepared amorphous CoOxCly catalysts for the CER reaction by electrodeposition in an acidic electrolyte containing Co2+ and Cl− ions, exhibiting about 100% selectivity for chlorine evolution in 0.5 mol/L NaCl solution with an overpotential of about 0.1 V at current density of 10 mA/cm2, which was far beyond the performance of commercial DSA [17]. Although these CER anode materials have been studied, difficulties such as complicated preparation procedures remain, which restrict their practical applications [12,18,19].
Due to their abundant reserves, flexible electronic structures and low cost, transition metals have been widely used as electrocatalytic materials, and Co3O4 with spinel structure is considered the most promising noble-metal alternative [20–22]. Co3O4 has a mixed cobalt-metal valence, where Co2+ occupies the tetrahedral coordination centers and Co3+ occupies the octahedral coordination centers [23]. These nanostructures with different growth directions show special stability and catalytic activity, which can effectively promote the reaction and reduce the overpotential and energy loss of the reaction [24–26]. The Co3O4 electrocatalysts can be prepared by thermal decomposition [27], chemical vapor deposition [28] and sol-gel method [29], among which the electrodeposition method [30,31] has the advantages of mild reaction conditions, controllable operation and good material coverage and stability [32]. Recent studies have demonstrated significant progress in cobalt-based chlorine evolution reaction (CER) catalysts. Xie et al. proposed an effective strategy to enhance ·Cl generation by incorporating electrophilic Cu2+ into Co3O4 nanowire anodes, which facilitated the conversion of Co2+ to Co3+. Their results indicated that Cu2+ doping increased the steady-state-·Cl concentration by approximately 1.5-fold, thereby promoting active chlorine production [33]. Tian et al. designed electron-deficient cobalt oxide catalysts (Sb2O5-Co3O4/Sn), where the Co3+ centers enhanced Cl− enrichment and amplified the potential gap between chlorine and oxygen evolution reactions. This design improved both CER activity and selectivity under low Cl− concentrations [34]. The scientific community currently recognized Co3+ as the primary active site for CER, where it adsorbed Cl− and mediated intermediate formation [23]. In addition, Co2+ served as an electron shuttle between Co2+/Co3+ redox cycles to preserve catalyst stability [35]. Although Co3O4-based CER catalysts have been reported, the CER process at low Cl− concentration and its application in practical environmental in-situ remediation have not been documented [23,24,36,37].
Herein, we report a non-noble metal Co3O4/Ti electrocatalyst prepared by a simple electrodeposition-calcination method using Ti plates as the substrate in view of environmental application. Co3O4 with interlaced nanosheet structure generates abundant surface area and active sites for CER reaction, and the porous structure on the nanosheets provides more opportunities for reaction mass transfer. Through parameters optimization of the electrode preparation, efficient and stable production of active chlorine (AC) in a wide pH range and at low Cl− concentration was achieved. Compared with the commercial noble metal-based DSA, this Co3O4/Ti anode is a reliable candidate for practical active chlorine production because of its advantages in high Faraday efficiency, low electric energy consumption and economic cost. In addition, we have demonstrated that this anode is not only advantageous for the treatment of organic pollutants, but also for the treatment of mariculture wastewater and rapid sterilization.
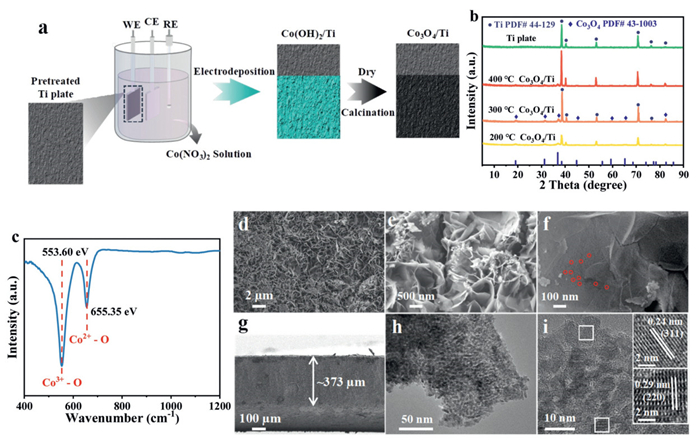
As shown in Fig. 1a, the preparation of Co3O4/Ti electrodes consisted of two steps: Electrodeposition and calcination. Firstly, Co(OH)2/Ti was obtained by electrodepositing Ti plates in the Co(NO3)2 solution, and then the Co(OH)2/Ti with green surface was converted to Co3O4/Ti by calcination at different temperatures. It has been reported that the temperature threshold for the conversion of Co(OH)2 to Co3O4 is located at 150–200 ℃, and the crystallinity of Co3O4 gradually increases without changing the physical phase after the calcination temperature is higher than 200 ℃ [31]. Fig. 1b showed the results of X-ray diffraction (XRD) analyses of Co3O4/Ti prepared at different calcination temperatures. Except for the peaks of Ti substrate, all Co3O4/Ti obtained by calcination at different temperatures belong to Co3O4 (PDF No. 43–1003), including (111), (220), (311), (400), etc., indicating that Co(OH)2 electrodeposited on Ti plates was completely converted to Co3O4 at a calcination temperature of 200 ℃, 300 ℃, and 400 ℃ (Fig. S1 in Supporting information). The lower the Full Width at Half Maximum (FWHM) in the XRD plot, the sharper the diffraction peaks and the better the crystallinity [38,39]. As shown in Fig. S2 (Supporting information), at 2θ = 36.8° (311), the FWHM was 0.92° for 200 ℃, whereas those for 300 ℃ and 400 ℃ were 0.84° and 0.49°, respectively. It was demonstrated that increased calcination temperature could enhance the crystallinity of Co3O4. In the FTIR spectrum of Co3O4/Ti (Fig. 1c), the intense absorption bands at 553.6 cm-1 and 655.35 cm-1 can be ascribed to the stretching vibrations of Co3+-O and Co2+-O bonds, respectively. This finding is in agreement with the outcomes of the XRD analyses obtained by calcining the electrode at 300 ℃.
Figure 1
Figure 1.
Preparation and characterization of samples. (a) Preparation flow chart of the Co3O4/Ti electrode. (b) XRD patterns and (c) FTIR spectrum of Co3O4/Ti. (d-f) SEM images of the surface. (g) SEM image of the cross-section. (h) TEM image and (i) HRTEM image of the Co3O4/Ti electrode. The Co3O4/Ti electrode was synthesized under optimized preparation conditions.
Figs. 1d and e showed the scanning electron microscope (SEM) characterization of Co3O4/Ti, which were grown on Ti plates, forming clusters with a unique spatially interlaced nanosheet network, and the nanosheets had uniform size and thickness (Fig. S3 in Supporting information). The interlaced nanosheet arrangement structure resulted in a larger specific surface area and a stronger backbone of the material. Moreover, the surface of Co3O4 nanosheets exhibited numerous nanopores with a diameter of about 10 nm (Fig. 1f), which facilitated the provision of more active sites in the electrochemical reactions. A cross-section of the Co3O4/Ti electrode was shown in Fig. 1g, observing that the Co3O4 deposited on the Ti plate surface was about 373 µm thick and tightly adhered to the Ti plate, and the Co3O4 also exhibited a nanosheet network structure in the cross-section (Fig. S4 in Supporting information). In addition, the energy-dispersive X-ray spectroscopy (EDS) elemental mapping of the material surface showed a uniform distribution of Co and O elements (Fig. S5 in Supporting information). The Brunauer-Emmett-Teller (BET) data for Co3O4/Ti and Ti plates was listed in Table S1 and Fig. S6 (Supporting information), including BET surface area, pore volume, and average pore size. The quantitative BET analysis confirmed that the Co3O4/Ti nanosheets exhibited a significantly enhanced specific surface area (51.80 vs. 0.14 m2/g) and a well-developed mesoporous structure (average pore size = 15.15 nm) compared to the bare Ti substrate. This finding strongly supports the observed superior catalytic activity.
As shown in Figs. 1h and i, the Co3O4/Ti was characterized by transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM), demonstrating obvious lattice stripes. The lattice spacings of 0.24 nm and 0.29 nm corresponded to the 311 and 220 crystalline planes of Co3O4 respectively, and these lattice spacings were in agreement with the XRD results. The above characterization results demonstrated the successful preparation of Co3O4/Ti electrodes.
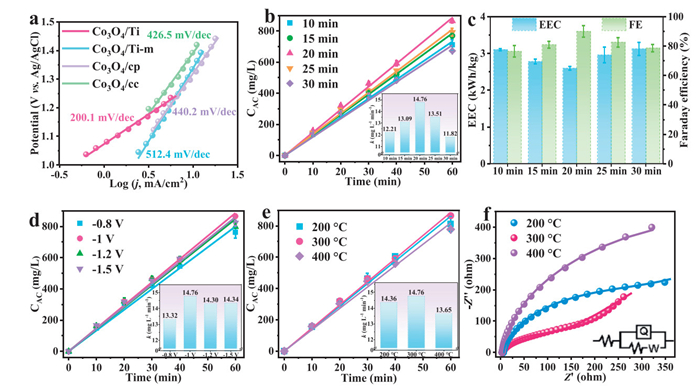
The performance of Co3O4 electrodeposited on different substrates (Ti plate, Ti mesh, carbon paper, and carbon cloth) was compared. The Co3O4/Ti plate, Co3O4/Ti mesh (Co3O4/Ti-m), Co3O4/carbon paper (Co3O4/cp), and Co3O4/carbon cloth (Co3O4/cc) were respectively used as the anode for active chlorine production in 2 h experiments. From the graph of active chlorine concentration per unit area of material (Fig. S7 in Supporting information), it could be seen that Co3O4/Ti has the highest chlorine concentration, which was better than that on carbon substrate and Ti mesh substrate. After that, LSV tests were conducted on the above four different substrate materials of the polar plates respectively. As shown in Fig. 2a, the Tafel slope of Co3O4/Ti (200.1 mV/dec), was much smaller than those of Co3O4/cc (426.5 mV/dec), Co3O4/cp (440.2 mV/dec), and Co3O4/Ti-m (512.4 mV/dec). This result implied that the kinetics for CER of Co3O4/Ti were the fastest, suggesting that it might possess superior electrocatalytic activity. It was proposed that the flat surface of the Ti plate facilitated uniform current distribution during electroplating, which promoted homogeneous deposition of Co3O4 after calcination. In contrast, the Ti mesh exhibited excessive porosity that hinders mass transport. Moreover, the uneven current distribution at the mesh pores during electroplating led to non-uniform coating [40,41]. Considering the poor stability of the carbon substrate material as an anode and the poor performance of the Ti mesh as a substrate, the Ti plate was chosen as the electrodeposition substrate material for the subsequent experiments.
Figure 2
Figure 2.
Optimization of preparation parameters of Co3O4/Ti electrode. (a) Tafel slope diagrams of Co3O4 electrodes electrodeposited on different substrates. (b) Active chlorine (AC) production on electrodes prepared with different electrodeposition times and (c) electric energy consumption and Faraday efficiency for a reaction of 10 min. (d) Active chlorine production on electrodes prepared at different deposition potentials. (e) Active chlorine production on electrodes prepared at different calcination temperatures and (f) EIS diagrams. Reaction conditions: Current density: 10 mA/cm2; Electrolyte: 50 mL of 0.5 mol/L NaCl solution.
Furthermore, the double layer capacitance (Cdl) of the Co3O4/Ti plate and the Ti substrate was calculated (Fig. S8 in Supporting information) by fitting a combined calculation at a potential of 0.4 V (vs. Ag/AgCl) based on the CV curves. The Cdl value of Co3O4/Ti was 0.097 µF/cm2, which was 3 times the Cdl value of the Ti plate (0.030 µF/cm2), demonstrating that the deposition of Co3O4 increased the active surface area of the Ti substrate. To highlight the catalytic contribution of Co3O4, we used a bare Ti plate as the anode for active chlorine generation. At a current density of 10 mA/cm2, the Ti plate underwent rapid electrochemical passivation when used, generating dense yellow oxides on the surface (Fig. S9 in Supporting information), while the anodic potential rose sharply by nearly 8 V in 430 s (Fig. S10 in Supporting information). More importantly, at 430 s the concentration of active chlorine in the solution was only 0.95 mg/L, which was much lower than that of the Co3O4/Ti electrode. In addition, linear scanning voltammetry tests (Fig. S11 in Supporting information) showed that the bare Ti plate possessed almost no CER activity compared to the Co3O4/Ti electrode. These results clearly indicated that the observed CER activity originates exclusively from the catalytic effect of the Co3O4-modified layer.
Subsequently, to determine the optimal conditions for electrodeposition preparation, a series of experiments were carried out on the factors of electrodeposition time, Co(NO3)2 concentration in the deposition solution, and the deposition potential. Five electrodeposition times (10, 15, 20, 25, and 30 min) were chosen to regulate the loading of Co3O4 on the Ti plate. As shown in Fig. 2b, as the electrodeposition time was extended, the active chlorine production rate exhibited the trend of initial increase followed by a decline. At the beginning, augmenting the Co3O4 loading could offer more reactive sites, facilitating the attachment of Cl− and thus promoting the production of active chlorine. Nevertheless, once the electrodeposition time surpassed 20 min, the growth of Co3O4 led to the blockage of active sites. The Co3O4/Ti electrode reached the peak active chlorine production rate of 14.76 mg L-1 min-1 at the electrodeposition time of 20 min. On the other hand, under different electrodeposition time conditions, the lowest electric energy consumption (EEC) and the highest Faraday efficiency were observed when the electrodeposition time was 20 min (Fig. 2c). As shown in Fig. 2d and Fig. S12 (Supporting information), the Co3O4/Ti electrode produced the highest active chlorine concentration when the electrodeposition potential was −1.0 V vs. the Ag/AgCl and the Co(NO3)2 concentration was 0.025 mol/L.
It was reported that the calcination temperature affected the ratio of Co2+ to Co3+ in Co3O4 loaded on the electrode plate, which in turn affected the electrocatalytic performance [32]. So, the effect of calcination temperature on the active chlorine production was investigated. As shown in Fig. 2e, the active chlorine production rate of Co3O4/Ti electrodes increased and then decreased with the increase of calcination temperature from 200 ℃ to 400 ℃. Meanwhile, due to the oxidation of the Ti plate surface to TiO2 at 400 ℃, the decrease in conductivity increased the EEC (Fig. S13). The analysis of Nyquist plots showed (Fig. 2f and Fig. S14 in Supporting information) that the Co3O4/Ti electrode calcined at 300 ℃ had the smallest Rct (113.3 Ω), which was smaller than that obtained at 200 ℃ (Rct = 317.2 Ω) and 400 ℃ (Rct = 797.6 Ω). Therefore, this electrode exhibited more efficient charge transfer and better conductivity. Moreover, different calcination temperatures may affect the crystallinity of Co3O4 [42–44]. The Co3O4/Ti electrode calcined at 200 ℃ emitted black powdery substances in the solution during the reaction. The ICP-MS test showed that the dissolved cobalt ions was higher (Fig. S15 in Supporting information), suggesting that the stability of the electrode calcined at 200 ℃ was poor. After optimization of preparation conditions, the Co3O4/Ti electrode fabricated with 0.025 mol/L Co(NO3)2 as the electrodeposition solution at −1.0 V for 20 min, followed by calcination at 300 ℃, was selected as the optimal material. All subsequent experiments were conducted based on this optimized Co3O4/Ti electrode.
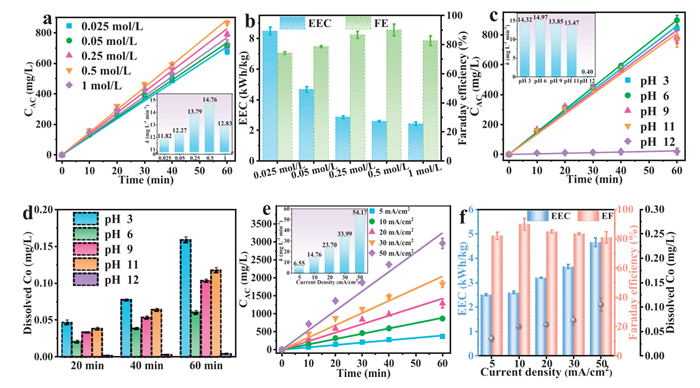
Fig. 3a depicted the dependence of active chlorine production and the corresponding apparent rate constant (k) with the different concentrations of NaCl electrolyte. Obviously, with the increased NaCl concentration less than 0.5 mol/L, the produced active chlorine gradually increased, however, further increased NaCl concentration led to the decrease of active chlorine production rate (insert figure). This is due to excess Na+ and Cl− in the solution forming a concentration polarization layer at the electrode interface, thereby covering the active sites on the surface of Co3O4 catalyst [45]. The corresponding EEC was found decreased with increasing Cl- concentration, but the Faraday efficiency demonstrated the highest at the NaCl concentration of 0.5 mol/L (Fig. 3b).
Figure 3
Figure 3.
Optimization of factors for the production of active chlorine by the Co3O4/Ti electrode. (a) The effect of electrolyte concentrations on active chlorine production, and (b) the electric energy consumption and Faraday efficiency. (c) The effect pH on the production of active chlorine. (d) The cobalt ions dissolution from the electrode. (e) The effect of current density on the production of active chlorine, and (f) the EEC, Faraday efficiency and cobalt ions dissolution.
From Fig. 3c, it could be seen that when the electrolyte pH was in range of 3–11, the electrode exhibited good active chlorine production. As shown in Fig. S16 (Supporting information), at pH 6, the Faraday efficiency reached the highest value (96.8%) with an energy consumption of 2.49 kWh/kg. However, almost no active chlorine was produced at pH 12. The reaction equation for the preparation of active chlorine from electrolyzed NaCl solution was shown in Eq. 1. Cl− loses electrons at the anode to produce chlorine gas (Eq. 2). In alkaline solution, the side reaction is aggravated (Eq. 3). The higher concentration of OH- promotes its participation in the reaction at the anode surface to produce oxygen, and OH− competed with Cl− for the loss of electrons at the anode, which made the original reaction Eq. 2 inhibited. In addition, since OH− participates in the reaction, it occupies the active sites on the electrode surface and consumes energy, further reducing the efficiency of chlorine generation [46]. We recorded the pH changes before and after 1 h energization reaction for each system (Fig. S17 in Supporting information), and found that for an initial pH ranging from 3 to 11, the final pH after reaction ranged from 8 to 11. Under acidic conditions, the significant pH increase during NaCl electrolysis primarily stemmed from the chlorine evolution reaction mechanism (Eq. 1) which generated NaOH. The amount of cobalt dissolved at pH 8–9 at the reaction endpoint was reduced compared to pH 3 (Fig. 3d), and this alkaline change enhanced the stability of the electrode. After the reaction under alkaline conditions, the endpoint pH was slightly decreased. Under highly alkaline conditions (pH 12), ClO− formed the intermediate activation complex Cl2O22−, which then decomposed rapidly according to Eq. 4 to produce O2 and Cl− [47]. In addition, we conducted a test on the dissolved cobalt ions by ICP-MS (Fig. 3d), and found that at pH 6 it was less than that at acidic and alkaline solution. In the pH range of 3 - 11, the dissolution of Co3O4/Ti was less than 0.2 mg/L, which was lower than the limit of 1 mg/L stipulated in the "Copper, Nickel, Cobalt Industrial Pollutant Emission Standards" (GB25467–2010) in China.
As an important parameter in electrochemical reactions, the electrode current density had a significant impact on the reaction rate. We thus investigated the effect of current density from 5 mA/cm2 to 50 mA/cm2 in 0.5 mol/L NaCl solution (pH 6). As shown in Fig. 3e, the generation rate of active chlorine accelerated with the increased current density. Meanwhile, it was found that the k for the production of active chlorine tended to increase linearly with the increase of current density (R2 = 0.996). As shown in Fig. 3e and Fig. S18 (Supporting information), the k increased from 6.55 mg L-1 min-1 to 54.17 mg L-1 min-1 when the current density increased from 5 mA/cm2 to 50 mA/cm2. This linear relationship suggested that adjusting the current density can optimize the productivity of active chlorine. On the other hand, we conducted a comprehensive investigation on the EEC and Faraday efficiency of the system at different current densities. As shown in Fig. 3f, the EEC gradually increased with the increase of the current density, and the Faraday efficiency showed an increasing-then-decreasing trend, which was in accordance with literature [48]. The highest Faraday efficiency of 90.0% was achieved at the current density of 10 mA/cm2, and its corresponding EEC was 2.60 kWh/kg. In addition, to evaluate the possible environmental impact of the Co3O4/Ti electrode in industrial applications, we tested the dissolution of cobalt ion (Fig. 3f and Fig. S19 in Supporting information), showing that it increased with the increase of current density. Nonetheless, even at a high current density of 50 mA/cm2, the leaching cobalt ion was only 0.105 mg/L after 1 h reaction, which was lower than the limit of 1 mg/L in China as mentioned before.
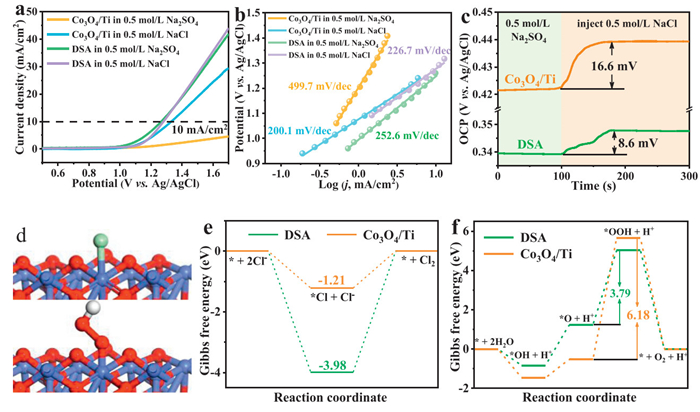
Considering that OER occurs on the anode during active chlorine production, the competition between OER and CER can seriously affect the effectiveness of active chlorine production. To evaluate the selectivity of the CER on the Co3O4/Ti electrode, we performed LSV tests in 0.5 mol/L NaCl and 0.5 mol/L Na2SO4 respectively to elucidate the OER and CER performances (Fig. 4a). The CER and OER activities on the commercial DSA are similar, indicating that the competition between OER and CER is intense and the CER selectivity is poor. In contrast, the CER activity on the Co3O4/Ti electrode without noble metals was significantly stronger than that of OER, indicating that the Co3O4/Ti electrode had higher selectivity for chlorine evolution than the commercial DSA. Next, we compared the Tafel slopes of these two electrodes (Fig. 4b), where the Tafel slope of the CER on Co3O4/Ti (200.1 mV/dec) was smaller than that of the commercial DSA (226.7 mV/dec), indicating that the CER kinetics of Co3O4/Ti is faster than that of the DSA. On the other hand, the Tafel slope of the OER on Co3O4/Ti (499.7 mV/dec) was much larger than that of the DSA (252.6 mV/dec), indicating that the OER kinetics of Co3O4/Ti was slower and the competing reactions were not easy to occur. In conclusion, the Co3O4/Ti had superior CER selectivity.
Figure 4
Figure 4.
Electrochemical characterization of the Co3O4/Ti electrode. (a) LSV curves of the commercial DSA electrode and the Co3O4/Ti electrode in Na2SO4 and NaCl solutions and (b) Tafel slope diagrams. (c) OCP measurements. (d) Schematic structures for Cl and OOH adsorption on Co3O4/Ti. Co: blue, O: red, Cl: green, and H: gray. Diagrams of Gibbs free energy of (e) CER on Co3O4/Ti and DSA, (f) Gibbs free energy of OER on Co3O4/Ti and DSA surface.
To evaluate the ability of Cl- adsorption on the surface of Co3O4/Ti electrode, we tested the open-circuit potential (OCP) in 0.5 mol/L Na2SO4. As shown in Fig. 4c, when the OCP was stabilized, 0.5 mol/L NaCl solution (1.46 g NaCl in 50 mL) was rapidly injected and the OCP changed immediately. After the potential was stabilized, the OCP of DSA and Co3O4/Ti electrodes increased by about 8.6 mV and 16.6 mV, respectively, indicating that more Cl- was adsorbed on the surface of Co3O4/Ti electrode, so that the adsorptive capacity for Cl− was stronger.
Density functional theory (DFT) calculations were employed to further elucidate the reaction mechanism of Co3O4/Ti in chlorine evolution reaction [49,50]. We calculated the Gibbs free energy of CER and OER on Co3O4/Ti and DSA, respectively. Fig. 4d illustrated the catalyst models for the adsorption of Cl and OOH by Co3O4. As shown in Fig. 4e, Co3O4/Ti (1.21 eV) had a lower ΔG than that of DSA (3.98 eV) in the generation of Cl2. It indicated that the energy barrier of Co3O4 was lower, which was favorable for the generation of Cl2. For comparison, we also compared the Gibbs free energy of the OER of Co3O4/Ti with that of DSA by DFT calculations. As shown in Fig. 4f, OER occurred through four steps, where the rate-determining step (RDS) was the transition from *O to *OOH. Our comparison of the rate-determining steps of Co3O4/Ti and DSA revealed that the energy barrier for the formation of *OOH on Co3O4 was 6.18 eV, which was higher than that of DSA (3.79 eV). This demonstrated that Co3O4/Ti exhibited lower OER activity compared to DSA, and further indicated that Co3O4/Ti had better selectivity for chlorine evolution.
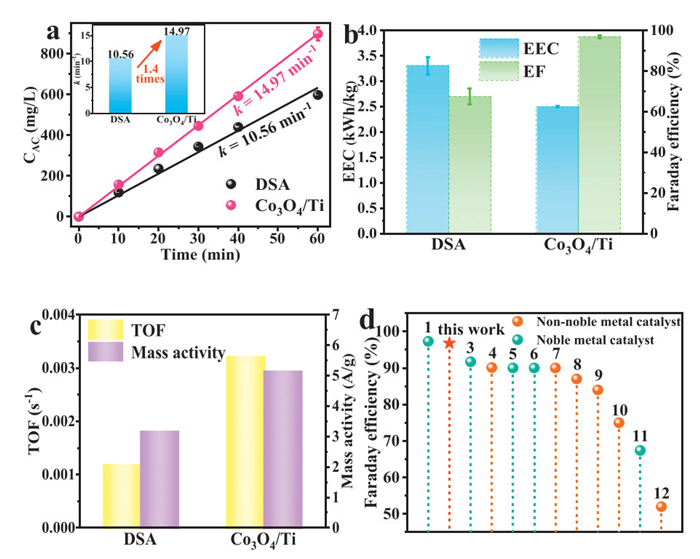
In addition to electrochemical tests, we carried out experiments on active chlorine production on these two electrodes. As shown in Fig. 5a, after 1 h reaction, the DSA produced an active chlorine concentration of 598 mg/L, while the Co3O4/Ti electrode produced concentration of 865 mg/L, which was 1.45 times higher than that of the DSA. Meanwhile, the k of the active chlorine reaction on the Co3O4/Ti electrode was 1.4 times higher than that of DSA.
Figure 5
Figure 5.
Comparison with other existing anodes. (a) Comparison of the effect of producing active chlorine between the commercial DSA electrode and the Co3O4/Ti electrode (Current density: 10 mA/cm2, Electrolyte: 50 mL of 0.5 mol/L NaCl solution, pH 6). (b) Comparison of EEC and Faraday efficiency. (c) Comparison of TOF value and mass activity. (d) Comparison of this work with literature.
Afterwards, we performed a comprehensive comparison of the EEC and Faraday efficiency of the Co3O4/Ti electrode and the DSA (Fig. 5b). The Co3O4/Ti electrode consumed only 75.4% of the EEC compared to the DSA for active chlorine production. Meanwhile, the Faraday efficiency of Co3O4/Ti electrode was 96.8%, which was also much higher than that of DSA electrode (67.4%). To further verify the economic feasibility of the Co3O4/Ti electrode, we estimated the costs of the two electrodes (Table S2 and Fig. S20 in Supporting information), showing that the cost of the Co3O4/Ti electrode (296.9 $/m2) was much lower than that of the commercial DSA. In addition, Turnover Frequency (TOF) and metal mass activity, which were widely recognized as key parameters for evaluating electrode activity [4], were also compared. As shown in Fig. 5c, the TOF (0.0032 s-1) and metal mass activity (5.13 A/gCo) of the Co3O4/Ti electrode were higher than those of the DSA (TOF = 0.0012 s-1, metal mass activity = 3.15 A/gRu+Ir). Taken together, the above results clearly showed that the Co3O4/Ti electrode had advantages in the active chlorine production rate, cost, electric energy consumption, and catalytic activity (Fig. S21 in Supporting information). To further elucidate the feasibility of Co3O4/Ti electrodes for industrial production of active chlorine, we compared the Faraday efficiency with recently reported anodes (Fig. 5d and Table S3 in Supporting information). The Faraday efficiency on the Co3O4/Ti electrodes was higher than those of the majority of the non-noble metal-based catalysts, and were comparable to those of most noble metal-based catalysts.
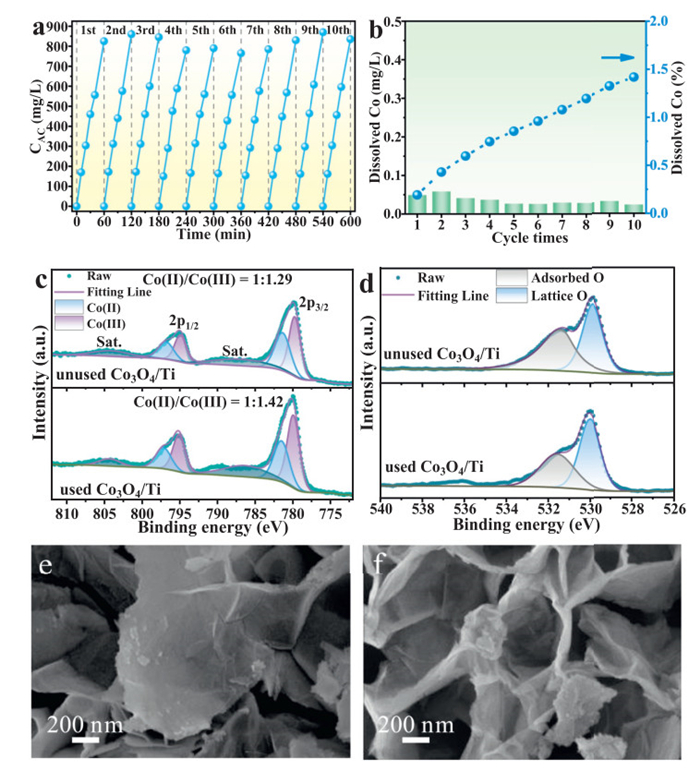
The electrode reusability and stability are crucial in real production [51,52]. Using the same electrode, we carried out 10 cycles under the optimal experimental conditions for active chlorine production. The Co3O4/Ti electrodes were reused in the next cycle by simply being rinsed with deionized water and drying. As presented in Fig. 6a, the electrode performance had slightly declined after 10 cycle tests. Meanwhile, the apparent rate constant of active chlorine production was maintained above 13.4 min-1 for 10 cycle tests (Fig. S22 in Supporting information). Moreover, the EEC and Faraday efficiency of this electrode did not change significantly (Fig. S23 in Supporting information). These results indicated that the catalytic activity of the electrode had not decreased with time. Moreover, at the end of each cycle, the concentration of cobalt ions in the solution was determined to evaluate the electrode stability. As shown in Fig. 6b, the leaching cobalt ions decreased with repeated times, reaching 0.023 mg/L at the end of the 10th experiment. After ten cycles, the total leaching cobalt ions from the electrode was only about 1.4% of the total amount of cobalt elements on the electrode (Fig. 6b), which proved that the electrodeposition-calcination method was effective in immobilizing Co3O4 on the Ti plate substrate, further demonstrating its reliability in practical applications. To evaluate the durability of the electrodes under acidic environments, we performed stability tests on Co3O4/Ti electrodes under pH 3 for 24 h of continuous operation using an electrochemical workstation (current density = 10 mA/cm2). As shown in Fig. S24 (Supporting information), the voltage across the Co3O4/Ti electrode only increased from 1.57 V to 1.80 V after 24 h of continuous operation. Comparing with the voltage change graph of the bare titanium plate (Fig. S10), it demonstrated that the adhesion between the Co3O4 and the Ti plate was good, and that the small change in voltage might be due to oxidization of the titanium plate and deterioration of its conductivity. In addition, testing the chlorine production and cobalt ion dissolution of the electrode in the long-term stability test (Fig. S25 in Supporting information) could prove that the Co3O4/Ti electrode still produced active chlorine linearly after 12 h of cycling, and the cobalt ion dissolution slowed down significantly, and the total cobalt ion dissolution was only 0.35 mg/L after 24 h of operation.
Figure 6
Figure 6.
Material stability tests. (a) The effect diagram of the Co3O4/Ti electrode producing active chlorine after 600 min of reaction and (b) the cobalt ions dissolution of the electrode after 10 cycle times. (c) Comparison of the Co 2p XPS spectra of the Co3O4/Ti electrode before and after the reaction and (d) the O 1 s XPS spectra. (e) SEM images of the electrode before the cyclic reaction and (f) SEM images of the electrode after the cyclic reaction.
To further understand the reasons underlying the excellent stability of the Co3O4/Ti electrode, we performed X-ray photoelectron spectroscopy (XPS) analysis and SEM tests on the electrodes before and after the cyclic tests. The high-resolution Co 2p spectra were shown in Fig. 6c, where the peaks at 781.4 and 797.0 eV are attributed to Co2+, while the peaks at 779.9 and 795.0 eV are attributed to Co3+. In addition, the fine spectrum of Co3O4/Ti O 1s (Fig. 6d) could be fitted to two peaks, centered at 531.5 and 529.9 eV respectively, corresponding to adsorbed oxygen (Oa) and lattice oxygen (OL). After 10 cycles, the Co3+/Co2+ ratio on the Co3O4/Ti electrode increased from 1.29 to 1.42. This might be due to the conversion of Co2+ to Co3+ caused by the reduction of the polar plate as an anode, and the OL/Oa ratio did not change significantly. These indicated that the properties of the Co3O4/Ti electrode remained largely unchanged after use. Similarly, the SEM results showed that the structure of the used Co3O4/Ti (Fig. 6e) was basically the same as that of the electrode before use (Fig. 6f). In summary, the Co3O4/Ti electrode has excellent stability, which makes it a reliable candidate for practical active chlorine production.
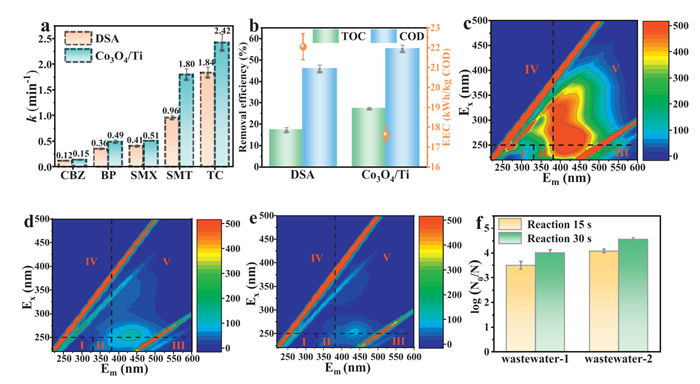
In order to examine the possible environmental application prospect of Co3O4/Ti electrode, we selected several bio-refractory organic pollutants (initial concentration: 20 mg/L each). It was exhibited that it had better degradation performance of carbamazepine (k = 0.15 min-1), phenol (k = 0.49 min-1), sulfamethoxazole (k = 0.51 min-1), sulfathiazole (k = 1.80 min-1), and tetracycline hydrochloride (k = 2.42 min-1) compared with that of the DSA (Fig. 7a), demonstrating its potential for environmental applications. Furthermore, the potential of the Co3O4/Ti electrode for application in real environments, was evaluate using high-salt mariculture wastewater (its quality parameters are detailed in Table S4 in Supporting information) for the degradation experiments. As shown in Fig. 7b, it removed much higher chemical oxygen demand (COD) and total organic carbon (TOC) within 30 min, reaching 1.20 and 1.58 times the removal on the DSA, respectively. Meanwhile, the EEC of the Co3O4/Ti electrode (17.6 kWh/kg COD) was also lower than that of the DSA (22.0 kWh/kg COD), which suggested that it had greater application potential from the perspective of economic cost. The Co3O4/Ti electrode maintained effective degradation performance in this authentic matrix, confirming stable catalytic activity in high-salinity environments with complex contaminants. Notably, adsorbed organics underwent rapid mineralization (Fig. 7a), completely preventing persistent organics accumulation. Most importantly, macroscopic inspection (Fig. S26 in Supporting information) revealed no detectable scaling after 2 h operation in actual aquaculture wastewater.
Figure 7
Figure 7.
Environmental applications. (a) Comparison of the rates of degradation of different pollutants by the Co3O4/Ti electrode and the commercial DSA electrode. (b) Comparison of the TOC and COD removal rates when degrading actual mariculture wastewater. (c) 3D EEM spectrogram of the Co3O4/Ti electrode degrading mariculture wastewater at 0 min, (d) 15 min and (e) 30 min. (f) Sterilization of the Co3O4/Ti electrode in two kinds of actual mariculture wastewater.
In addition, we evaluated the changes in fluorescence properties of the Co3O4/Ti electrode during the treatment of mariculture wastewater using the three-dimensional excitation-emission matrix spectroscopy technique (3D EEMs) [53]. Moreover, three distinct fluorescence peaks (Ⅲ, Ⅳ and Ⅴ) emerged in the raw wastewater (Fig. 7c), indicating that it contained a large amount of fulvic acid-like, soluble microbial byproduct and humic acid-like substances. After treatment for 15 min (Fig. 7d) and 30 min (Fig. 7e), it could be seen that the fluorescence intensities of all regions of the EEM spectra were significantly decreased, which was attributed to the high reactivity of active chlorine in the treatment of humic acid-like substances. The change in fluorescence properties after treatment on DSA was shown in Fig. S27 (Supporting information), confirming that the removal of fluorescent substances was significantly worse than that on Co3O4/Ti. The change in peak intensity of the above process was consistent with the removal rates of COD and TOC.
Meanwhile, as an excellent disinfectant, active chlorine could sterilize the mariculture wastewater. The samples were taken at 15 s and 30 s of the energization reaction, respectively. The 1000-fold diluted raw water and disinfected samples were uniformly coated in solid medium and incubated. As shown in Fig. 7f and Fig. S28 (Supporting information), the active chlorine produced by the Co3O4/Ti electrode was sufficient to achieve 100% sterilization in this mariculture wastewater within 30 s (log(N0/N) = 4). Similar results (log(N0/N) = 4.6) were observed for another mariculture wastewater. Compared to other electrodes, this Co3O4/Ti electrode sterilized mariculture wastewater faster and produced lower residual chlorine after sterilization [4,23].
In summary, the Co3O4/Ti electrode had the advantage of efficiently utilizing Cl- in real wastewater, and showed great potential in the field of chlorine-mediated treatment of highly mineralized water, so that it had broad application prospects in other real water treatments.
In conclusion, a stable non-noble metal Co3O4/Ti anode was developed by a simple electrodeposition-calcination method to realize the efficient production of active chlorine at low Cl- concentration. The surface of the Co3O4/Ti anode featured a spatially interspersed porous nanosheet structure. Specifically, due to the robust backbone structure, the electrode had excellent stability and maintained the high Faraday efficiency about 92.2% without deactivation after 10 cycling tests. It proved that the Co3O4/Ti anode was superior to the noble metal DSA in active chlorine production including Faraday efficiency, electric energy consumption, and economic cost. The electrode demonstrated advantages not only in treating organic pollutants (CBZ, BP, SMX, SMT, TC), but also in mariculture wastewater and sterilize rapidly. Overall, this electrode provides a new idea for constructing highly efficient, low electric energy consumption, and wide pH range anodes of producing active chlorine, which is found to be efficient in treating real wastewater and disinfection with the help of chlorine-mediated oxidation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We acknowledge financial support from the National Natural Science Foundation of China (No. U23B20165), and Tianjin Science and Technology Project (Nos. 23YDTPJC00870 and 22YFYSHZ00300).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111857.
[1]
S. Hong, G. Choi, N. Thi Yen Phan, H. Shin, J. Lim, Chem. Eng. J. 493 (2024) 152698. doi: 10.1016/j.cej.2024.152698
[2]
M. Xue, J. Zhao, X. Yu, et al., Adv. Funct. Mater. 34 (2023) 2308567.
[3]
B. Li, Y. Duan, L. Wei, et al., Adv. Funct. Mater. 34 (2024) 2314150. doi: 10.1002/adfm.202314150
J. Liu, R. Liang, Z. Hu, X. Zhang, M. Zhou, Chem. Eng. J. 491 (2024) 152088.
[53]
S. Li, J. Xie, J. Gu, M. Zhou, Chin. Chem. Lett. 34 (2023) 108204.
Figure 1
Preparation and characterization of samples. (a) Preparation flow chart of the Co3O4/Ti electrode. (b) XRD patterns and (c) FTIR spectrum of Co3O4/Ti. (d-f) SEM images of the surface. (g) SEM image of the cross-section. (h) TEM image and (i) HRTEM image of the Co3O4/Ti electrode. The Co3O4/Ti electrode was synthesized under optimized preparation conditions.
Figure 2
Optimization of preparation parameters of Co3O4/Ti electrode. (a) Tafel slope diagrams of Co3O4 electrodes electrodeposited on different substrates. (b) Active chlorine (AC) production on electrodes prepared with different electrodeposition times and (c) electric energy consumption and Faraday efficiency for a reaction of 10 min. (d) Active chlorine production on electrodes prepared at different deposition potentials. (e) Active chlorine production on electrodes prepared at different calcination temperatures and (f) EIS diagrams. Reaction conditions: Current density: 10 mA/cm2; Electrolyte: 50 mL of 0.5 mol/L NaCl solution.
Figure 3
Optimization of factors for the production of active chlorine by the Co3O4/Ti electrode. (a) The effect of electrolyte concentrations on active chlorine production, and (b) the electric energy consumption and Faraday efficiency. (c) The effect pH on the production of active chlorine. (d) The cobalt ions dissolution from the electrode. (e) The effect of current density on the production of active chlorine, and (f) the EEC, Faraday efficiency and cobalt ions dissolution.
Figure 4
Electrochemical characterization of the Co3O4/Ti electrode. (a) LSV curves of the commercial DSA electrode and the Co3O4/Ti electrode in Na2SO4 and NaCl solutions and (b) Tafel slope diagrams. (c) OCP measurements. (d) Schematic structures for Cl and OOH adsorption on Co3O4/Ti. Co: blue, O: red, Cl: green, and H: gray. Diagrams of Gibbs free energy of (e) CER on Co3O4/Ti and DSA, (f) Gibbs free energy of OER on Co3O4/Ti and DSA surface.
Figure 5
Comparison with other existing anodes. (a) Comparison of the effect of producing active chlorine between the commercial DSA electrode and the Co3O4/Ti electrode (Current density: 10 mA/cm2, Electrolyte: 50 mL of 0.5 mol/L NaCl solution, pH 6). (b) Comparison of EEC and Faraday efficiency. (c) Comparison of TOF value and mass activity. (d) Comparison of this work with literature.
Figure 6
Material stability tests. (a) The effect diagram of the Co3O4/Ti electrode producing active chlorine after 600 min of reaction and (b) the cobalt ions dissolution of the electrode after 10 cycle times. (c) Comparison of the Co 2p XPS spectra of the Co3O4/Ti electrode before and after the reaction and (d) the O 1 s XPS spectra. (e) SEM images of the electrode before the cyclic reaction and (f) SEM images of the electrode after the cyclic reaction.
Figure 7
Environmental applications. (a) Comparison of the rates of degradation of different pollutants by the Co3O4/Ti electrode and the commercial DSA electrode. (b) Comparison of the TOC and COD removal rates when degrading actual mariculture wastewater. (c) 3D EEM spectrogram of the Co3O4/Ti electrode degrading mariculture wastewater at 0 min, (d) 15 min and (e) 30 min. (f) Sterilization of the Co3O4/Ti electrode in two kinds of actual mariculture wastewater.

 DownLoad:
DownLoad:

 下载:
下载:








 下载:
下载:
