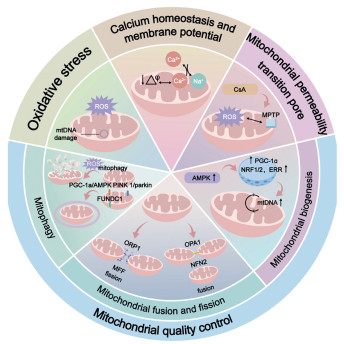
Figure 1.
The effects of mitochondrial homeostasis on MI and MI/RI.
Research progress on nanobiomedicine targeting mitochondrial homeostasis for improving myocardial ischemia
Jing Qian , Guoxing Ling , Yue Li , Yan Liu , Xiaoxuan Guan , Zuyuan Huang , Ming Gao , Cheng Luo , Baoshi Zheng

Myocardial infarction (MI) remains one of the leading causes of death among cardiovascular diseases, posing a serious threat to the health of middle-aged and elderly individuals, and increasingly, to younger populations as well [1]. MI happens due to ischemic and hypoxic conditions that injure the heart muscle, where long-term and repeated ischemia leads to a significant loss of heart muscle cells, causing irreversible damage and scar tissue formation [2]. The best way to treat MI is through early coronary revascularization, which quickly restores blood flow to save healthy heart tissue. Although reperfusion is crucial for myocardial protection and functional recovery, it has long been recognized that reperfusion itself after MI can lead to acute myocardial dysfunction and necrosis, causing MI-reperfusion injury (MI/RI). MI/RI is defined as the paradoxical worsening of myocardial injury after rapid restoration of blood flow in the ischemic area. These damaging processes include fatal myocardial cell injury, myocardial stunning, vascular damage and dysfunction, and tissue edema, which can exacerbate the condition of patients with MI [3,4]. Among the various pathological mechanisms, mitochondrial dysfunction plays a critical role in the onset and progression of myocardial ischemia diseases like MI and MI/RI [5,6]. Normally, more than 95% of the adenosine triphosphate (ATP) that heart cells need is produced by mitochondria, which are the main places where energy and reactive oxygen species (ROS) are made in eukaryotic cells [7]. Healthy mitochondrial function is essential for mitigating oxidative stress and preventing cell death. MI and MI/RI induce oxidative stress, damaging mitochondria and disrupting their function in heart cells. Ischemia prevents both aerobic and anaerobic glycolysis, resulting in a lack of energy and cellular function imbalance. This results in a buildup of ROS in both the cytoplasm and mitochondria, calcium overload, and repeated acidosis. Studying how mitochondrial damage and dysfunction occur in myocardial ischemia, along with related treatment options, is crucial [8,9].
Despite the existence of various traditional treatment strategies, and the widespread use of conventional medicines for MI and MI/RI patients, their treatments remain a significant challenge [10]. Recent advances in nanotechnology have introduced new possibilities in biomedicine and healthcare, and have been extensively applied in the treatment of myocardial ischemia [11,12]. Modern medicine places great emphasis on the application of nanotechnology in targeting mitochondrial function in myocardial ischemia, providing a promising drug delivery method that overcomes the limitations of traditional treatments. Nanomaterials can simultaneously load multiple functional molecules (such as antioxidants, anti-apoptotic drugs, and angiogenic factors), delivering multiple therapeutic benefits [13]. While reperfusion therapy restores blood flow, it can also cause bursts of mitochondrial ROS and excessive opening of the mitochondrial permeability transition pore. Nanomaterials can be tailored to react to ischemic microenvironments (such as low pH or high ROS levels) to precisely target mitochondria, and release drugs during the reperfusion phase, thereby inhibiting secondary damage [14].
The use of nanomaterials to target mitochondrial balance in the treatment of heart attacks holds great promise. By targeting mitochondria, nanomaterials can effectively improve the survival and function of myocardial cells, improve myocardial ischemia, and promote myocardial repair [15-17]. As such, this article provides a comprehensive overview of the role of mitochondrial homeostasis in MI and MI/RI, then reviews recent advances in the use of nanomaterials to modulate mitochondrial function, focusing on aspects such as oxidative stress, mitophagy, and mitochondrial fusion and fission. We conclude by discussing the prospects of using nanomaterials targeting mitochondrial homeostasis in the treatment of myocardial ischemia.
The heart beats about 100,000 times a day, requiring approximately 6 kg of ATP to maintain the contraction and relaxation of cardiac muscle. Mitochondria continuously supply this ATP through oxidative phosphorylation [18]. MI and MI/RI are leading causes of death and disability worldwide [19]. Cardiac mitochondria act as both protectors and destroyers of cells after MI [6]. During acute myocardial ischemia, due to hypoxia and insufficient supply of nutrients to myocardial cells, a series of severe biochemical metabolic disorders are triggered in myocardial cells, many of which have adverse effects on mitochondrial function and ATP production [5]. Researchers widely believe that mitochondrial dysfunction is a significant marker of early myocardial suppression [18].
Mitochondria maintain a relatively constant number and functional state through adaptive changes, known as mitochondrial homeostasis [20]. This involves regulatory mechanisms at multiple levels, including regulating oxidative stress, including ROS levels, calcium balance [21-23], and membrane potential, mitochondrial quality control, which includes mitophagy, the dynamics of fusion and fission, and mitochondrial biogenesis [24,25], as well as changes in mitochondrial DNA (mtDNA) (Fig. 1) [26]. Mitochondrial dysfunction in damaged myocardial cells often manifests as mitochondrial homeostasis disruption, such as insufficient energy production, excessive ROS generation, and decreased mitochondrial membrane potential (MMP), all of which directly affect the survival and function of myocardial cells [27]. Therefore, maintaining mitochondrial homeostasis is a crucial therapeutic strategy in MI and MI/RI.
Mitochondrial reactive oxygen species (mtROS) are a key component of intracellular ROS, and serve as crucial signals that help regulate physiological functions, playing a significant role in maintaining redox balance [28]. The production of mtROS is primarily influenced by oxidative phosphorylation (OXPHOS) capacity, the oxidation of nicotinamide adenine dinucleotide phosphate/nicotinamide adenine dinucleotide (NADPH/NADH), and the biosynthesis of heme and iron-sulfur centers [29]. Excessive production of mtROS can lead to oxidative stress in lipids, proteins, and DNA. To prevent further cell damage, mitochondria eliminate excess ROS using antioxidant systems to maintain redox balance [30,31]. Myocardial ischemia triggers a series of severe biochemical and metabolic disturbances in cardiomyocytes, which adversely affect mitochondrial function and ATP production [32,33]. During myocardial ischemia, the way cells produce energy changes from using oxygen to a process that doesn't require oxygen, leading to the accumulation of intracellular lactate and a decrease in pH, which creates an acidic environment in cardiomyocytes. The accumulation of intracellular protons activates the Na+/H+ exchanger, depleting ATP and reducing Na+/K+ ATPase activity, resulting in intracellular Na+ overload. The Na+/Ca2+ exchanger increases Na+ efflux, leading to intracellular and mitochondrial Ca2+ overload [34]. Early in acute MI/RI, further mitochondrial Ca2+ overload, oxidative stress, and the opening of the mPTP occur [35]. These changes exacerbate the harmful effects caused by acute myocardial ischemia, and synergistically induce mitochondrial dysfunction and cardiomyocyte death, resulting in acute myocardial injury. The chemical reactivity of ROS can lead to the loss of function of proteins, enzymes, and organelles. The accumulation of ROS produced by mitochondria is a measure of dysfunction and disease [36,37]. An ideal therapeutic strategy must selectively target damaged mitochondria to prevent the production of pathological ROS, while maintaining normal ROS levels necessary for cardiomyocyte physiological function [38]. Mitochondrial oxidative stress and its interaction with endoplasmic reticulum stress may also contribute to the endothelial cell damage during MI and MI/RI [39].
The MMP arises from redox reactions linked to tricarboxylic acid cycle (TCA) activity, serving as an intermediate energy reservoir harnessed by ATP synthase for ATP generation. These redox reactions yield both an electric potential (due to charge separation) and a proton gradient, which together constitute the transmembrane potential of hydrogen ions. Under physiological conditions, cells maintain homeostasis of intracellular ATP and MMP levels, a balance deemed essential for preserving normal cellular function [40].
As a second messenger, Ca2+ participates in excitation-contraction coupling, impulse transmission, and regulation of numerous ion transport systems, serving as the foundation of cell fate. Mitochondria contribute to Ca2+ homeostasis regulation through chelation and release of Ca2+ [41]. Ca2+ activates multiple enzymes to drive the tricarboxylic acid (TCA) cycle, regulates mitochondrial OXPHOS rates (reducing NAD+ accumulation), and participates in inflammation by controlling the opening of mPTP [42,43]. During MI, Ca2+ overload accelerates electron transfer in the electron transport chain (ETC), leading to massive electron leakage and mtROS production, which in turn impedes ATP synthesis [34,44].
The outer mitochondrial membrane (OMM) primarily serves as a physical barrier for molecules over 5000 daltons, mediating cellular signaling pathways through numerous integral membrane proteins and contact sites. The inner mitochondrial membrane (IMM) exhibits high impermeability, with most molecules and ions entering the matrix via specialized membrane transporters located in the inner boundary membrane parallel to the outer membrane [45]. With the high expression of voltage-dependent anion-selective channel proteins (VDAC), OMM shows high permeability to Ca2+ [46]. The mitochondrial calcium uniporter (MCU) and Na+/Ca2+ exchanger (NCLX) or H+/Ca2+ exchanger (HCX) on the IMM are crucial for regulating mitochondrial Ca2+ influx and efflux [47]. The proton electrochemical gradient potential generated during OXPHOS serves as the essential driving force for Ca2+ entry into the mitochondrial matrix via the MCU [48].
Mitochondrial outer membrane permeabilization (MOMP) leads to the release or extrusion of mitochondrial damage associated molecular patterns (DAMPs) into the cytoplasm and even the extracellular environment. Pro-apoptotic proteins BAX and BCL-2 antagonist/killer (BAK1) translocate to the OMM upon apoptotic stimuli, forming lipid pores via oligomerization on the OMM to induce MOMP [49]. The strong selectivity, nonpolarity, and negative potential (−160 mV to −180 mV) of the IMM dictate that most mitochondrial targeting modules are lipophilic and positively charged. These include lipophilic cationic small molecules and positively charged peptides, which also facilitate endocytosis and escape from endosomes/lysosomes [50].
The mPTP is a large non-selective channel that opens during reperfusion, allowing ions and solutes with molecular weights up to 1.5 kDa to cross the IMM. This leads to mitochondrial swelling, mitochondrial membrane depolarization, uncoupling of OXPHOS, ATP depletion, and cell death (primarily necrosis) [51]. Available evidence suggests that the F0F1 ATPase may directly participate in the formation of mPTP, with two distinct models proposed: one based on the c-subunit of F0F1 ATPase, and the other suggesting that the oligomers and dimers of F0F1 ATPase, playing a role in mPTP formation [52].
During acute MI/RI, the mPTP remains closed during ischemia due to the acidic environment within cardiomyocytes. The inhibitory effect of H+ on mPTP appears to be mediated by a highly conserved histidine residue (H112) in the oligomycin-sensitive conferring protein (OSCP) subunit of mitochondrial F0F1 ATPase [53]. The mPTP opens only within the first few minutes of reperfusion, in response to mitochondrial calcium overload, oxidative stress, ATP depletion, and rapid pH recovery [54]. Therefore, during acute MI/RI, formulating therapeutic strategies targeting these factors that induce mPTP opening can indirectly inhibit mPTP opening during reperfusion, and reduce the area of MI. Blocking ROS release by regulating the closure of mPTP can improve myocardial function, suppress myocardial apoptosis and fibrosis, and ultimately attenuate MI area by maintaining mitochondrial function [55].
At the onset of MI/RI, the use of known pharmacological inhibitors of mPTP, such as cyclosporine A (CsA) targeting cyclophilin D, can reduce the MI area by 40%–50% in animal MI models [56]. To discover and test more effective and specific mPTP inhibitors as novel mitochondrial protective therapies, further research is needed to clarify the molecular composition of mPTP. In summary, experimental studies support mPTP as an important therapeutic target for preventing fatal MI/RI injury. Indirect inhibition of mPTP opening during reperfusion can also be achieved by improving cellular bioenergetics, limiting oxidative stress, and increasing ATP availability through elevating creatine and phosphocreatine levels. This has been validated in mice with cardiac overexpression of creatine transporters [57].
Mitophagy is crucial for maintaining mitochondrial quality, helping to selectively remove damaged or dysfunctional mitochondria through mechanisms such as reduced MMP and metabolic stress [58]. The activation of mitophagy has a protective role during ischemia, but excessive mitochondrial engulfment during reperfusion can induce cell death and impair cardiac function, which makes mitophagy a bit of a double-edged sword [59]. In the mechanisms of acute MI, a moderate inflammatory response facilitates the recovery of cardiomyocyte function and vascular regeneration, whereas excessive inflammatory responses may lead to irreversible cellular damage. Moderate mitophagy can suppress excessive inflammatory reactions, and protect cardiomyocytes [60]. However, when mitochondrial function is disrupted, such as insufficient clearance of damaged mitochondria or excessive activation of mitophagy, MI and MI/RI can be aggravated [61]. The most typical pathway for maintaining a healthy mitochondrial network by promoting the autophagic clearance of damaged mitochondria is the PTEN induced kinase 1 (PINK1)/Parkin pathway [62]. After 30 min of ischemia and 24 h of reperfusion, MI/RI can induce cardiac dysfunction, inflammation, apoptosis, and increased autophagy and mitophagy [63]. The absence of Parkin leads to increased infarct size, ROS production, and accumulation of dysfunctional mitochondria, while overexpression of Parkin protects the heart from MI/RI [64]. Studies show that using PR-364 to target Parkin mediated mitophagy can help protect heart tissue after MI [65].
In addition to affecting the classical PINK1/Parkin pathway, other related receptor pathways have been proposed. Many studies aim to reduce damage in MI by intervening in mitophagy. Melatonin preconditioning improves MI/RI by regulating the apelin/sirtuin 3 (SIRT3) pathway to inhibit excessive mitophagy [66]. Other studies have investigated the role of the zinc transporter ZIP7, which regulates Zn2+ levels, in inhibiting mitophagy, a key feature of MI/RI. Effective mitophagy plays a crucial role in cell survival and differentiation [67]. Researchers have also analyzed the roles of general autophagy and specific mitophagy in regulating cardiac function after MI, demonstrating that mitophagy, rather than general autophagy, aids in cardiac protection by modulating mitochondrial function [68]. Non-coding RNAs (microRNAs (miRNAs) and long non-coding RNAs (lncRNAs)) also participate in the regulation of autophagy in cardiomyocytes during and after MI [69]. In addition to post-translational modifications, proteins involved in mitophagy are also regulated by non-coding RNAs. For example, knockdown of miR-130a significantly activates FUNDC1 mediated mitophagy [70]. Traditional Chinese medicine components, such as paeoniflorin, have also become a research hotspot in the treatment of MI, with studies indicating that paeoniflorin may protect against oxidative damage and autophagy by activating the SIRT1-PINK1/Parkin pathway [71].
Current research suggests that the balance between moderate mitophagy and the inhibition of the NOD-like receptor protein 3 (NLRP3) inflammasome may have significant clinical implications for the treatment of MI/RI [72]. The relationship between autophagy and NLRP3 inflammation is complex, with both processes exhibiting bidirectional effects and sometimes producing opposing outcomes under certain conditions. Autophagy generally inhibits NLRP3 inflammation under most conditions, and the interaction between autophagy and the NLRP3 inflammasome during MI/RI warrants further investigation [60]. Mitophagy primarily belongs to macroautophagy, an intracellular degradation pathway mediated by lysosomes that breaks down damaged proteins and organelles, playing a crucial role in maintaining intracellular homeostasis [73]. Mitophagy selectively targets and clears damaged mitochondria, playing a crucial role in cellular energy supply [60]. The mechanisms of mitophagy require further research to provide more valuable therapeutic pathways for MI and MI/RI.
Mitochondrial fission can increase the number of mitochondria, and render cells more susceptible to damage. Its functions include isolating damaged mitochondrial regions through fission, facilitating the clearance of damaged parts via mitophagy, and increasing mitochondrial numbers to enhance ATP production efficiency in response to changes in energy demand [74]. Mitochondrial fission in cardiomyocytes is primarily mediated by guanosine triphosphatases (GTPase) and dynamin related protein 1 (Drp1) [75]. Studies have found that after MI, the expression and activity of Drp1 significantly increase, resulting in shortened and rounded mitochondrial shapes and increased mitochondrial numbers, and ultimately inducing increased ROS production and cardiomyocyte apoptosis [75]. The mechanism of MI induced mitochondrial fission is not clear, but it may be related to calcium overload and the generation of oxidative stress, which again leads to mitochondrial permeability transition pore (MPTP) opening [76]. Several basic studies have verified that targeting downregulation of Drp1 can reduce mitochondrial fission, thereby improving MI and MI/RI [77]. Research exploring the potential mechanisms of MI induced mitochondrial fission has shown that intervention with adenosine monophosphate-activated protein kinase (AMPK) activators improves MMP, reduces ROS production, and inhibits mitochondrial damage, confirming that AMPK activation plays a protective role in MI/RI, largely dependent on inhibiting Drp1 mediated mitochondrial fission [78]. In a rat model of MI/RI, administration of the mitochondrial fission inhibitor (Mdivi-1) effectively alleviated ROS production, swelling, and dynamic imbalance, thereby improving MI/RI injury [79]. Thus, reducing phosphorylation of Drp1 Ser616 through multiple pathways can inhibit mitochondrial fission, protecting against MI and MI/RI [80]. This mechanism has also been applied in the treatment of mitochondrial fission targeting using nanoparticles, with studies evaluating the potential of ceria nanoparticles (CNPs) coated with different ligands in treating MI/RI rats. Results indicate that CNPs can effectively modulate oxidative stress in cardiac tissue by eliminating ROS and stimulating endogenous antioxidant systems. Inhibition of oxidative stress results in a reduction of p-Drp1 (Ser616), which is crucial for driving mitochondrial fission and fragmentation. These findings provide useful information for the future preparation of inorganic antioxidant nanomedicines for treating MI/RI [81].
Additionally, the regulation of other proteins involved in mitochondrial fission may also play an important role. Studies have shown that the expression level of mitochondrial fission factor (Mff) significantly increases in cardiomyocytes after MI/RI [82]. Mff gene deletion can reduce infarct size, improve endothelial damage, and enhance mitochondrial morphology and function. Other studies have verified that the deficiency of dual specificity protein phosphatase 1 (DUSP1) induced by MI/RI promotes JNK activation, thereby upregulating Mff expression, with higher Mff expression levels associated with increased mitochondrial fission and mitochondrial apoptosis [80]. Mff mediated mitochondrial fission plays a significant role in cardiac MI/RI [83]. In addition to classical fission related factors, new pathways involved in mitochondrial fission continue to be proposed. Furthermore, studies have found that GJA1–20k-mediated fission is independent of typical Drp1, with stress responsive, internally translated GJA1–20k stabilizing actin filaments to stimulate non-classical mitochondrial fission, thereby limiting MI and MI/RI [84]. Various pathways influencing myocardial injury after MI or MI/RI have been identified, providing more avenues for mitochondrial targeted therapy for MI.
Mitochondrial fusion helps keep mitochondria's structure and function normal, even when fission happens. Fusion relies on mitochondrial transmembrane GTPases, including mitochondrial fusion protein 1 (Mfn1), which mediates OMM fusion, and Mfn2, which mediates IMM fusion, as well as optic atrophy 1 (OPA1), which also mediates IMM fusion [85]. Mitochondrial fusion proteins (Mfn, Mfn1 and Mfn2) are highly conserved transmembrane GTPases encoded by Mfn genes, and abundantly expressed in the myocardium. Additionally, Mfn is located in the outer mitochondrial membrane, and participates in outer membrane fusion [86]. After infarction, the expression of OPA1 and Mfn2 in the heart decreases, and during MI/RI, the expression of OPA1 also decreases, along with downregulation of mitochondrial respiratory complex expression [87]. Pre-treatment with drugs or increasing OPA1 expression can reduce mitochondrial fission, increase mitochondrial respiratory complex expression, and improve mitochondrial function, thereby alleviating myocardial ischemia injury [85]. Mitochondria are dynamic organelles that continuously change their shape between a fragmented disconnected phenotype achieved through fission and a fused state. Thus, mitochondrial fusion plays a crucial role in the exchange of genetic material between mitochondria, enhancing their function and resilience [88]. And mitochondrial fission is a fundamental process that enables the fragmentation of organelles, and maintains their quality through mitophagy, thereby ensuring optimal cellular function. In acute MI/RI, the imbalance between mitochondrial fusion and fission affects mitochondrial respiratory function, mitochondrial quality control, and susceptibility to cell death, making mitochondrial fusion and fission proteins important targets for cardiac protection.
Mitochondrial biogenesis is the process of creating new mitochondria in cells, including their proliferation, fusion, fission, and the restoration and maintenance of function. It is a complex biological process involving the regulation of various signaling pathways and gene expression, aiming at increasing the number of mitochondria and enhancing their function to meet cellular energy demands and maintain normal physiological functions [25,89]. Mitophagy is responsible for clearing damaged or useless mitochondria. There is a finely tuned balance between mitochondrial biogenesis and mitophagy, which is crucial for maintaining mitochondrial quantity and function, ensuring cellular homeostasis, and adapting to metabolic demands and extracellular stimuli [90]. Peroxisome proliferator activated receptor gamma coactivator 1-alpha (PGC-1α) is considered the primary regulatory factor of mitochondrial biogenesis, capable of regulating various transcription factors related to mitochondrial biogenesis, such as estrogen-related receptor (ERR), peroxisome proliferator-activated receptor γ (PPARγ), and nuclear respiratory factor-1/2 (NRF-1/2), thereby promoting mitochondrial generation and functional enhancement [91]. It activates these transcription factors, thereby increasing the replication and transcription of mtDNA, which enhances mitochondrial quantity and function. Mitochondrial biogenesis can increase OXPHOS capacity, reduce pathological oxidative stress, and repair mitochondrial related functional impairments. The induction of mitochondrial biogenesis is associated with the activation of transcription factors that regulate the expression of mitochondrial genes and enhance the local translation of mitochondrial proteins. Studies have suggested that in MI, oxidative stress and mitochondrial biogenesis can be regulated through PGC-1α, making targeting mitochondrial biogenesis a promising new treatment option [92]. By increasing PGC-1α expression, mitochondrial biogenesis pathways can mitigate isoproterenol induced MI [93]. In animal models, mitochondrial dysfunction has been observed in MI rat models, characterized by increased mitochondrial oxidative stress, mitochondrial membrane depolarization, and mitochondrial swelling, with impaired mitochondrial biogenesis reflected by decreased PGC-1α expression. Additionally, research has shown that in MI animal models, activation of AMPK can increase mtDNA copy number and PGC-1α expression, promoting mitochondrial biogenesis. Thus, increasing PGC-1α expression to regulate mitochondrial biogenesis may be one of the pathways for treating MI [94]. By promoting mitochondrial biogenesis, it is possible to improve energy metabolism in cardiomyocytes, reduce oxidative stress damage, decrease apoptosis, and mitigate cardiac remodeling, thereby offering potential benefits for heart function recovery after a heart attack.
Myocardial ischemia is one of the leading causes of heart failure and death worldwide, with the main issue being the irreversible damage to myocardial cells caused by ischemia and hypoxia [95]. In recent years, with a deeper understanding of mitochondrial function, treatment strategies targeting mitochondria have emerged as a new approach to treating MI and MI/RI. Mitochondria, as the powerhouses of cells, not only regulate ATP production but also participate in key processes such as calcium homeostasis, ROS balance, and apoptosis [96,97]. Therefore, helping myocardial cells survive better by protecting or repairing mitochondrial function has become an important research direction in the field of cardiovascular disease treatment [98]. Although traditional treatment methods such as coronary thrombolysis and stent implantation can restore blood supply to the heart, they cannot reverse the damage to already injured myocardial cells, and reperfusion injury may exacerbate mitochondrial damage, which is a major reason for myocardial cell damage [99]. Due to the unique structure of mitochondria, there are still various obstacles in delivering biomacromolecules to mitochondria. Additionally, the transfer of some exogenous substances into the mitochondrial matrix may interfere with the intrinsic pathways of mitochondria, leading to damage and toxicity to mitochondria and cells. Therefore, special transport pathways that deliver specific substances into mitochondria to maintain mitochondrial homeostasis are particularly important [100]. In recent years, nanomaterials, because of their unique properties and precise targeting abilities, have provided new technological avenues for mitochondrial therapy [100,101]. By designing functionalized nanocarriers, drugs, genes, or healthy mitochondria can be efficiently delivered to repair damaged myocardium and improve cardiac function [102]. Nanoparticles (such as liposomes, polymer nanoparticles, gold nanoparticles) can actively accumulate in mitochondria through surface modifications (such as mitochondrial targeting peptides, triphenylphosphonium (TPP) cation TPP+, thereby enhancing drug utilization. Nanocarriers can encapsulate easily degradable antioxidants (such as MitoQ), mPTP inhibitors (such as CsA), or drugs that promote mitochondrial biogenesis to prevent their clearance in circulation [103,104]. Some nanomaterials (such as carbon based nanomaterials, metal organic frameworks) also exhibit antioxidant, anti-inflammatory, and pro-angiogenic effects, providing hope for the prevention and treatment of MI and MI/RI with novel multifunctional nanoparticles [105-109]. Targeting mitochondrial therapy for myocardial ischemia, through precise delivery, multi-mechanism synergy, and efficient repair, offers a groundbreaking strategy for cardiovascular regenerative medicine. With the continuous advancement of nanotechnology and in-depth preclinical research, this field is expected to achieve translation from the laboratory to the clinic in the future, bringing new treatment hopes to patients with MI or MI/RI.
Mitochondria are the energy factories within cells, and the dysregulation of mitochondrial stability is a key pathological mechanism in myocardial ischemia. Therefore, targeting nanomaterials to mitochondria can achieve precise treatment and enhance the efficacy of drugs. The mechanisms of mitochondrial targeting by nanomaterials can be divided into active targeting and passive targeting.
Active targeting involves modifying the surface of nanomaterials with specific ligands or molecules, so they can specifically find and attach to mitochondria [110]. Mitochondrial inner membrane's unique transmembrane potential and low permeability impede most drugs from entering. Recently, mitochondrial targeted delivery systems using functional peptides to modify drugs have gained attentions. These peptides alter the physicochemical properties of drugs to actively target mitochondria. The common mitochondrial targeting ligands include TPP, imidazole groups, etc. [111]. TPP is a commonly used mitochondrial targeting molecule [112], which can specifically deliver nanomaterials to mitochondria through interactions with MMP [113]. TPP modified nanoparticles can efficiently accumulate within mitochondria. One study coupled a trivalent compound (TRV3) with a peptide carrier via redox disulfide bonds, and combined it with TPP and Cy5 fluorophore, resulting in a nanoconjugate (C-TRV3-A) that demonstrated effective endosomal escape and mitochondrial localization, constructing a multifunctional nanocarrier for targeted subcellular delivery and efficient gene therapy [114]. Another study constructed CsA@Dex-Gal/TPP by assembling CsA with dextran grafted galactose (Dex-Gal) and dextrorotatory TPP (Dex-TPP), where the exposed TPP portion guided the nanotherapeutic drug to escape from lysosomes and target mitochondria through enhanced positive charge, achieving precise in situ drug delivery and restoring mitochondrial autophagy [115]. In the treatment of ischemic stroke, cerium dioxide nanomaterials targeting mitochondria, after TPP modification, can regulate mitochondrial oxidative stress levels, protecting neurons from ischemic injury [116]. The targeting capability of TPP can directly intervene in the occurrence and development of diseases by targeting mitochondria and regulating their biological functions [117,118]. Additionally, some studies have also achieved high specificity targeting of mitochondria by modifying nanomaterials with peptides, proteins, antibodies, and other biomolecules [119,120]. Specific protein markers exist on the surface of mitochondria, and by modifying nanomaterials with aptamers, antibodies, or peptide segments that can specifically recognize these markers, precise targeting can be achieved. For example, the team of Wang developed a lysosome-mitochondria cascade targeting nanostrategy, achieving efficient drug delivery from lysosomes to mitochondria through a modular acid/enzyme dual-gated nanotechnology platform (TAEN) [121]. SS31 is a mitochondria targeting peptide that has gained widespread attention in recent years due to its research value in the treatment of MI and other diseases [122,123]. SS31 can directly repair defects in the IMM by targeting the IMM, significantly improving mitochondrial function, reducing oxidative stress and apoptosis, thereby improving the energy metabolism and functional recovery of myocardial cells [124]. Nanomaterials have utilized this by developing a peptide-drug conjugate OI-FFG-ss-SS31 (ISP), which can self-assemble into nanofibers in response to acidic microenvironments, and bind with high affinity to Keap-1, thereby activating Nrf2 and enhancing antioxidant capacity [125]. At the same time, the release of SS31 can improve mitochondrial function, and reduce ROS, ultimately restoring redox homeostasis and effectively alleviating MI/RI.
Passive targeting of nanomaterials to mitochondria mainly relies on the physicochemical properties of the nanomaterials, such as size, surface charge, and hydrophilicity, allowing them to accumulate at target sites during distribution in the body naturally [126]. The size of nanomaterials typically ranges from 10 nm to 200 nm, enabling them to enter tissues through the gaps between vascular endothelial cells. The size and shape of nanomaterials affect their distribution and targeting within cells [127]. Smaller sized nanomaterials (e.g., less than 100 nm) are more likely to penetrate cell membranes and further target mitochondria. The shape of nanomaterials can also influence their interactions with mitochondria. For example, spherical, rod-shaped, or sheet-like nanomaterials may exhibit different targeting efficiencies due to varying physical properties [128]. Some studies have reported that ultra-small tungsten based nanoparticles (TWNDs) with strong ROS scavenging capabilities can be effectively passively targeted to renal tubular mitochondria without the need for complex active targeting strategies, marking the first use of ultra-small negatively charged nanoparticles for passive targeting of mitochondria in treatment [128]. This provides insight into the passive targeting of mitochondria in the treatment of MI. These nanomaterials have shown potentials in the treatment of various diseases; size-uniform black phosphorus/cerium dioxide nanomaterials (TBP@CeO2) can precisely target mitochondria, promoting ATP production by restoring the activity of mitochondrial respiratory chain complex Ⅱ, and effectively repairing mitochondrial function including mitochondrial structure and membrane potential [129,130].
In recent years, many researchers have been dedicated to developing nanomedicine materials aimed at targeting mitochondrial homeostasis to improve myocardial ischemia (Table S1 in Supporting information).
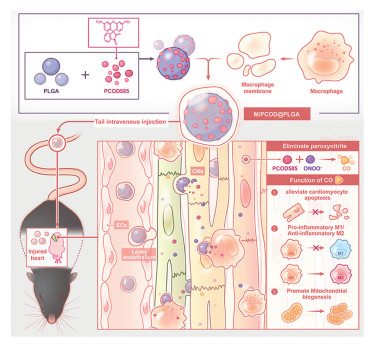
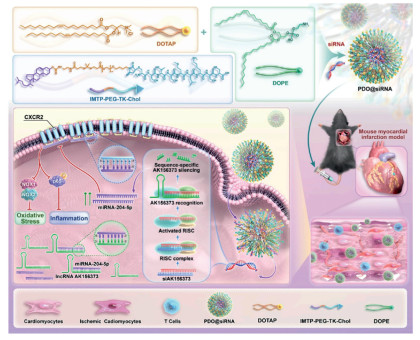
Oxidative stress is characterized by an excessive amount of ROS. It is linked to a bunch of health issues, including heart attacks, cancer, and brain diseases. Dealing with the damage caused by ROS has become a big deal in biomedical research [131]. After myocardial ischemia, oxidative stress is initiated in the cells, producing ROS and exacerbating mitochondrial damage, thereby disrupting mitochondrial balance. Therefore, using nanomaterials to target mitochondria, reduce ROS, and manage oxidative stress is a key mechanism in treating MI and MI/RI [123]. Antioxidants eliminate ROS, and show great potentials in the treatment of MI/RI. Cerium vanadate nanorods (CNVRs) can serve as an effective ROS scavenger for the treatment of MI/RI, and antioxidant nanoenzymes preserve the morphology and function of mitochondria [132]. Nanoparticles synthesized by incorporating selenium elements into a carbon framework through C-Se bonds (SSSe NPs) have also been used to treat MI. SSSe NPs can efficiently eliminate ROS on demand at different pathological stages of MI, showcasing excellent therapeutic effects for MI and effectively reversing heart failure back to normal cardiac function [133]. To improve the biocompatibility and targeting of nanoparticles, immune cell membrane coated nanoparticles also have potential applications. Platelet membrane encapsulated ROS responsive drug release nanoparticles (PMN@NIC-MalNPs) deliver dimethyl malonate and niclosamide (NIC), reducing oxidative stress levels and restoring mitochondrial function by increasing the in vitro baseline mitochondrial oxygen consumption rate (OCR), thereby preventing damage caused by MI [134]. Carbon monoxide (CO) has been shown to improve mitochondrial energy metabolism and, enhance mitochondrial biogenesis. Therefore, research has explored its therapeutic potentials in MI, proposing a new biomimetic CO nanogenerator (M/PCOD@PLGA) that was coated with macrophage membranes to target ischemic areas, and neutralized pro-inflammatory cytokines. CO was continuously released locally in the ischemic area, improving mitochondrial dysfunction and oxidative stress. M/PCOD@PLGA enhanced the capacity for mitochondrial biogenesis and reduced the number of damaged mitochondria (Fig. 2) [135]. Nitric oxide (NO) is oxidized by the burst of ROS into harmful peroxynitrite during MI, causing secondary damage to cardiomyocytes. Study had synthesized a peptide drug conjugate targeting damaged myocardium (CTP-PBA-ISN). This compound alleviated oxidative stress injury, activated NO related signaling pathways to promote angiogenesis, effectively inhibited cardiomyocyte apoptosis, improved myocardial function, restored mitochondrial homeostasis, and reduced adverse cardiac remodeling after myocardial infarction [136]. Multiple lncRNAs can target miRNAs to inhibit the occurrence and development of MI. However, free siRNAs, due to their hydrophilicity and electronegativity, have difficulty effectively penetrating the cell membrane and are easily degraded by nucleases [137]. Research had modified the ischemic heart targeting peptide cstsmlkac (IMTP) and successfully prepared a nanosystem, PDO@siRNA, loaded with siAK156373. These NPs demonstrated ischemic myocardial targeting ability and responsiveness to ROS. The results indicated that NPs mediated delivery of siAK156373 effectively silenced the expression of lncRNA AK156373 in myocardial tissue, thereby enhancing the viability and mitochondrial function of hypoxic myocardial cells, and reducing the levels of intracellular inflammatory cytokines and the production of ROS (Fig. 3) [138]. Recently, drug delivery systems carrying natural small molecule formulations have garnered significant attention due to their low side effects, high bioavailability, and sustained release properties. Many researchers have applied them to the treatment of MI. For example, Yu et al. successfully synthesized ellagic acid functionalized gold nanoparticles (EA-AuNPs) which had high ROS scavenging activity. They applied these nanoparticles to an MI animal model, and found that they effectively inhibited the expression of apoptosis related proteins and the elevation of cardiac enzyme activity, thereby improving oxidative stress damage in MI mice. Further studies suggested that EA-AuNPs might also alleviate myocardial injury by inhibiting ROS induced changes in oxidized lipids and restoring disrupted anti-inflammatory oxidized lipids [139]. A new nanoengineering therapy is used to enhance the therapeutic effect of mesenchymal stem cells (MSCs) on MI/RI, utilizing anti-inflammatory and antioxidant nanoparticles TPCD, which were encapsulated by MSCs' natural endocytosis (tn-MSC). Tn-MSC alleviated MI/RI by increasing MMP, and protecting myocardial cell mitochondria from ROS induced cytotoxicity and apoptosis [140]. Research had developed a peptide-drug conjugate ISP by integrating the Nrf2 activator 4-octylitaconate (OI) and SS31. ISP could self-assemble into nanofibers in response to an acidic microenvironment, and the release of SS31 could improve mitochondrial function and reduce ROS, ultimately restoring redox homeostasis and effectively alleviating MI/RI [125]. A PEGylated allomelanin nanoparticles medicine (AMNPs@PEG) exhibited excellent capabilities in clearing intracellular free radicals, inhibiting MMP depolarization, and enhancing cell survival rates. Experimental results indicated that AMNPs@PEG could reduce pro-inflammatory gene expression and increase anti-inflammatory gene expression, leading to a significant reduction in MI area and markedly improving heart function after MI/RI [141]. Research also reports that tannic acid coated Mn-Co3O4 (MCT) nanoparticles, when used in combination with injected collagen hydrogel, could inhibit the surge of ROS after MI, playing an anti-apoptotic and anti-inflammatory role to improve ventricular remodeling in early MI [142]. Additionally, Guo et al. developed a Zr metal organic framework combined with SOD (SOD-ZrMOF) for the treatment of MI in animal models. Long term research results indicated that SOD-ZrMOF could alleviate ROS induced myocardial damage, protect mitochondrial function, thereby reducing infarct size, protecting cardiac function, promoting angiogenesis, and inhibiting pathological myocardial remodeling, demonstrating significant potential for safe and effective treatment of acute MI [103].
Nanomaterials possess highly efficient ROS clearance capabilities, precise targeted delivery capabilities, and powerful mitochondrial protection and functional recovery effects. They can achieve multi-functional synergistic treatment while overcoming the inherent defects of drugs such as siRNA and natural small molecules. These advantages eventually translate into therapeutic effects such as improving cardiac function, reducing infarct area and inhibiting poor remodeling. However, some drugs have inherent flaws, such as the difficulty in cell penetration and easy degradation of siRNA, and peptide drug-coupled molecules need to modify to enhance biocompatibility. These are the key challenges to be solved in the future. In summary, nanomaterials can target mitochondria through multiple pathways to reduce ROS levels, achieving the therapeutic goal of alleviating MI and MI/RI damage.
The stability of MMP plays a crucial role in myocardial ischemia by regulating mitochondrial function and protective mechanisms, thereby providing new targets and strategies for treating MI and MI/RI. After MI, the MMP rapidly decreases, leading to mitochondrial dysfunction, which further affects cellular energy metabolism, reduces ATP production, and ultimately results in weakened contractile function of cardiomyocytes [143]. The loss of MMP is one of the early events in apoptosis. During the process of MI, the instability of the MMP can lead to the release of cytochrome c, activating the apoptotic pathway and ultimately resulting in cardiomyocyte apoptosis [144]. There are various nanobiological platforms that can target the maintenance of MMP in cells, becoming a new strategy for treating MI/RI. Spherical selenium nanoparticles (SeNPs) had a protective mechanism for cardiomyocytes, and studies had verified that SeNPs could reduce mitochondrial ROS levels and stabilize MMP [145]. A nanobioplatform of LDH@miR-182 based on layered double hydroxides (LDH) and coated with macrophage membranes (MM-LDH@miR-182) had been reported, which utilized the properties of macrophage membranes to prolong blood circulation. This platform regulated the IP3R pathway, reduced calcium release in the endoplasmic reticulum, protected MMP, and inhibited the production of mitochondrial ROS, thereby alleviating myocardial fibrosis and improving cardiac function [146]. Research had reported that a mature pro-angiogenic nanorods of terbium hydroxide (THNR) could improve MI/RI. Experimental results indicated that the mechanism of THNR was related to the reduction of oxidative stress, lipid peroxidation, calcium overload, restoration of cytoskeletal integrity, and MMP [147]. Additionally, research had proposed an ultra-small PtIr bimetallic nanoenzyme for the treatment of MI. The PtIr nanoenzyme could maintain the MMP within cells, increase mitochondrial activity to protect mitochondrial structure, and improve cardiac function to remodel the infarct microenvironment, making it a promising strategy for MI treatment [148]. Additionally, scholars have synthesized selenium coated gold nanocages (AAS) loaded with l-arginine, and modified them with PCM (WLSEAGPVVTVRALRGTGSW) to obtain AASP, which could enhance MMP, restore ATP synthase activity, prevent ROS generation, and inhibit NO oxidation. NO prevented ROS release by regulating the closure of the mPTP, ultimately alleviating MI/RI in rats by maintaining mitochondrial function and regulating NO signaling [55]. Research had reported that biomimetic nanoparticles (PTPN) have thrombus targeting properties for the treatment of acute MI. PTPN consisted of a thrombus microenvironment responsive phenylboronic acid (PBA) nanocarrier, the antioxidant molecule protocatechuic aldehyde (PC), and tissue plasminogen activator (tPA) with thrombolytic effects, which was encapsulated by platelet membranes. The treatment with PTPN significantly increased the MMP, and restored mitochondrial function, thereby improving MI (Fig. S1 in Supporting information) [149]. Gold-selenium core-shell nanostructure (AS-I/S NCs) with excellent near-infrared (NIR)-Ⅱ photoacoustic imaging have also been designed for MI/RI therapy recently. AS-I/S NCs were nanocomposites modified by SS31, which could significantly inhibit MMP depletion, and alleviate MI/RI by suppressing ROS mediated oxidative damage and regulating the MAPK and PI3K/AKT pathways [124].
The application of nanomaterials in the treatment of MI and MI/RI has demonstrated significant advantages, especially in precisely targeting and stabilizing the core pathological link of MMP, and thereby giving rise to multiple protective effects such as powerful antioxidation, protection of mitochondrial, anti-apoptosis, and improvement of energy metabolism and cardiac function.
Inhibition of mPTP opening enhances mitochondrial stability, and can serve as a crucial therapeutic target for preventing MI and MI/RI. MiR-222–3p loaded stem cell nanovesicles could reduce the opening of mPTP, inhibited MOS, improve mitochondrial function, and played a protective role for myocardium in MI/RI [150]. A study developed a biomimetic, mitochondria targeted nanoenzyme system (B-Ce@D-P@M), modified with triphenylphosphine for mitochondrial targeting, and further coated with macrophage derived membranes to enhance myocardial accumulation. B-Ce@D-P@M reduced mPTP opening, and improved mitochondrial stability, thereby effectively maintaining mitochondrial integrity and providing a promising nano-therapeutic platform for MI/RI (Fig. S2 in Supporting information) [35]. Some scholars had proposed a unique snowflake-like ultrathin manganese doped ruthenium branched nanosheet RuMn BNSP, which could maintain mitochondrial function, and improve MI/RI by preventing MMP depolarization and the opening of mPTPs [151]. To better apply nanoenzymes for targeted mitochondrial therapy of MI, research had utilized genetically engineered ferritin nanocages (i.e., imFTn) to develop mitochondrial targeted nanoenzymes. Using imFTn as a nanoenzyme platform, a nanoenzyme capable of effectively catalyzing the production of NO from the l-Arg substrate had been developed. This nanoenzyme reduced the production of mitochondrial ROS, inhibited the opening of the mPTP, and enhanced the MMP [152]. Li et al. developed porous silica nanoparticles loaded with selenium quantum dots (Se@PSN), which could reduce MR/RI induced mPTP opening, decrease mitochondrial ROS production, and protect the integrity of OXPHOS in cardiomyocytes, showing promising applications in the treatment of MR/RI injury [153]. CsA has a significant inhibitory effect on the opening of the mPTP [115]. Nanomaterials can utilize this mechanism to target the delivery of CsA for the treatment of MI. Research had proposed nanoparticles that targeted mitochondrial delivery of CsA (CsA@PLGA-PEG-SS31), which precisely delivered CsA to the mitochondria of ischemic myocardial cells. These nanoparticles protected the myocardium from damage by reducing the recruitment of inflammatory cells and maintaining mitochondrial functional integrity in MI/RI rats, thereby reducing the apoptosis of myocardial cells and the area of MI, and demonstrating significant cardioprotective effects in rats with MI/RI [56]. Some researchers also pointed out that using a combined administration of polymeric nanoparticles composed of poly-lactic/glycolic acid and CsA enhanced myocardial protection more effectively than using CsA-containing nanoparticles alone [154]. Additionally, scholars had developed an injectable mitochondrial targeted nanocarrier hydrogel, which was used to restore mitochondrial function, and alleviate oxidative stress layer by layer, thereby reducing MI/RI. The mitochondrial targeted SS31 modified amphiphilic polymer PTPS could self-assemble into nanomicelles (PTPSC) for loading CsA. Targeted delivery of CsA to mitochondria inhibited the mitochondrial mediated apoptosis signaling pathway to prevent cardiomyocyte apoptosis, while reducing mitochondrial ROS output to lower cytoplasmic ROS levels [123]. Whether it was the multifunctionality of the nanozyme itself or the mechanism of targeting mitochondrial delivery using loaded CsA to inhibit the opening of MPTP, this mechanism can serve as a means for nanomaterials to target mitochondrial homeostasis.
In conclusion, some nanoparticles can protect drugs, control drugs release and improve the stability of drug treatment. If multiple nanoparticle drugs are used in combination, they can synergistically inhibit the abnormal opening of mPTP, abd improve mitochondrial dysfunction and cell death.
After MI and MI/RI, mitochondrial autophagy can accurately identify and eliminate damaged or dysfunctional mitochondria, thereby maintaining mitochondrial function homeostasis within cells and reducing the risk of apoptosis [155]. Mitochondrial autophagy plays a crucial role in both normal physiological and pathological states in cardiovascular diseases. Numerous studies have confirmed that promoting mitochondrial autophagy can significantly alleviate myocardial injury, thereby providing protection to the heart [156]. In recent years, MSCs therapy has emerged as a promising approach for treating MI [157]. However, the challenge of low cell survival and retention rates after injection, especially in the context of high levels of ROS and oxidative stress at the infarct site, hinders clinical application. Some studies had utilized nanotechnology to synthesize Prussian blue nanomaterials rich in indocyanine green, referred to as PB@PEI@ICG. The experiment engineered MSCs with PB@PEI@ICG through co-culture, and the introduction of the nanomaterial significantly enhanced mitochondrial autophagy via the BNIP3 and PINK1 pathways, thereby enhancing the therapeutic potential of stem cells. This represented a novel strategy for MSCs therapy in MI [158]. In addition to the application of nanoenzymes, the delivery system based on biodegradable nanoparticles, namely bMSNs-Mel@PDMC–CHP nanoparticles, also achieved targeted therapy of mitochondria in MI. This system exhibited excellent macrophage evasion, cardiac targeting, and drug release characteristics, with its molecular mechanism attributed to enhanced mitophagy and reduced mitochondrial damage (Fig. S3 in Supporting information) [159]. Geniposide (GP) can alleviate cell apoptosis by restoring mitochondrial function and has certain therapeutic potential in MI/RI. Research had developed a mitochondria targeted nanoliposome delivery system (GP@Lip-TPP) based on the mitochondrial targeting molecule triphenylphosphine combined with GP. This system exerted therapeutic effects on MI/RI by enhancing mitochondrial fusion and inhibiting mitochondrial fission [160]. A novel therapeutic cerium dioxide nanoparticle (CeO2@Tf) had been developed based on the Tf protein through biomineralization, which improved mitochondrial autophagy and cardiac remodeling in mice [161]. Research had also combined an excellent antioxidant melanin (Mel) with a metformin mimic poly (dimethylsiloxane) through electrostatic interactions to obtain a nanocomposite (Met-Mel), which was then encapsulated with platelet membranes (PMN) to form biomimetic nanoparticles PMN@Met-Mel, capable of targeting damaged myocardium and exerting cardioprotective effects by activating the autophagy pathway [162].
The proposal of these nanomaterials offers a promising prospect for effective targeted mitochondrial autophagy therapy in MI and MI/RI, with unique advantages in precise intervention of mitochondrial function, enhanced cell protection, and breakthrough of delivery barriers. But the low survival rate and retention rate of certain molecules, such as MSCs, after injection are the main obstacles in clinical application. Although nanomaterials have attempted to solve these problems, their actual effects still need to be further verified. The synthesis and functionalization processes of some other nanomaterials are complex, and further improvement of synthesis techniques is still needed for their application in clinical practice.
The application of nanobiomedicines targeting mitochondria in myocardial ischemia treatment holds significant research value and clinical prospects, but it also faces several challenges, primarily including the following aspects: (1) The biocompatibility and safety of nanobiomedicines. Such factors like composition, shape, size, and surface modification can affect their biocompatibility. Some nanomaterials may have potential toxicity. Therefore, developing low toxicity and highly biocompatible nanobiomedicines is crucial. We also need to consider the stability of nanomaterials in the body. (2) Difficulty in establishing uniform quality control standards. Some nanobiomedicines need to be coated with exosomes, platelet membranes and stem cell membranes. These techniques are difficult to control the quality of cell products due to low cell culture yield and complex purification technology. For example, exosomes contain a large number of biologically active molecules, and their levels can easily be influenced by the state of the cells. Utilizing exosomes to coat nanobiomedicines requires consideration of its stability [159]. (3) Precision of targeted delivery. Nanobiomedicines need to target myocardial mitochondria precisely, but current targeting strategies may be limited. Furthermore, the distribution and metabolism of nanobiomedicines in vivo are complex, making it difficult to ensure their efficient accumulation in myocardial mitochondria. (4) Complex pathological mechanisms. The processes that regulate MI/RI are complicated, involving oxidative stress, mitochondrial dysfunction, inflammatory responses, and other aspects. A single nanomaterial may struggle to address these issues simultaneously, necessitating the development of multifunctional synergistic nanomaterials. (5) Difficulty in clinical translation. Although nanobiomedicines have shown promising therapeutic potential in laboratory studies, translating them into clinical applications remains a significant challenge. Most nanosized multifunctional systems have defects such as complex formulations, ambiguous chemical structures, variations between batches, and questionable stability in vivo. These issues undoubtedly increase the difficulty of future translation [137]. Moreover, systematic safety assessments must be conducted before clinical applications to ensure the safety of these nanomaterials in clinical use. Current research primarily focuses on in vitro experiments and animal models, with a lack of large-scale clinical trials to validate their safety.
Future research directions should focus on several aspects: First, creating new kinds of nanomaterials to enhance their targeting ability and biocompatibility. For example, using biomaterials or natural products as the basis for nanobiomedicines may enhance their safety and efficacy [163]. Secondly, in-depth research on the behavior of nanobiomedicines in vivo, including their metabolic pathways, excretion mechanisms, and interactions with biological systems, is necessary to better understand their pharmacokinetic characteristics [164]. Additionally, combining various therapeutic approaches, such as gene therapy and stem cell therapy, has led to research establishing magnetic guidance for the transplantation of ex vivo lentivirus transduced and magnetic nanoparticle loaded embryonic cardiac fibroblasts into mouse hearts, effectively targeting and transplanting cells into cardiac scars, providing strong protection against ventricular arrhythmias, with a 50% reduction in incidence at 2 and 8 weeks' post-MI. Cell therapy, combined with magnetic guidance of cardiac fibroblasts, could effectively target cardiac scars and regulate their functional characteristics [165]. Finally, artificial intelligence (AI) has been applied to the diagnosis and treatment of MI and MI/RI [166]. Future applications of AI in mitochondrial targeted therapy can be explored. Currently, AI has enabled the customization of precision nanobiomedicines for cancer treatment. By adjusting the properties of nanobiomedicines, the design process can be optimized to achieve effective drug synergy, reduce nanotoxicity, and thereby enhance the targeting capability, personalized dosing, and therapeutic efficacy of nanobiomedicines [167]. AI models based screening tools can rapidly predict the delivery efficiency of NPs based on their physicochemical properties, without relying on animal training datasets [168]. It can become an important direction for the application of nanotechnology in myocardial ischemia.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jing Qian: Methodology. Guoxing Ling: Project administration. Yue Li: Investigation. Yan Liu: Validation. Xiaoxuan Guan: Data curation. Zuyuan Huang: Software. Ming Gao: Software, Resources. Cheng Luo: Investigation, Funding acquisition. Baoshi Zheng: Formal analysis, Data curation.
This study was financially supported by the Key Research & Development Program of Guangxi (No. GuiKeAB24010153), the Guangxi Natural Science Foundation (No. 2025GXNSFAA069056), the National Natural Science Foundation of China (No. 82060082), the Youth Science Foundation of Guangxi Medical University (No. GXMUYSF202334), and the Major Talent Project of Guangxi Autonomous Region.
Supplementary material associated with this article can be found, in the online version, at doi:
GBD 2019 Stroke Collaborators, Lancet Neurol. 20 (2021) 795–820. doi: 10.1016/S1474-4422(21)00252-0
A. Kumar, K. Connelly, K. Vora, et al., Can. J. Cardiol. 40 (2024) 1–14.
Y. Mastoor, E. Murphy, B. Roman, J. Clin. Invest. 135 (2025) e184134. doi: 10.1172/JCI184134
F.G.P. Welt, W. Batchelor, J.R. Spears, et al., J. Am. Coll. Cardiol. 83 (2024) 2196–2213.
C.J.A. Ramachandra, S. Hernandez-Resendiz, G.E. Crespo-Avilan, et al., EBioMedicine 57 (2020) 102884. doi: 10.1016/j.ebiom.2020.102884
S. Cai, M. Zhao, B. Zhou, et al., J. Clin. Invest. 133 (2023) e159498. doi: 10.1172/JCI159498
F. Waltz, R.D. Righetto, L. Lamm, et al., Science 387 (2025) 1296–1301. doi: 10.1126/science.ads8738
G. Heusch, I. Andreadou, R. Bell, et al., Redox Biol. 67 (2023) 102894. doi: 10.1016/j.redox.2023.102894
E. Murphy, J.C. Liu, Cardiovasc. Res. 119 (2023) 1105–1116. doi: 10.1093/cvr/cvac134
Y. Saito, K. Oyama, K. Tsujita, et al., J. Cardiol. 81 (2023) 168–178. doi: 10.1016/j.jjcc.2022.07.003
Q. Lv, J. Lin, H. Huang, et al., Adv. Sci. 11 (2024) e2305895. doi: 10.1002/advs.202305895
H. Peng, F. Yao, J. Zhao, et al., Exploration 3 (2023) 20220115. doi: 10.1002/EXP.20220115
Z. Zhang, R. Dalan, Z. Hu, et al., Adv. Mater. 34 (2022) e2202169. doi: 10.1002/adma.202202169
Y. Yao, J. Ding, Z. Wang, et al., Biomaterials 232 (2020) 119726. doi: 10.1016/j.biomaterials.2019.119726
K. Wang, J. Wen, W.Y. Wang, et al., Bioact. Mater. 48 (2025) 43–54.
X. Zheng, T. Wang, J. Gong, et al., Mater. Horiz. 11 (2024) 4998–5016. doi: 10.1039/d4mh00552j
S. Masoudi Asil, E.D. Guerrero, G. Bugarini, et al., View 4 (2023) 20220056. doi: 10.1002/VIW.20220056
H. Zhou, J. Ren, S. Toan, D. Mui, Ageing Res. Rev. 66 (2021) 101250. doi: 10.1016/j.arr.2020.101250
K. Jiang, J. Hwa, Y. Xiang, Pharmacol. Res. 205 (2024) 107256. doi: 10.1016/j.phrs.2024.107256
H. Xu, X. Chen, S. Luo, et al., Redox Biol. 79 (2025) 103471. doi: 10.1016/j.redox.2024.103471
J.S. Harrington, S.W. Ryter, M. Plataki, et al., Physio. Rev. 103 (2023) 2349–2422. doi: 10.1152/physrev.00058.2021
E. Balderas, S.H.J. Lee, N.K. Rai, et al., Physiology 39 (2024) 247–268. doi: 10.1152/physiol.00014.2024
Y. Guo, T. Guan, K. Shafiq, et al., Ageing Res. Rev. 88 (2023) 101955. doi: 10.1016/j.arr.2023.101955
H. Zhang, C.K. Tsui, G. Garcia, et al., Cell 187 (2024) 4289-4304.e26. doi: 10.1016/j.cell.2024.05.057
H. Jiao, D. Jiang, X. Hu, et al., Cell 184 (2021) 2896-2910.e13. doi: 10.1016/j.cell.2021.04.027
Y. Fu, O. Sacco, E. DeBitetto, et al., Mol. Cell 83 (2023) 3740-3753.e9. doi: 10.1016/j.molcel.2023.09.026
L. Na, W. Ying, Y. Ling, et al., Chem. Eng. J. 519 (2025) 165274. doi: 10.1016/j.cej.2025.165274
H. Sies, D.P. Jones, Nat. Rev. Mol. Cell Biol. 21 (2020) 363–383. doi: 10.1038/s41580-020-0230-3
A.D. Read, R.E. Bentley, S.L. Archer, et al., Redox Biol. 47 (2021) 102164. doi: 10.1016/j.redox.2021.102164
L. Su, J. Zhang, H. Gomez, et al., Autophagy 19 (2022) 401–414.
X. Wu, B. Wang, Y. Li, et al., Chin. Chem. Lett. 36 (2025) 110211. doi: 10.1016/j.cclet.2024.110211
J. Wang, H. Zhou, Acta Pharm. Sin. B 10 (2020) 1866–1879. doi: 10.1016/j.apsb.2020.03.004
Q. Xiang, X. Yi, X.H. Zhu, et al., Trends Endocrinol. Metab. 35 (2024) 219–234. doi: 10.1016/j.tem.2023.10.010
T. Zhao, W. Wu, L. Sui, et al., Bioact. Mater. 7 (2022) 47–72.
K. Wang, K. Zhu, Z. Yuan, et al., Chem. Eng. J. 517 (2025) 164177. doi: 10.1016/j.cej.2025.164177
K. Kaarniranta, H. Uusitalo, J. Blasiak, et al., Prog. Retin. Eye Res. 79 (2020) 100858. doi: 10.1016/j.preteyeres.2020.100858
P. Wen, Z. Sun, F. Gou, et al., Ageing Res. Rev. 104 (2025) 102667. doi: 10.1016/j.arr.2025.102667
Z. Zheng, C. Lei, H. Liu, et al., Adv. Healthc. Mater. 11 (2022) e2200990. doi: 10.1002/adhm.202200990
J. Li, J. Zhang, P. Yu, et al., Bio. Mater. 37 (2024) 239–252. doi: 10.3847/1538-4357/ad3af2
X. Long, M. Liu, Y. Nan, et al., Adv. Mater. 36 (2024) e2308239. doi: 10.1002/adma.202308239
B. Wacquier, L. Combettes, G. Dupont, Cold Spring Harb. Perspect. Biol. 11 (2019) a035139. doi: 10.1101/cshperspect.a035139
A.A. Lombardi, A.A. Gibb, E. Arif, et al., Nat. Commun. 10 (2019) 4509. doi: 10.1038/s41467-019-12103-x
K.M. Koo, S.J. Seo, C.D. Kim, et al., Adv. Compos. Hybrid Mater. 8 (2025) 256. doi: 10.1007/s42114-025-01331-z
M. Yu, M. Song, M. Zhang, et al., Cell Prolif. 58 (2025) e70002. doi: 10.1111/cpr.70002
L. Zhang, R. Guo, C. Xiao, et al., hLife 2 (2024) 189–200. doi: 10.1016/j.hlife.2024.03.003
C.W. Kontchou, I.E. Gentle, A. Weber, et al., Cell Death Differ. 29 (2022) 2046–2059. doi: 10.1038/s41418-022-00995-0
J.F. Garbincius, J.W. Elrod, Physiol. Rev. 102 (2022) 893–992. doi: 10.1152/physrev.00041.2020
T. Zhao, W. Wu, L. Sui, et al., Bioact. Mater. 7 (2022) 47–72.
S. Marchi, E. Guilbaud, S.W.G. Tait, et al., Nat. Rev. Immunol. 23 (2023) 159–173. doi: 10.1038/s41577-022-00760-x
L. Jiang, S. Zhou, X. Zhang, et al., Nat. Commun. 12 (2021) 2390. doi: 10.1038/s41467-021-22594-2
P. Bernardi, C. Gerle, A.P. Halestrap, et al., Cell Death Differ. 30 (2023) 1869–1885. doi: 10.1038/s41418-023-01187-0
Z. Chen, X. Tan, T. Jin, et al., Adv. Sci. 11 (2024) e2307880. doi: 10.1002/advs.202307880
M. Paillard, M. Abdellatif, I. Andreadou, et al., Eur. J. Heart Fail. 27 (2025) 1720–1736. doi: 10.1002/ejhf.3674
E. Bertero, T.A. Popoiu, C. Maack, Basic Res. Cardiol. 119 (2024) 569–585. doi: 10.1007/s00395-024-01060-2
Z. Wang, N. Yang, Y. Hou, et al., Adv. Sci. 10 (2023) e2302123. doi: 10.1002/advs.202302123
C.X. Zhang, Y. Cheng, D.Z. Liu, et al., J. Nanobiotechnology 17 (2019) 18. doi: 10.1117/12.2542489
Q. Zhang, Q. Song, R. Yu, et al., Adv. Sci. 10 (2023) e2204596. doi: 10.1002/advs.202204596
A.T. Moehlman, R.J. Youle, Annu. Rev. Cell Dev. Biol. 36 (2020) 265–289. doi: 10.1146/annurev-cellbio-021820-101354
C. Gao, R. Wang, B. Li, et al., Cardiovasc. Res. 116 (2020) 645–657. doi: 10.1093/cvr/cvz152
M.A. Matter, F. Paneni, P. Libby, et al., Eur. Heart J. 45 (2024) 89–103. doi: 10.1093/eurheartj/ehad486
X. Ding, C. Zhu, W. Wang, et al., Pharmacol. Res. 199 (2024) 106957. doi: 10.1016/j.phrs.2023.106957
D.P. Narendra, R.J. Youle, Nat. Cell Biol. 26 (2024) 1639–1651. doi: 10.1038/s41556-024-01513-9
M. Chen, G. Zhong, M. Liu, et al., Pharmacol. Res. 189 (2023) 106682. doi: 10.1016/j.phrs.2023.106682
M. Song, G. Gong, Y. Burelle, et al., Circ. Res. 117 (2015) 346–351. doi: 10.1161/CIRCRESAHA.117.306859
L. Ai, J. de Freitas Germano, C. Huang, et al., Eur. Heart J. 46 (2025) 380–393. doi: 10.1093/eurheartj/ehae782
Z. Wang, D. Lin, B. Cui, et al., Eur. J. Pharmacol. 963 (2024) 176292. doi: 10.1016/j.ejphar.2023.176292
N. Yang, R. Zhang, H. Zhang, et al., Cardiovas. Diabeto. 23 (2024) 399. doi: 10.1186/s12933-024-02499-2
C. Xu, Y. Cao, R. Liu, et al., J. Cell Mol. Med. 26 (2022) 1315–1326. doi: 10.1111/jcmm.17190
S. Ghafouri-Fard, H. Shoorei, M. Mohaqiq, et al., Autophagy 18 (2022) 949–970. doi: 10.1080/15548627.2021.1883881
Y. Yan, L.Y. Tian, Q. Jia, et al., Cell Death Discov. 9 (2023) 77.
X. Chen, T. Sun, Y. Qi, et al., Mol. Immunol. 177 (2025) 32–43. doi: 10.1016/j.molimm.2024.12.003
T. Luo, X. Jia, W.D. Feng, et al., Acta Pharm. Sin. B 44 (2023) 1867–1878. doi: 10.1038/s41401-023-01094-7
C.J. Griffey, A. Yamamoto, Nat. Rev. Neurosci. 23 (2022) 411–427. doi: 10.1038/s41583-022-00588-3
A. Liu, F. Kage, A.F. Abdulkareem, et al., Nat. Cell Biol. 26 (2024) 731–744. doi: 10.1038/s41556-024-01400-3
X. Niu, J. Zhang, S. Hu, et al., Cell Mol. Biol. Lett. 29 (2024) 72. doi: 10.1186/s11658-024-00588-4
T. Zhang, Q. Liu, W. Gao, et al., Autophagy 18 (2021) 1216–1239.
H.X. Su, L.L. Xu, P.B. Li, et al., Cell Death Dis. 15 (2024) 803. doi: 10.1038/s41419-024-07189-1
J. Du, H. Li, J. Song, et al., Front. Pharmacol. 13 (2022) 862204. doi: 10.3389/fphar.2022.862204
X. Zeng, Y.D. Zhang, R.Y. Ma, et al., Mil. Med. Res. 9 (2022) 25. doi: 10.18653/v1/2022.autosimtrans-1.5
Q. Jin, R. Li, N. Hu, et al., Redox Biol. 14 (2018) 576–587. doi: 10.1016/j.redox.2017.11.004
Y. Sun, J. Xu, L. Zou, et al., Mater. Today Bio. 32 (2025) 101770. doi: 10.1016/j.mtbio.2025.101770
J. Wang, H. Zhuang, L. Jia, et al., Int. J. Biol. Sci. 20 (2024) 4458–4475. doi: 10.7150/ijbs.95853
G. Chen, J. Gan, F. Wu, et al., Eur. Heart J. 46 (2025) 2579–2594. doi: 10.1093/eurheartj/ehaf032
D. Shimura, E. Nuebel, R. Baum, et al., eLife 10 (2021) e69207. doi: 10.7554/eLife.69207
R. Quintana-Cabrera, L. Scorrano, Mol. Cell 83 (2023) 857–876. doi: 10.1016/j.molcel.2023.02.012
L. Hu, D. Tang, B. Qi, et al., Adv. Sci. 11 (2024) e2307749. doi: 10.1002/advs.202307749
M. Semenzato, M.J. Kohr, C. Quirin, et al., Redox Biol. 63 (2023) 102755. doi: 10.1016/j.redox.2023.102755
P. Gatti, C. Schiavon, J. Cicero, et al., Nat. Commun. 16 (2025) 451.
L. Liu, Y. Li, G. Chen, et al., J. Biomed. Sci. 30 (2023) 86. doi: 10.1186/s12929-023-00975-7
S.C. da Silva Rosa, M.D. Martens, J.T. Field, et al., Autophagy 17 (2020) 2257–2272.
S. Fan, X. Yan, X. Hu, et al., J. Exp. Clin. Cancer Res. 43 (2024) 180. doi: 10.1049/icp.2023.3329
X. Chang, A. Lochner, H.H. Wang, et al., Theranostics 11 (2021) 6766–6785. doi: 10.7150/thno.60143
D.S.E. Mahmoud, M.A. Kamel, I.E. El-Sayed, et al., J. Biochem. Mol. Toxicol. 38 (2024) e23804. doi: 10.1002/jbt.23804
S. Wan, Z. Cui, L. Wu, et al., Redox Biol. 60 (2023) 102610. doi: 10.1016/j.redox.2023.102610
J. Boeddinghaus, A. Bularga, C. Taggart, et al., Eur. Heart J. Acute Cardiovasc. Care 14 (2025) 131–141. doi: 10.1093/ehjacc/zuaf002
R. Song, C. Dasgupta, C. Mulder, et al., Circulation 145 (2022) 1140–1153. doi: 10.1161/circulationaha.121.056929
Z. Li, S. Li, W. Chen, et al., Interdiscip. Med. 3 (2025) e20250005. doi: 10.1002/inmd.20250005
A.S. Manolis, A.A. Manolis, T.A. Manolis, et al., Med. Res. Rev. 41 (2021) 275–313. doi: 10.1002/med.21732
C. Liu, D. Zhang, K. Long, et al., Pharmacol. Res. 209 (2024) 107468. doi: 10.1016/j.phrs.2024.107468
W. Yi, Y. Jing-Song, Z. Min, et al., Adv. Drug Deliv. Rev. 211 (2024) 115355. doi: 10.1016/j.addr.2024.115355
Q. Ye, Y. Lin, R. Li, et al., Semin. Cancer Biol. 86 (2022) 607–623. doi: 10.1016/j.semcancer.2022.03.016
H.T. Shi, Z.H. Huang, T.Z. Xu, et al., eBioMedicine 78 (2022) 103968. doi: 10.1016/j.ebiom.2022.103968
J. Guo, Z. Yang, Y. Lu, et al., Bioact. Mater. 10 (2022) 56–67.
E. Hamdi, S. Hidouri, A.B. Muniz-Gonzalez, et al., Nano TransMed 4 (2025) 100074. doi: 10.1016/j.ntm.2025.100074
B. Tan, Y. Zheng, J. Hao, et al., Adv. Compos. Hybrid Mater. 8 (2025) 126. doi: 10.1007/s42114-025-01219-y
W. Wang, P. Gao, H. Gui, et al., Coord. Chem. Rev. 522 (2025) 216205. doi: 10.1016/j.ccr.2024.216205
M. Zhou, Y. Tang, Y. Lu, et al., ACS Nano 19 (2025) 3455–3469. doi: 10.1021/acsnano.4c12917
A. Pu, W.S. Sim, Y. Ji, et al., Bioact. Mater. 53 (2025) 366–385.
Y. Sun, Y. Zhu, J. Si, et al., Chin. Chem. Lett. 36 (2025) 110012. doi: 10.1016/j.cclet.2024.110012
W.L. Hong, H. Huang, X. Zeng, et al., Mil. Med. Res. 11 (2024) 59.
M. Tang, T. Duan, Y. Lu, et al., Adv. Mater. 36 (2024) e2411906. doi: 10.1002/adma.202411906
Z. Yu, X. Luo, C. Zhang, et al., Chin. Chem. Lett. 35 (2024) 109519. doi: 10.1016/j.cclet.2024.109519
X. Sun, H. Chen, R. Gao, et al., ACS Nano 17 (2023) 896–909. doi: 10.1021/acsnano.2c05286
C. Pegoraro, E. Masiá Sanchis, S. Đorđević, et al., Chem. Mater. 37 (2025) 1457–1467. doi: 10.1021/acs.chemmater.4c02742
J. Zhang, W. Yang, Y. Zhu, et al., Acta Biomater. 194 (2025) 323–335. doi: 10.1016/j.actbio.2025.01.019
J. Liao, Y. Li, L. Fan, et al., ACS Nano 18 (2024) 5510–5529.
N. Guo, W. Zhou, Z. Zhang, et al., Chem. Eng. J. 503 (2025) 158468. doi: 10.1016/j.cej.2024.158468
L. Huang, Z. Sun, Q. Shen, et al., Chin. Chem. Lett. 33 (2022) 4146–4156. doi: 10.1016/j.cclet.2022.02.047
J. Jin, P. Yuan, W. Yu, et al., ACS Nano 16 (2022) 10327–10340. doi: 10.1021/acsnano.2c00192
J. Yang, Z. Yang, H. Wang, et al., Angew. Chem. Int. Ed. 63 (2024) e202402291. doi: 10.1002/anie.202402291
J. Liu, Y. Yan, Y. Zhang, et al., J. Am. Chem. Soc. 146 (2024) 34568–34582. doi: 10.1021/jacs.4c12264
K. Qian, P. Yang, Y. Li, et al., Asian J. Pharm. Sci. 19 (2024) 100938.
X. Zhang, Y. Sun, R. Yang, et al., Biomaterials 287 (2022) 121656. doi: 10.1016/j.biomaterials.2022.121656
Y. Sun, P. Zhang, Y. Li, et al., ACS Nano 16 (2022) 18667–18681. doi: 10.1021/acsnano.2c07311
X. Liao, M. Tang, J. Li, et al., Adv. Healthc. Mater. 14 (2025) e2404319. doi: 10.1002/adhm.202404319
S. Zhang, X. Zhao, Y. Xue, et al., J. Nanobiotechnology 22 (2024) 342. doi: 10.1186/s12951-024-02615-0
J.L.Y. Wu, Q. Ji, C. Blackadar, et al., Nat. Nanotechnol. 20 (2025) 672–682. doi: 10.1038/s41565-025-01877-5
Q. Huang, Y. Yang, T. Zhao, et al., Bioact. Mater. 21 (2023) 381–393.
X. Gao, X. Yang, C. Deng, et al., Biomaterials 318 (2025) 123133. doi: 10.1016/j.biomaterials.2025.123133
Q. Huang, J. Liao, J. Li, et al., Chin. Chem. Lett. 36 (2025) 109914. doi: 10.1016/j.cclet.2024.109914
Q. Luo, J. Sun, Z. Li, et al., Chin. Chem. Lett. 36 (2025) 110433. doi: 10.1016/j.cclet.2024.110433
L. Ding, S. Zhang, Y. Li, et al., Chem. Eng. J. 486 (2024) 150177. doi: 10.1016/j.cej.2024.150177
Q. Sun, H. Ma, J. Zhang, et al., Adv. Sci. 10 (2022) e2204999.
N. Hu, M. Sun, N. Lv, et al., ACS Appl. Mater. Interfaces 16 (2024) 12188–12201. doi: 10.1021/acsami.3c16735
J. Zhang, L. Liu, Z. Dong, et al., Bioact. Mater. 28 (2023) 480–494.
Z. Lu, Q. Chai, W. Dai, et al., J. Nanobiotechnology 23 (2025) 496. doi: 10.1186/s12951-025-03578-6
J.M. Kneuer, I.A. Grajek, M. Winkler, et al., Circulation 150 (2024) 1101–1120. doi: 10.1161/circulationaha.124.069315
M. Gao, L. Yin, B. Zhang, et al., ACS Nano 19 (2025) 18475–18491. doi: 10.1021/acsnano.5c01641
X. Yu, J. Wang, T. Wang, et al., J. Nanobiotechnology 22 (2024) 554. doi: 10.1186/s12951-024-02796-8
L. Pan, X. Hu, H. Yang, et al., Adv. Funct. Mater. 35 (2025) 2417338. doi: 10.1002/adfm.202417338
M. Xinhai, X. Huijing, W. Lu, et al., Nano Today 46 (2022) 101589. doi: 10.1016/j.nantod.2022.101589
H. Xu, Z. Zhao, P. She, et al., J. Control. Release 375 (2024) 788–801. doi: 10.1016/j.jconrel.2024.09.038
J. Wu, K.C.V. Subbaiah, O. Hedaya, et al., Cardiovasc. Res. 119 (2023) 2441–2457. doi: 10.1093/cvr/cvad124
M.Y. Chen, B.C. Tsai, W.W. Kuo, et al., Am. J. Chin. Med. 51 (2023) 1211–1232. doi: 10.1142/s0192415x23500556
C. Chen, J. Ma, S. Duan, et al., Biomaterials 318 (2025) 123119. doi: 10.1016/j.biomaterials.2025.123119
C. Wu, H. Wang, J. Niu, et al., Chem. Eng. J. 508 (2025) 160833. doi: 10.1016/j.cej.2025.160833
B. Papia, R. Arpita, P. Chitta Ranjan, et al., Biomater. Adv. 153 (2023) 213531. doi: 10.1016/j.bioadv.2023.213531
Y. Gong, Y. Xiao, C. Zhao, et al., ACS Nano 19 (2025) 13723–13739. doi: 10.1021/acsnano.4c14869
X. Guo, T. Hong, J. Zang, et al., J. Nanobiotechnology 20 (2022) 531. doi: 10.1186/s12951-022-01686-1
M. Liu, L. Wang, Z. Liu, et al., Life Sci. 365 (2025) 123447. doi: 10.1016/j.lfs.2025.123447
X. Mo, L. Liu, L. Zhang, et al., Adv. Funct. Mater. 35 (2025) 2415084. doi: 10.1002/adfm.202415084
X. Zhang, Q. Liu, R. Zhao, et al., Nano Lett. 25 (2025) 663–672. doi: 10.1021/acs.nanolett.4c04462
T. Li, B. Yang, X. Liu, et al., Int. J. Nanomedicine 20 (2025) 1843–1864. doi: 10.2147/ijn.s500810
G. Ikeda, T. Matoba, A. Ishikita, et al., J. Am. Heart Assoc. 10 (2021) e019521. doi: 10.1161/JAHA.120.019521
M. Jin, C. Li, Z. Wu, et al., Adv. Sci. 12 (2025) e2414830. doi: 10.1002/advs.202414830
J. Yang, G.W. CHO, L. He, et al., Circ. Res. 142 (2020) 14412.
K. Lv, T. Wu, S. Liu, et al., Acta Pharm. Sin. B 14 (2024) 4526–4543. doi: 10.1016/j.apsb.2024.06.027
Z. Sun, T. Mei, Y. Xin, et al., Appl. Mater. Today 39 (2024) 102333. doi: 10.1016/j.apmt.2024.102333
Y. Jiao, H. Wang, X. Weng, et al., Aggregate 5 (2024) e563. doi: 10.1002/agt2.563
Q. Guo, X. Shang, Y. Liu, et al., ACS Appl. Nano Mater. 8 (2025) 12639–12652. doi: 10.1021/acsanm.5c01093
Q. Zhang, Y. Zhang, S. Cheng, et al., Eur. Heart J. 44 (2023) ehad655.795. doi: 10.1093/eurheartj/ehad655.795
M. Sun, N. Hu, Y. Gao, et al., Adv. Healthc. Mater. 13 (2024) e2303101. doi: 10.1002/adhm.202303101
N.H. Kamal, L.A. Heikal, O.Y. Abdallah, Drug Deliv. Transl. Res. 15 (2025) 2253–2271. doi: 10.1007/s13346-024-01763-y
M. Lv, Q. Sun, Y. Yu, et al., Front. Bioeng. Biotechnol. 12 (2024) 1511331.
M. Schiffer, K. Wagner, E. Carls, et al., Theranostics 15 (2025) 4287–4307. doi: 10.7150/thno.103816
M.S. Lee, T.G. Shin, Y. Lee, et al., Eur. Heart J. 46 (2025) 1917–1929. doi: 10.1093/eurheartj/ehaf004
P. Tan, X. Chen, H. Zhang, et al., Semin. Cancer Biol. 89 (2023) 61–75. doi: 10.1016/j.semcancer.2023.01.005
W.C. Chou, Q. Chen, L. Yuan, et al., J. Control. Release 361 (2023) 53–63. doi: 10.1016/j.jconrel.2023.07.040

Figure 2 Illustrations displaying the preparation process of the macrophage cell membrane camouflaged M/PCOD@PLGA and the mechanism of M/PCOD@PLGA to treat MI/RI. The process is divided into 3 steps: (1) macrophage membrane coating promotes accumulation of nanoparticles (PCOD@PLGA) in ischemic area of heart. (2) M/PCOD@PLGA is internalized by nearby cells and release PCOD585, which react with ONOO− to release CO. (3) CO inhibits apoptosis, decreases pro-inflammatory macrophage, and facilitates the mitochondrial biogenesis. Copied with permission [135]. Copyright 2023, Elsevier.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: