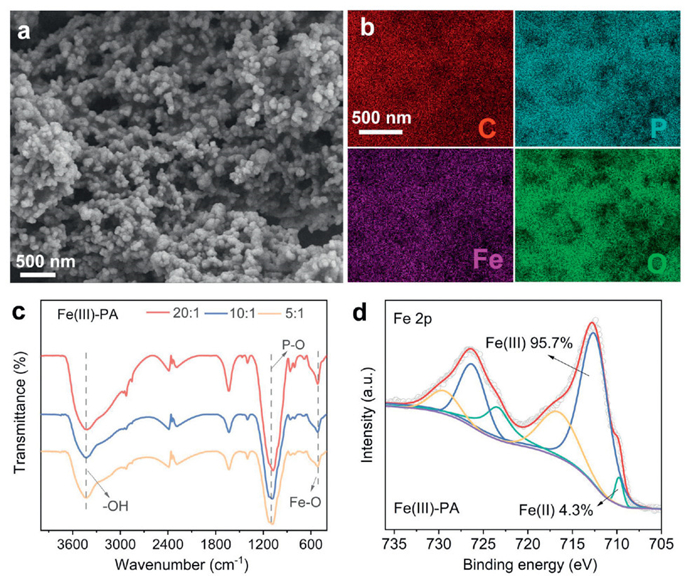
Figure 1.
(a) SEM images of Fe(Ⅲ)-PA. (b) EDS mapping. (c) FT-IR spectra of Fe(Ⅲ)-PA. (d) XPS of Fe 2p spectrum for Fe(Ⅲ)-PA.
Peroxymonosulfate activation by Fe(Ⅲ)-phytate co-precipitation for efficient water treatment at circumneutral pH: The critical role of direct electron transfer
Lijuan Huang , Rui Gan , Xin Han , Meilin Sun , Li Chen , Wen Liu , Xiaoxin Zhang , Fei Pan

Advanced oxidation processes (AOPs) have garnered significant attention in water purification research due to their strong oxidizing capacity and rapid reaction rates [1–4]. Specifically, peroxymonosulfate (PMS)-based AOPs present several advantages, such as ease of storage, readily activated oxidation potentials, and a broader pH application range, making them a promising alternative to traditional Fenton processes [5–8]. PMS can be activated by transition metal ions, metal oxides, carbon materials, and energy, generating various reactive oxygen species (ROS), including hydroxyl radicals, sulfate radicals, and singlet oxygen [9–12]. Following reports of organic pollutant degradation through the addition of Fe(Ⅱ) to PMS [13], there has been increased interest in both homogeneous and heterogeneous activation using Fe ions and Fe oxides in PMS-based AOPs [14,15]. However, similar to the Fenton process, the low rate of Fe(Ⅱ) production during the reaction leads to a greater tendency for generated Fe(Ⅲ) to undergo hydrolytic precipitation [16,17]. This results in the cessation of the Fe(Ⅱ)/Fe(Ⅲ) cycle, depleting Fe(Ⅱ) and generating significant amounts of iron sludge [18]. Therefore, enhancing the reactivity of Fe(Ⅲ) is crucial for maintaining catalytic activity in Fenton-like chemistry [19].
To address this, researchers have focused on promoting the conversion of Fe(Ⅲ) to Fe(Ⅱ) by applying additional reducing agents. For instance, hydroxylamine, ascorbic acid, sodium sulfite, and L-cysteine have been employed to reduce Fe(Ⅲ), thereby enhancing the removal of target pollutants in Fenton-like systems [20–25]. Notably, the reductants inevitably consume ROS during the reaction, and this self-quenching effect interferes with the electron selectivity of ROS for the target pollutants [26,27]. Consequently, this leads to the overuse of the oxidizing agent in practice. Additionally, ligands such as oxalic acid, citric acid, ethylenediaminetetraacetic acid (EDTA), and N,N'-ethylenedi-L-aspartic acid (EDDS) have been utilized to enhance the solubility of Fe(Ⅲ) and further slow down its deactivation [28–30]. However, challenges persist in employing ligand strategies to enhance the oxidation rate of Fenton-like systems. Yang et al. demonstrated that using pyridine carboxylic acid as a ligand could enhance the reaction rate of Fe(Ⅲ)-catalyzed PMS and revealed the crucial role of metal-PMS complexes in promoting ROS generation in the system [31]. Nevertheless, the oxidation pathways mediated by various organic ligands in Fenton-like systems remain significantly different. Thus, it is essential to rationally introduce new organic ligands to optimize key parameters, such as enhancing electron selectivity and the apparent activity of Fenton-like catalysis.
Phytate (PA), derived from plant tissue, is a natural, non-toxic, saturated cyclic acid containing six phosphate groups, which confer strong chelating properties [32]. Our previous study demonstrated that modifying the zero-valent iron (ZVI) surface with phytates accelerates electron migration from the iron core to the surface layer. Additionally, the ZVI surface enhances its coordination ability to target molecules by adsorbing phytate molecules [33]. The properties of phytic acid make it an ideal candidate for ligand-enhanced Fenton-like systems. To the best of our knowledge, the enhancement of Fe(Ⅲ) reactivity by phytate ligands has not been reported, and the mechanism of its mediated Fenton-like oxidation requires further elucidation. Furthermore, it is believed that ROS are more likely to interact with the ligand or Fe(Ⅲ) during homogeneous processes, resulting in self-quenching phenomena. Therefore, designing the reaction as a non-homogeneous process would be beneficial for further improving electron utilization efficiency [34,35].
In this study, we prepared a catalyst called Fe(Ⅲ)-PA by co-precipitating phytate with iron ions to explore how the phytate ligand modulates the Fenton-like reaction pathway. Tetracycline hydrochloride (TCH) was chosen as a model contaminant to investigate the enhancement of Fe(Ⅲ) activation of PMS by phytate ligands. Batch experiments and electron paramagnetic resonance (EPR) revealed the mechanism of active substance generation during the reaction. The mechanism of phytate ligand-enhanced Fenton-like reactivity was elucidated through density functional theory (DFT) calculations. Additionally, the TCH removal efficacy of the Fe(Ⅲ)-PA/PMS system under complex aqueous environmental conditions was examined. Finally, the transformation pathways of the target pollutants were analyzed using ultra-performance liquid chromatography-mass spectrometry (UPLC-MS), and the ecotoxicity of the intermediates was further evaluated.
The morphology of Fe(Ⅲ)-PA was characterized by scanning electron microscopy (SEM). As shown in Fig. 1a, Fe(Ⅲ)-PA exhibited a dispersed spherical particle structure. The particle size of Fe(Ⅲ)-PA decreased with increasing initial concentration of the PA solution, with average particle sizes of 40 nm, 60 nm and 0.8 µm for Fe(Ⅲ)-PA(5:1), Fe(Ⅲ)-PA(10:1) and Fe(Ⅲ)-PA(20:1) respectively (Fig. S1 in Supporting inforamtion). The results of energy dispersive X-ray spectroscopy (EDS) analysis of Fe(Ⅲ)-PA showed a uniform distribution of Fe and P elements on its surface (Fig. 1b). This indicated that PA was adsorbed on the surface of Fe(Ⅲ)-PA during the co-precipitation of Fe(Ⅱ) with PA. In addition, the Fourier transform infrared spectroscopy (FTIR) results showed that the peak of Fe(Ⅲ)-PA at 1180 cm-1 could be attributed to the stretching vibration of P-O (Fig. 1c) [36,37]. The chemical compositions of the elements on the surface of Fe(Ⅲ)-PA were further detected using X-ray photoelectron spectroscopy (XPS). The P 2p signal at peak near 133.5 eV was detected in the XPS survey of Fe(Ⅲ)-PA (Fig. S2a in Supporting information) [38], which corresponded to the EDS results and confirmed the successful adsorption of PA on the Fe(Ⅲ)-PA surface. The high-resolution profile of Fe 2p was subjected to further analysis, resulting in the identification of peaks at 713 cm-1 and 709 cm-1 (Fig. 1d), which correspond to Fe(Ⅱ) and Fe(Ⅲ) [39], respectively. Through the calculation of peak areas, it was determined that Fe(Ⅲ) constituted up to 95.7% of the total content. Furthermore, no significantly change were observed in the characteristic peak areas corresponding to Fe(Ⅱ) and Fe(Ⅲ) of the reacted Fe(Ⅲ)-PA, which demonstrated that the primary participant in the reaction is Fe(Ⅲ).
The catalytic performance of Fe(Ⅲ)-PA in the Fenton-like system was investigated using TCH as a model contaminant. As shown in Fig. 2a, the TCH degradation rate was only 9.9% when only Fe(Ⅲ)-PA was present, indicating that the TCH removal was not dependent on adsorption of Fe(Ⅲ)-PA. Comparative experiments were conducted to evaluate the degradation efficiency of TCH by PMS activated with different catalysts. Among them, Fe(Ⅲ)-PA demonstrated the highest catalytic activity. The TCH removal rate was 42.8% within 60 min in the presence of PMS only, suggesting that PMS itself is incapable of complete degradation of TCH. The efficiency of Fe(Ⅲ)/PMS system in removing TCH was not significantly improved compared with PMS alone, indicating that Fe(Ⅲ) could not activate PMS effectively. In addition, the removal of TCH by the PA/PMS system was negligible. Conversely, the Fe(Ⅲ)-PA/PMS system exhibited remarkable catalytic efficacy, achieving a TCH removal of 93.3% within 60 min. It is noteworthy that the rate constant (k) for TCH removal in the Fe(Ⅲ)-PA/PMS system reached 0.064 min-1. The catalytic reaction removed TCH at a faster rate in the first 20 min, with 21% degradation of TCH by PMS alone in 20 min, while the degradation rate of TCH by Fe(Ⅲ)-PA/PMS was enhanced to 79.5%. Comparison with PMS system, the reaction kinetics of Fe(Ⅲ)-PA/PMS system was increased by 6.4-fold within 60 min, which demonstrated that Fe(Ⅲ)-PA could effectively promote the activation of PMS to effectively degrade TCH.
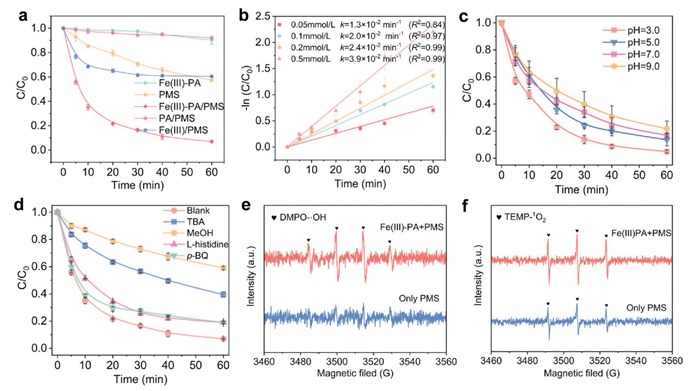
In order to optimize the catalyst dosage and explore the removal rate of TCH with different concentrations of Fe(Ⅲ)-PA. As shown in Fig. S3 (Supporting information), the degradation rate of TCH in the system increased with the increase of Fe(Ⅲ)-PA dosage from 0.1 g/L to 0.4 g/L, which was attributed to the addition of catalyst to provide more active sites for the reaction. The degradation effect was not significantly enhanced when the Fe(Ⅲ)-PA dosage was greater than 0.3 g/L. Furthermore, the optimal dosage of PMS has been investigated. As the PMS concentration increased from 0.05 mmol/L to 0.5 mmol/L, the corresponding kobs value increased from 0.013 min-1 to 0.039 min-1 (Fig. 2b). When the initial concentration of PMS exceeded 0.1 mmol/L, the enhancement of the degradation efficiency of TCH was no longer significant. Therefore, the Fe(Ⅲ)-PA dosage of 0.3 g/L and PMS concentration of 0.1 mmol/L were selected for the subsequent experiments.
The pH is a critical factor in reactions, particularly in iron-based material-mediated advanced oxidation processes (AOPs). As shown in Fig. 2c, the degradation efficiency of the system for TCH consistently remained above 78.2% within the initial pH range of 3.0 to 9.0. As the initial pH of the solution increased from 3.0 to 9.0, the removal rate of TCH decreased from 95.1% to 78.2%. The rate constants were 0.065, 0.045, 0.035, and 0.031 min-1 for pH values of 3.0, 5.0, 7.0, and 9.0, respectively. To investigate whether the leaching of metal ions affected the reaction results, we examined the leaching of iron ions at pH 3.0, and the results indicated that almost no iron ions were leached. Overall, Fe(Ⅲ)-PA /PMS system demonstrated satisfactory degradation efficacy over a wide pH range (Fig. S4 in Supporting information).
The quenching experiments and electron paramagnetic resonance (EPR) were conducted to identify the reactive oxygen species (ROS) in Fe(Ⅲ)-PA/PMS/TCH system. In this study, methanol (MeOH: k[SO4•–] = 1.1 × 107 L mol-1 s-1, k[•OH] = 9.7 × 108 L mol-1 s-1) [40] and tert–butanol (TBA: k[SO4•–] = 7.6 × 105 L mol-1 s-1, k[•OH] = 6.0 × 108 L mol-1 s-1) [40] were used to confirm the contribution of SO4•– and •OH. The degradation efficiency of TCH was reduced to 41.0% and 60.5% by the addition of 100 mmol/L MeOH and TBA (1000-fold higher concentration than that of PMS) to the reaction system. The quenching experiments results indicate that the main ROS were SO4•– and •OH in the Fe(Ⅲ)-PA/PMS/TCH system. EPR spectra revealed that the characteristic signal peaks of SO4•– and •OH were not observed in the presence of PMS alone, suggesting that PMS itself cannot be degraded to produce free radicals (SO4•–and •OH). Notably, the characteristic signal of DMPO-•OH was observed in Fe(Ⅲ)-PA/PMS/TCH system (Fig. 2e). However, when benzoic acid was used as the probe of •OH, no significant degradation of benzoic acid was observed in the Fe(Ⅲ)-PA/PMS system (Fig. S5 in Supporting information). Therefore, the inhibitory effect of MeOH and BA on TCH removal efficiency may be attributed to the direct reaction of MeOH and TBA with PMS, which in turn limits the oxidation of TCH by PMS. In brief, SO4•– and •OH are not major ROS in the Fe(Ⅲ)-PA/PMS system. p-Benzoquinone (p-BQ: k[O2•-] = (0.9–1.0) × 109 L mol-1 s-1) [41] act as a scavenger of O2•-, resulting in a TCH degradation rate of 81.0% upon the incorporation of 5 mmol/L p-BQ (500 times the concentration of PMS), exhibiting a minimal inhibition effect. Furthermore, the EPR spectra did not show any signal of DMPO- O2•-, thus confirming that O2•- was not generated in the Fe(Ⅲ)-PA system.
The unique advantages of the non-radical pathway make it play a key role in non-homogeneous persulfate activations. To further explore the non-radical pathway in the Fe(Ⅲ)-PA/PMS system, L-histidine (k[1O2] = 3.2 × 107 L mol-1 s-1) [42] was used as 1O2 scavenger into the Fe(Ⅲ)-PA/PMS/TCH system. As shown in Fig. 2d, the addition of L-histidine at a ratio of 1:50 exhibited 12% inhibition of TCH removal. The TEMP-1O2 signal was detected within the reaction system in the presence of only PMS, which was attributable to the 1O2 signal generated by the oxidation of TEMP by PMS itself [43]. Although the identical signal was also detected within the Fe(Ⅲ)-PA/PMS system, it was not significantly enhanced compared to the PMS system alone. The observed changes in the weak TEMP-1O2 EPR signal and the burst results indicate that 1O2 is not the primary agent responsible for TCH degradation in the Fe(Ⅲ)-PA/PMS system (Fig. 2f).
It has been demonstrated by previous studies that high-valent metal-oxo species (HVMO) also contribute to the oxidation of organic matter [44]. To determine the role of HVMO in the system, DMSO and were used as scavenger for HVMO. Furthermore, methyl benzylidene phenol (PMSO) can be selectively oxidized to methylbenzene sulfone (PMSO2) by HVMO [44,45]. Consequently, PMSO was selected as a chemical probe for HVMO to further ascertain the contribution of Fe(Ⅳ)=O in the Fe(Ⅲ)-PA/PMS system. The addition of DMPO significantly inhibited the degradation of TCH, and the TCH degradation rate decreased to 38.6% within 60 min (Fig. S7 in Supporting information). However, in the Fe(Ⅲ)-PA/PMS/PMSO system, no significant change in the concentration of PMSO was observed, while the amount of PMSO2 generated were not significantly different from that of PMS alone. (Fig. S8 in Supporting information). Therefore, the inhibitory effect of DMSO could be attributed to the direct depletion of PMS in the system. The above results indicate that HVMO is not the main ROS of TCH degradation in the Fe(Ⅲ)-PA/PMS/TCH system.
Notable that in non-homogeneous persulfate systems, the catalyst-mediated electron transfer process (ETP) may be the primary cause of contaminant degradation [46,47]. In this process, the catalyst act as an electron shuttle, thereby facilitating the transfer of electrons from the pollutant to the oxidant. Moreover, the catalyst has the potential to first form an intermediate state complex with PMS, which can directly oxidize the degraded pollutant [48]. In order to elucidate the electron transfer process in the Fe(Ⅲ)-PA/PMS/TCH system, open-circuit potential (OCP) analysis was performed to determine the interaction between Fe(Ⅲ)-PA, PMS and TCH. As shown in Fig. 3a, the potential increased rapidly from 0.458 V to 0.69 V when PMS was added to the solution containing Fe(Ⅲ)-PA. Subsequently, TCH was added to the Fe(Ⅲ)-PA/PMS system, and it was clearly observed that the potential of the Fe(Ⅲ)-PA/PMS system decreased from 0.69 V to 0.61 V in the following 600 s, which suggests that TCH acts as an electron donor, offsetting part of the oxidation potential [48]. This result provided direct evidence that an electron transfer process existed within Fe(Ⅲ)-PA/PMS/TCH system and mediated TCH degradation. In situ Raman spectroscopy was employed to further characterize the changes in chemical bonding during the reaction. As displayed in Fig. 3b, with the peak at 1059 cm-1 corresponding to the HSO5- of the PMS and the peak at 980 cm-1 corresponding to the SO42- [49], which can be observed in the PMS system alone. The addition of PMS to the Fe(Ⅲ)-PA surface resulted in the observation of a distinct signal of HSO5-, while the signal at SO42- was weak. This finding suggested that the adsorption of Fe(Ⅲ)-PA on HSO5- was selective, thereby favoring the activation of PMS, which is consistent with the experimental results of OCP. Subsequent to the addition of TCH, the peak of HSO5- disappeared and the peak of SO42- increased, that is, the PMS adsorbed on the surface of Fe(Ⅲ)-PA was directly involved in the degradation of TCH.
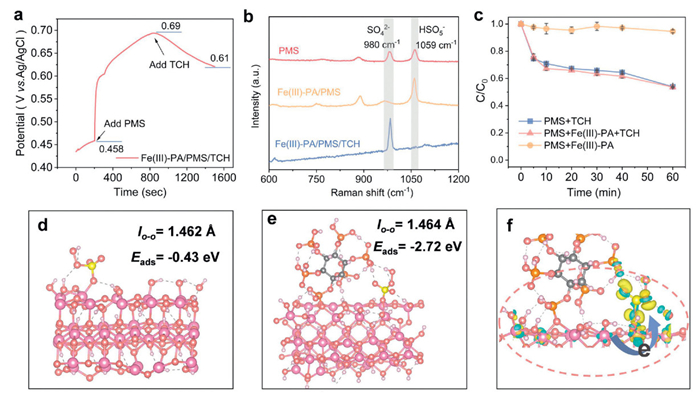
Next, the PMS consumption of the three systems was investigated. In the PMS/TCH system, a PMS consumption of 46.2% was observed over a 60-min period, suggesting that TCH would directly consume PMS as an electron donor (Fig. 3c). This finding is consistent with the observation that TCH degrades when PMS is used alone. In the radical activation process, PMS is consumed by decomposition in the presence of a catalyst, regardless of the presence or absence of contaminants [48]. Moreover, the presence of contaminants could even decelerate the decomposition process of PMS [50]. However, in the Fe(Ⅲ)-PA/PMS system, there was no significant decomposition of PMS when TCH was not added, which is typical of PMS consumption kinetics during ETP process. Furthermore, in the Fe(Ⅲ)-PA/PMS/TCH system, the consumption of PMS did not increase significantly in comparison with the PMS/TCH system, while the degradation efficiency of TCH increased by 42.8% from 93.3%. These phenomena demonstrated that a greater quantity of TCH could be degraded by consuming an equivalent chemical dose of PMS in the presence of Fe(Ⅲ)-PA. That is, the Fe(Ⅲ)-PA-mediated ETP process enhanced the electron selectivity in the degradation of TCH by PMS, thereby increasing the utilization of PMS.
To further elucidate the process by which the phytate ligand regulates the activation of PMS, the coordination mode of PMS on Fe(Ⅲ)-PA was simulated using DFT calculations. Two reaction models were constructed: hydro ferrite (Model Ⅰ: a typical hydrolysis product of iron ions) and hydro ferrite after the adsorption of phytic acid (Model Ⅱ). Based on the optimized structure, it was determined that the phytic acid molecules are coordinately bonded to two Fe atoms on the surface of hydrotalcite. Similarly, PMS undergoes monodentate coordination with Fe atoms on the surface of hydrotalcite. Subsequently, the adsorption energy of PMS on both models was calculated. The adsorption energy of PMS on Model Ⅱ (−2.72 eV) was significantly lower than that of Model Ⅰ (−0.43 eV), suggesting that the adsorption of phytate onto hydrotalcite is more favorable for PMS adsorption. Additionally, the O–O bond length of PMS in Model Ⅱ (1.464 Å) is longer than that in Model Ⅰ (1.462 Å), indicating that the adsorption of phytate to hydrotalcite is more conducive to the activation of PMS (Figs. 3d and e). The results of the differential charge analysis show that electrons are transferred from the catalyst to PMS (Fig. 3f). However, PMS decomposition experiments have demonstrated that the decomposition of PMS is not significant when the catalyst is present alone with PMS. Therefore, in the Fe(Ⅲ)-PA/PMS/TCH system, Fe(Ⅲ)-PA acted more as an electron shuttle to accelerate the electron transfer process between TCH and PMS. In conclusion, the Fe(Ⅲ)-PA/PMS system optimized the two-step activation process of PMS, namely the adsorption of PMS on Fe(Ⅲ)-PA and the direct electron transfer process between TCH and PMS.
The degradation of target pollutants via the ETP pathway is typically selective. Hence, the effect of inorganic ions (Cl-, NO3-, SO42-, HCO3-, H2PO4-, Ca2+ and Mg2+) on the removal of TCH by Fe(Ⅲ)-PA/PMS system was investigated. As shown in Fig. 4a, all of them, with the exception of HCO3-, exhibited varying degrees of inhibition on the degradation effect of TCH. It is noteworthy that the addition of HCO3- enhanced the degradation performance. The similar situation was found in other studies, which could be attributed to the fact that the asymmetric structure of PMS is susceptible to decomposition by HCO3- attack [51]. Natural organic matter (NOMs) in water frequently competed for ROS, thereby affecting the oxidative degradation of the target pollutants. In this study, humic acid (HA) was chosen as NOMs to investigate the effect of Fe(Ⅲ)-PA/PMS system on TCH removal. Satisfactorily, the removal of TCH was still 72.3% in 60 min in the presence of HA. As shown in Fig. S9 (Supporting information), the application of Fe(Ⅲ)-PA in natural water matrix, including tap water and lake water were further evaluated (The lake water used in the experiment was obtained from Tangxun Lake located in Wuhan, Hubei, China). The basic properties of water were displayed in Table S1 in Supporting information). Interestingly, the removal rate of Fe(Ⅲ)-PA/PMS for TCH reached 91.8% and 95.1% in tap water and lake water, respectively. In addition, the reaction rate was slightly faster than that of ultrapure water within 30 min. To recapitulate, coexisting ions and NOMs interfere less with the Fe(Ⅲ)-PA/PMS/TCH system, showing selective removal of target contaminants by the ETP pathway.
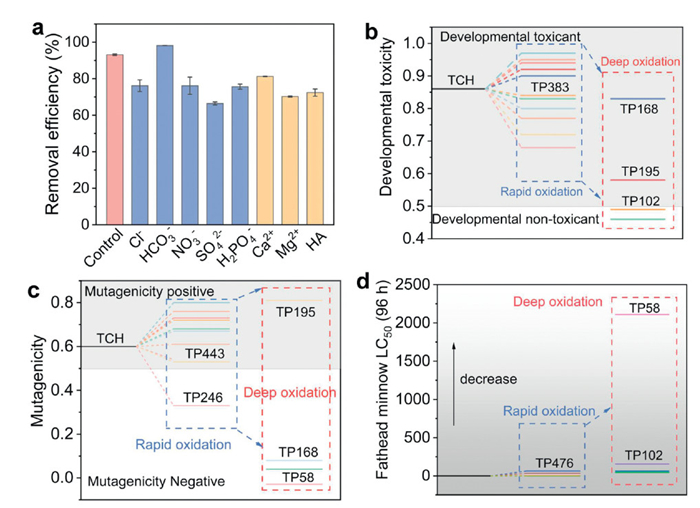
Finally, the transformation products (TPs) of TCH in the Fe(Ⅲ)-PA/PMS/TCH system were detected using ultra-high performance liquid chromatography-mass spectrometry (UPLC-MS), and their three possible transformation pathways were analyzed (Figs. S10 and S11 in Supporting information). In Pathway I, TCH was first hydroxylated to form the intermediate P2 (m/z 476.14). Subsequently, P2 underwent dehydration and deamination to yield P3 (m/z 443.12). In Pathway Ⅱ, P6 (m/z 425.11) was produced through the deamination and hydroxylation of P1, followed by the formation of P9 (m/z 272.06) via a ring-opening reaction. Moreover, in Pathway Ⅲ, the intermediate P10 (m/z 400.12) was generated through demethylation, followed by the formation of P11 (m/z 401.11) via deamidation. Ultimately, these intermediates were further oxidized to yield small molecule products such as P13 (m/z 246.05), P14 (m/z 168.11), P15 (m/z 102.06), and P16 (m/z 58.04). The toxicity of TCH and its TPs, including developmental toxicity, mutagenicity, and blackhead minnow LC50 (96 h), was evaluated by T.E.S.T. As shown in Fig. 4b, the developmental toxicity of TCH underwent a gradual transformation, with an initial value of 0.86 decreasing to non-toxic or low-toxicity products, such as P15 or P16. Regarding mutagenicity, TCH was considered mutagenic; however, the final small molecule products (P14 and P16) exhibited detoxified mutagenicity (Fig. 4c). The LC50 (96 h) value for blackhead minnow from TCH was 0.9 mg/L, with some intermediate products having even lower values (Fig. 4d). Fortunately, with further deep oxidation, the final small-molecule product achieved an LC50 value of 2109.0 mg/L, indicating a significant reduction in biotoxicity.
In this study, we propose a phytate ligand strategy to enhance the Fe(Ⅲ) activation of PMS, focusing on the rate-limiting step in the Fenton-like system. It is demonstrated that the coordination ability of the catalyst with PMS can be enhanced by adsorbing phytate on the surface of ferrihydrite, thus favoring the activation process of PMS on Fe(Ⅲ)-PA surface. In addition, the PMS-Fe(Ⅲ)-PA mediated electron transfer pathway played a key role in the degradation of the target pollutant TCH. ETP as a typical non-radical pathway, is selective for the removal of target pollutants, which was also verified in the Fe(Ⅲ)-PA/PMS/TCH system. Finally, the application in natural water matrix and the toxicity of TCH and its TPs from Fe(Ⅲ)-PA/PMS/TCH system were investigated. This work provides a feasible strategy for ligand-enhanced catalytic activity of Fe(Ⅲ) in Fenton-like chemistry.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Lijuan Huang: Writing – original draft, Visualization, Validation, Investigation, Formal analysis. Rui Gan: Writing – original draft, Methodology, Conceptualization. Xin Han: Validation, Investigation. Meilin Sun: Visualization, Investigation. Li Chen: Investigation, Formal analysis. Wen Liu: Writing – review & editing, Software, Funding acquisition. Xiaoxin Zhang: Investigation. Fei Pan: Writing – review & editing, Supervision, Project administration, Methodology.
This work was financially supported by the National Key Research and Development Program of China (No. 2021YFA1202500), the National Natural Science Foundation of China (No. 52270053), the Beijing Natural Science Foundation (No. 8232035), the Beijing National Laboratory for Molecular Sciences (No. BNLMS2023011), the Science and Technology Project of Beijing Municipal Ecology and Environment (BJST20250207), the Major Science and Technology Projects in Yunnan Province (202502AQ080002), the PKU-NUS Center for Applied Sciences-Joint Research Funding, the State Key Laboratory of Clean Energy Utilization (Open Fund Project No ZJUCEU2023014), the State Key Laboratory of New Textile Materials and Advanced Processing, Wuhan Textile University (No. FZ2025006), and the Graduate Student Innovated Foundation of Wuhan Textile University.
Supplementary material associated with this article can be found, in the online version, at doi:
M. Yang, Z. Hou, X. Zhang, et al., Environ. Sci. Technol. 56 (2022) 11635–11645. doi: 10.1021/acs.est.2c01261
Z. Wu, B. Huang, X. Wang, et al., Environ. Sci. Technol. 57 (2023) 14046–14057. doi: 10.1021/acs.est.3c04343
M. Ao, J. Wei, C.-S. He, et al., Chin. Chem. Lett. 36 (2025) 109882. doi: 10.1016/j.cclet.2024.109882
P. Li, M. Guo, Q. Wang, et al., Appl. Catal. B 259 (2019) 118107. doi: 10.1016/j.apcatb.2019.118107
W.D. Oh, Z. Dong, T.T. Lim, Appl. Catal. B 194 (2016) 169–201. doi: 10.1016/j.apcatb.2016.04.003
H. Li, N. Yuan, J. Qian, B. Pan, Environ. Sci. Technol. 56 (2022) 4498–4506. doi: 10.1021/acs.est.1c08790
Y. Zhuang, S. Spahr, H.V. Lutze, et al., Water Res. 265 (2024) 122267. doi: 10.1016/j.watres.2024.122267
J. Lee, U. von Gunten, J.-H. Kim, Environ. Sci. Technol. 54 (2020) 3064–3081. doi: 10.1021/acs.est.9b07082
Y. Yao, J. Zhang, Y. Wang, et al., Chin. Chem. Lett. 35 (2024) 109633. doi: 10.1016/j.cclet.2024.109633
J. Lu, Q. Lu, L. Di, Y. Zhou, Y. Zhou, Chin. Chem. Lett. 34 (2023) 108357. doi: 10.1016/j.cclet.2023.108357
M.Y. Lan, Y.H. Li, C.C. Wang, et al., Nat. Commun. 15 (2024) 7208. doi: 10.1038/s41467-024-51525-0
Y. Yan, Z. Wei, X. Duan, et al., Environ. Sci. Technol. 57 (2023) 12153–12179. doi: 10.1021/acs.est.3c05153
G.P. Anipsitakis, D.D. Dionysiou, Environ. Sci. Technol. 38 (2004) 3705–3712. doi: 10.1021/es035121o
S. Xiao, M. Cheng, H. Zhong, et al., Chem. Eng. J. 384 (2020) 123265. doi: 10.1016/j.cej.2019.123265
H. Dong, Q. Xu, L. Lian, et al., Environ. Sci. Technol. 55 (2021) 15390–15399. doi: 10.1021/acs.est.1c04563
S. Meng, P. Zhou, Y. Sun, et al., Water Res. 218 (2022) 118412. doi: 10.1016/j.watres.2022.118412
Z. Liu, S. Pan, F. Xu, et al., Water Res. 225 (2022) 119142. doi: 10.1016/j.watres.2022.119142
Z.Y. Li, L. Wang, Y.L. Liu, et al., Water Res. 195 (2021) 116973. doi: 10.1016/j.watres.2021.116973
M. Sun, P. Zhou, S. Meng, et al., Environ. Sci. Technol. 58 (2024) 10817–10827. doi: 10.1021/acs.est.4c00472
Y. Li, H. Dong, J. Xiao, et al., Chem. Eng. J. 455 (2023) 140773. doi: 10.1016/j.cej.2022.140773
H. Zhang, L. Li, N. Chen, et al., Appl. Catal. B 312 (2022) 121410. doi: 10.1016/j.apcatb.2022.121410
X. Wu, X. Gu, S. Lu, et al., Sep. Purif. Technol. 147 (2015) 186–193. doi: 10.1016/j.seppur.2015.04.031
C. Qi, Y. Wen, Y. Zhao, et al., Chin. Chem. Lett. 33 (2022) 2125–2128. doi: 10.1016/j.cclet.2021.10.087
Y. Dai, H. Cao, C. Qi, et al., Chin. Chem. Lett. 451 (2023) 138588.
Y. Dai, S. Yang, L. Wu, et al., J. Hazard. Mater. 465 (2024) 133303. doi: 10.1016/j.jhazmat.2023.133303
D.Q. He, Y.J. Zhang, D.N. Pei, et al., J. Hazard. Mater. 382 (2020) 121090. doi: 10.1016/j.jhazmat.2019.121090
H. Zhou, J. Peng, X. Duan, et al., Environ. Sci. Technol. 57 (2023) 3334–3344. doi: 10.1021/acs.est.2c07447
D. Han, J. Wan, Y. Ma, et al., Chem. Eng. J. 269 (2015) 425–433. doi: 10.1016/j.cej.2015.01.106
H. Dong, Z. Qiang, J. Hu, C. Sans, Chem. Eng. J. 316 (2017) 288–295. doi: 10.1016/j.cej.2017.01.099
A. Rastogi, S.R. Al-Abed, D.D. Dionysiou, Water Res. 43 (2009) 684–694. doi: 10.1016/j.watres.2008.10.045
Z. Yang, Y. Cui, B. Pan, J.J. Pignatello, Environ. Sci. Technol. 57 (2023) 18918–18928. doi: 10.1021/acs.est.3c00777
A. Chen, Y. Li, J. Shang, Y. Arai, Environ. Sci. Technol. 54 (2020) 8837–8847. doi: 10.1021/acs.est.0c02465
R. Gan, Y. Ye, Z. Zhan, et al., J. Hazard. Mater. 466 (2024) 133636. doi: 10.1016/j.jhazmat.2024.133636
X. Duan, H. Sun, J. Kang, et al., ACS Catal. 5 (2015) 4629–4636. doi: 10.1021/acscatal.5b00774
Y. Gao, F. Wang, J. Tang, et al., Chem. Eng. J. 495 (2024) 153651. doi: 10.1016/j.cej.2024.153651
A.M. Puziy, O.I. Poddubnaya, A. Martıńez-Alonso, F. Suárez-Garcıá, J.M.D. Tascón, Carbon 40 (2002) 1493–1505.
Y. Deng, L. Xiao, H. Zhou, et al., Sep. Purif. Technol. 342 (2024) 126976.
F. Yuan, C. Wu, Y. Cai, et al., Chem. Eng. J. 322 (2017) 353–365.
X.B. Ding, L. Zhang, Y.H. Qin, et al., Chem. Commun. 57 (2021) 6935–6938. doi: 10.1039/d1cc02047a
Y. Yang, G. Banerjee, G.W. Brudvig, J.H. Kim, J.J. Pignatello, Environ. Sci. Technol. 52 (2018) 5911–5919. doi: 10.1021/acs.est.8b00735
Y. Zhao, L. Yu, C. Song, et al., Environ. Sci. Technol. 56 (2022) 10710–10720. doi: 10.1021/acs.est.2c01759
E.T. Yun, J.H. Lee, J. Kim, H.D. Park, J. Lee, Environ. Sci. Technol. 52 (2018) 7032–7042. doi: 10.1021/acs.est.8b00959
Y. Zong, L. Chen, Y. Zeng, J. Xu, et al., Environ. Sci. Technol. 57 (2023) 9394–9404. doi: 10.1021/acs.est.3c01553
Z. Wang, W. Qiu, S. Pang, et al., Environ. Sci. Technol. 56 (2022) 1492–1509. doi: 10.1021/acs.est.1c04530
Z. Wang, W. Qiu, S. Pang, et al., Water Res. 172 (2020) 115504.
W. Ren, L. Xiong, G. Nie, et al., Environ. Sci. Technol. 54 (2020) 1267–1275. doi: 10.1021/acs.est.9b06208
W. Ren, L. Xiong, X. Yuan, et al., Environ. Sci. Technol. 53 (2019) 14595–14603. doi: 10.1021/acs.est.9b05475
W. Ren, C. Cheng, P. Shao, et al., Environ. Sci. Technol. 56 (2022) 78–97. doi: 10.1021/acs.est.1c05374
S. Liu, J. Du, H. Wang, et al., Water Res. 254 (2024) 121417.
A. Jawad, K. Zhan, H. Wang, et al., Environ. Sci. Technol. 54 (2020) 2476–2488. doi: 10.1021/acs.est.9b04696
M. Jiang, J. Lu, Y. Ji, D. Kong, Water Res. 116 (2017) 324–331.

Figure 1 (a) SEM images of Fe(Ⅲ)-PA. (b) EDS mapping. (c) FT-IR spectra of Fe(Ⅲ)-PA. (d) XPS of Fe 2p spectrum for Fe(Ⅲ)-PA.

Figure 2 (a) TCH degradation in different systems. (b) kobs values obtained via pseudo-first-order kinetic model fitting. (c) TCH removal by Fe(Ⅲ)-PA/PMS system under different solution pH conditions. (d) TCH Degradation performance in the presence of different quenchers. (e, f) EPR spectra. Experiment conditions: [TCH]0 = 10 mg/L, [PMS]0 = 0.1 mmol/L, [catalyst] = 0.3 g/L, [MeOH] = [TBA] = 100 mmol/L, [p-BQ] = [L-his] = 5 mmol/L, [DMPO] = [TEMP] = 100 mg/L.

Figure 3 (a) OCP curves of Fe(Ⅲ)-PA. (b) Raman spectra. (c) consumption of PMS in the different system, the favorite adsorption configurations of PMS molecule adsorbed on ferrihydrite (d) and Fe(Ⅲ)-PA. (e, f) Charge density (the yellow and blue isosurface represent the accumulation and the depletion of electrons, respectively) in PMS adsorption configurations on Fe(Ⅲ)-PA, the level of the isosurface is set to 0.005 eÅ-3).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: