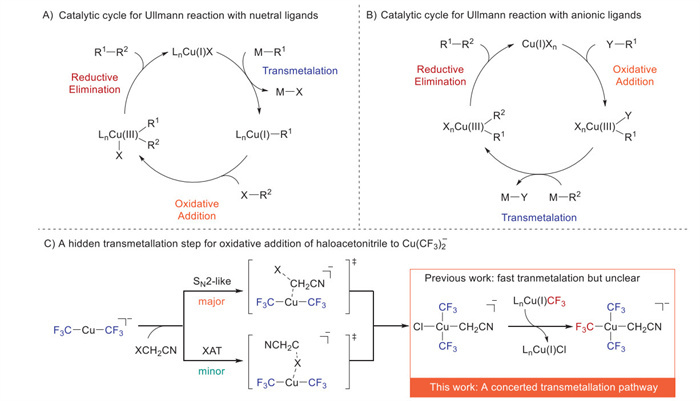
Scheme 1.
Transmetalation in copper-mediated coupling reaction.
Unraveling the mechanism of trifluoromethyl group transfer from Cu(Ⅰ) to Cu(Ⅲ)
Yifa Yang , Sheng-Ye Zhang , Yongrui Luo , Jian Wu , Xuebing Leng , Qilong Shen

Copper, a versatile coinage metal, continues to drive innovations in synthetic methodologies, ranging from classic Ullmann [1-4] and Chan-Evans-Lam [5-7] couplings to modern asymmetric reactions [8-16]. While its unparalleled capacity to forge carbon-carbon and carbon-heteroatom bonds is widely acknowledged [17,18], the mechanistic intricacies of copper-mediated processes persist as subjects of intense debate (Schemes 1A and B) [19-23]. A simplified catalytic cycle comprises three elementary steps: oxidative addition [24], transmetalation [25,26], and reductive elimination [27]. However, despite having various Cu(Ⅲ) complexes known [28-33], details of the transformation of the copper species in the Cu(Ⅰ)/Cu(Ⅲ) redox pair were difficult to trace. Current mechanistic studies have largely focused on oxidative addition [34-40] and reductive elimination [38,41-51], where trifluoromethyl groups act both as coupling partners and stabilizing ligands for high-valent copper species. These studies have systematically mapped the reactivity of trifluoromethyl-copper(Ⅰ/Ⅲ) complexes under various activation strategies. Paradoxically, transmetalation, a pivotal step that delivers two functional groups from different coupling partners to the key Cu(Ⅲ) intermediate for the subsequent product-forming reductive elimination [50,51], has been surprisingly overlooked, despite extensive studies on Pd [52,53], Ni [54-57], and other transition metals [53,58,59].
In 2023, our group disclosed the oxidative addition of haloacetonitrile (XCH2CN; X = Cl, Br, I) to the ionic Cu(Ⅰ) complex Q+[Cu(CF3)2]− (Q = Ph4P) [39], marking a key advancement in understanding high-valent copper reactivity. This transformation proceeded through the generation of a highly reactive Cu(Ⅲ) intermediate, trans-[Cu(CF3)2(X)(CH2CN)]−Q+, which subsequently participated in rapid transmetalation with a second equivalent of Q+[Cu(CF3)2]− to afford the stable, fully substituted Cu(Ⅲ) product Q+[Cu(CF3)3(CH2CN)]−. Kinetic studies suggested that this transmetalation step occurred significantly faster than the initial oxidative addition, implying a low barrier for trifluoromethyl group transfer between copper centers in different oxidation states (Scheme 1C). Despite these advances, the fundamental mechanism of this transmetalation process remains unresolved. In particular, the precise pathway by which the trifluoromethyl group migrates from the anionic Cu(Ⅰ) species [Cu(CF3)2]− to the Cu(Ⅲ) intermediate trans-[Cu(CF3)2(X)(CH2CN)]− has not been elucidated, primarily due to the inability to isolate or spectroscopically characterize the key halogenated intermediate trans-[Cu(CF3)2(X)(CH2CN)]−Q+. Herein, we describe the successful isolation and structural characterization of the pivotal halogenated Cu(Ⅲ) intermediate trans-[Cu(CF3)2(Cl)(CH2CN)]−Q+, enabling direct mechanistic investigation of its transmetalation with Q+[Cu(CF3)2]− (Scheme 1C). These findings provide unprecedented experimental evidence for the proposed Cu(Ⅰ)-to-Cu(Ⅲ) transmetalation pathway, resolving long-standing uncertainties in copper-mediated trifluoromethylation reactions.
Previous studies by Nebra and coworkers demonstrated that neutral, pyridine-ligated Cu(Ⅲ) complexes could be converted to stable ionic species through ligand exchange [50]. Specifically, they observed that [(Py)Cu(CF3)3] reacted with Ph4P+Cl− to form the isolable five-coordinate ionic complex Q+[Cu(CF3)3(Cl)(Py)]−. Subsequent treatment with AgF led to the formation of Q+[Cu(CF3)3(F)]−, demonstrating halide exchange at the Cu(Ⅲ) center. In parallel work, our group showed that [(Py)Cu(CF3)3] reacted with Me4NF in THF/CH3CN to produce Me4N+[Cu(CF3)3(F)]− (64% yield, 10 min, room temperature), with complete pyridine displacement [51]. These precedents established that the pyridine ligand in neutral four-coordinate Cu(Ⅲ)-CF3 complexes could be directly or indirectly replaced by a halogen anion. Building on these insights, we envisioned that synthesizing a related acetonitrile-derived analogue, trans-[(Py)Cu(CF3)2(CH2CN)] 1-Py, might enable access to our target intermediate trans-[Cu(CF3)2(Cl)(CH2CN)]−Q+ through analogous anion metathesis.
Our initial attempt to synthesize 1-Py by direct oxidative addition of ICH2CN to Q+[Cu(CF3)2]− in the presence of pyridine, generating a few unisolable species. Instead, employing 2, 2′-bipyridine as alternative ligand generated trans-[(bpy)Cu(CF3)2(CH2CN)] in 84% yield [39]. In addition, treatment of trans-[(bpy)Cu(CF3)2(CH2CN)] with Ph4P+Cl− in dichloromethane for 1 h at room temperature did not generate the target trans-[Cu(CF3)2(Cl)(CH2CN)]−Q+ 2-Cl. Notably, when trans-[(bpy)Cu(CF3)2(CH2CN)] was treated with 3.0 equiv. of HBF4·Et2O in 1, 2-dichloroethane (DCE), an immediate reaction occurred within 30 s, producing two distinct changes: (1) Formation of a white precipitate identified as bpy·2HBF₄, and (2) complete disappearance of the original resonance at −39.4 ppm in 19F NMR spectroscopy with concurrent appearance of a new signal at −34.6 ppm [60]. We attributed this new signal to the formation of a ligandless dimeric Cu(Ⅲ) intermediate, [Cu(CF3)2(CH2CN)]2 (Scheme 2, Eq. 1). This assignment was unequivocally confirmed when rapid filtration and subsequent addition of 1.0 equiv. of bpy quantitatively regenerated the original signal at −39.4 ppm in 95% yield, demonstrating the reversible nature of this decomplexation/ligation process.
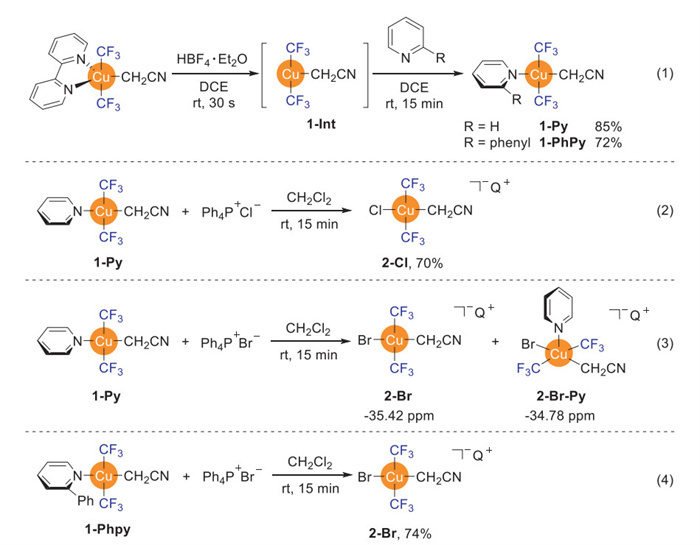
Subsequent addition of 1.0 equiv. of Ph4P+Cl− to the DCE solution did generate the desired ionic Cu(Ⅲ) complex trans-[Cu(CF3)2(Cl)(CH2CN)]−Q+ 2-Cl in 55% yield, as evidenced by the appearance of a new signal −34.5 ppm in the 19F NMR spectroscopy (Fig. S4 in Supporting information for details). Nevertheless, the presence of HBF4 complicated the isolation and purification process. On the other hand, addition of 4.0 equiv. of pyridine or 2-phenylpyridine to the DCE solution, followed by flash column chromatography, afforded target complexes 1-Py (85%) and 1-PhPy (72%), respectively (Scheme 2, Eq. 1). The target ionic Cu(Ⅲ) complexes trans-[Cu(CF3)2(X)(CH2CN)]−Q+ (X = Cl or Br) could be easily synthesized by ligand exchange of the pyridine-derived ligand in 1-Py and 1-PhPy with chloride and bromide salts. Treatment of 1-Py with 1.0 equiv. of Ph4P+Cl− in CH2Cl2 generated trans-[Cu(CF3)2(Cl)(CH2CN)]−Q+ 2-Cl in 70% isolated yield after 15 min at room temperature (Scheme 2, Eq. 2). The same approach using Ph4P+Br−, however, generated a mixture with the desired trans-[Cu(CF3)2(Br)(CH2CN)]−Q+ (2-Br, δ = −35.42 ppm in 19F NMR spectroscopy) and another species with chemical shift at −34.78 ppm in a ratio of 1:0.3, which was tentatively assigned as [Cu(CF3)2(Br)(CH2CN)(Py)]−Q+ (2-Br-Py) (Scheme 2, Eq. 3). Notably, using 1-PhPy instead of 1-Py gave the desired 2-Br in 74% yield (Scheme 2, Eq. 4).
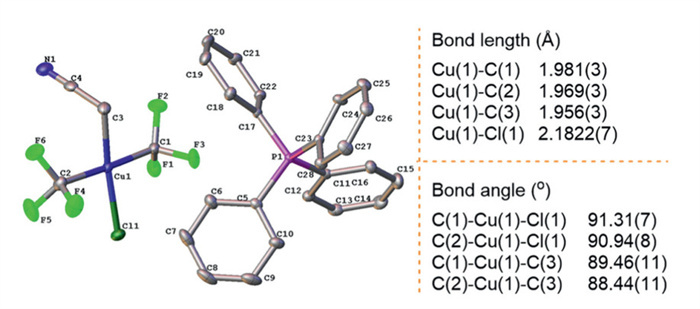
Single crystals of 2-Cl suitable for X-ray diffraction analysis were obtained via vapor diffusion using a CH2Cl2/pentane solvent system (Fig. 1). Likewise, single crystals of 2-Br were obtained by employing the same approach (Fig. S19 in Supporting information for details). Structural determination of 2-Cl revealed a square-planar geometry at the Cu(Ⅲ) center, with C1, C2, C3, and Cl atoms lying approximately coplanar (Σbond angles = 360.15°). The two trifluoromethyl groups occupied trans positions with nearly identical Cu-C bond lengths (1.981(3) Å vs. 1.969(3) Å), consistent with the single peak observed in the 19F NMR spectrum. The Cu-Cl bond length of 2.1822(7) Å confirmed strong coordination of the chloride ligand. Notably, the Cu-CH2CN bond length in 2-Cl (1.956(3) Å) closely matched that in 1-Py (1.954(2) Å), while no oligomerization was observed as 1-Py.
With well-defined complexes 2-Cl and 2-Br in hand, we next investigated their transmetalation with Q+[Cu(CF3)2]−. We first evaluated the influence of solvent on the transmetalation reaction. As summarized in Table 1, it was found that the polarity of the solvent significantly affected both the yields and the rates of the reaction. Full conversion of 2-Cl was observed after 20 min at 25 ℃ when the reaction was conducted in moderately polar solvent THF. However, the transmetalation product Q+[Cu(CF3)3(CH2CN)]− 3 was obtained in only 5% yield, along with the formation of Cu(Ⅰ) species Q+[Cu(CF3)(Cl)]− in 8% yield (Table 1, entry 1). When the reaction was carried out in CH2Cl2, high yielding formation of 3 (91%) was observed after 20 min at 25 ℃, along with the formation of Q+[Cu(CF3)(Cl)]− in 27% yield (Table 1, entry 2). The lower yield of Q+[Cu(CF3)(Cl)]− compared to 3 suggested that Q+[Cu(CF3)(Cl)]− could also transfer the trifluoromethyl group to 2-Cl. Switching to more polar solvent DMF slightly slowed the reaction but generated 3 and Q+[Cu(CF3)(Cl)]− in an approximate 1:1 ratio (41% vs. 45% after 10 min; 81% vs. 87% after 120 min), indicating that in polar solvent, transmetalation of Q+[Cu(CF3)(Cl)]− was much slower than that of Q+[Cu(CF3)2]− (Table 1, entries 3 and 4). Notably, in highly polar solvents such as DMSO and CH3CN, the reaction proceeded extremely rapidly, affording 3 and Q+[Cu(CF3)(Cl)]− in quantitative yields within 1.0 min at room temperature (Table 1, entries 5 and 6).
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | Solvent | Time (min) | Yield (%) | |
| 3 | [ClCuCF3]− | |||
| 1 | THF | 20 | 5 | 8 |
| 2 | CH2Cl2 | 20 | 91 | 27 |
| 3 | DMF | 10 | 41 | 45 |
| 4 | DMF | 120 | 87 | 81 |
| 5 | DMSO | < 1 | > 99 | > 99 |
| 6 | CH3CN | < 1 | > 99 | > 99 |
| a Conditions: Q +[Cu(CF 3) 2] − (0.02 mmol) and 2-Cl (0.02 mmol) in solvent (0.5 mL) at room temperature. | ||||
After determined DMF as the suitable solvent for kinetic study, we investigated the kinetic behavior during transmetalation. Under pseudo-first-order conditions at −20 ℃, we determined that the reaction follows second-order kinetics (first-order with respect to each reactant, Figs. 2A and B, see pages S14-S19 in Supporting information for more details). Furthermore, Eyring analysis revealed activation parameters ΔH‡ = 10.6 ± 0.8 kcal/mol and ΔS‡ = −26.8 ± 3.1 e.u., with an activation Gibbs free energy (ΔG‡) of 17.4 kcal/mol at 253 K (Fig. 2C).
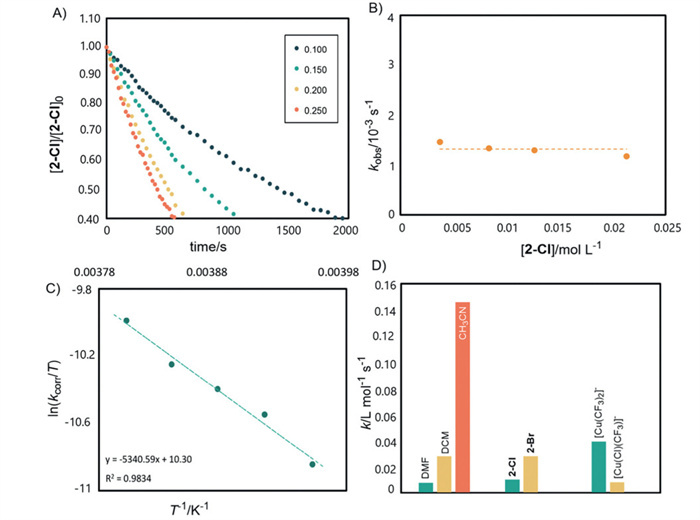
To quantitatively compare reaction rates across different solvents, we performed kinetic studies at −10 ℃, where the slower reaction rates facilitated more accurate measurements (Fig. 2D, the first set of data). Kinetic analysis revealed that reaction in DMF (kDMF = (7.61 ± 0.98) × 10–3 L mol−1 s−1) proceeded approximately 4 times slower than in CH2Cl2 (kCH2Cl2 = (2.78 ± 0.34) × 10–2 L mol−1 s−1) and 20 times slower than in CH3CN (kCH3CN = (1.46 ± 0.01) × 10–1 L mol−1 s−1).
To quantitatively determine the effect of Q+[Cu(CF3)(Cl)]− on the transmetalation process, we developed a synthetic route for the preparation of Q+[Cu(CF3)(Cl)]− by mixing 1.0 equiv. of Q+[Cu(CF3)2]− with Q+[CuCl2]− in CH2Cl2. We then studied the kinetics of the reaction of Q+[Cu(CF3)(Cl)]− with trans-[Cu(CF3)2(Cl)(CH2CN)]−Q+ 2-Cl in CH2Cl2 at −10 ℃ (Fig. 2D, the third set of data). Kinetic analysis revealed that transmetalation Q+[Cu(CF3)(Cl)]− to 2-Cl proceeded roughly 2.5 times slower than that of Q+[Cu(CF3)2]− ((1.01 ± 0.08) × 10–2 L mol−1 s−1 vs. (2.78 ± 0.34) × 10–2 L mol−1 s−1). This difference in reactivity aligns with our experimental observations during kinetic studies.
To evaluate the influence of the halogen ligand on the Cu(Ⅲ) complex, we also studied the kinetics of transmetalation of Q+[Cu(CF3)2]− to trans-[Cu(CF3)2(Br)(CH2CN)]−Q+ 2-Br in DMF (Fig. 2D, the second set of data). It was found that transmetalation with 2-Br (k2-Br ≈ (3.93 ± 0.40) × 10–2 L mol−1 s−1 in DMF at −10 ℃) is roughly 5 times faster than with 2-Cl (k2–Cl = (0.79 ± 0.01) × 10–2 L mol−1 s−1 in DMF at −10 ℃). This enhanced reactivity can be attributed to the stronger leaving group ability of the bromide anion and the weaker Cu-Br bond compared to Cu-Cl (BDE: 15.3 kcal/mol vs. 16.8 kcal/mol), which promotes faster ligand exchange.
Based on the kinetic data and prior mechanistic understanding of transmetalation, several plausible pathways can be proposed. First, the transmetalation could proceed via a concerted metathesis between Q+[Cu(CF3)2]− and 2-Cl without discrete intermediates (pathway A in Scheme 3). Alternatively, the reaction might initiate by dissociation of chloride from 2-Cl to form a neutral three-coordinate intermediate, followed by trifluoromethyl group transfer from Q+[Cu(CF3)2]− either via SN2-tpye or concerted mechanism (pathway B in Scheme 3). A third pathway involves the dissociation of the trifluoromethyl group from Q+[Cu(CF3)2]− to generate a neutral Cu(Ⅰ) species [CuICF3] and CF3⁻, both of which could subsequently transfer the CF3 group to 2-Cl through an SN2-tpye or concerted pathway (pathway C in Scheme 3). Finally, an electron-transfer from Q+[Cu(CF3)2]− to 2-Cl could take place, followed by inner-sphere or outer-sphere halogen-atom transfer (XAT) to trifluoromethyl radical (CF3·) transfer (pathway D in Scheme 3).
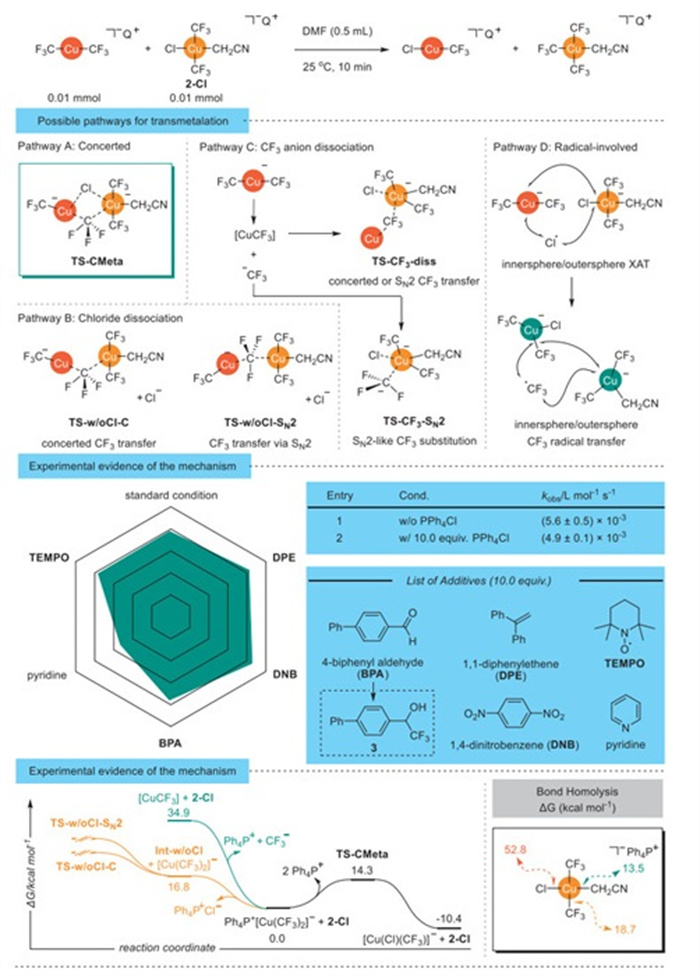
One approach to probe whether a chloride dissociation is proceeded with the addition of chloride to the reaction mixture (Scheme 3, pathway B). If the chloride dissociation occurs before the rate limiting step, the addition of chloride could significantly retard the transmetalation process. Experimentally, addition of 10.0 equiv. of Ph4P+Cl⁻ as a chloride source had neglectable effect on the rate of the transmetalation (kw/Cl, corr = (5.6 ± 0.5) × 10–3 L mol−1 s−1 vs. kw/oCl, corr = (4.9 ± 0.1) × 10–3 L mol−1 s−1). DFT calculation showed that while chloride dissociation from the Cu(Ⅲ) center of 2-Cl is thermodynamically feasible (ΔG‡ = 16.8 kcal/mol), subsequent CF3 transfer did not yield a viable reaction trajectory. Thus, pathway B was ruled out.
To determine whether the reaction proceeds through the dissociation of CF3⁻ from Q+[Cu(CF3)2]−, followed by rate-limiting SN2-like substitution via transition state TS-CF3-SN2, or via a concerted or SN2 transfer from neutral CuCF3 via transition state TS-CF3-diss (Scheme 3, pathway C), we conducted a control experiment using 4-biphenyl aldehyde as a CF3− trap. Given the strong nucleophilicity of CF3−, its reaction with an aldehyde is a fast process. However, the formation of 3 was not detected, ruling out the generation of free CF3− and disfavoring pathway C.
To evaluate whether a radical is involved either through Cu-Cl or Cu-CF3 bond homolytic cleavage or single-electron-transfer (SET) process (Scheme 3, pathway D), several sets of experiments were conducted. If the transmetalation involves a free radical, the reaction should be prohibited by radical scavengers. However, even with 10.0 equiv. of 2, 2, 6, 6-tetramethylpiperidin-1-oxyl (TEMPO) or 1, 1-diphenylethene (DPE), transmetalation yields remained unchanged. Similarly, adding 10.0 equiv. of 1, 4-dinitrobenzene (DNB), a strong electron acceptor, did not alter yields, thereby excluding single-electron transfer mechanisms. Additionally, analysis of the bond homolysis energy revealed that the Cu-CH2CN bond is the weakest (ΔG = 13.5 kcal/mol) among all Cu-C bonds in 2-Cl. However, its dissociation does not lead to a productive transmetalation pathway. Overall, pathway D is disfavored.
Thus, the combined experimental and computational evidences strongly support a concerted metathesis mechanism (pathway A in Scheme 3). This conclusion is further reinforced by density functional theory (DFT) calculations, which identify the concerted pathway as having the lowest activation barrier (ΔG‡ = 14.3 kcal/mol), in agreement with the parameters derived from the Eyring equation.
This study presents the first mechanistic investigation on the transmetalation of the trifluoromethyl group from an ionic Cu(Ⅰ) species Q+[Cu(CF3)2]− to a well-defined halogenated Cu(Ⅲ) trans-[Cu(CF3)2(Cl)(CH2CN)]−Q+ 2-Cl. Kinetic and thermodynamic analyses, supported by DFT calculations, reveal that the transmetalation occurs through a bimolecular pathway involving direct ligand exchange between Cu(Ⅰ) and Cu(Ⅲ) species, featuring by a highly ordered transition state. The observed solvent-dependent reactivity and ligand electronic effects provide key insights for copper catalyst optimization. Future studies should expand to diverse transmetalation systems to develop general principles governing ligand exchange kinetics and selectivity. Additionally, integrating these transmetalation steps with oxidative addition/reductive elimination processes will be crucial for establishing complete catalytic cycles, addressing the current gap in understanding copper-mediated bond-forming sequences, an active focus of our ongoing research.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yifa Yang: Writing – review & editing, Writing – original draft, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Sheng-Ye Zhang: Writing – review & editing, Writing – original draft, Methodology, Investigation, Data curation. Yongrui Luo: Writing – review & editing, Investigation, Formal analysis, Conceptualization. Jian Wu: Writing – review & editing, Investigation. Xuebing Leng: Writing – review & editing, Investigation. Qilong Shen: Writing – review & editing, Writing – original draft, Resources, Project administration, Funding acquisition, Formal analysis, Conceptualization.
We gratefully acknowledge the financial support from the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB0590000) and the National Natural Science Foundation of China (No. 22531010). We acknowledge the Beijing Beilong Super Cloud Computing Co., Ltd (URL:
Supplementary material associated with this article can be found, in the online version, at doi:
S. Perveen, G. Zhang, P. Li, Org. Biomol. Chem. 23 (2025) 4006–4023. doi: 10.1039/d5ob00392j
A.Th. Abdulghaffar, H. Zhang, Q. Zhang, et al., Org. Chem. Front. 12 (2025) 346–367. doi: 10.1039/d4qo01814a
Q. Yang, Y. Zhao, D. Ma, Org. Process Res. Dev. 26 (2022) 1690–1750. doi: 10.1021/acs.oprd.2c00050
I. Beletskaya, A.V. Cheprakov, Coord. Chem. Rev. 248 (2004) 2337–2364. doi: 10.1016/j.ccr.2004.09.014
F. Abedinifar, M. Mahdavi, E.B. Rezaei, M. Asadi, B. Larijani, Curr. Org. Synth. 19 (2022) 16–30. doi: 10.2174/1570179417666210105120706
X. Jia, X. Tong, Chin. J. Org. Chem. 42 (2002) 2640–2658.
A. Vijayan, D.N. Rao, K.V. Radhakrishnan, P.Y.S. Lam, P. Das, Synthesis 53 (2021) 805–847. doi: 10.1055/s-0040-1705971
G. Evano, N. Blanchard, Copper-mediated Cross-coupling Reactions, John Wiley & Sons Itd, Oxford, 2013.
S.R. Chemler, Copper Catalysis in Organic Synthesis, Beilstein-Institut, Frankfurt am Main, 2015.
F. Wang, P. Chen, G. Liu, Acc. Chem. Res. 51 (2018) 2036–2046. doi: 10.1021/acs.accounts.8b00265
A. Hossain, A. Bhattacharyya, O. Reiser, Science 364 (2019) eaav9713. doi: 10.1126/science.aav9713
Z.L. Li, G.C. Fang, Q.S. Gu, X.Y. Liu, Chem. Soc. Rev. 49 (2020) 32–48. doi: 10.1039/c9cs00681h
L.J. Cheng, N.P. Mankad, Chem. Soc. Rev. 49 (2020) 8036–8064. doi: 10.1039/d0cs00316f
Z. Zhang, P. Chen, G. Liu, Chem. Soc. Rev. 51 (2022) 1640–1658. doi: 10.1039/d1cs00727k
M. Soleiman-Beigi, M. Mohammadi, H. Kohzadi, Coord. Chem. Rev. 529 (2025) 216438. doi: 10.1016/j.ccr.2025.216438
M. Kim, G. Kim, D. Kim, J.H. Lee, S.H. Chi, Beilstein J. Org. Chem. 21 (2025) 639–658. doi: 10.3762/bjoc.21.51
F. Monnier, M. Taillefer, Angew. Chem. Int. Ed. 48 (2009) 6954–6971. doi: 10.1002/anie.200804497
S. Bhunia, G.G. Pawar, S.V. Kumar, Y. Jiang, D. Ma, Angew. Chem. Int. Ed. 56 (2017) 16136–16179. doi: 10.1002/anie.201701690
N. Yoshikai, E. Nakamura, Chem. Rev. 112 (2012) 2339–2372. doi: 10.1021/cr200241f
C. Sambiagio, S.P. Marsden, A.J. Blacker, P.C. McGowan, Chem. Soc. Rev. 43 (2014) 3525–3550. doi: 10.1039/C3CS60289C
S.J. Li, Y. Lan, Chem. Commun. 56 (2020) 6609‒6619. doi: 10.1039/d0cc01946a
J.M. Ahn, T. Ratani, K.I. Hannoun, G.C. Fu, J.C. Peters, J. Am. Chem. Soc. 139 (2017) 12717–12723.
H. Lee, J.M. Ahn, P.H. Oyala, et al., J. Am. Chem. Soc. 144 (2022) 4114–4123. doi: 10.1021/jacs.1c13151
J.A. Labinger, Organometallics 34 (2015) 4784–4795. doi: 10.1021/acs.organomet.5b00565
D.V. Partyka, Chem. Rev. 111 (2011) 1529–1595. doi: 10.1021/cr1002276
S.C. Rasmussen, ChemTexts 7 (2021) 1. doi: 10.1093/ije/dyaa221
J.F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, New York, 2010.
H. Liu, Q. Shen, Coord. Chem. Rev. 442 (2021) 213923. doi: 10.1016/j.ccr.2021.213923
M.A.P. Ball, P.J. Myers, G.D. Ritch, et al., Angew. Chem. Int. Ed. 64 (2024) e202420677.
M.S. Reese, M.G. Bonanno, J.K. Bower, C.E. Moore, S. Zhang, J. Am. Chem. Soc. 145 (2023) 26810–26816. doi: 10.1021/jacs.3c09260
J.K. Bower, M. Reese, I.M. Mazina, et al., Chem. Sci. 14 (2023) 1301–1307. doi: 10.1039/d2sc06573h
H. Zhang, C. Feng, N. Chen, S. Zhang, Angew. Chem. Int. Ed. 134 (2022) e202209029. doi: 10.1002/ange.202209029
J.K. Bower, A.D. Cypcar, B. Henriquez, S.C.E. Stieber, S. Zhang, J. Am. Chem. Soc. (2020) 8514–8521. doi: 10.1021/jacs.0c02583
T. Cohen, I. Cristea, J. Am. Chem. Soc. 98 (1976) 748–753. doi: 10.1021/ja00419a018
S.H. Bertz, S. Cope, M. Murphy, C.A. Ogle, B.J. Taylor, J. Am. Chem. Soc. 129 (2007) 7208–7209. doi: 10.1021/ja067533d
E.R. Bartholomew, S.H. Bertz, S. Cope, et al., Chem. Commun. (2008) 1176–1177. doi: 10.1039/b717290g
T. Gärtner, W. Henze, R.M. Gschwind, J. Am. Chem. Soc. 129 (2007) 11362–11363. doi: 10.1021/ja074788y
A. Casitas, A.E. King, T. Parella, et al., Chem. Sci. 1 (2010) 326–330. doi: 10.1039/c0sc00245c
Y. Luo, Y. Li, J. Wu, et al., Science 381 (2023) 1072–1079. doi: 10.1126/science.adg9232
W. Yan, A.T. Poore, L. Yin, et al., J. Am. Chem. Soc. 146 (2024) 15176–15185. doi: 10.1021/jacs.4c01668
L. Liu, M. Zhu, H.T. Yu, W.X. Zhang, Z. Xi, J. Am. Chem. Soc. 139 (2017) 13688–13691. doi: 10.1021/jacs.7b08803
H.M. Bergman, D.D. Beattie, R.C. Handford, et al., J. Am. Chem. Soc. 144 (2022) 9853–9858. doi: 10.1021/jacs.2c02581
X. Tan, Z. Liu, H. Shen, et al., J. Am. Chem. Soc. 139 (2017) 12430–12433. doi: 10.1021/jacs.7b07944
M. Paeth, S.B. Tyndall, L.Y. Chen, et al., J. Am. Chem. Soc. 141 (2019) 3153–3159. doi: 10.1021/jacs.8b12632
W. Yan, S. Carter, C.T. Hsieh, et al., J. Am. Chem. Soc. 145 (2024) 26152–26159.
S. Liu, H. Liu, S. Liu, et al., J. Am. Chem. Soc. 142 (2020) 9785–9791. doi: 10.1021/jacs.0c03304
Y.C. Weng, D. Pan, J. Wu, et al., Chem. Eur. J. (2025) e202403620.
Z.H. Lu, H. Liu, X.B. Leng, Y. Lan, Q. Shen, Angew. Chem. Int. Ed. 58 (2019) 8510–8514. doi: 10.1002/anie.201904041
G. Wang, M. Li, X. Leng, X. Xue, Q. Shen, Chin. J. Chem. 40 (2022) 1924–1930. doi: 10.1002/cjoc.202200230
D. Joven-Sanco, A. Echeverri, N. Saffon-Merceron, J. Contreras-García, N. Nebra, Angew. Chem. Int. Ed. 63 (2024) e202319412. doi: 10.1002/anie.202319412
G. Wang, H. Li, X. Leng, L. Lu, Q. Shen, Chin. J. Chem. 42 (2024) 1107–1113. doi: 10.1002/cjoc.202400041
A.J.J. Lennox, G.C. Lloyd-Jones, Angew. Chem. Int. Ed. 52 (2013) 7362–7370. doi: 10.1002/anie.201301737
K. Osakada, Y. Nishihara, Dalton Trans. 51 (2022) 777–796. doi: 10.1039/d1dt02986j
J. Yang, M.C. Neary, T. Diao, J. Am. Chem. Soc. 146 (2024) 6360–6368. doi: 10.1021/jacs.4c00370
P.-A. Payard, L.A. Perego, I. Ciofini, L. Grimaud, ACS Catal. 8 (2018) 4812–4823. doi: 10.1021/acscatal.8b00933
A.H. Christian, P. Müller, S. Monfette, Organometallics 33 (2014) 2134–2137. doi: 10.1021/om5001327
K.W. Quasdorf, A. Antoft-Finch, P. Liu, et al., J. Am. Chem. Soc. 133 (2011) 6352–6363. doi: 10.1021/ja200398c
K. Koszinowski, Organometallics 43 (2024) 205–218. doi: 10.1021/acs.organomet.3c00415
V.N. Mikhaylov, I.A. Balova, Russ. J. Gen. Chem. 91 (2021) 2194–2248. doi: 10.1134/s1070363221110098
A.M. Romine, N. Nebra, A.I. Konovalov, et al., Angew. Chem. Int. Ed. 54 (2015) 2745–2749. doi: 10.1002/anie.201411348

Figure 1 ORTEP diagram of trans-[Cu(CF3)2(Cl)(CH2CN)]−Q+ 2-Cl. Ellipsoids are shown at the 30% level, and selected bond lengths and bond angles are listed.

Figure 2 (A) Pseudo-first-order kinetics for transmetalation of Q+[Cu(CF3)2]− (0.100-0.250 mmol) to 2-Cl (0.005 mmol). (B) Pseudo-first-order kinetics for transmetalation from Q+[Cu(CF3)2]− (0.100 mmol) to 2-Cl (0.005-0.020 mmol). (C) Effect of temperature. (D) Effect of solvents, halide ligand in 2 and structure of Cu(Ⅰ) complexes.

Scheme 3 Mechanistic hypothesis, investigations, and DFT calculations for transmetalation of Q+[Cu(CF3)2]− to 2-Cl.
Table 1. Transmetalation of CF3 from Q+[Cu(CF3)2]− to 2-Cl in different solvents.a
|
||||
| Entry | Solvent | Time (min) | Yield (%) | |
| 3 | [ClCuCF3]− | |||
| 1 | THF | 20 | 5 | 8 |
| 2 | CH2Cl2 | 20 | 91 | 27 |
| 3 | DMF | 10 | 41 | 45 |
| 4 | DMF | 120 | 87 | 81 |
| 5 | DMSO | < 1 | > 99 | > 99 |
| 6 | CH3CN | < 1 | > 99 | > 99 |
| a Conditions: Q +[Cu(CF 3) 2] − (0.02 mmol) and 2-Cl (0.02 mmol) in solvent (0.5 mL) at room temperature. | ||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: