jll@fzu.edu.cn (L. Jiang). 1 These authors contributed equally to this work.
Received Date:
20 May 2025 Accepted Date:
10 September 2025 Revised Date:
23 August 2025 Available Online:
15 July 2026
Abstract:
The potential use of ammonia (NH3) as a hydrogen energy carrier has generated significant interest in developing efficient catalysts for producing NH3 under mild conditions. The main obstacles for NH3 synthesis are the activation of the N≡N bond and the desorption of NH3 from the catalyst surface. Here, we report the use of C60 to overcome these challenges. Through electron transfer, migration, and feedback between C60 and Ru, there is a balance of electronic density at the Ru active sites and a shift in the d-band center. This simultaneously satisfies the electronic requirements for enhancing N2 activation while weakening NH3 adsorption, thereby circumventing the bottlenecks in NH3 synthesis under mild conditions. Meanwhile, anchoring of C60 accelerates hydrogen spillover and enhances the exchangeability of hydrogen species in the ZrH2 support, as well as expose a greater number of B5 sites of Ru entities, resulting in the co-optimization of hydrogen migration and nitrogen activation. As a consequence, the NH3 synthesis rate of the C60-Ru/ZrH2 catalyst is approximately twice that of the Ru/ZrH2 catalyst at 400 ℃ and 1 MPa. This study shows that doping C60 represents a fundamentally different approach compared to traditional promoters for catalytic NH3 synthesis. We anticipate that this strategy may be generalized to generate widespread interest in the catalysis of NH3 synthesis.
Ammonia (NH3) is a major nitrogen source for the production of fertilizers and other nitrogen-containing chemicals [1,2]. Furthermore, it is also increasingly recognized as an alternative hydrogen energy vector [3-5]. The current industrial NH3 production heavily relies on the well-established Haber-Bosch (H-B) process using iron-based catalyst, which requires high temperatures and pressures (400−500 ℃, 10−30 MPa) [6-8]. Additionally, the supply of hydrogen (H2) primarily comes from the steam reforming of natural gas or coal accompanying by high energy consumption, accounting for 1%−2% of the global energy supply and at least 500 million tons of greenhouse gas emissions per year [1,9,10]. Therefore, under the background of energy conservation and carbon neutrality, it is essential to develop an energy-saving and environmentally friendly NH3 synthesis process [11]. Currently, the technology for producing H2via water electrolysis using renewable energy is becoming increasingly mature. Coupling water electrolysis with the H-B process for NH3 synthesis can significantly reduce energy consumption and carbon emissions [12,13]. To achieve this, the development advanced catalysts that can produce NH3 efficiently under mild conditions is crucial for NH3 synthesis.
It is well known that the NH3 synthesis reaction consists of several key steps, such as the activation of the reaction molecules N2 and H2, the formation of the intermediate NHx species and the desorption of NH3 [14]. Among them, the dissociation of N2 is generally considered to be the rate-determining step (RDS) for NH3 synthesis. Therefore, extensive efforts have been dedicated to the development of novel catalysts aimed at investigating N2 activation pathways to reduce the reaction energy barrier. In recent years, various types of support materials, including electride materials [15], nitrides [16-18], hydrides [19,20], perovskite oxynitride-hydrides [21], and intermetallic compounds [22], supported transition metal catalysts have been reported to exhibit excellent catalytic activities for NH3 synthesis under mild conditions. Compared to oxide supports, these hydrides and nitrides provide advantages in NH3 synthesis due to their anion vacancies, which facilitate the activation or reaction with N2. For example, Zhou et al. [23] investigated the impact of metal-anion interactions on NH3 synthesis performance in different Zr materials (i.e., ZrH2, ZrN, and ZrO2)-supported Ru catalysts. The results indicated that the moderate Ru-H interaction and easy hydrogen spillover in ZrH2, which facilitates the activation of N2 at the Ru sites. In addition to modifying the support composition, promoters also play a crucial role in enhancing the catalytic activity of metal catalysts. In the absence of electronic promoters, even industrially utilized Ru or Fe-based catalysts exhibit negligible catalytic performance [24,25]. At present, the promoters that are extensively studied in NH3 synthesis primarily include alkali metals, alkaline earth metals, and rare earth elements [26]. These element promoters facilitate unidirectional electron transfer to the surfaces of active metals, thereby increasing their electron density and enhancing catalytic performance. Unlike elemental promoters, fullerene C60 has been reported to function as an "electron buffer" in metal-based catalysts, which can dynamically store and release electrons, thereby helping to balance the electron density of active metal species [27,28]. Additionally, the C60 possesses a unique geometric structure and the ability to reversibly store and release hydrogen, which leads to the development of C60-modified, oxide-supported Ru-based catalysts that mitigate the effects of hydrogen poisoning [28]. Anchoring a controllable C60 layer on Mo2CTx as a second H2 adsorption site enhances their electronic interaction, substantially improving NH3 synthesis performance [29]. These unique properties enable C60 to overcome the limitations of traditional element promoters and therefore exhibit potential for NH3 synthesis under mild conditions.
Herein, C60-modified Ru/ZrH2 catalysts were prepared by a straightforward solvent-free ball milling method. Experimental characterizations demonstrate that the introduction of C60 decreases the size and increases the dispersion of Ru species, leading to the formation of more B5 active sites, which is beneficial to the activation of N2. Meanwhile, the interaction between Ru and C60 results in a significant decrease in the work function and an increase in the electron density of Ru atoms, as well as induces a shift in the d-band center. This promotes the activation of N2 and the desorption of produced NH3. Moreover, the presence of C60 promotes the migration of hydrogen atoms and accelerates the exchange of hydrogen species in ZrH2 with gaseous H2. Therefore, the C60-modified Ru/ZrH2 catalyst shows a higher NH3 synthesis rate than free-C60 sample. Our findings could stimulate broad interest in using C60 for NH3 synthesis under mild conditions.
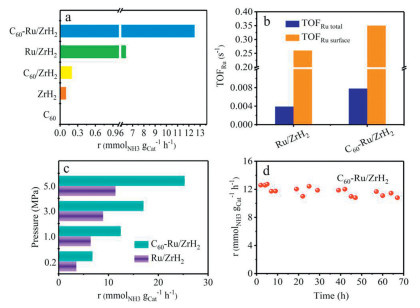
Initially, pristine C60 was investigated for NH3 synthesis, and the corresponding result is depicted in Fig. 1a. At 400 ℃ and 1 MPa, pristine C60 exhibits inactivity, resulting in a negligible rate of NH3 synthesis. Meanwhile, the NH3 synthesis rate of pure ZrH2 is only 0.1 mmolNH3 gcat-1 h-1. Anchoring Ru onto ZrH2 support (Table S1 in Supporting information) increases the NH3 synthesis rate to 6.4 mmolNH3 gcat-1 h-1. Further anchoring C60 onto Ru/ZrH2 results in an NH3 synthesis rate of 12.5 mmolNH3 gcat-1 h-1, which is about ~2 times as high as that of the Ru/ZrH2 catalyst (Fig. 1a). Moreover, a volcano-type dependence between N2/H2 vol ratio and NH3 synthesis rate over C60-Ru/ZrH2 is observed, exhibiting the maximum of 15.6 mmolNH3 gcat-1 h-1 at ~1/1 (Fig. S1 in Supporting information). To evaluate the intrinsic activity of Ru/ZrH2 and C60-Ru/ZrH2 catalysts, the total turnover frequency (TOFRu total) and the surface turnover frequency (TOFRu surface) were calculated based on the total and surface Ru atom counts, respectively. Remarkably, the TOFRu total and TOFRu surface of C60-Ru/ZrH2 reach values as high as 7.80 × 10–3 and 0.35 s-1 at 400 ℃ and 1 MPa (Fig. 1b), respectively. These values are significantly higher than those of Ru/ZrH2, highlighting the crucial role of C60 in enhancing intrinsic activity.
Figure 1
Figure 1.
(a) NH3 synthesis rate and (b) TOFRu of Ru/ZrH2 catalysts with or without C60 promotion. (c) Dependence of NH3 synthesis rate on the reaction pressure at 400 ℃ for Ru/ZrH2 catalysts with or without C60 promotion. (d) Catalytic stability of C60-mediated Ru/ZrH2 catalyst (conditions: N2: H2 = 1:3; 400 ℃ and 1 MPa).
Fig. 1c displays the variation in the NH3 synthesis rate as a function of reaction pressure for both Ru/ZrH2 and C60-Ru/ZrH2 at 400 ℃. Clearly, the NH3 synthesis rate over Ru/ZrH2 and C60-Ru/ZrH2 exhibits an approximately linear increase with pressure rises from 0.2 MPa to 5.0 MPa, implying that utilizing ZrH2 hydride as a support can mitigate the hydrogen poisoning effect caused by strong hydrogen absorption on Ru-based catalysts. Stability is a crucial factor for evaluating the catalytic activity of the C60-promoted Ru/ZrH2 catalyst in NH3 synthesis. No significant deactivation of the NH3 synthesis rate was observed during continuous operation for 67 h at 400 ℃ and 1 MPa (Fig. 1d), confirming the long-term stability of the C60-Ru/ZrH2 catalyst. Moreover, the concentration of methane discerned in the exhaust is negligible and quite low (Fig. S2 in Supporting information), indicating that C60 is stable under the adopted conditions for NH3 synthesis. Additionally, high-performance liquid chromatography-atmospheric pressure chemical ionization-mass spectrometry (HPLC-APCI-MS) analysis of the used C60-Ru/ZrH2 catalyst shows a sharp peak located at ~720 m/z (Fig. S3 in Supporting information), which is attributed to the C60 molecule [30]. Notably, most of the C60 in the used C60-Ru/ZrH2 sample can be recovered after the reaction through the condensation reflux of the toluene solution. Subsequent XRD analysis further confirms that the structure of the recycled C60 is similar to that of the fresh sample (Fig. S4 in Supporting information). All these observations indicate that the C60-modified Ru/ZrH2 sample is highly active and stable under the adopted NH3 synthesis conditions.
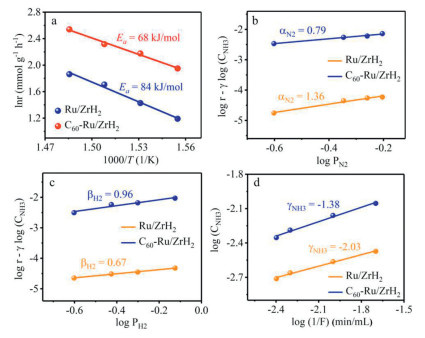
To gain the mechanism information for NH3 synthesis, kinetic analyses were conducted over Ru/ZrH2 and C60-Ru/ZrH2. The apparent activation energy (Ea) of the Ru/ZrH2 catalyst is estimated to be 84 kJ/mol (Fig. 2a), which decreases to 68 kJ/mol after the addition of C60. Such low Ea value for the C60-Ru/ZrH2 catalyst is significantly lower than that of conventional Ru-based catalysts (90–120 kJ/mol, Table S2 in Supporting information). These observations reveal that the introduction of C60 could decrease the reaction barrier for NH3 synthesis. The N2 reaction order (α) of the C60-Ru/ZrH2 catalyst is 0.79 (Fig. 2b), which is remarkably lower than that of the Ru/ZrH2 (1.36), proving that the addition of C60 can promote the adsorption and activation of N2 [31]. The distinct differences in the α value observed between the Ru/ZrH2 and C60-Ru/ZrH2 catalysts can be ascribed to the variation of the size and electronic structure of the Ru entities after C60 doping (vide infra). Interestingly, both Ru/ZrH2 and C60-Ru/ZrH2 catalysts exhibit a positive reaction order with respect to H2 (Fig. 2c). This observation suggests that the ZrH2 support effectively avoids hydrogen poisoning effect on Ru catalysts [32], which are consistent with the finding that the NH3 synthesis rate increases monotonically with reaction pressure (Fig. 1c). The absence of hydrogen poisoning effect may result from ZrH2 is a strong electron-donating support material that has reversible hydrogen storage-and-release ability [33,34].
Figure 2
Figure 2.
(a) Arrhenius plot of Ru/ZrH2 and C60-Ru/ZrH2 at 1 MPa. Reaction orders of (b) N2, (c) H2, and (d) NH3 over the Ru/ZrH2 and C60-Ru/ZrH2 at 400 ℃ and 1 MPa.
Moreover, the reaction order of NH3 over Ru/ZrH2 is −2.03 (Fig. 2d), implying that the adsorbed NHx species are too strong to desorb from the catalyst surface at low temperature, resulting in the requirement for high temperature to desorb the adsorbed NH3 [16,35]. The addition of C60 to the Ru/ZrH2 catalyst results in the increase of the NH3 reaction order to −1.38, indicating that C60 induces weak adsorption of NH3 on the catalyst surface, possibly due to its electronic effect (vide infra). Overall, these kinetic analyses indicate that the introduction of C60 could simultaneously enhance the activation of N2 and weaken the adsorption of the product NH3 on the catalyst surface.
To elucidate the impact of C60 on the chemical state over the Ru/ZrH2, the physicochemical properties of Ru/ZrH2 and C60-Ru/ZrH2 were investigated. X-ray diffraction (XRD) was used to analyze the crystal structure of the prepared catalysts. As shown in Fig. S5a (Supporting information), the XRD patterns of the Ru/ZrH2 and C60-Ru/ZrH2 mainly show the characteristic diffraction peak of ZrH2 (JCPDS No. 073–2076). Two additional diffraction peaks at 38.38° and 44.02° for the Ru/ZrH2 catalyst are attributed to the hexagonal Ru (100) and (101) crystal planes (JCPDS No. 089–4903) (Fig. S5b in Supporting information). However, the diffraction peaks related to Ru species disappear after the doping of C60, indicating that the addition of C60 decreases the size and promotes the dispersion of Ru species. Moreover, no diffraction peaks related to C60 are observed, primarily due to the high dispersion of C60 on the support or because the size of the C60 layer falls below the detection limit of the XRD technique. Besides, the 13C-labeled solid-state nuclear magnetic resonance (ssNMR) spectrum of the C60-Ru/ZrH2 shows a peak at 143.8 ppm, corresponding to the characteristics of underivatized C60 with Ih symmetry [36,37], indicating that C60 has been successfully loaded onto the catalyst (Fig. S6 in Supporting information).
Raman spectrum of pure C60 shows three distinct bands at 267, 493 and 1457 cm-1, which are attributed to the Ag(1), Hg(1) and Ag(2) vibrational modes of the fullerene C60 [38], respectively (Fig. S7a in Supporting information). All of the Raman bands of C60 are also observed in the case of C60-Ru/ZrH2, proving that C60 has been successfully incorporated into the Ru/ZrH2 catalyst. Notably, the position of the Ag(2) peak is an important index of the charge transfer between C60 and transition metals [39,40]. Compared to pure C60 (1457 cm-1), the Ag(2) peak of C60-Ru/ZrH2 shifts to 1462 cm-1 (Fig. S7b in Supporting information), implying that the existence of a strong electronic interaction between Ru atom and C60 clusters. The specific surface area of Ru/ZrH2 is 5.8 m2/g, which increases to 33.8 m2/g for C60-Ru/ZrH2 (Table S1 in Supporting information). The increase of the specific surface area can be attributed to the addition of C60, which improves the pore structure of the catalyst.
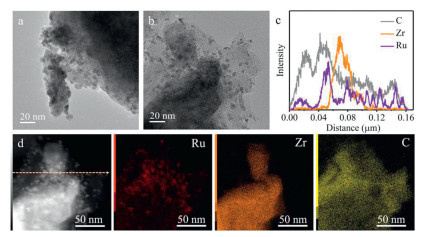
Transmission electron microscopy (TEM, Fig. 3a) and high-angle annular dark-field scanning-TEM (HAADF-STEM, Figs. S8a-c in Supporting information) images of the Ru/ZrH2 catalyst reveal the presence of large agglomerated Ru nanoparticles, with an average particle size of ~6.5 nm on the ZrH2 support. Interestingly, the addition of C60 leads to a decrease in the size of Ru, resulting in a uniform size of approximately 3.6 nm (Fig. 3b and Figs. S8d-f in Supporting information). This observation further indicates that the introduction of C60 facilitates the dispersion of the Ru nanoparticles, as confirmed by the results of the CO pulse experiment (Table S1). The TEM images of Ru/ZrH2 and C60-Ru/ZrH2 primarily display two distinct lattice fringes with spacings of 0.21 and 0.28 nm (Figs. S9 and S10 in Supporting information), corresponding to the (101) crystal plane of metallic Ru and (101) crystal plane of ZrH2 [41,42], respectively. Notably, the line scan profiles of C60-Ru/ZrH2 display that C60 layers are wrapped around the periphery of the Ru/ZrH2 (Fig. 3c). Such continuous carbon shells could accelerate the charge transport [43]. Besides, the energy-dispersive X-ray spectroscopy (EDX) mappings of C60-Ru/ZrH2 reveal the relatively homogeneous dispersion of Ru, Zr, and C elements throughout the catalyst (Fig. 3d).
Figure 3
Figure 3.
TEM images and the corresponding Ru particle size distributions of (a) Ru/ZrH2 and (b) C60-Ru/ZrH2. (c) Line scan profiles of C60-Ru/ZrH2 corresponding to the position in picture d. (d) HAADF−STEM image of C60-Ru/ZrH2 and related EDX mappings of Ru, Zr and C elements.
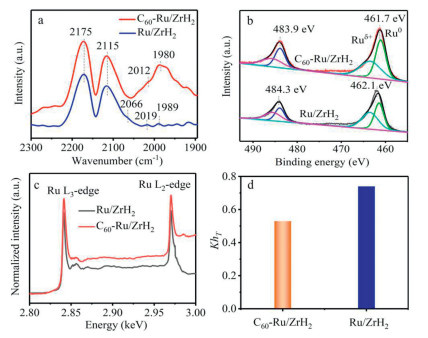
Previous studies have shown that the amount of exposed surface-active sites, such as B5 sites, gradually increases as the size of Ru particles decreases [44-46]. Compared to Ru/ZrH2, C60-Ru/ZrH2 exhibits a smaller Ru particle size, suggesting that more active sites are available for N2 activation. To investigate the effect of C60 on Ru adsorption sites, in situ diffuse reflectance infrared Fourier transform spectroscopic (DRIFTS) measurement using CO as a probe over Ru/ZrH2 and C60-Ru/ZrH2 was performed. As shown in Fig. 4a, in addition to the two gaseous CO adsorption peaks at 2175 and 2115 cm-1, a shoulder peak at 2066 cm-1 observed over Ru/ZrH2 can be attributed to multicarbonyl species adsorbed on oxidized Ru sites ([Ruδ⁺-(CO)x]) [45,46]. Two peaks located at 2019 and 1989 cm-1 are attributed to the mono-adsorption of CO at the step site [12] and the bonding of CO to the Ru bridge site at the Ru-support interface [47,48], respectively. Interestingly, the intensity of the absorption peaks for C60-Ru/ZrH2 is significantly higher than that of Ru/ZrH2. This observation indicates that the addition of C60 results in a significant increase in the number of exposed step and interface Ru sites, which can be attributed to the reduction in Ru particle size after C60 doping. Compared to Ru/ZrH2, the disappearance of the peak at 2066 cm-1 and the slight redshift of the other two peaks (2012 and 1980 cm-1) in the C60-Ru/ZrH2 catalyst indicate that the addition of C60 is favourable for stabilizing low-valence state Ru0 species [49].
Figure 4
Figure 4.
(a) In situ DRIFTS spectra of CO adsorption at 50 ℃. (b) Ru 3p XPS and (c) Ru L-edge NEXAFS spectra of Ru/ZrH2 and C60-Ru/ZrH2 catalysts. (d) KhT calculation results from the NEXAFS spectra.
Moreover, the chemical states of the Ru species on Ru/ZrH2 and C60-Ru/ZrH2 were investigated using X-ray photoelectron spectroscopy (XPS). The Ru 3p XPS spectrum of Ru/ZrH2 shows two peaks at ~462.1 and 484.3 eV, corresponding to Ru 3p3/2 and Ru 3p1/2, respectively (Fig. 4b) [50]. Clearly, the Ru 3p spectrum of C60-Ru/ZrH2 is negatively shifted by 0.4 eV compared to that of Ru/ZrH2, indicating that the Ru species in C60-Ru/ZrH2 exhibit a state of electron enrichment. Furthermore, the Ru 3p XPS spectra of these two catalysts can be further deconvoluted into multiple peaks, which can be assigned to Ru0 and Ruδ+ [51]. Notably, the ratio of Ru0/(Ru0+Ruδ+) for C60-Ru/ZrH2 is 52.4%, which is higher than the ratio for Ru/ZrH2 at 46.7%. This indicates that the addition of C60 leads to the formation of a greater number of metallic Ru sites.
To further identify the electronic structure of the Ru sites, synchrotron radiation-based near-edge X-ray absorption fine structure spectroscopy (NEXAFS) was conducted. The white lines observed at the Ru L3- and L2-edges in the NEXAFS primarily reflect electron transitions from the Ru core 2p3/2 and 2p1/2 to the unoccupied 4d5/2 and 4d3/2 orbitals [52,53], respectively. Therefore, the intensity of the white line can be utilized as a metric for assessing the empty state density of the Ru d orbitals or the quantity of 4d holes [54]. As shown in Fig. 4c, the C60-promoted Ru/ZrH2 catalyst exhibits a significantly lower white-line intensity compared to that of Ru/ZrH2, indicating that more occupied states are formed in the Ru d orbitals after C60 doping. This phenomenon arises from the strong electronic coupling interaction between the C60 and Ru atoms, resulting in the formation of fewer 4d holes, which is consistent with the XPS results. The extent of electron transfer is influenced by the number of unoccupied d orbitals (KhT) at the Ru sites, which can be semi-quantitatively assessed using the Ru L-edge NEXAFS spectra [54]. The KhT value for C60-Ru/ZrH2 (0.53) is obviously lower than that of Ru/ZrH2 (0.74) (Fig. 4d and Table S3 in Supporting information). This indicates that the introduction of C60 induces a strong interaction between the C60π-orbitals and the Ru 4d orbitals, resulting in a substantial decrease in the amount of unpaired Ru 4d charge.
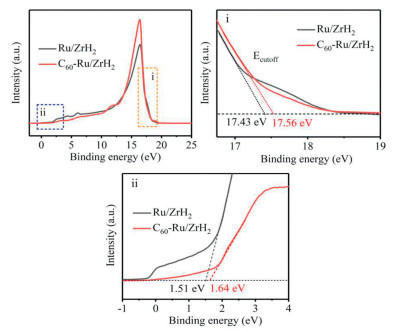
To further investigate the impact of C60 addition on the electronic structure of these catalysts, ultraviolet photoelectron spectroscopy (UPS) measurements were conducted (Fig. 5). The work function (ΦWF), determined from the cutoff of the secondary electron emission (magnified image i), is 3.79 eV for Ru/ZrH2 and 3.66 eV for C60-Ru/ZrH2, respectively. The doping of C60 decreases the work function, enabling that the trapped electrons at the C60-Ru/ZrH2 interface are loosely bound. This results in a decreased barrier for electron donation from the Ru sites to the anti-bonding π orbitals of the adsorbed N2 [55,56]. Consequently, this weakens the N≡N bond and promotes the activation of N2. Moreover, upon the addition of C60, the d-band center (εd, as shown in the magnified image ii) of the Ru/ZrH2 catalyst shifts downward from −1.51 eV to −1.64 eV with respect to the Fermi energy. This downshift in εd for the C60-Ru/ZrH2 indicates a decrease in the antibonding energy state, which results in a weakened interaction between the adsorbed intermediate species NHx (where x = 0–3) and Ru metal. This weakening is beneficial for the NHx desorption process, as evidenced by the significantly higher positive NH3 reaction order observed with C60-Ru/ZrH2 compared to Ru/ZrH2. These findings suggest that the introduction of C60 plays a crucial role in reducing the work function and lowering the d-band center of the Ru/ZrH2 catalyst. This adjustment simultaneously meets the electronic requirements for promoting N2 activation and facilitating NHx desorption. This effect may be attributed to the function of C60 as an electron buffer, which helps balance the electron density of the active metal [27,28].
Figure 5
Figure 5.
UPS spectra and the magnified images of corresponding positions ⅰ and ⅱ over Ru/ZrH2 and C60-Ru/ZrH2 catalysts.
Previous studies have shown that negative hydrogen (H-) ions in hydrides are reactive during NH3 synthesis process [57-59]. To determine whether H- ions in ZrH2 can participate in the formation of NH3, we exposed the C60-Ru/ZrH2 catalyst to pure N2 to detect the NH3 yield at 400 ℃ and 1 MPa. With the progress of the reaction time, the accumulated amount of NH3 at the outlet gradually increased and approached a steady state after 12 h (Fig. S11 in Supporting information), indicating that the H- ions in ZrH2 can combine with the N species to form NH3. To further investigate the reactivity of H species in C60-Ru/ZrH2, an isothermal surface reaction experiment was performed at 400 ℃. As shown in Fig. S12 (Supporting information), the NH3 signal gradually decreases with the introduction of 5%N2/Ar gas, indicating the depletion of H species and the subsequent formation of H vacancies. When the gas was switched to 10%H2/Ar, the NH3 signal was largely restored, indicating that the H species in ZrH2 can be replenished and react with the adsorbed N species to form NH3. A similar isothermal surface reaction experiment was performed with isotopic D2 to further verify that H species in the catalyst can be supplemented (Fig. S13 in Supporting information). When D2 gas was introduced, signals for ND3 (m/z = 20) and ND2H (m/z = 19) were detected, revealing that the D species in the replenished C60-Ru/ZrH2 originated from gas-phase D2 [60]. Additionally, a stronger HD (m/z = 3) signal is observed from the C60-modified Ru/ZrH2 catalyst compared to that from the Ru/ZrH2 (Fig. S14 in Supporting information). This observation demonstrates that the presence of C60 enhances the activation of H2 and facilitates the exchange of lattice H species with gaseous H2. H2-temperature programmed desorption (H2-TPD) curve (Fig. S15 in Supporting information) for Ru/ZrH2 shows two distinct H2 desorption peaks: one in the range of 420–600 ℃ (α) and another in the range of 600–800 ℃ (β), corresponding to the desorption of H2 from the Ru surface or subsurface sites and the ZrH2 support, respectively. Notably, the introduction of C60 into Ru/ZrH2 increases the intensity of the two H2 desorption peaks and shifts these peaks to lower temperature (α peak), indicating that the presence of C60 enhances the spillover effect of hydrogen species while weakening the adsorption strength at the subsurface sites. Moreover, the presence of C60 accelerates the migration and spillover of hydrogen species, as further demonstrated by the WO3 color change experiment [28,61]. As shown in Fig. S16 (Supporting information), pure WO3 does not exhibit significant color changes. However, a more pronounced color change is observed in the C60-modified Ru/ZrH2 compared to the unmodified Ru/ZrH2 during hydrogen treatment. This indicates that the addition of C60 enhances the migration of adsorbed hydrogen species. Moreover, the addition of C60 significantly increases the amount of NH3 desorption compared to the C60-free Ru sample (Fig. S17 in Supporting information). This suggests that the presence of C60 facilitates the formation of more active sites with weaker NH3 adsorption, which is consistent with the remarkable increase in the NH3 reaction order after C60 doping (Fig. 2d).
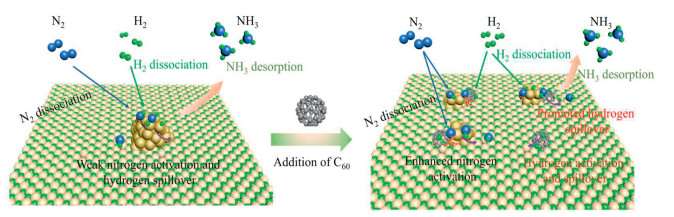
To elucidate the reaction pathway for NH3 synthesis, we conducted isotope reaction experiments using a C60-Ru/ZrH2 catalyst with a feedstock gas mixture of 15N2–14N2H2 at a temperature of 400 ℃. If the dissociation of N2 is the rate-determining step (RDS), then the dissociated nitrogen (N) atoms can immediately react with hydrogen species to form NH3. Conversely, if the formation of N—Hx is the RDS, the dissociated N atoms may accumulate and exchange to form 14N15N species [21,42]. As shown in Fig. S18 (Supporting information), a distinct signal for 14N15N (m/z = 29) is observed in the C60-Ru/ZrH2 catalyst compare to that of Ru/ZrH2, which proves the shift of RDS from the dissociation of N2 to the formation of the N—Hx bond. This may be attributed to the lower activation energy (Fig. 2a) and the smaller reaction order for N2 (Fig. 2b) associated with the C60 modification. As a result, the C60-Ru/ZrH2 catalyst can efficiently activate N2, leading to outstanding NH3 synthesis performance (Scheme 1).
Scheme 1
Scheme 1.
Reaction pathways of Ru-based catalysts with or without C60 modification for NH3 synthesis.
To summarize, we developed a C60-modified Ru/ZrH2 catalyst that serves as an efficient catalyst for NH3 synthesis at mild conditions. Our results demonstrate that the strong interaction between the C60 and Ru entities not only regulates the charge density of Ru atoms but also promotes the dispersion of Ru species to expose more step sites. Meanwhile, the presence of C60 facilitates the exchange of lattice H species in ZrH2 with gaseous H2 and accelerates the hydrogen spillover from Ru metal to the catalyst surface. Therefore, the NH3 synthesis rate of the C60-modified Ru/ZrH2 catalyst is approximately 2 times as high as that of the C60-free Ru sample at 400 ℃ and 1 MPa. This enhancement is attributed to the co-optimization of nitrogen activation and hydrogen migration resulting from the addition of C60. Moreover, the incorporation of C60 adjusts the d-band center, facilitating the desorption of NH3 from the catalyst surface and thereby freeing up additional active sites for the continuous activation of N2. These findings broaden the range of promoters available for the NH3 synthesis reaction.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The work was supported by the National Key Research and Development Program (No. 2022YFA1604101), and National Natural Science Foundation of China (Nos. 22222801, 22221005, 92361303, 22038002, 22478075, 22472028, 22108037), and the Key R & D plan of Shanghai Science and Technology Commission (No. 21DZ1209002), and the China Postdoctoral Science Foundation (No. 2025T180317).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111820.
Y. Baik, M. Kwen, K. Lee, et al., J. Am. Chem. Soc. 145 (2023) 11364–11374. doi: 10.1021/jacs.3c02529
[33]
O.S. Morozova, Global Atmospheric Change and Its Impact on Regional air Quality, in: I. Barnes (Ed.), Hydrogen technology: H2 Storage and Release, Springer, 2002, pp. 303–308.
[34]
Y.J. Choi, Y. Xu, W.J. Shaw, et al., J. Phys. Chem. C 116 (2012) 8349–8358. doi: 10.1021/jp210460w
[35]
B. Fang, C. Zhang, Z. Qi, et al., AIChE J. 68 (2022) e17849. doi: 10.1002/aic.17849
[36]
R.D. Johnson, G. Meijer, D.S. Bethune, J. Am. Chem. Soc. 112 (1990) 8983–8984. doi: 10.1021/ja00180a055
[37]
J.D. Fortner, D.I. Kim, A.M. Boyd, et al., Environ. Sci. Technol. 41 (2007) 7497–7502. doi: 10.1021/es0708058
[38]
D. Saha, S. Deng, Langmuir 27 (2011) 6780–6786. doi: 10.1021/la200091s
Y. Guan, C. Liu, Q. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202205805. doi: 10.1002/anie.202205805
[59]
F. Chang, Y. Guan, X. Chang, et al., J. Am. Chem. Soc. 140 (2018) 14799–14806. doi: 10.1021/jacs.8b08334
[60]
L. Li, T. Zhang, J. Cai, et al., J. Catal. 389 (2020) 218–228.
[61]
M. Xiong, Z. Gao, P. Zhao, et al., Nat. Commun. 11 (2020) 4773.
Figure 1
(a) NH3 synthesis rate and (b) TOFRu of Ru/ZrH2 catalysts with or without C60 promotion. (c) Dependence of NH3 synthesis rate on the reaction pressure at 400 ℃ for Ru/ZrH2 catalysts with or without C60 promotion. (d) Catalytic stability of C60-mediated Ru/ZrH2 catalyst (conditions: N2: H2 = 1:3; 400 ℃ and 1 MPa).
Figure 2
(a) Arrhenius plot of Ru/ZrH2 and C60-Ru/ZrH2 at 1 MPa. Reaction orders of (b) N2, (c) H2, and (d) NH3 over the Ru/ZrH2 and C60-Ru/ZrH2 at 400 ℃ and 1 MPa.
Figure 3
TEM images and the corresponding Ru particle size distributions of (a) Ru/ZrH2 and (b) C60-Ru/ZrH2. (c) Line scan profiles of C60-Ru/ZrH2 corresponding to the position in picture d. (d) HAADF−STEM image of C60-Ru/ZrH2 and related EDX mappings of Ru, Zr and C elements.
Figure 4
(a) In situ DRIFTS spectra of CO adsorption at 50 ℃. (b) Ru 3p XPS and (c) Ru L-edge NEXAFS spectra of Ru/ZrH2 and C60-Ru/ZrH2 catalysts. (d) KhT calculation results from the NEXAFS spectra.

 DownLoad:
DownLoad:

 下载:
下载:







 下载:
下载:
