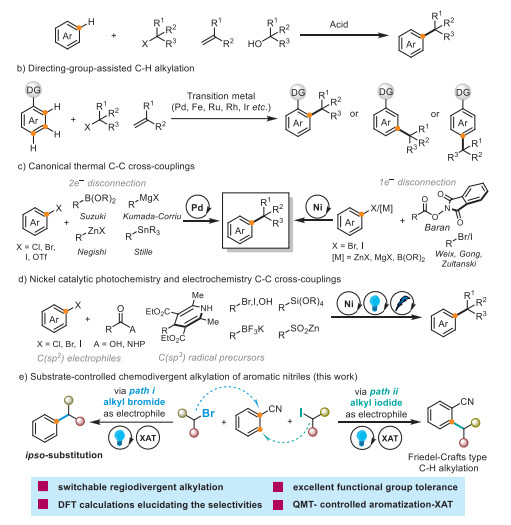
Scheme 1.
Arene alkylation strategies and impetus.
Alkyl halides dictate site selectivity in aromatic nitrile alkylations
Yuchuan Zhu , Kaili Xie , Yufei Li , Fang Liu , Yucheng Gu , Qingmin Wang , Weihua Zhang , Qing Xia

Arene alkylations are among the most fundamental transformations in organic synthesis. Indeed, alkylated aromatic hydrocarbons are widely used as fine chemicals, bioactive natural products, pharmaceuticals, and functional materials [1,2]. Controlling the regioselectivity is crucial to ensuring synthetic value in direct alkylation reactions that functionalize aromatic C–H bonds [3,4]. State-of-the-art techniques for regioselectively alkylating arenes primarily employ the Friedel–Crafts alkylation method [5,6], which uses an alkyl halide, alkene, or alcohol as the alkylating agents in the presence of a strong acid (Scheme 1a) [7]. However, this traditional approach has several drawbacks, including harsh reaction conditions, rearrangements, and the poor reactivities of electron-deficient arenes. Recently, transition-metal-catalyzed direct C–H bond alkylations using alkenes or alkyl halides as the alkylating reagents has emerged as robust alternatives to traditional alkylation. Such reactions provide alkylation products whose regioselectivities depend on the transition metals and ligands used (Scheme 1b) [8-17]. However, this strategy often requires the assistance of a directing group to enhance reactivity and maintain regioselectivity. In addition, the directing group is inherently difficult to remove if undesired in the final alkylated arene, which somewhat limits the scope and synthetic utility of such a process. Moreover, the selective transition-metal-catalyzed cross-coupling of two fragments to form a new C(sp2)–C(sp3) bond has become an indispensable tool for modern synthetic chemists (Scheme 1c). For example, the Suzuki, Kumada–Corriu, Negishi, and Stille coupling reactions are most often mediated by palladium catalysts [18,19]. Alternatively, radical-based nickel-catalyzed cross-couplings utilize building blocks such as carboxylic acids and their activated alkyl esters; this reaction was pioneered by Baran [20], while Zultanski, Weix and Gong popularized a similar reaction using alkyl halides [21-23]. Despite extraordinary advances in cross-coupling chemistry, some intrinsic obstacles remain unsolved, including the use of expensive metal-ligand catalysts and stoichiometric metal reductants. More recently, nickel-catalyzed photochemical and electrochemical cross-couplings have become popular strategies for unlocking a new C(sp2)–C(sp3) cross-coupling paradigm (Scheme 1d) [24-31]. These approaches greatly expand the variety of intricate substrates and highlight the benefits of metallaphotoredox reactions for the late-stage functionalization of complicated molecules. The practical utility of redox control displayed by photochemistry and electrochemistry is compelling.
Based on the above background, we envisaged the development of a new reaction for producing both ipso-substitution and Friedel–Crafts-type C—H alkylation products via the substrate-controlled regiodivergent alkylations of aromatic nitriles. Halogen atom transfer (XAT) is a mature technique in which the strong polar effects in the transition state involving the alkyl halide benefit the formation of free radicals and can be used as part of a C–C bond–forming strategy. Typically, Mn2(CO)10 [32], tin [33,34], silicon reagents [35-44], and trialkyl borane–O2 systems [45] can act as halogen extraction reagents to transform unactivated alkyl halides into their corresponding carbon radicals. More recently, XAT has also been incorporated into photocatalytic strategies [46-61] and tertiary amines have been utilized as halogen extraction reagents [62,63]. Tertiary amines, which are sustainable, low-toxic and abundant, and highly useful for organic synthesis as a consequence, may be used as alternatives to environmentally unfriendly metals or silicon reagents [64-72].
Herein, we designed synergistically regiodivergent alkylation chemistry for aromatic nitriles that involves XAT and photocatalysis. The two reactivity manifolds, which require the same photoredox catalyst, reaction conditions, and additives, can be selected to suit requirements by choosing appropriate alkyl halides that produce ipso-substitution products or Friedel–Crafts-type alkylation products (Scheme 1e). We decided that a detailed investigation is worth pursuing because regioselectivity is controlled by simply changing the alkyl halide, thereby dictating the regioisomeric outcome of the process.
We used 1 4-dicyanobenzene (1) as the model substrate (Table 1). A solution of bromocyclohexane (2, 2.0 equiv.), Ir(dFppy)3 (1 mol%), triethylamine (3.0 equiv.), and K3PO4 (2.0 equiv.) in CH3CN (0.1 mol/L) was irradiated by blue LED light for 24 h to afford ipso-substitution product 26 in 88% yield (entry 1, path ⅰ). After carefully investigating the reaction parameters, we discovered that differently substituted products were obtained when alkyl bromides or alkyl iodides were used. Friedel–Crafts-type alkylation products were obtained by substituting the alkyl bromide for the alkyl iodide, to afford product 44 in 79% yield (entry 11, path ⅱ). The yield of 26 decreased substantially when Ir(dFppy)3 was replaced with other photocatalysts (entry 2 and Table S1 in Supporting information for details). Among various solvents, CH3CN proved to be the most effective, whereas low reactivities were observed in DMF, DMSO, acetone, DMA, DCE, and MeOH (entry 3 and Table S2 in Supporting information for details). Substituting Et3N for N,N-diisopropylethylamine (iPr2NEt) afforded a slightly lower yield (entry 4), whereas other XAT reagents exhibited much lower reactivities (entry 5 and Table S3 in Supporting information for details). K3PO4 was crucial for the reaction as evidenced by other bases resulting in a marked decrease in reaction efficiency (Table S4 in Supporting information). Commonly used light sources were screened, with white or purple LED irradiation affording much lower conversions of the desired product 26 (entry 6). Using 1.0 or 2.0 equiv. of triethylamine resulted in a notable decrease in efficiency, demonstrating that the amount of triethylamine used also impacts reaction efficiency (entry 7). Shortening the reaction time to 12 h afforded the desired product in 49% yield (entry 8). Control experiments revealed that light is essential for the reaction (entry 9). Finally, a reaction-condition-based sensitivity analysis revealed that the reaction is sensitive to oxygen (entry 10). In the Friedel–Crafts-type alkylation process, substituting Et3N for iPr2NEt afforded a marginally lower yield (entry 12), while the other XAT reagents exhibited much lower reactivities (entry 13). Using 1.0 or 2.0 equiv. of triethylamine resulted in a slight decrease in efficiency, demonstrating that the amount of triethylamine used impacts reaction efficiency (entry 14). Reducing the reaction time to 12 h afforded the desired product in 45% yield (entry 15). Control experiments revealed that light is essential for this reaction (entry 16). Finally, a reaction-condition-based sensitivity analysis show ed that the reaction is sensitive to oxygen (entry 17). Additionally, alkyl chlorides and alkyl fluorides are not applicable to this reaction, due to the excessively large bond dissociation energies (BDEs) of R-F and R−Cl bonds [73].
 DownLoad:
CSV
DownLoad:
CSV
 | ||
| Entry | Variations from standard conditions | Yield of 26/44 (%)a |
| Ipso-substitution (path ⅰ) | ||
| 1 | No deviationb | 88 |
| 2 | Other photocatalysts | < 20 |
| 3 | DMF, DMSO, Acetone, DMA, DCE, MeOH as solvent | < 40 |
| 4 | iPr2NEt as XAT reagent | 82 |
| 5 | nBu3N, TMEDA, (TMS)3SiH as XAT reagent | < 30 |
| 6 | 395 nm Purple LEDs or White LEDs | < 35 |
| 7 | 1.0 or 2.0 equiv. of triethylamine | < 63 |
| 8 | 12 h reaction | 49 |
| 9 | In the dark | 0 |
| 10 | In the air | 40 |
| Friedel−Crafts-type C–H alkylation (path ⅱ) | ||
| 11 | No deviationc | 79 |
| 12 | iPr2NEt as XAT reagent | 75 |
| 13 | nBu3N, TMEDA, (TMS)3SiH as XAT reagent | Trace |
| 14 | 1.0 or 2.0 equiv. of triethylamine | < 70 |
| 15 | 12 h reaction | 45 |
| 16 | In the dark | 0 |
| 17 | In the air | 60 |
| a Isolated yield based on 1. b 1 (0.2 mmol), 2 (0.4 mmol, 2 equiv.), Ir(dFppy)3 (0.002 mmol), K3PO4 (0.4 mmol), triethylamine (0.6 mmol), CH3CN (2 mL), room temperature, N2 atmosphere, 12 W blue LEDs, 24 h. c 1 (0.2 mmol), 3 (0.3 mmol, 1.5 equiv.), Ir(dFppy)3 (0.002 mmol), K3PO4 (0.4 mmol), triethylamine (0.6 mmol), CH3CN (2 mL), room temperature, N2 atmosphere, 12 W blue LEDs, 24 h. | ||
Having identified the optimal conditions for the ipso-substitution reaction between an alkyl bromide and 1,4-dicyanobenzene (1), we next evaluated the scope of the process (Scheme 2). We explored the alkyl-bromide substrate range by initiating reactions between primary alkyl bromides and 1, which afforded alkylated products 4–13 in 39%–58% yield. This outcome is attributable to the lower stability and nucleophilicity of primary radicals compared to their secondary counterparts [74]. Notably, this protocol can selectively abstract bromide instead of chloride; hence 1‑bromo-6-chlorohexane is a suitable substrate, affording the target product 10 in moderate yield. Unsurprisingly, under our standard conditions, acyclic secondary alkyl bromides afforded corresponding products 14–18 in excellent yields (up to 92%). Notably, 1,3-dibromobutane was compatible with the reaction conditions, affording target product 19 in moderate yield. In addition, alicyclic alkyl bromides were suited to the reaction and cyclic and heterocyclic alkyl bromides 20–28 afforded the desired products in good yields. In particular, the reaction proceeded with Boc-protected piperidine motifs to afford 23 in 85% yield; this type of product is an important precursor and synthetic block for the synthesis of drugs [75]. Owing to spatial hindrance, the tertiary alkyl bromide afforded the desired product 29 in moderate yield. Subsequently, we explored the scope of the reaction with respect to the aryl nitrile using 2 as the alkyl bromide. Methyl 4-cyanobenzoate and ethyl 4-cyanobenzoate afforded moderate-to-good yields (27%–73%) of the corresponding products. Moreover, the lactone group on the aryl ring was accommodated under these reaction conditions, with a moderate yield (58%) of 34 obtained. As expected, polysubstituted benzonitriles, such as 2-methyl-1,4-benzenedicarbonitrile and 2,5-dimethylbenzene-1,4-dicarbonitrile, were viable reagents to afford 35–37 in 73%–79% yield. The scalability of the method was assessed by reacting 1.28 g of 1 and 2.0 equiv. of 2, which resulted in the production of 1.33 g (72% yield) of 26.
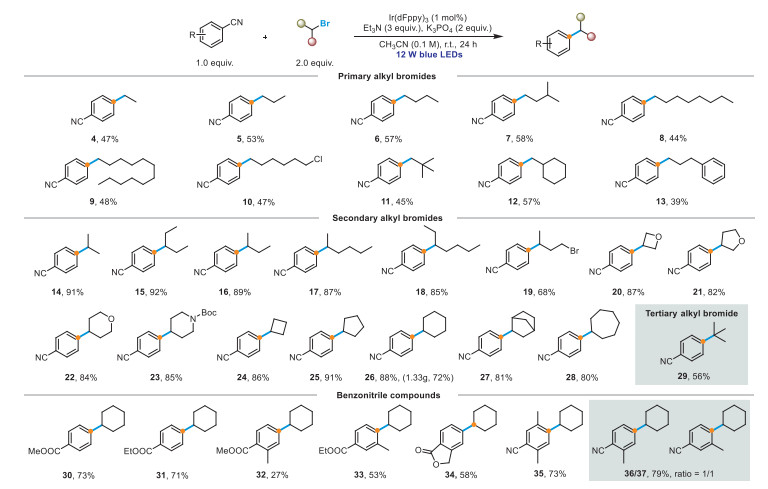
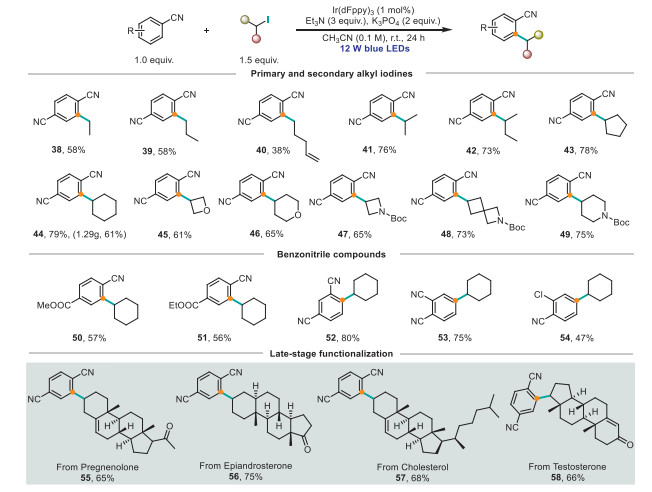
Next, we explored the scope of the Friedel−Crafts-type C–H alkylation reaction (Scheme 3). Acyclic primary alkyl iodides afforded the corresponding products 38–40 in moderate yields (38%–58%). Unsurprisingly, secondary alkyl iodides, under the standard conditions listed herein, afforded the corresponding products 41 and 42 in good yields (76% and 73%, respectively). Alicyclic alkyl iodides were also suited to the reaction, carbocyclic and heterocyclic alkyl iodides 43–49 afforded the desired products in good yields. For the alkyl iodide coupling partners, methyl 4-cyanobenzoate and ethyl 4-cyanobenzoate afforded the corresponding products in moderate yields (50 and 51, 57%, and 56%, respectively). 1,3-Dicyanobenzene and 1,2-dicyanobenzene afforded the corresponding products 52 and 53 in good yields (80%, and 75%, respectively). In addition, 2-chlorobenzonitrile was a suitable substrate (54, 47% yield). To showcase the versatility of this protocol, we successfully applied it to four polycyclic alkyl iodides derived from steroidal natural products containing olefins and carbonyl groups to afford target compounds 55–58 in high yields (65%–75%). This outcome underscores the practical synthetic potential of the developed process. The scalability of the method was assessed by reacting 1.28 g of 1 and 1.5 equiv. of 3, which afforded 1.29 g (61% yield) of 44.
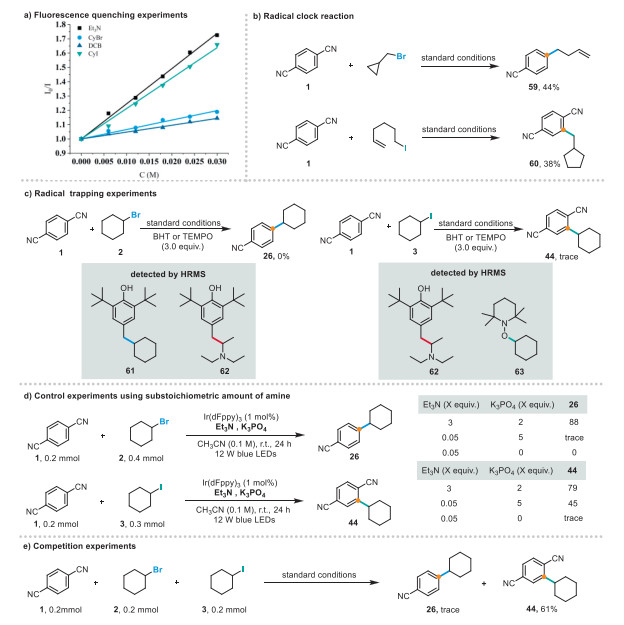
Next, we investigated the reaction mechanisms. To understand the sequence of each step, we performed a series of fluorescence-quenching experiments using a photocatalyst (Scheme 4a). Fluorescence quenching and Stern–Volmer studies suggested that the excited photocatalyst Ir(dFppy)3 initially reacts with triethylamine via single-electron transfer (Figs. S1–S4 in Supporting information for details). Subsequently, a radical-clock experiment was conducted. We selected (bromomethyl)cyclopropane and 6-iodo-1-hexene as substrates that react with 1, with ring-cleavage products 59 and cyclized products 60 observed, respectively, which indicates that both reaction processes generate alkyl radicals (Scheme 4b). The reaction of 1 with alkyl halide 2 or 3 was annihilated when a radical scavenger such as BHT (butylated hydroxytoluene, 3.0 equiv.), or TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy, 3.0 equiv.) was used; these reactions delivered trace amounts of adducts derived from the α-aminoalkyl radical and cyclohexyl radical, which suggests that the reactions follow radical pathways and that α-aminoalkyl radical and cyclohexyl radical are generated and serve as intermediates for both reactions (Scheme 4c). Next, a loading of only 5 mol% triethylamine and 5.0 equiv. of K3PO4 afforded desired product 44 in 45% yield and a trace amount of desired product 26; almost no reaction took place in the absence of K3PO4 (Scheme 4d). These results indicate that 1 and 3 are involved in a radical chain propagation reaction, and that K3PO4, as a secondary base, serves to quench the HI formed in this process. A light/dark experiment revealed that products 26 and 44 are only formed under light irradiation, which indicates that the chain-propagation process is short-lived and that light is essential for product formation (Fig. S5 in Supporting information). A competition experiment between 2 and 3 was conducted using a 1:1 mixture of 2 and 3 under standard conditions (Scheme 4e). Interestingly, 44 was obtained in 61% yield along with trace amounts of 26. These results further demonstrate the distinct reactivity differences between the two substrates. While the mechanism of the ipso-substitution reaction (path ⅰ) can be elucidated on the basis of our experimental observations and literature reports [76-79], that of the Friedel−Crafts-type C–H alkylation reaction remains unclear; therefore, the mechanism associated with path ⅱ requires further exploration.
To gain further insight into the observed selectivity, density functional theory (DFT) calculations (see Supporting information for computational details) were performed on the model reaction to explore the proposed arene alkylation mechanism. Our calculations revealed that substrate 2 can progress along path ⅰ (Scheme 5a). α-Aminoalkyl radical 66 initiates the XAT reaction with 2 via transition state TS1 (∆Gǂ = 23.1 kcal/mol), which affords the nucleophilic alkyl radical 67; this reaction is exergonic by 3.9 kcal/mol. The free-radical addition of 68 to radical 67 via transition state TS3 (∆Gǂ = 27.8 kcal/mol) affords INT1. Subsequently, the carbanion intermediate 69 is generated via the transition state for free naphthenic base migration (TS4; ∆Gǂ = 26.6 kcal/mol) in INT1. Thereafter, elimination of CN– via transition state TS5 (∆Gǂ = 12.1 kcal/mol), affords the thermodynamically stable product 26.
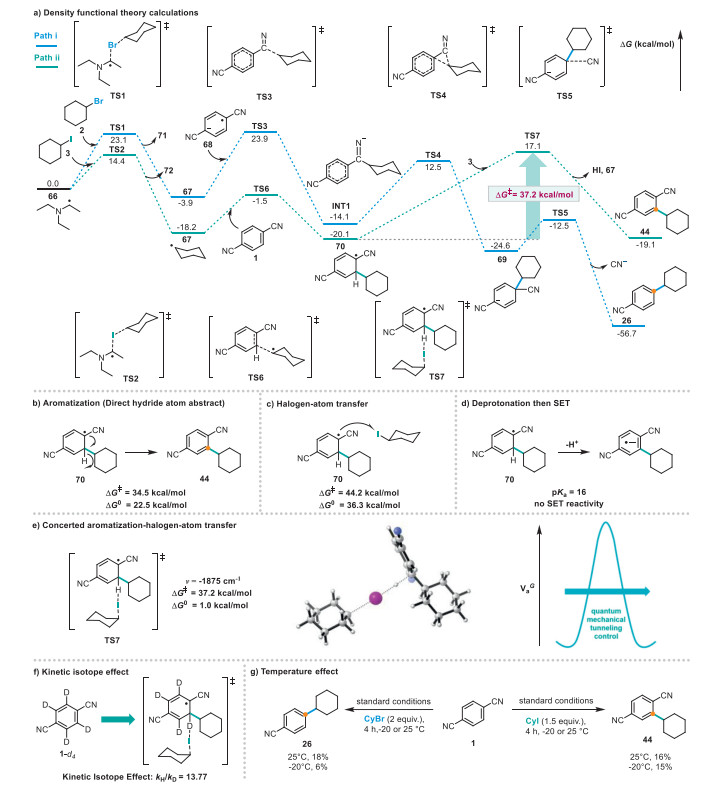
We initially attributed the outcome of path ⅱ to aromatization followed by faster hydrogen atom abstraction [49]. However, computational analysis of this reaction profile revealed that the hydrogen atom abstraction is energetically uphill (> 22 kcal/mol) with a high kinetic barrier (Scheme 5b), which indicates that a distinct mechanistic pathway operates under these reaction conditions. We also eliminated the possibility of iodine-atom transfer occurring from 70 because this process is highly endergonic (∆G0 = 36.3 kcal/mol) and kinetically unfavorable (∆Gǂ = 44.2 kcal/mol) (Scheme 5c). Deprotonation of 70 to the corresponding phenyl radical anion, followed by SET activation of the substrate, was also excluded based on the calculated high acid dissociation constant pKa = 16 [acetonitrile (CH3CN)] for the proton in 70 (Scheme 5d).
Substrate 3 can progress along path ⅱ by combining aromatization and halogen transfer (Scheme 5a). α-Aminoalkyl radical 66 initiates the XAT reaction with 3, which occurs via transition state TS2 (∆Gǂ = 14.4 kcal/mol); this reaction affords the nucleophilic alkyl radical 67 and is exergonic by 18.2 kcal/mol. Thereafter, radical 67 adds to 1 via transition state TS6 (∆Gǂ = 16.7 kcal/mol) to form intermediate 70. Compounds 70 and 3 are typically close to thermoneutral (∆G0 = −1.0 kcal/mol) but still have a significantly high kinetic barrier (∆Gǂ = 37.2 kcal/mol), which is expected to thwart reactivity. However, the very large imaginary frequency observed (ν = −1875 cm-1) implies that the barrier is very narrow (Scheme 5e) [80-82]. The energy barrier for the reaction between 67 and 68 is higher than that for 67 and 1, leading 67 to react preferentially with 1, thereby yielding product 44 (Fig. S7 in Supporting information). Under these circumstances, hydrogen atoms are generally considered to transfer via quantum mechanical tunneling, leading to large H/D KIEs [49,83-86]. Accordingly, kinetic isotope effect experiments were performed to validate this mechanistic scenario. An intermolecular competition reaction between 1,4-dicyanobenzene (1) and (2H4)benzene-1,4-dicarbonitrile was carried out in the same pot, which showed a clear isotope effect (kH/kD = 13.77, Scheme 5f), as calculated from the product ratios of 44 and 64. These results provide evidence that the C–H bond breaking step in 1,4-dicyanobenzene, in which 3 participates, is governed by tunneling and also indicate that tunneling is important to the rate-determining step [87-89]. Next, we repeated the reaction for 2 in the presence of 1 at a lower temperature (T = −20 ℃) and observed a significant loss of yield (18% at T = 25 ℃ versus 6% at T = −20 ℃) (Scheme 5g). However, the reaction for 3 in the presence of 1 at a lower temperature (T = −20 ℃) afforded an almost identical yield (16% at 25 ℃ vs. 15% at −20 ℃). These results demonstrate that the rate of hydrogen transfer by 3 is insensitive to temperature, providing additional evidence that the reaction occurs via quantum mechanical tunneling [90].
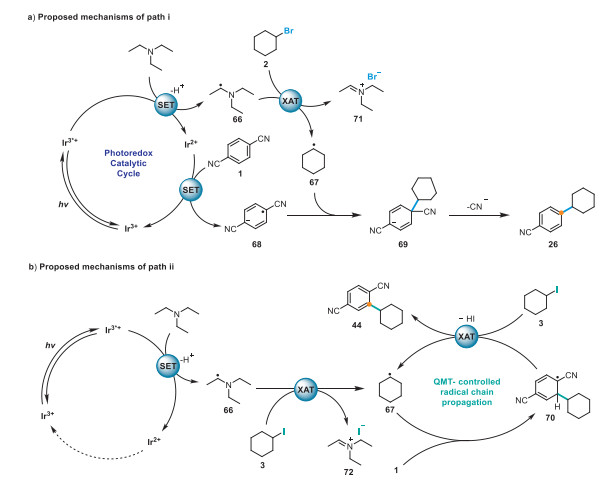
Finally, we propose a mechanism for path ⅰ (Scheme 6a), in which the Ir(dFppy)3 photocatalyst is excited by blue light, and the resulting excited Ir3*+ state (Eox 1/2 PC*/PC− = +0.36 V vs. SCE) [91] oxidizes Et3N (Eox 1/2 = +0.77 V vs. SCE), followed by deprotonation to produce α-aminoalkyl radical 66 and the Ir2+ species. Single-electron reduction of 1 4-dicyanobenzene (1, Ered 1/2 = −1.61 V vs. SCE) [92,93] by Ir2+ (Ered 1/2 PC/PC− = −1.87 V vs. SCE) [91] generates radical carbanion intermediate 68, which completes the photoredox cycle. Thereafter, radical cation 66 cleaves the C–Br bond in 2 to generate nucleophilic alkyl radical 67, which adds to the carbanion intermediate 69 and eliminates CN– to form product 26. In path ⅱ (Scheme 6b), radical 67 then adds to 1 via a Friedel–Crafts-type alkylation pathway to afford radical 70, which subsequently triggers the quantum-mechanical-tunneling-enabled aromatization–halogen atom transfer (QMT-XAT) process to produce desired product 44 and regenerate radical 67 to facilitate the radical chain process.
In summary, we demonstrated an unprecedented substrate-controlled regiodivergent alkylation strategy for aromatic nitriles. Remarkably, site selectivity during alkyl radical trapping with an aromatic nitrile can be controlled by judicious choice of alkyl halide. While the use of an alkyl bromide preferentially affords the ipso-substitution product, site selectivity can be switched to Friedel−Crafts-type C–H alkylation by swapping to an alkyl iodide. Mechanistic studies and DFT calculations established that quantum-mechanical-tunneling-enabled aromatization–halogen atom transfer directs the process toward the C–H alkylation pathway, while photocatalytic radical coupling leads to the ipso-substitution pathway. The protocol permits aromatic nitriles with cyclic and acyclic alkyl halides to be efficiently and selectively functionalized, and is scalable to the gram level. We believe that this work provides a valuable conceptual extension to existing aromatic nitrile alkylations, with improved efficiencies, reactivities, and synthetic utilities.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yuchuan Zhu: Writing – original draft, Investigation. Kaili Xie: Investigation. Yufei Li: Investigation. Fang Liu: Conceptualization. Yucheng Gu: Supervision. Qingmin Wang: Supervision. Weihua Zhang: Supervision. Qing Xia: Writing – review & editing, Conceptualization.
This study was supported by the Outstanding Youth Science Fund Project of Natural Science Foundation of Jiangsu Province (No. BK20240196), the National Natural Science Foundation of China (No. 32372581), the Fundamental Research Funds for the Central Universities (No. KJYQ2025010).
Supplementary material associated with this article can be found, in the online version, at doi:
J.L. Segura, N. Martín, J. Mater. Chem. 10 (2000) 2403–2435. doi: 10.1039/b004407p
R.D. Taylor, M. MacCoss, A.D.G. Lawson, J. Med. Chem. 57 (2014) 5845–5859. doi: 10.1021/jm4017625
A.S.S. Wilson, M.S. Hill, M.F. Mahon, et al., Science 358 (2017) 1168–1171. doi: 10.1126/science.aao5923
J. Dong, F. Yue, W. Xu, et al., Green Chem. 22 (2020) 5599–5604. doi: 10.1039/d0gc02111c
S.L. You, Q. Cai, M. Zeng, Chem. Soc. Rev. 38 (2009) 2190–2201. doi: 10.1039/b817310a
R.R. Naredla, D.A. Klumpp, Chem. Rev. 113 (2013) 6905–6948. doi: 10.1021/cr4001385
G. Evano, C. Theunissen, Angew. Chem. Int. Ed. 58 (2019) 7202–7236. doi: 10.1002/anie.201806629
P.S. Thuy-Boun, G. Villa, D. Dang, et al., J. Am. Chem. Soc. 135 (2013) 17508–17513. doi: 10.1021/ja409014v
D.R. Armstrong, J.A. Garden, A.R. Kennedy, et al., Angew. Chem. Int. Ed. 52 (2013) 7190–7193. doi: 10.1002/anie.201302426
P.X. Shen, X.C. Wang, P. Wang, et al., J. Am. Chem. Soc. 137 (2015) 11574–11577. doi: 10.1021/jacs.5b08914
F. Yu, T. Wang, H. Zhou, et al., Org. Lett. 19 (2017) 6538–6541. doi: 10.1021/acs.orglett.7b03244
R.Y. Zhu, L.Y. Liu, H.S. Park, et al., J. Am. Chem. Soc. 139 (2017) 16080–16083. doi: 10.1021/jacs.7b09761
H. Shi, A.N. Herron, Y. Shao, et al., Nature 558 (2018) 581–585. doi: 10.1038/s41586-018-0220-1
P. Liu, C. Chen, X. Cong, et al., Nat. Commun. 9 (2018) 4637. doi: 10.1038/s41467-018-07069-1
S. Rej, Y. Ano, N. Chatani, Chem. Rev. 120 (2020) 1788–1887. doi: 10.1021/acs.chemrev.9b00495
W. Zhu, T.B. Gunnoe, J. Am. Chem. Soc. 143 (2021) 6746–6766. doi: 10.1021/jacs.1c01810
L. Winfrey, L. Yun, G. Passeri, et al., Chem. Eur. J. 30 (2024) e202303130. doi: 10.1002/chem.202303130
A. Biffis, P. Centomo, A. Del Zotto, et al., Chem. Rev. 118 (2018) 2249–2295. doi: 10.1021/acs.chemrev.7b00443
R. Jana, T.P. Pathak, M.S. Sigman, Chem. Rev. 111 (2011) 1417–1492. doi: 10.1021/cr100327p
J. Cornella, J.T. Edwards, T. Qin, et al., J. Am. Chem. Soc. 138 (2016) 2174–2177. doi: 10.1021/jacs.6b00250
S.L. Zultanski, G.C. Fu, J. Am. Chem. Soc. 135 (2013) 624–627. doi: 10.1021/ja311669p
D.J. Weix, Acc. Chem. Res. 48 (2015) 1767–1775. doi: 10.1021/acs.accounts.5b00057
Y. Gong, J. Hu, C. Qiu, et al., Acc. Chem. Res. 57 (2024) 1149–1162. doi: 10.1021/acs.accounts.3c00810
J.C. Tellis, C.B. Kelly, D.N. Primer, et al., Acc. Chem. Res. 49 (2016) 1429–1439. doi: 10.1021/acs.accounts.6b00214
J.A. Milligan, J.P. Phelan, S.O. Badir, et al., Angew. Chem. Int. Ed. 58 (2019) 6152–6163. doi: 10.1002/anie.201809431
A.Y. Chan, I.B. Perry, N.B. Bissonnette, et al., Chem. Rev. 122 (2021) 1485–1542.
L.F.T. Novaes, J. Liu, Y. Shen, et al., Chem. Soc. Rev. 50 (2021) 7941–8002. doi: 10.1039/d1cs00223f
Y. Liu, P. Li, Y. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202306679. doi: 10.1002/anie.202306679
M.D. Palkowitz, M.A. Emmanuel, M.S. Oderinde, Acc. Chem. Res. 56 (2023) 2851–2865. doi: 10.1021/acs.accounts.3c00479
W. Xu, T. Xu, Acc. Chem. Res. 57 (2024) 1997–2011. doi: 10.1021/acs.accounts.4c00309
F. Yue, M. Li, F. Yuan, et al., Chin. Chem. Lett. (2025) 111053. doi: 10.1016/j.cclet.2025.111053
R.S. Herrick, T.R. Herrinton, H.W. Walker, et al., Organometallics 4 (1985) 42–45. doi: 10.1021/om00120a008
W.P. Neumann, Synthesis (Mass) 1987 (1987) 665–683. doi: 10.1055/s-1987-28044
J.J. Chen, H.M. Huang, Tetrahedron Lett. 102 (2022) 153945. doi: 10.1016/j.tetlet.2022.153945
J.J. Devery, J.D. Nguyen, C. Dai, et al., ACS Catal. 6 (2016) 5962–5967. doi: 10.1021/acscatal.6b01914
R.T. Smith, X. Zhang, J.A. Rincon, et al., J. Am. Chem. Soc. 140 (2018) 17433–17438. doi: 10.1021/jacs.8b12025
C. Chatgilialoglu, C. Ferreri, Y. Landais, et al., Chem. Rev. 118 (2018) 6516–6572. doi: 10.1021/acs.chemrev.8b00109
G.H. Lovett, S. Chen, X.S. Xue, et al., J. Am. Chem. Soc. 141 (2019) 20031–20036. doi: 10.1021/jacs.9b11434
J. Dong, X. Lyu, Z. Wang, et al., Chem. Sci. 10 (2019) 976–982. doi: 10.1039/c8sc04892d
N.W. Dow, A. Cabre, D.W.C. MacMillan, Chem 7 (2021) 1827–1842. doi: 10.1016/j.chempr.2021.05.005
P.J. Deneny, R. Kumar, M.J. Gaunt, Chem. Sci. 12 (2021) 12812–12818. doi: 10.1039/d1sc04554g
A. Luridiana, D. Mazzarella, L. Capaldo, et al., ACS Catal. 12 (2022) 11216–11225. doi: 10.1021/acscatal.2c03805
S. Mistry, R. Kumar, A. Lister, et al., Chem. Sci. 13 (2022) 13241–13247. doi: 10.1039/d2sc03516b
H.W. Du, Y.D. Du, X.W. Zeng, et al., Angew. Chem. Int. Ed. 62 (2023) e202308732. doi: 10.1002/anie.202308732
C. Ollivier, P. Renaud, Chem. Rev. 101 (2001) 3415–3434. doi: 10.1021/cr010001p
C. Le, T.Q. Chen, T. Liang, et al., Science 360 (2018) 1010–1014. doi: 10.1126/science.aat4133
X.S. Zhou, D.M. Yan, J.R. Chen, Chem 6 (2020) 823–825. doi: 10.1016/j.chempr.2020.03.007
Z.Q. Zhang, Y.Q. Sang, C.Q. Wang, et al., J. Am. Chem. Soc. 144 (2022) 14288–14296. doi: 10.1021/jacs.2c05356
T. Constantin, B. Górski, M.J. Tilby, et al., Science 377 (2022) 1323–1328. doi: 10.1126/science.abq8663
P. Dai, Y. Li, Y. Chen, et al., Org. Lett. 24 (2022) 1357–1361. doi: 10.1021/acs.orglett.2c00048
T. Wan, L. Capaldo, D. Ravelli, et al., J. Am. Chem. Soc. 145 (2023) 991–999. doi: 10.1021/jacs.2c10444
G. Levitre, A. Granados, G.A. Molander, Green Chem. 25 (2023) 560–565. doi: 10.1039/d2gc04578h
S. Zhai, R. Wang, Q. Dong, et al., Org. Chem. Front. 10 (2023) 4816–4820. doi: 10.1039/d3qo01012k
T. Singh, S.R. Nasireddy, G.C. Upreti, et al., Org. Lett. 25 (2023) 5558–5562. doi: 10.1021/acs.orglett.3c01800
X. Jiao, Z. Huang, W. Meng, et al., Org. Chem. Front. 10 (2023) 4542–4549. doi: 10.1039/d3qo00870c
L.Q. Huang, D.Y. Yang, C.L. Dong, et al., Adv. Synth. Catal. 365 (2023) 2553–2559. doi: 10.1002/adsc.202300519
L. Caiger, H. Zhao, T. Constantin, et al., ACS Catal. 13 (2023) 4985–4991. doi: 10.1021/acscatal.3c00571
M.G. Pizzio, E.G. Mata, P. Dauban, et al., Eur. J. Org. Chem. 26 (2023) e202300616. doi: 10.1002/ejoc.202300616
Y. -L. Tu, B. -B. Zhang, B. -S. Qiu, et al., Angew. Chem. Int. Ed. 62 (2023) e202310764. doi: 10.1002/anie.202310764
Y. Li, K. Xie, S. Yang, et al., ChemCatChem 16 (2024) e202400675. doi: 10.1002/cctc.202400675
H. Yang, L.C. Wang, X.F. Wu, Chin. Chem. Lett. 36 (2025) 110843. doi: 10.1016/j.cclet.2025.110843
T. Constantin, F. Julia, N.S. Sheikh, et al., Chem. Sci. 11 (2020) 12822–12828. doi: 10.1039/d0sc04387g
V.S. Kostromitin, A.O. Sorokin, V.V. Levin, et al., Chem. Sci. 14 (2023) 3229–3234. doi: 10.1039/d3sc00027c
F. Yue, J. Dong, Y. Liu, et al., Org. Lett. 23 (2021) 7306–7310. doi: 10.1021/acs.orglett.1c02905
C.Y. Li, Y. Ma, Z.W. Lei, et al., Org. Lett. 23 (2021) 8899–8904. doi: 10.1021/acs.orglett.1c03390
B. Górski, A.L. Barthelemy, J.J. Douglas, et al., Nat. Catal. 4 (2021) 623–630. doi: 10.1038/s41929-021-00652-8
H. Zhao, A.J. McMillan, T. Constantin, et al., J. Am. Chem. Soc. 143 (2021) 14806–14813. doi: 10.1021/jacs.1c06768
L. Caiger, C. Sinton, T. Constantin, et al., Chem. Sci. 12 (2021) 10448–10454. doi: 10.1039/d1sc03083c
B. Niu, K. Sachidanandan, B.G. Blackburn, et al., Org. Lett. 24 (2022) 916–920. doi: 10.1021/acs.orglett.1c04267
X. Tian, J. Kaur, S. Yakubov, et al., ChemSusChem 15 (2022) e202200906. doi: 10.1002/cssc.202200906
S. Govaerts, K. Nakamura, T. Constantin, et al., Org. Lett. 24 (2022) 7883–7887. doi: 10.1021/acs.orglett.2c02840
J. Zhang, M. Jiang, C.S. Wang, et al., Nat. Commun. 13 (2022) 7961. doi: 10.1038/s41467-022-35613-7
F. Juliá, T. Constantin, D. Leonori, Chem. Rev. 122 (2022) 2292–2352. doi: 10.1021/acs.chemrev.1c00558
A. Velloth, P. Kumar, S. Butt, et al., Asian J. Org. Chem. 14 (2025) e202400686.
M.M. Abdelshaheed, I.M. Fawzy, H.I. El-Subbagh, et al., Future J. Pharm. Sci. 7 (2021) 188. doi: 10.1186/s43094-021-00335-y
A. McNally, C.K. Prier, D.W.C. MacMillan, Science 334 (2011) 1114–1117. doi: 10.1126/science.1213920
M.T. Pirnot, D.A. Rankic, D.B.C. Martin, et al., Science 339 (2013) 1593–1596. doi: 10.1126/science.1232993
J.D. Cuthbertson, D.W.C. MacMillan, Nature 519 (2015) 74–77. doi: 10.1038/nature14255
M.M. Walker, B. Koronkiewicz, S. Chen, et al., J. Am. Chem. Soc. 142 (2020) 8194–8202. doi: 10.1021/jacs.9b13165
P.R. Schreiner, H.P. Reisenauer, D. Ley, et al., Science 332 (2011) 1300–1303. doi: 10.1126/science.1203761
D. Ley, D. Gerbig, P.R. Schreiner, Org. Biomol. Chem. 10 (2012) 3781–3790. doi: 10.1039/c2ob07170c
A. Nandi, Z. Alassad, A. Milo, et al., ACS Catal. 11 (2021) 14836–14841. doi: 10.1021/acscatal.1c04475
L.M. Slaughter, P.T. Wolczanski, T.R. Klinckman, et al., J. Am. Chem. Soc. 122 (2000) 7953–7975. doi: 10.1021/ja000112q
S. Fukuzumi, T. Kobayashi, T. Suenobu, J. Am. Chem. Soc. 132 (2010) 1496–1497. doi: 10.1021/ja910349w
J. Ho, J. Zheng, R. Meana-Pañeda, et al., J. Org. Chem. 78 (2013) 6677–6687. doi: 10.1021/jo400927y
P.R. Schreiner, Trends Chem. 2 (2020) 980–989.
K. Hiraoka, T. Sato, S. Sato, et al., Astrophys. J. 577 (2002) 265.
H. Hidaka, M. Watanabe, A. Kouchi, et al., Astrophys. J. 702 (2009) 291.
S. Andersson, T.P.M. Goumans, A. Arnaldsson, Chem. Phys. Lett. 513 (2011) 31–36.
S.H. Bae, X. -X. Li, M.S. Seo, et al., J. Am. Chem. Soc. 141 (2019) 7675–7679. doi: 10.1021/jacs.9b02272
K. Teegardin, J.I. Day, J. Chan, et al., Org. Process Res. Dev. 20 (2016) 1156–1163. doi: 10.1021/acs.oprd.6b00101
Y. Mori, Y. Sakaguchi, H. Hayashi, J. Phys. Chem. A 104 (2000) 4896–4905.
C. Miao, J. Yao, Chin. J. Org. Chem. 43 (2023) 1341–1364. doi: 10.6023/cjoc202209007

Scheme 2 Substrate scope of the ipso-substitution. See Table 1, entry 1 for detailed conditions.

Scheme 3 Substrate scope of the Friedel−Crafts-type C–H alkylation. See Table 1, entry 11 for detailed conditions.

Scheme 5 DFT calculations. Outlining possible mechanistic scenarios leading to quantum-tunneling-controlled concerted halogen-atom-transfer. Potential energies and Gibbs free energies (kcal/mol) were calculated at the wB97-XD/def2qzvpp/SMD (acetonitrile)//wB97-XD/def2svp/SMD(acetonitrile)) level relative to 66 and 2/3.
Table 1. Optimizing the reaction conditions.
| ||
| Entry | Variations from standard conditions | Yield of 26/44 (%)a |
| Ipso-substitution (path ⅰ) | ||
| 1 | No deviationb | 88 |
| 2 | Other photocatalysts | < 20 |
| 3 | DMF, DMSO, Acetone, DMA, DCE, MeOH as solvent | < 40 |
| 4 | iPr2NEt as XAT reagent | 82 |
| 5 | nBu3N, TMEDA, (TMS)3SiH as XAT reagent | < 30 |
| 6 | 395 nm Purple LEDs or White LEDs | < 35 |
| 7 | 1.0 or 2.0 equiv. of triethylamine | < 63 |
| 8 | 12 h reaction | 49 |
| 9 | In the dark | 0 |
| 10 | In the air | 40 |
| Friedel−Crafts-type C–H alkylation (path ⅱ) | ||
| 11 | No deviationc | 79 |
| 12 | iPr2NEt as XAT reagent | 75 |
| 13 | nBu3N, TMEDA, (TMS)3SiH as XAT reagent | Trace |
| 14 | 1.0 or 2.0 equiv. of triethylamine | < 70 |
| 15 | 12 h reaction | 45 |
| 16 | In the dark | 0 |
| 17 | In the air | 60 |
| a Isolated yield based on 1. b 1 (0.2 mmol), 2 (0.4 mmol, 2 equiv.), Ir(dFppy)3 (0.002 mmol), K3PO4 (0.4 mmol), triethylamine (0.6 mmol), CH3CN (2 mL), room temperature, N2 atmosphere, 12 W blue LEDs, 24 h. c 1 (0.2 mmol), 3 (0.3 mmol, 1.5 equiv.), Ir(dFppy)3 (0.002 mmol), K3PO4 (0.4 mmol), triethylamine (0.6 mmol), CH3CN (2 mL), room temperature, N2 atmosphere, 12 W blue LEDs, 24 h. | ||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: