Received Date:
29 June 2025 Accepted Date:
08 September 2025 Revised Date:
29 August 2025 Available Online:
15 January 2026
Abstract:
Rational design of nanozymes with enhanced catalytic efficiency remains a central challenge in the development of artificial enzymes. Herein, we report the construction of ultrasmall gold nanocluster-based nanoassemblies (Dp-AuNCs@Fe2+) through the coordination of Fe2+ ions by a dopa-containing peptidomimetic ligand (DpCDp). This nanoarchitecture simultaneously integrates catalytically active gold cores and redox-active Fe2+ centers, bridged by DpCDp to facilitate directional electron transfer. Comprehensive spectroscopic and kinetic analyses reveal that DpCDp promotes efficient charge migration from the Au core to surface-bound Fe2+, significantly enhancing H2O2-mediated peroxidase-like activity. Compared to bare Dp-AuNCs, Dp-AuNCs@Fe2+ display a 4.3-fold improvement in detection sensitivity, a 6.7-fold increase in catalytic efficiency, and markedly stronger hydroxyl radical generation. Mechanistically, this activity stems from a synergistic triad: direct H2O2 oxidation at gold surfaces, radical generation at Fe2+ sites, and DpCDp-facilitated electron shuttling. This work presents a robust strategy for nanozyme enhancement via electronic and structural co-engineering, offering valuable insights for the future design of bioinspired catalytic systems.
Natural enzymes demonstrate precise catalytic specificity through optimal spatial complementarity between active sites and substrates [1]. However, their protein/RNA nature imposes intrinsic constraints: Limited stability, narrow operational parameters, and prohibitive production costs from intricate purification protocols [2,3]. Nanozymes, engineered nanomaterials mimicking enzymatic functions, overcome these limitations by offering exceptional stability, environmental robustness, and scalable synthesis [4,5]. Their catalytic activity and selectivity are tunable through tailoring size, morphology, and composition [6]. This emerging class of artificial catalysts not only addresses the partial bottlenecks of traditional enzymatic systems but also establishes a novel pathway for overcoming technological barriers in catalysis-driven fields, thereby propelling transformative innovations through functional customization [7].
Peroxidase (POD)-mimic have garnered significant attention due to their unique capacity to catalytically decompose hydrogen peroxide (H2O2) into reactive oxygen species [8,9]. These exceptional catalytic properties enable diverse applications in biosensing [10,11], tumor therapeutics [12], and environmental monitoring [13]. The catalytic performance of nanomaterial-based POD-mimic can be precisely optimized through strategic modulation of composition and structural parameters. Researchers have engineered various nanomaterial systems for POD-mimic, including transition metal oxides [14], carbon-based architectures [15], and noble metal clusters [16,17]. Notably, gold nanoclusters (AuNCs) exhibit molecular-like characteristics, ultrahigh surface-to-volume ratios, and exceptional optical properties, coupled with stable catalytic capabilities [18-20]. Such advantages establish AuNCs as a transformative material platform, driving innovations in analytical diagnostics and biomedical technologies while emerging as a focal point in advanced nanozyme research [21-23].
To advance the practical utility of AuNCs, strategic paradigms have been developed to enhance their POD-mimic activity. Kernel engineering approaches involve alloying with transition metals (Pt, Pd) to modulate electronic structure [24,25], thereby improving H2O2 activation capacity. Surface modification methodologies have also demonstrated efficacy, exemplified by Zhang's synthesis [26] of AuNCs using amine-terminated thiol ligands with truncated carbon chains, which increased substrate affinity and reduced steric hindrance. Post-synthetic functionalization with catalytic cofactors (aptamers [27,28], imidazole derivatives [29]) further amplifies substrate affinity through surface adsorption mechanisms, achieving catalytic enhancement for biosensing applications. These orthogonal optimization strategies effectively address the activity issue in nanozyme design while expanding the applicability of AuNCs in practical application.
While notable progress has been achieved in augmenting the POD-mimetic activity of AuNCs through core structural modulation and surface functionalization, translational implementation confronts potential barriers. Crucial limitations involve synthesis protocol complexity, unstable catalytic amplification efficiency, and inadequate spatial coupling precision between heterocomponents. These challenges highlight the imperative for establishing facile operational methodologies with precisely regulated catalytic outputs, which would significantly advance AuNCs-based nanozyme applications in next-generation bio-detection systems and targeted therapeutic interventions.
Based on the preceding analysis, a nanoassembly termed Dp-AuNCs@Fe2+ (AuNCs@Fe2+) was constructed by integrating two catalytically active components, Dp-AuNCs (AuNCs) and ferrous ions (Fe2+), via precise self-assembly bridged by the ligand molecules. Notably, electron transfer within the nanoassembly, as well as charge transport between the assembly and hydrogen peroxide (H2O2), was efficiently facilitated through coordination among the gold core, tailored ligands, and chelated iron ions. This synergistic interaction significantly enhanced the catalytic performance, enabling efficient conversion of H2O2 into hydroxyl radicals (•OH).
To endow the system with the desired functionality, the design of the gold nanoclusters ligand was of central importance. There ligand was required to fulfill multiple roles: stabilizing the gold nanoclusters during synthesis and assembly, precisely chelating Fe2+ ions, and promoting effective intracluster electron transfer. To meet these criteria, l-dopa and cysteine were selected as the core structural units. l-dopa provides phenolic hydroxyl groups capable of chelating metal ions, while cysteine introduces a thiol group that forms robust Au-S covalent bonds, ensuring nanocluster stability. Two peptidomimetic ligands, DpCDp and CDpE, were synthesized via standard peptide-coupling protocols, wherein dopa and natural amino acids were covalently linked through amide bonds. Their sequences, chemical structures, and nomenclature are detailed in Table S1 (Supporting information).
Short peptides have previously been utilized for the synthesis of gold nanoclusters [30-32]. However, many such ligands serve singular purposes, such as stabilizing the nanoclusters, lacking additional functional groups for further interaction or catalytic enhancement. In contrast, our peptidomimetic ligands were engineered to offer multiple functions. Under physiological pH (7.4), their phenolic hydroxyls groups readily deprotonate to form phenoxide anions [33], which serve as effective chelation sites for Fe2+. Upon coordination, these phenoxide ligands enable electron delocalization within the Fe2+–catechol complex, producing enhanced absorption in the visible region and generating a characteristic purple Dopa/Fe2+ chelate with a 2:1 stoichiometry. The ligands were deliberately designed to align with this coordination behavior (Fig. 1A).
Figure 1
Figure 1.
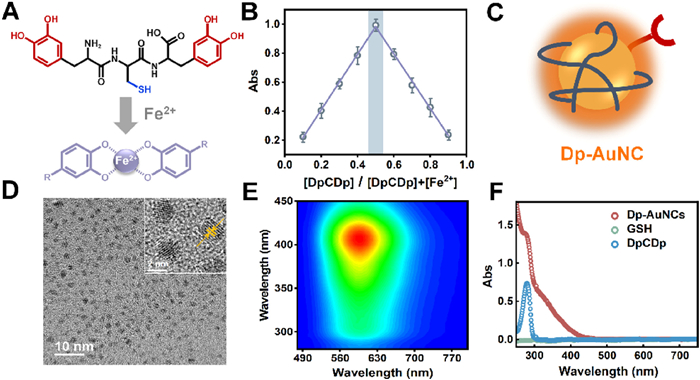
(A) Schema of chelation of DpCDp with Fe2+. (B) Job's plot indicating the binding ratio between DpCDp and Fe2+. (C) Schematic diagram of Dp-AuNC. (D) TEM image of Dp-AuNCs. Inset: high-resolution TEM image showing lattice fringes. (E) Three-dimensional fluorescence spectrum of Dp-AuNCs. (F) UV–vis absorption spectra of Dp-AuNCs, GSH (0.6 mmol/L) and DpCDp (0.16 mmol/L).
The distinct Fe2+-chelating properties of the free peptidomimetics were initially validated via titration experiments in solution (Fig. S1 in Supporting information). UV–vis absorption spectroscopy revealed that DpCDp and CDpE exhibited characteristic ligand-to-metal charge-transfer bands at 560 and 580 nm, respectively, consistent with previously reported catechol-Fe2+ complexes [34]. As a control, the wildly used ligand glutathione (GSH) failed to induce any new absorption peaks (Fig. S1E), underscoring the indispensable role of ortho-diphenolic groups in metal coordination. This finding was further supported by the visual color changes observed upon Fe2+ addition (Fig. S1F), Job's plot analyses determined a binding stoichiometry of 1:1 for DpCDp: Fe2+ (Fig. 1B) and 2:1 for CDpE: Fe2+ (Fig. S2 in Supporting information), indicating that Fe2+-chelating capacity was closely correlated with the number of ortho-diphenol moieties. The presence of bis-dopa units in DpCDp conferred a superior Fe2+ chelating ability compared to CDpE, which contains a single dopa group. The binding constant (K = 806.45 L/mol) and molar extinction coefficient (ε = 21,034.9 L mol−1 cm−1) for the DpCDp/Fe2+ complex were obtained by fitting the absorbance data (Fig. S3 in Supporting information) using equations derived from the law of mass conservation and chemical equilibrium principles. To validate the Fe2+-binding specificity of DpCDp, a competitive chelation assay was performed using EDTA [35]. Upon addition of EDTA to the pre-formed DpCDp/Fe2+ complex, a significant decrease in the absorbance at 560 nm was observed (Fig. S4 in Supporting information), confirming that Fe2+ was chelated by DpCDp.
In addition to Fe2+ chelation, the ligand must also stabilize the gold nanoclusters and modulate their electronic structure to achieve the desired physicochemical properties [36]. However, the use of dopa-containing peptidomimetics in gold nanoclusters synthesis poses two primary challenges: (1) The hydrophobic aromatic moieties are liable to inter-cluster aggregation, compromising colloid stability; (2) The ortho-bisphenol groups are prone to oxidation, which can diminish their metal-chelating efficiency. To address these limitations, GSH, a ligand known for its hydrophilicity and reducing capability, was introduced as a co-ligand [37]. This strategy enabled the facile, one-pot hydrothermal synthesis of orange-fluorescent Dp-AuNCs (Fig. 1C), with imoproved stability and functionality.
The transmission electron microscopy (TEM) analysis (Fig. 1D) demonstrated that the Dp-AuNC products were spherical, uniformly distributed, and well dispersed in aqueous media. The observed lattice fringes exhibited a spacing of approximately 0.22 nm, consistent with the crystalline structure of metallic gold reported in the literature [38,39]. Subsequent characterization of the synthesized product revealed a distinct fluorescence emission peak centered at 600 nm under 404 nm excitation (Fig. 1E). The UV–vis absorption spectrum (Fig. 1F) lacked the characteristic 520 nm plasmon resonance peak [40], indicating absence of large gold particles. A notable absorption feature at 280 nm was assigned to the catechol groups of peptidomimetic ligand bound to on the Au core. X-ray photoelectron spectroscopy (XPS) analysis confirmed the surface elemental composition and valence states. The survey spectrum (Fig. S5A in Supporting information) revealed signals corresponding to both the gold core and organic ligands, while the high-resolution Au 4f spectrum (Fig. S5B in Supporting information) showed the coexistence of Au0 and Au+ species [41], corroborating successful formation of ultrasmall gold nanocluster.
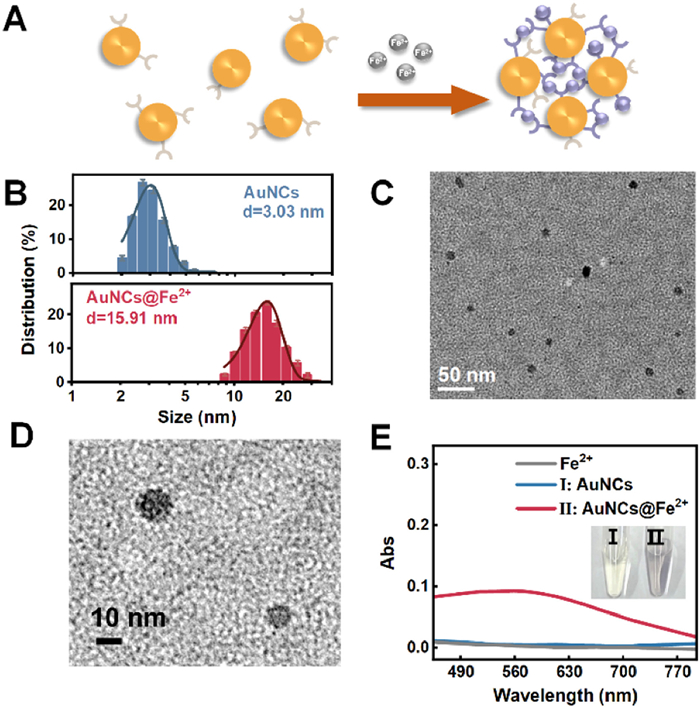
With DpCDp ligands covalently anchored to gold nanoclusters via Au–S bonds, their catechol moieties were subsequently employed to coordinate Fe2+ ions, yielding the nanoassemblies Dp-AuNCs@Fe2+. Unlike previously reported multicomponent complexes stabilized by weak interactions such as electrostatics or physisorption [42,43], which are often plagued by constituent leaching and poor stability, our system leverages dopa-containing peptidomimetic ligands as bridges to achieve precise and robust assembly. Owing to the multidentate nature of DpCDp, Fe2+ ions were able to simultaneously coordinate with catechol groups on different ligands across separate nanoclusters, thereby forming interparticle cross-links (Fig. 2A). This coordination-driven assembly strategy effectively enables the construction of higher-order AuNC structures. Size and morphology analyses confirmed substantial growth upon Fe2+ coordination: the hydrated diameter increased from 3.03 nm to 15.91 nm (Fig. 2B), while TEM images showed expansion from 1.35 nm to 12.25 nm (Fig. 2C and Fig. S6 in Supporting information). Notably, high-resolution TEM image (Fig. 2D) clearly reveals that individual nanoassemblies are composed of multiple small-sized cluster cores, providing strong evidence that Dp-AuNCs@Fe2+ formation results from Fe2+-mediated bridging of Dp-AuNCs via DpCDp ligands. Furthermore, a reduction in zeta potential indicated decreased electrostatic repulsion among clusters, which also facilitated aggregation through DpCDp/Fe2+ coordination (Fig. S7 in Supporting information).
Figure 2
Figure 2.
(A) Schematic illustration of Dp-AuNCs chelating Fe2+. (B) TEM-based particle size distributions demonstrating the size increase from Dp-AuNCs to Dp-AuNCs@Fe2+ upon Fe2+-mediated assembly. (C) TEM image of Dp-AuNCs@Fe2+ assemblies. (D) High-resolution TEM image of Dp-AuNCs@Fe2+ nanoassemblies. (E) UV–vis absorption spectra and photograph showing the color change of Dp-AuNCs before and after chelation with Fe2+ (25 µmol/L).
In addition to structural characterization, optical analyses revealed substantial changes in the electronic properties of the nanoclusters upon Fe2+-induced assembly. UV–vis spectroscopy showed the emergence of a distinct absorption peak at 580 nm following Fe2+ addition, with intensity increasing proportionally to Fe2+ concentration and reaching a plateau above 100 µmol/L ([Dp-AuNCs] = 296.8 µg/mL) (Fig. 2E, Figs. S8A and B in Supporting information). This rapid development is completed within 120 s (Fig. S8C in Supporting information), indicating the formation of a visible-light-absorbing DpCDp–Fe2+ chelate. Notably, the 580 nm peak is red-shifted by ~20 nm compared to the free DpCDp/Fe2+ complex (560 nm), suggesting that interactions with the gold surface may alter the local charge distribution or steric configuration of the ligands, thereby modulating Fe2+ coordination behavior [44]. Inductively coupled plasma mass spectrometry (ICP-MS) analysis yielded consistent results, showing that at a total Dp-AuNCs concentration of 296.8 µg/mL, the maximum concentration of Fe2+ bound to the nanoclusters reached 100 µmol/L, suggesting that the surface-bound DpCDp ligand concentration was also approximately 100 µmol/L (Fig. S9 in Supporting information). Control experiments employing GSH-AuNCs, which lack the DpCDp ligand, exhibited no spectral change upon Fe2+ addition (Fig. S10A in Supporting information), confirming DpCDp as the essential chelating moiety. Endowed with thiol and vicinal bisphenol groups, DpCDp serves as a "bridge" to connect Dp-AuNCs and Fe2+, thus promoting the formation of nanoassemblies. After treatment with EDTA significantly reduced the 580 nm absorbance, verifying that Fe2+ was bound to AuNCs-anchored DpCDp ligands rather than remaining free in solution (Fig. S10B in Supporting information). Moreover, a notable size reduction was observed in the system, which was comparable to that of Dp-AuNCs alone, further underscoring the bridging function of DpCDp (Fig. S11 in Supporting information). These results suggest that the size and optical properties of the Fe2+-bridged Dp-AuNC assemblies Dp-AuNCs@Fe2+ are tunable through precise modulation of the DpCDp ligand density and the Fe2+/Dp-AuNC ratio.
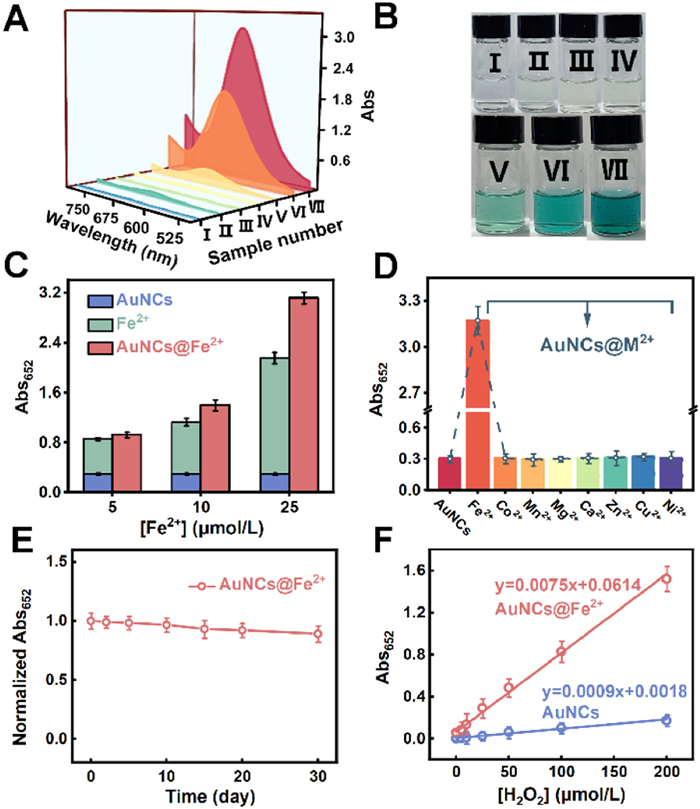
To evaluate the catalytic performance of the nanoassemblies, we assessed the peroxidase (POD)-mimicking activity of Dp-AuNCs and Dp-AuNCs@Fe2+ using 3,3′,5,5′-tetramethylbenzidine (TMB) as a chromogenic substrate, monitored by absorbance at 652 nm (Fig. 3A). In the absence of H2O2, neither TMB alone nor its mixtures with Dp-AuNCs or Dp-AuNCs@Fe2+ showed appreciable absorbance, indicating no oxidase-like activity. In contrast, when TMB and H2O2 were co-incubated with Dp-AuNCs, Fe2+, or Dp-AuNCs@Fe2+, a significant increase in absorbance was observed, accompanied by a clear blue coloration of the solution (Fig. 3B). These results confirm that the catalytic activity requires both the presence of H2O2 and the nanomaterials or metal ions, consistent with a POD-like reaction mechanism. Critically, the catalytic performance of Dp-AuNCs@Fe2+ exceeded that of Dp-AuNCs or Fe2+ alone, suggesting a synergistic rather than additive effect. To quantify this enhancement, a series of nanoassemblies with varying Fe2+ chelation densities was prepared (Fig. 3C). All variants exhibited catalytic activities surpassing the theoretical sum of their individual components, with activity increasing proportionally with Fe2+ loading. This trend suggests that Fe2+ ions, through coordination with the DpCDp ligands, modulate the electron density of the Dp-AuNCs cores and enhance the catalytic turnover.
Figure 3
Figure 3.
(A) UV–vis absorption spectra of different reaction components. The concentrations of TMB, H2O2, Dp-AuNCs, Fe2+ and Dp-AuNCs@Fe2+are 3.0 mmol/L, 2.0 mmol/L, 98 µg/mL, 5.0 µmol/L and 100 µg/mL, respectively. (B) Photographs of the reaction mixtures under daylight (Ⅰ: TMB, Ⅱ: TMB + H2O2, Ⅲ: TMB + Dp-AuNCs, Ⅳ: TMB + Dp-AuNCs@Fe2+, Ⅴ: TMB + H2O2 + Dp-AuNCs, Ⅵ: TMB + H2O2 + Fe2+, Ⅶ: TMB + H2O2 + Dp-AuNCs@Fe2+). (C) Comparison of catalytic activities of Dp-AuNCs@Fe2+ at varying Fe2+ chelation concentrations, with those of Dp-AuNCs alone and Fe2+ alone at corresponding concentrations. (D) Catalytic activities of Dp-AuNCs coordinated with various metal ions, showing the highest activity for Fe2+-bound assemblies. (E) Catalytic stability of Dp-AuNCs@Fe2+ over a period of 0–30 days. (F) Calibration curves of Dp-AuNCs and Dp-AuNCs@Fe2+ for H2O2 detection in the range of 0–200 µmol/L.
The enhanced activity of Dp-AuNCs@Fe2+ was further validated using a range of additional peroxidase substrates, confirming the generalizability of the observed catalytic behavior (Figs. S12A–C in Supporting information). Gold nanoclusters are known to function as peroxidase mimics, with Au(Ⅰ) sites serving as catalytic centers [45], and their efficiency is intimately linked to interfacial electron density and mobility [46]. Fe2+, due to its reversible redox behavior, can effectively accept electrons from the Dp-AuNC core upon chelation, thereby promoting interfacial charge transfer and enhancing the electron-rich environment at the catalytic interface [47]. This charge redistribution not only enhances the decomposition of H2O2 but also improves overall catalytic performance. Furthermore, Fe2+ itself contributes intrinsic catalytic activity via Fenton-like reactions, generating hydroxyl radicals that directly promote substrate oxidation. The dual role of Fe2+, as both an electron mediator and a catalytic species, creates a synergistic effect that exceeds the simple additive contributions of Dp-AuNCs and Fe2+ alone. In contrast, other metal ions tested (e.g., Co2+, Mn2+, Mg2+, Ca2+, Zn2+, Cu2+, Ni2+) either lack sufficient redox activity or fail to mediate efficient charge transfer, and thus do not significantly improve the catalytic activity of Dp-AuNCs (Fig. 3D) [48-50]. This specificity underscores the unique and indispensable role of Fe2+ in the observed POD-mimicking catalysis.
In addition to catalytic efficiency, the operational stability of the system was evaluated. Benefiting from the stabilizing effect of DpCDp-mediated Fe2+ chelation, Dp-AuNCs@Fe2+ retained approximately 90% of its initial activity after 30 days of storage at 4 ℃ (Fig. 3E), demonstrating excellent long-term stability of the nanoassemblies. Moreover, we observed that the catalytic efficiency of Dp-AuNCs@Fe2+ retained over 85% after three successive recovery cycles (Fig. S13 in Supporting information). These findings collectively confirm the structural robustness of Dp-AuNCs@Fe2+.
The Dp-AuNCs@Fe2+ nanoassemblies exhibit superior peroxidase-mimicking activity. Comparative catalytic studies were performed under identical H2O2 concentrations to assess their responsiveness. Absorption spectral analysis revealed that Dp-AuNCs@Fe2+ catalyzed TMB-H2O2 reactions with markedly stronger signal changes than bare Dp-AuNCs, especially as H2O2 concentrations increased (Figs. S14A and B in Supporting information). Quantification at 652 nm confirmed this enhanced responsiveness (Fig. S14C in Supporting information), and the resulting color gradients were clearly discernible by the naked eye (Fig. S14D in Supporting information). Both systems displayed linear responses to H2O2 concentrations ranging from 0 to 200 µmol/L; however, Dp-AuNCs@Fe2+ achieved a detection limit of 0.27 µmol/L, representing a 4.3-fold improvement over bare Dp-AuNCs (1.17 µmol/L, Fig. 3F). This heightened sensitivity reflects the catalytic enhancement imparted by the Fe2+–Dp-AuNC synergism mediated through DpCDp coordination.
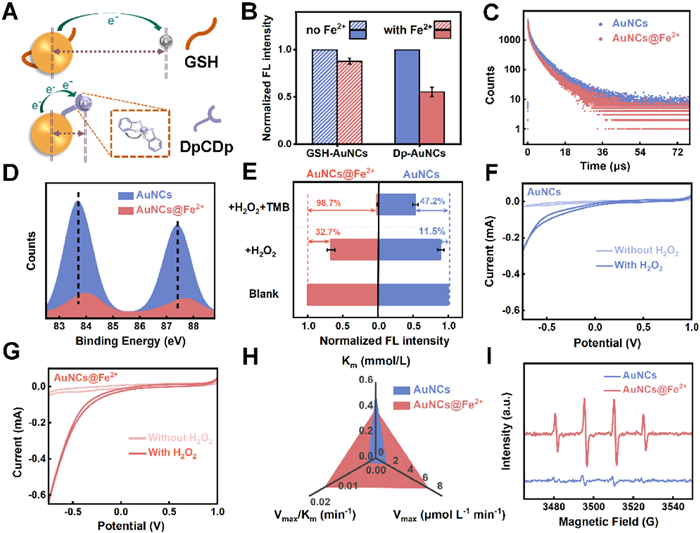
To elucidate the origin of this enhancement, we investigated the electronic interactions within the nanoassembly. In catalytic H2O2 reactions, the mechanism generally involves three sequential steps: substrate adsorption, activation, and product desorption [51,52]. The activation step is typically governed by charge transfer between the catalytic center and the substrate, and a high electron density at the catalytic interface is crucial for enhancing this process [53]. Gold nanoclusters, with delocalized valence electrons, serve as efficient electron donors [54], while Fe2+, possessing incomplete 3d orbitals, acts as an electron acceptor [55]. Upon coordination through the peptidomimetic ligand, partial electron transfer from Fe2+ to the π* orbitals of DpCDp results in the formation of back-donated π bonds [56]. Simultaneously, DpCDp bridges gold cores and Fe2+ centers with sub-nanometer precision, minimizing donor-acceptor distance and facilitating electron flow (Fig. 4A). This dual role of DpCDp—as both electronic mediator and structural linker—amplifies interfacial charge migration, enabling superior peroxidase-like activity.
Figure 4
Figure 4.
(A) Schematic diagram of electron transfer process between DpCDp-enhanced Dp-AuNCs and Fe2+. (B) Fluorescence intensity at 600 nm of GSH-AuNCs and Dp-AuNCs mixed with Fe2+(25 µmol/L). (Ex = 404 nm). (C) Time-resolved fluorescence lifetime decay spectra of Dp-AuNCs after addition of Fe2+ (25 µmol/L). (D) XPS binding energy changes of Dp-AuNCs chelating Fe2+(25 µmol/L). (E) Fluorescence intensity changes of Dp-AuNCs and Dp-AuNCs@Fe2+ after treatment with H2O2 (2.0 mmol/L) and with a mixture of H2O2 (2.0 mmol/L) and TMB (3.0 mmol/L). (F, G) Cyclic voltammetry (CV) curves of (F) Dp-AuNCs and (G) Dp-AuNCs@Fe2+ in the presence and absence of H2O2. (H) Michaelis−Menten kinetic analysis of Dp-AuNCs and Dp-AuNCs@Fe2+ toward H2O2. (I) EPR spectroscopy for the detection of ·OH generated by Dp-AuNCs and Dp-AuNCs@Fe2+.
Fluorescence studies further confirmed the electron transfer. A noTable 45% fluorescence quenching occurred upon Fe2+ binding to Dp-AuNCs, in stark contrast to only 12% quenching in DpCDp-free GSH-AuNCs (Fig. 4B). Spectral analysis showed progressive quenching with increasing Fe2+ levels (Figs. S15A and B in Supporting information), accompanied by a significant decrease in fluorescence lifetime (Fig. 4C and Fig. S16 in Supporting information), indicating accelerated electron transfer from the Au core to Fe2+.
XPS provided additional insight [57]. A 0.3 eV positive shift in the Au 4f binding energy of Dp-AuNCs@Fe2+ compared to pristine Dp-AuNCs was observed (Fig. 4D), attributed to electron redistribution from the Au core to the Fe2+ coordination sites via DpCDp. Collectively, these findings demonstrate that the enhanced peroxidase-like activity of Dp-AuNCs@Fe2+ arises from efficient charge transfer mediated by DpCDp.
Given that the peroxidase-mimicking mechanism involves single-electron [58] oxidation of H2O2 to generate •OH (POD → e− + h+, e− + H2O2 → •OH + OH−), the superior electron-shuttling capacity of Dp-AuNCs@Fe2+ was further validated. Upon exposure to H2O2, Dp-AuNCs@Fe2+ displayed a 32.7% decrease in fluorescence intensity, significantly higher than the 11.5% drop in Dp-AuNCs alone (Fig. 4E), demonstrating that Dp-AuNCs@Fe2+ transfer electrons more efficiently to H2O2. Moreover, fluorescence resonance energy transfer (FRET) [59] analysis leveraging spectral overlap between Dp-AuNCs emission and oxidized TMB absorption revealed a quenching efficiency of 98.7% for Dp-AuNCs@Fe2+, over twice that of the control system (47.2%). These results indicate superior electron donation to H2O2, facilitating •OH generation.
To further elucidate the working mechanism of Dp-AuNCs@Fe2+, electrochemical assays were employed to compare the catalytic activities of Dp-AuNCs and Dp-AuNCs@Fe2+ toward H2O2. Following their immobilization onto the glassy carbon electrode, both systems exhibited reduction currents in N2-saturated acetic acid-sodium acetate electrolyte (Figs. 4F and G), confirming electron transfer from donors to H2O2 as the acceptor. Notably, Dp-AuNCs@Fe2+ demonstrated substantially higher reductive activity than Dp-AuNCs, indicating its capacity to transfer more electrons to H2O2, validating the marked enhancement in its catalytic capacity. As catalyst charge transfer capability generally correlates with impedance [60], electrochemical impedance spectroscopy (EIS) was used to compare resistances between the two. The latter showed smaller radius than the former, corresponding to lower resistance, thus enabling Dp-AuNCs@Fe2+ to achieve faster electron transfer rates (Fig. S17 in Supporting information), which matches its strong catalytic capacity.
On the other side, chelated Fe2+ contributed to the POD-mimicking activity through a typical action pathway (Fe2+ + H2O2 → Fe3+ + ·OH + OH−) [61] and the optimized adsorption geometries coupled with the spillover of core electrons in Dp-AuNCs boosted its affinity with the substrate. To quantify this activity, Michaelis-Menten kinetic analyses were conducted (Fig. S18 in Supporting information). Dp-AuNCs@Fe2+ showed a reduced Km for H2O2 (0.387 mmol/L vs. 0.525 mmol/L for Dp-AuNCs) and an elevated Vmax (7.163 µmol L-1 min-1vs. 1.451 µmol L-1 min-1), corresponding to a 6.7-fold increase in catalytic efficiency (Vmax/Km, Fig. 4H). Similar kinetic advantages were observed for TMB oxidation (Fig. S19 in Supporting information). When benchmarked against previously reported peroxidase-mimicking nanozymes, many of which rely on physical adsorption or electrostatic interactions, the DpCDp-bridged Dp-AuNCs@Fe2+ system demonstrated superior catalytic activity (Table S2 in Supporting information). This enhancement is attributed to the stable spatial coordination and improved interfacial electron transfer between the two functional components.
Furthermore, electron paramagnetic resonance (EPR) spectroscopy provided direct evidence of •OH generation. Dp-AuNCs@Fe2+ produced a characteristic 1:2:2:1 quartet signal corresponding to DMPO—OH adducts [62], with signal intensity exceeding that of bare Dp-AuNCs by more than fourfold (Fig. 4I).
We thus delineate a tripartite catalytic mechanism for Dp-AuNCs@Fe2+: (1) Direct oxidation of H2O2 at gold surfaces, (2) Fe2+-driven Fenton-like •OH generation, and (3) DpCDp-mediated electron transfer bridging gold and Fe2+. This multifunctional integration leads to a synergistic enhancement of peroxidase-mimicking activity far exceeding that of individual components.
In summary, Dp-AuNCs@Fe2+ nanoassemblies were rationally designed using a dopa-containing peptidomimetic ligand (DpCDp) to bridge ultrasmall Au nanoclusters with Fe2+ ions. The resulting assemblies exhibit significantly enhanced peroxidase-like activity due to efficient electron transfer from the Au core to catalytic interfaces. This work demonstrates a versatile strategy for constructing highly active artificial enzymes and provides a conceptual framework for future nanozyme development via electronic and structural integration.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
N. Alizadeh, A. Salimi, T.K. Sham, et al., Chem. Eng. J. 417 (2021) 129134. doi: 10.1016/j.cej.2021.129134
Figure 1
(A) Schema of chelation of DpCDp with Fe2+. (B) Job's plot indicating the binding ratio between DpCDp and Fe2+. (C) Schematic diagram of Dp-AuNC. (D) TEM image of Dp-AuNCs. Inset: high-resolution TEM image showing lattice fringes. (E) Three-dimensional fluorescence spectrum of Dp-AuNCs. (F) UV–vis absorption spectra of Dp-AuNCs, GSH (0.6 mmol/L) and DpCDp (0.16 mmol/L).
Figure 2
(A) Schematic illustration of Dp-AuNCs chelating Fe2+. (B) TEM-based particle size distributions demonstrating the size increase from Dp-AuNCs to Dp-AuNCs@Fe2+ upon Fe2+-mediated assembly. (C) TEM image of Dp-AuNCs@Fe2+ assemblies. (D) High-resolution TEM image of Dp-AuNCs@Fe2+ nanoassemblies. (E) UV–vis absorption spectra and photograph showing the color change of Dp-AuNCs before and after chelation with Fe2+ (25 µmol/L).
Figure 3
(A) UV–vis absorption spectra of different reaction components. The concentrations of TMB, H2O2, Dp-AuNCs, Fe2+ and Dp-AuNCs@Fe2+are 3.0 mmol/L, 2.0 mmol/L, 98 µg/mL, 5.0 µmol/L and 100 µg/mL, respectively. (B) Photographs of the reaction mixtures under daylight (Ⅰ: TMB, Ⅱ: TMB + H2O2, Ⅲ: TMB + Dp-AuNCs, Ⅳ: TMB + Dp-AuNCs@Fe2+, Ⅴ: TMB + H2O2 + Dp-AuNCs, Ⅵ: TMB + H2O2 + Fe2+, Ⅶ: TMB + H2O2 + Dp-AuNCs@Fe2+). (C) Comparison of catalytic activities of Dp-AuNCs@Fe2+ at varying Fe2+ chelation concentrations, with those of Dp-AuNCs alone and Fe2+ alone at corresponding concentrations. (D) Catalytic activities of Dp-AuNCs coordinated with various metal ions, showing the highest activity for Fe2+-bound assemblies. (E) Catalytic stability of Dp-AuNCs@Fe2+ over a period of 0–30 days. (F) Calibration curves of Dp-AuNCs and Dp-AuNCs@Fe2+ for H2O2 detection in the range of 0–200 µmol/L.
Figure 4
(A) Schematic diagram of electron transfer process between DpCDp-enhanced Dp-AuNCs and Fe2+. (B) Fluorescence intensity at 600 nm of GSH-AuNCs and Dp-AuNCs mixed with Fe2+(25 µmol/L). (Ex = 404 nm). (C) Time-resolved fluorescence lifetime decay spectra of Dp-AuNCs after addition of Fe2+ (25 µmol/L). (D) XPS binding energy changes of Dp-AuNCs chelating Fe2+(25 µmol/L). (E) Fluorescence intensity changes of Dp-AuNCs and Dp-AuNCs@Fe2+ after treatment with H2O2 (2.0 mmol/L) and with a mixture of H2O2 (2.0 mmol/L) and TMB (3.0 mmol/L). (F, G) Cyclic voltammetry (CV) curves of (F) Dp-AuNCs and (G) Dp-AuNCs@Fe2+ in the presence and absence of H2O2. (H) Michaelis−Menten kinetic analysis of Dp-AuNCs and Dp-AuNCs@Fe2+ toward H2O2. (I) EPR spectroscopy for the detection of ·OH generated by Dp-AuNCs and Dp-AuNCs@Fe2+.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
